

Abstract
N 6-Methyladenine (N6-mA or 6 mA) is an epigenetic DNA modification in eukaryotic genomes. In contrast to the well-established roles of 5-methylcytosine for epigenetic regulation of gene expression, the functional roles of N6-mA remain elusive. In particular, the impact of N6-mA modification of the DNA template on RNA polymerase II (pol II) transcription elongation is not known. In this work, using the Saccharomyces cerevisiae pol II transcriptional elongation system as a model, we investigated the molecular mechanism of pol II recognition and processing of N6-mA sites via both biochemical and structural approaches. We found that N6-mA causes site-specific pol II pausing/stalling. Structural analysis revealed that while N6-mA can reach the +1 template position, the stability of the N6-mA and UTP base pairing is compromised. Taken together, we reveal that the presence of the 6-methyl group on adenine reduces incorporation efficiency and promotes backtracking translocation. Our studies with yeast pol II provide molecular insights into understanding the impacts of N6-mA on pol II transcription dynamics in different organisms.
Graphical Abstract
INTRODUCTION
Epigenetic DNA methylation plays important roles in various biological processes.1 It is well-established that epigenetic mark 5-methylcytosine (5mC) plays crucial roles in regulating gene expression and affects X chromosome inactivation, cellular differentiation, and tumor development.2–4 Another naturally occurring DNA modification, N6-methyladenine (N6-mA or 6mA) (Figure 1a), was previously thought to be absent in metazoans and only present in prokaryotes and a limited number of unicellular eukaryotes.5,6 Recently, several new reports described the genomic distributions of N6-mA in many phylogenetically distinct eukaryotes including fungi,7 green algae,8 insects,9 nematodes,10 frogs,11 fish,12 and mammals.11–13 Interestingly, the genomic abundance of N6-mA varies from ~0.0001–0.0003% of adenines in plants and mammals to as high as 2.8% of adenines in early diverging fungi.14 Mounting evidence from different laboratories suggested intriguing functional relevance between N6-mA and gene expression in vivo, despite the underlying mechanism still not being fully understood. The genomic distribution and effects of N6-mA on gene expression have been found to differ among different eukaryotes. In particular, for certain species associated with high abundance of N6-mA, such as green algae and fungi, N6-mA is associated with active gene expression.8 We previously showed that in Chlamydomonas reinhardtii N6-mA is enriched around the transcription start site (TSS) of more than 14 000 genes (approximately 84% of all genes in this organism), most of which are actively transcribed.8 Furthermore, RNA-Seq analysis suggested that the presence of N6-mA at TSS is well correlated with high transcriptional activity.8 Intriguingly, another recent paper revealed that up to 2.8% of all adenines were methylated in early diverging fungi and these N6-mAs are clustered at or immediately downstream of the transcription start site (TSS) (~500 bp) of active genes.7 In Drosophila melanogaster, N6-mA is enriched in transposon bodies and is associated with increased transcriptional activity of transposons. 9 In Caenorhabditis elegans, N6-mA levels were also found to be elevated in mutants with elevated levels of histone H3 lysine 4 dimethylation (H3K4me2), which is a mark associated with active transcription.10 Taken together, these studies from different species strongly indicate the functional relevance between N6-mA and gene expression in vivo; however, the molecular mechanisms of N6-mA in gene expression regulation remain elusive.15,16
Figure 1.

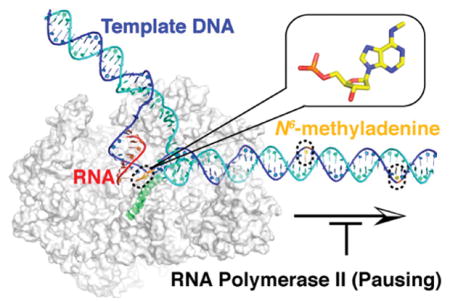
N6-Methyladenine (N6-mA) causes RNA pol II transcriptional pausing. (a) Chemical structure of adenine and N6-mA. 6-Methyl group is in red. (b) Transcription scaffold containing N6-mA at DNA template strand. (c, d) N6-mA cause site-specific pol II pausing (highlighted with *) in the absence of TFIIS (c) and presence of 200 nM TFIIS (d). The concentration of NTPs is 25 μM. Time points are 0, 10 s, 30 s, 1 min, 5 min, 20 min, 1 h, and 3 h, respectively. 0* refers time zero for transcription from N6-mA template. All experiments were repeated at least 4 times with similar results.
RNA polymerase II (pol II) is responsible for pre-mRNA transcription and is sensitive to various DNA modifications. 17–19 During transcription elongation, RNA pol II travels along the DNA template strand and may encounter all kinds of transcriptional obstructions, including covalent DNA lesions and epigenetic DNA modifications.17–19 These DNA modifications can affect the template recognition by RNA pol II and lead to distinct transcriptional outcomes (transcriptional pausing/stalling, lesion bypass, arrest) and downstream biological processes.20–24 Previously, we revealed that RNA pol II is able to detect 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) via a conserved epi-DNA loop, suggesting the functional role of these oxidized 5-methylcytosines (oxi-mCs) in fine-tuning the transcriptional dynamics of RNA pol II.25,26 It is not clear whether N6-mA, a different eukaryotic DNA modification, can be recognized by RNA pol II and influences the transcriptional elongation in a similar manner.
In this study, we systematically investigated the molecular mechanism of how N6-mA affects RNA pol II transcriptional elongation using biochemical and structural approaches. Our studies revealed that the presence of N6-mA in the DNA template causes site-specific RNA pol II transcriptional pausing. Based on our combined biochemical and structural data, we propose a unified mechanism of how different epigenetic DNA modifications are recognized by pol II during transcription. This study provides new insights into understanding the potential functional roles of N6-mA in gene expression.
RESULTS AND DISCUSSION
N6-mA-Induced Site-Specific pol II Transcriptional Pausing
To investigate whether the presence of N6-mA affects pol II transcription elongation, we performed in vitro transcription assays with purified RNA pol II and scaffolds containing multiple site-specifically modified N6-mA sites in the DNA template strand (Figure 1b). As shown in Figure 1c, we observed specific transcriptional pausing bands induced by N6-mA, in comparison with the unmodified DNA template. The pol II pausing sites are located right before the N6-mA sites (for example, 10mer and 13mer, Figure 1c and Figure S1). Consistently, we only observed a single N6-mA-induced pol II pausing/stalling for the template containing a single N6-mA in the DNA template strand (10mer position, Figure S2a). Interestingly, pol II is able to bypass these pausing sites with prolonged incubation in the presence of NTPs, leading to the generation of full-length RNA transcripts.
Transcription factor S-II (TFIIS) facilitates pol II bypass of a variety of obstacles, including certain DNA lesions, pausing sequences, and nucleosomes.27–29 The apparent stimulatory role of TFIIS on pol II bypass is achieved by its capability to facilitate the cleavage of the backtracking RNA transcript, reactivating backtracked RNA pol II. We then tested whether TFIIS can help RNA pol II bypass the N6-mA site. Intriguingly, in the presence of TFIIS, we observed prolonged pol II stalling right before it reaches N6-mA sites (e.g., 10mer position), whereas no obvious TFIIS effect was observed at the same position for control A template (10mer position, Figure 1d and S2c). This result suggests that the forward translocation driven by nucleotide addition opposite the N6-mA site is much slower than backtracking and TFIIS-stimulated transcript cleavage. Consequently, we observed that TFIIS delays pol II transcriptional bypass of N6-mA site.
N6-mA Reduces UTP Incorporation and Promotes pol II Backtracking
To further understand the molecular basis of N6-mA-induced pol II transcriptional pausing and its impact on transcriptional fidelity, we systematically investigated the effect of N6-mA on three transcriptional fidelity checkpoints: insertion, extension, and proofreading. For the insertion step, we performed single nucleotide incorporation into the single N6-mA site and observed ~6-fold slow down of UMP incorporation opposite N6-mA in comparison with unmodified DNA template (Figures 2a and S3). We also tested the effect of N6-mA on nucleotide selectivity and found that UTP is still the most efficient substrate to be incorporated opposite the N6-mA template among all four nucleotide triphosphates (NTPs) (Figure S4). Taken together, these results suggest that while the methylation of adenosine substantially reduces the nucleotide incorporation efficiency, there is no notable effect on nucleotide selectivity. For the extension step, we did not observe any notable effects between the N6-mA template and unmodified template (Figure S2b). For the proofreading step, we compared TFIIS-stimulated transcript cleavage activity in the N6-mA modified and unmodified A template. We found that the presence of N6-mA affects both cleavage rates and translocation register. The cleavage rate in the N6-mA template is ~2–4-fold faster than that for the unmodified template (Figures 2b and S5). Importantly, the presence of N6-mA significantly alters the cleavage pattern of RNA transcript. For the scaffold containing a 3′-rU:dA pair, we observed that a majority of cleaved transcript is the n – 1 cleavage product (black arrow in Figure 2b) derived from pol II at a pretranslocation register (47% ± 1%, 10mer) (Figure S5b), whereas only a minor portion of the cleaved transcript is the n – 2 cleavage product (blue arrow in Figure 2b) presumably derived from a backtracked pol II (14% ± 0.4%, 9mer) (Figure S5c). In sharp contrast, for the scaffold containing a 3′-rU:N6-mA pair, we observed that the backtracked-pol II-derived n – 2 cleavage product became dominant (~46% ± 1%, 9mer) (Figure S5c), whereas n – 1 cleavage product (derived from pol II at pretranslocation register) dropped to ~31% (Figure S5b). These results suggest that an rU:N6-mA pair is less stable than its unmodified counterpart (an rU:dA pair). For the N6-mA template, pol II is more prone to backtracking and subsequent cleavage to generate a shorter RNA transcript (n – 2). After cleavage, pol II can rapidly incorporate one more nucleotide and pause/stall at n – 1 position because nucleotide insertion opposite the N6-mA template is relatively slow. Pol II may experience several cycles of addition–backtracking–cleavage before it can eventually bypass the N6-mA site.
Figure 2.
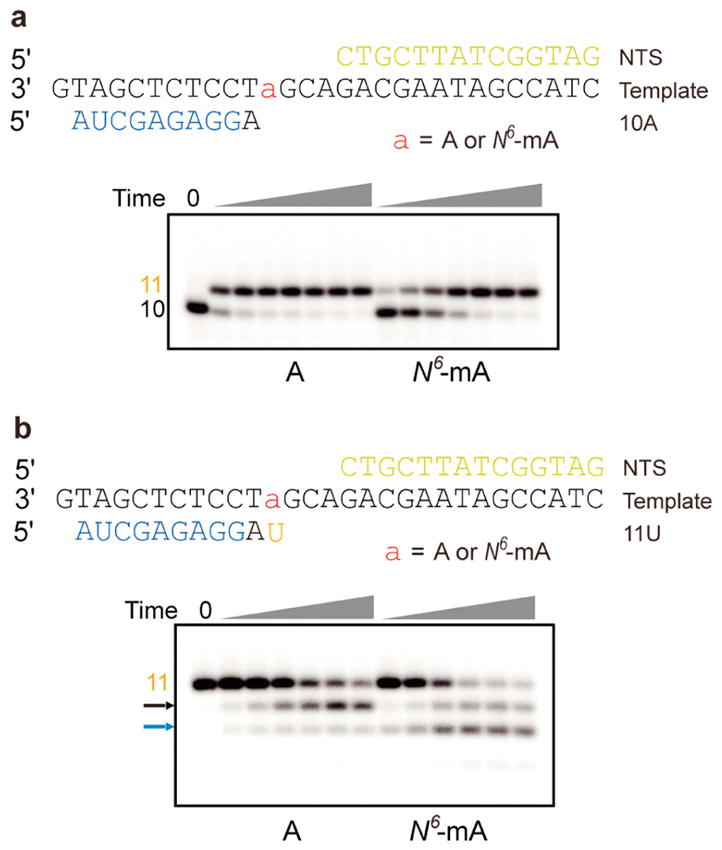
N6-mA slows down UMP incorporation and promotes pol II backtracking. (a) The presence of N6-mA in the DNA template strand leads to slow UMP addition. The concentration of UTP is 25 μM. Time points are 0, 15 s, 30 s, 1 min, 2 min, 5 min, 20 min, and 1 h. (b) The presence of N6-mA in the DNA template promotes further backtracking revealed by TFIIS-stimulated transcript cleavage assay. Time points are 0, 15s, 1 min, 5 min, 20 min, 1 h, and 3 h, respectively. The DNA/RNA scaffolds are listed with i + 1 position highlighted. The n – 1 and n – 2 cleavage products are highlighted by black and blue arrows, respectively.
Structural Basis of the Impacts of N6-mA on pol II Transcription
To reveal the structural basis of how N6-mA is accommodated at the pol II active site, we first determined the crystal structure of pol II elongation complex containing a site-specific N6-mA at the i + 1 position (EC-I, Table S1). We found that the N6-mA is located at the canonical i + 1 template site in a similar manner to the unmodified A template (PDB code 2NVZ)30 (Figure S6), suggesting that the methylation on the N6 position of adenine does not alter the template positioning. To further understand how N6-mA affects UTP incorporation, we solved the crystal structure of pol II elongation complex containing a nonhydrolyzable UTP analogue (UMPNPP) paired with N6-mA (EC-II, Figures 3 and S7, Table S1). We built UMPNPP into the canonical addition site to fit the electron density in the pol II active site (Figure S7a). Interestingly, while UMPNPP maintained a Watson–Crick base pair with N6-mA, the UMPNPP base ring rotated ~30° in comparison with its unmodified counterpart UTP:dA pair (PDB code 2NVZ). This observation suggests a weakened UTP:N6-mA pair in comparison with canonical UTP:dA pair, which is consistent with previously reported NMR studies.31 In addition, we noticed additional electron density nearby the UMPNPP triphosphate moiety. An alternative conformation of UTP triphosphate moiety, which can best fit the redundant density, was built into the final model, though other potential interpretations may also exist (Figure S7).
Figure 3.

Crystal structure of N6-mA-containing RNA pol II elongation complex with UMPNPP soaked (EC-II). (a) Overall structure of pol II EC-II containing an N6-mA template and UMPNPP. Pol II is shown from the side view as a ribbon model in silver, with the bridge helix (BH) highlighted in green and Rpb2 omitted for clarity. The nucleic acids are shown as stick models: template DNA (TS, blue), nontemplate DNA (NTS, cyan), and RNA (red). The N6-mA at i + 1 site and one conformation of UMPNPP are highlighted as yellow and orange, respectively. (b) Detailed interaction of UMPNPP:N6-mA from top view. There exist two conformations of UMPNPP due to the flexibility of triphosphate moiety (more details in Figure S7), but their base groups adopt similar orientation, so the other conformation is omitted for clarity here. Hydrogen bonds are indicated as black dotted lines. (c) UTP and corresponding unmodified dA template from canonical pol II EC (PDB code 2NVZ) are superimposed, which are shown as silver sticks and labeled as black. The potential rotational range of the methyl group of N6-mA is represented as tiny cyan dots, in which the syn-form is shown as dashed stick.
Mechanistic Insights into the Interplay between pol II and Epigenetic DNA Modifications
Several novel mechanistic insights can be obtained by comparing the functional interplays between pol II transcription and two important classes of epigenetic DNA marks: N6-mA and 5mC (and its oxi-mC derivatives). First, all of these DNA modifications are small and are located at the major groove of DNA duplex, and therefore are better tolerated by pol II active site than bulky DNA modifications or minor-groove modifications. The structural analysis revealed that the matched incoming nucleotide can still form a Watson–Crick base pair with modified DNA template to maintain the correct nucleotide selection. Second, while none of these epigenetic DNA modifications permanently block pol II elongation, they have distinct effects on pol II elongation dynamics and pausing. For example, the methyl group of N6-mA, but not 5mC, can cause pol II pausing. Among the modifications at the same 5-position of cytosine, 5fC and 5caC but not 5mC and 5hmC lead to pol II pausing.26 Third, we revealed a novel pol II pausing mechanism by N6-mA, which is different from 5caC/5fC-induced pol II pausing. Previous study revealed that a conserved epi-DNA loop at the Rpb2 subunit of pol II directly recognizes 5caC/5fC by hydrogen-bonding interactions and has an important role in 5caC/5fC-induced pol II pausing.26 In contrast, N6-mA can freely translocate into the canonical i + 1 template position, escaping the detection by epi-DNA loop.
Three-Step Model for the Processing of Epigenetic DNA Modifications by pol II
Comparing the molecular mechanisms of 5fC/5caC-induced pol II pausing26 and N6-mA-induced pol II pausing, we propose the following three-step model for pol II recognition and processing of epigenetic DNA modifications (Figure 4). The first step of recognition is “template positioning”. In this step, the downstream template base (i + 2) crosses over the bridge helix and reaches the canonical i + 1 template position. The conserved epi-DNA loop can detect specific epigenetic DNA modifications via specific hydrogen-bonding interactions (such as 5fC/5caC). On the other hand, 5mC and N6-mA can escape epi-DNA loop detection as their hydrophobic methyl group cannot form hydrogen-bonding interactions. The second step of recognition is termed “substrate positioning and active site assembly”. In this step, the correct substrate is positioned into the addition site to form a Watson–Crick base pair with template base, with the trigger loop and other active site pol II residues undergoing conformational changes to reorganize the pol II active site in order to commit effective nucleotide addition. In this step, 5caC/5fC and N6-mA compromise substrate positioning and full-assembly of pol II active site through distinct mechanisms. The 5caC:GTP pair has a positional shift from the canonical position of a C:GTP base pair (due to the 5caC–epi-DNA loop interaction) and consequently slows down GMP incorporation. 26 In contrast, the methyl group of the N6-mA directly weakens the base pairing and consequently slows down UMP incorporation. Notably, the methyl group of 5mC has minimum effect on hydrogen-bonds within base pair, substrate positioning, and active site assembly in this step; therefore, the GTP incorporation opposite 5mC is as fast as that with unmodified C. The third step of recognition is “post-insertion translocation”. In this step, the newly formed base pair undergoes either forward translocation for the next round of NTP addition or rearward translocation for backtracking. Because backtracking requires the disruption of the 3′-base pair, the hydrogen-bonding strength of the 3′-base pair is crucial. For the N6-mA template, the weakened base pair between U:N6-mA promotes backtracking. The presence of N6-mA increases cleavage rate and changes the cleavage pattern in comparison with unmodified A template (Figure 2b). Interestingly, the presence of 5fC and 5caC also promotes the backtracking population of pol II complex. The TFIIS-mediated cleavage rates for scaffolds containing 5fC and 5caC increased by 5.0- and 2.6-fold, respectively, whereas no noticeable changes were observed in the case of 5hmC and 5mC compared with unmodified C template.25 Taken together, these results indicate that the presence of 5fC/5caC or N6-mA greatly shifts pol II from an active population (poised for elongation) to a paused population and promotes pol II stalling and RNA backtracking.
Figure 4.

Model of epigenetic mark N6-mA recognition and processing by RNA pol II. The upper panel shows the side view, and the lower panel shows the top view. (a) The residue Q531 of Rpb2 (colored as salmon) can capture 5caC (colored as magenta) (PDB code 4Y52) but not N6-mA of template during template translocation. Addition site (A site) is shown as dotted oval. (b) UTP entry is slowed down due to impaired stability of UTP:N6-mA pair. (c) After UMP addition, the extended RNA is prone to backtracking due to the impaired stability of UMP/N6-mA pair.
Functional Interplays between N6-mA and Transcription
Previous studies indicated that N6-mA can impact transcription indirectly by modifying transcription factor binding or altering chromatin structure. Indeed, the presence of N6-mA has been shown to modulate transcription factor binding to its target sequence.32 Furthermore, N6-mA can also affect pol II transcription by affecting nucleosome positioning, which has a profound effect on every phase of pol II transcription process including initiation and elongation. We previously reported that N6-mA occurrence at TSS exhibits a periodic pattern with maxima that are complementary to nucleosome binding sites, suggesting that N6-mA potentially is involved in nucleosome positioning in C. reinhardtii,8 which is also consistent with early observation in Tetrahymena thermophile.33 In this study, we presented that N6-mA can directly interact with RNA pol II and cause transcriptional pausing/stalling. Considering the distribution patterns of N6-mA across the genome in different organisms, it is possible that these stalling effects may add a new layer of fine-tuned regulation of Pol II transcription elongation dynamics. The transient Pol II pausing caused by N6-mA may also provide signals for the recruitment of various transcription elongation factors or chromatin remodeling complexes to induce additional functional consequences. Thus, the mechanisms of N6-mA in controlling in vivo RNA pol II transcription could be multilayer, and future studies are warranted.
CONCLUSION
In summary, here we investigated the effect of N6-mA on core pol II transcriptional machinery using both biochemical and structural approaches. We found that N6-mA can cause specific pol II pausing and revealed a novel mechanism of N6-mA-induced pol II pausing that is different from the previous reported 5caC-induced pol II pausing. We also found that N6-mA can escape the detection of epi-DNA loop and reach the canonical template position. However, the methyl group of N6-mA weakens the Watson–Crick base pair with incoming UTP and slows down incorporation opposite N6-mA. This weakened pairing between UMP:N6-mA also promotes pol II backtracking. Our results indicate that pol II can read and distinguish different epigenetic marks, and we propose a three-step recognition mechanism for pol II processing of epigenetic DNA modifications. These findings provide an important new perspective on the functional interplay between epigenetic DNA modification and transcription elongation dynamics.
MATERIALS AND METHODS
Materials
RNA pol II was purified from Saccharomyces cerevisiae as previously described.30,34 The template and nontemplate DNA oligonucleotides were purchased from IDT. The N6-mA templates and RNA primers were purchased from TriLink Biotechnologies (Figure S8). RNA primers were radiolabeled using [γ-32P]ATP (PerkinElmer) and T4 Polynucleotide Kinase (NEB). The ultrapure NTPs were purchased from Affymetrix.
In Vitro Transcription Assays
The pol II elongation complexes (pol II EC) for the in vitro transcription assays were assembled using established methods.35,36 Briefly, an aliquot of 5′-32P-labeled RNA was annealed with a 1.5-fold amount of template DNA and 2-fold amount of nontemplate DNA to form the RNA/DNA scaffold (final stock concentration 200 nM, defined by RNA concentration) in elongation buffer (20 mM Tris-HCl (pH 7.5), 40 mM KCl, 5 mM MgCl2). An aliquot of the annealed scaffold of RNA/DNA was then incubated with a 4-fold amount of pol II at room temperature for 10 min to ensure the formation of pol II elongation complex. The pol II EC is ready for in vitro transcription upon mixing with equal volumes of NTP solution of various concentrations. Final reaction concentrations after mixing were 25 nM scaffold, 100 nM pol II, 5 mM DTT, 5 mM MgCl2, 40 mM KCl, 20 mM Tris-HCl (pH 7.5), and NTP. For transcription assay in the presence of TFIIS, the pol II elongation complex was mixed with equal volume of NTP solution containing 400 nM or 2 μM TFIIS. Final reaction concentrations after mixing were 25 nM scaffold, 100 nM pol II, 5 mM DTT, 5 mM MgCl2, 40 mM KCl, 20 mM Tris-HCl (pH 7.5), and 200 nM or 1 μM TFIIS with NTP in the elongation buffer. The products were quenched by 0.5 M EDTA (pH 8.0) and subsequently analyzed by 16% denaturing urea/TBE PAGE.
Single Turnover Nucleotide Incorporation Assays
The assays were carried out as previously described.35,36 Briefly, nucleotide incorporation assays were conducted by preincubating 50 nM scaffold with 200 nM pol II for 10 min in elongation buffer to form pol II elongation complex at 22 °C. The preincubated pol II EC was then mixed with an equal volume of solution containing 40 mM KCl, 20 mM Tris-HCl (pH 7.5), 10 mM DTT, 10 mM MgCl2, and 2-fold concentrations of NTP. Final reaction concentrations after mixing were 25 nM scaffold, 100 nM pol II, 5 mM MgCl2, and NTP in elongation buffer. Reactions were quenched at various time points by the addition of one volume of 0.5 M EDTA (pH 8.0) and analyzed as described above. Nonlinear-regression data fitting was performed using Prism 6. The time dependence of product formation was fit to a one-phase association equation to determine the observed rate (kobs).
TFIIS-Stimulated Cleavage Assay
Recombinant TFIIS was purified as described.25,36 Cleavage reactions were performed by preincubating pol II with various scaffolds as previously described with slight modification. The solution was then mixed with an equal volume of solution containing TFIIS and MgCl2 in elongation buffer. Final reaction conditions were 100 nM pol II, 25 nM scaffold, 200 nM (or 1 μM) TFIIS, and 5 mM MgCl2. Reactions were quenched at various time points by the addition of one volume of 0.5 M EDTA (pH 8.0). Products were separated by denaturing PAGE as described above. The observed cleavage rates (kobs) were determined with two-phase decay fitting equations.
X-ray Crystallography Setup and Analysis
The template DNA, nontemplate DNA, and RNA oligonucleotides were annealed to form the scaffold at a ratio of 1:2:2. The pol II EC was assembled as previously described.30 The pol II EC crystals were grown using the hanging drop method in solutions containing 390 mM (NH4)2HPO4/NaH2PO4 (pH 6.0), 50 mM dioxane, 10 mM DTT, and 10–12.5% (w/v) PEG6000. Crystals were transferred in a stepwise manner to cryo buffer as previously described.30 For the pol II EC with UMP analogue, pol II EC crystals were soaked with 3.5 mM UMPNPP (Jena Bioscience) and 5 mM MgCl2 overnight.
Diffraction data were collected at beamline 8.2.1 at the Advanced Light Source, Lawrence Berkeley National Laboratory, as well as beamline 7.1 at the Stanford Synchrotron Radiation Lightsource. Data were processed with the program MOSFLM37 and scaled with SCALA from the CCP4 program suite,38 and then the structure factors were further analyzed by UCLA Diffraction Anisotropy Server.39 Molecular replacement was done with PHASER of the CCP4 program suite using unmodified pol II EC (PDB code 2NVZ) as a search model. Model building and structural refinement were performed with COOT40 and PHENIX,41 respectively. Notably, given the potential rotation flexibility of 6-methyl group and current resolution limitation (3.4 and 3.6 Å resolution), the conformation of the 6-methyl group of N6-mA cannot be traced from the electron density map unambiguously. We built in the 6-methyl group of N6-mA based on reported high-resolution structures containing N6-mA. For the unpaired N6-mA, the 6-methyl group of N6-mA was built in a syn-conformation, which is energetically preferred.42–44 For the N6-mA paired with UMPNPP, the 6-methyl group of N6-mA was built in an anti-conformation (Figure 3c) based on previous high-resolution structures of N6-mA RNA duplex (PDB code 2MVS)31 and the complex of DpnI with N6-mA double-stranded DNA (PDB code 4ESJ and 4KYW).45 The statistics of data collection and refinement are summarized in Table S1. The quality of these final models was checked with MolProbity.46 All of the structural figures were rendered in PyMOL (http://www.pymol.org).
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (Grant R01 GM102362 to D.W. and Grant R01 HG006827 to C.H.). L.H. is a Chicago Fellow, and C.H. is a Howard Hughes Medical Institute Investigator. This research used resources of the Advanced Light Source (8.2.1), which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH. We appreciate beamline staff for their support during our data collection.
Footnotes
Accession Codes
The atomic coordinates and structure factors have been deposited in Protein Data Bank with accession codes of 5W4U (EC-I) and 5W51 (EC-II).
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b06381.
Additional data of pol II pausing, quantitative analysis of observed kinetic rates for single UMP incorporation, data verifying UMP incorporation opposite N6-mA site, analysis of TFIIS-stimulated transcript cleavage rates and patterns, crystal structure of EC-I complex, electron density maps of pol II EC-II and two conformations of UMPNPP, deconvoluted ESI mass spectrum characterization of synthetic N6-mA containing DNA oligo, and X-ray diffraction data and refinement statistics (PDF)
References
- 1.Smith ZD, Meissner A. Nat Rev Genet. 2013;14:204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 2.Bestor TH, Edwards JR, Boulard M. Proc Natl Acad Sci U S A. 2015;112:6796–6799. doi: 10.1073/pnas.1415301111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schubeler D. Nature. 2015;517:321–326. doi: 10.1038/nature14192. [DOI] [PubMed] [Google Scholar]
- 4.Klose RJ, Bird AP. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 5.Ratel D, Ravanat JL, Berger F, Wion D. BioEssays. 2006;28:309–315. doi: 10.1002/bies.20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wion D, Casadesus J. Nat Rev Microbiol. 2006;4:183–192. doi: 10.1038/nrmicro1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mondo SJ, Dannebaum RO, Kuo RC, Louie KB, Bewick AJ, LaButti K, Haridas S, Kuo A, Salamov A, Ahrendt SR, Lau R, Bowen BP, Lipzen A, Sullivan W, Andreopoulos BB, Clum A, Lindquist E, Daum C, Northen TR, Kunde-Ramamoorthy G, Schmitz RJ, Gryganskyi A, Culley D, Magnuson J, James TY, O’Malley MA, Stajich JE, Spatafora JW, Visel A, Grigoriev IV. Nat Genet. 2017;49:964–968. doi: 10.1038/ng.3859. [DOI] [PubMed] [Google Scholar]
- 8.Fu Y, Luo GZ, Chen K, Deng X, Yu M, Han D, Hao Z, Liu J, Lu X, Dore LC, Weng X, Ji Q, Mets L, He C. Cell. 2015;161:879–892. doi: 10.1016/j.cell.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang G, Huang H, Liu D, Cheng Y, Liu X, Zhang W, Yin R, Zhang D, Zhang P, Liu J, Li C, Liu B, Luo Y, Zhu Y, Zhang N, He S, He C, Wang H, Chen D. Cell. 2015;161:893–906. doi: 10.1016/j.cell.2015.04.018. [DOI] [PubMed] [Google Scholar]
- 10.Greer EL, Blanco MA, Gu L, Sendinc E, Liu J, Aristizabal-Corrales D, Hsu CH, Aravind L, He C, Shi Y. Cell. 2015;161:868–878. doi: 10.1016/j.cell.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koziol MJ, Bradshaw CR, Allen GE, Costa AS, Frezza C, Gurdon JB. Nat Struct Mol Biol. 2016;23:24–30. doi: 10.1038/nsmb.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu TP, Wang T, Seetin MG, Lai Y, Zhu S, Lin K, Liu Y, Byrum SD, Mackintosh SG, Zhong M, Tackett A, Wang G, Hon LS, Fang G, Swenberg JA, Xiao AZ. Nature. 2016;532:329–333. doi: 10.1038/nature17640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma L, Zhao B, Chen K, Thomas A, Tuteja JH, He X, He C, White KP. Genome Res. 2017;27:385–392. doi: 10.1101/gr.212563.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Summerer D. Angew Chem, Int Ed. 2015;54:10714–10716. doi: 10.1002/anie.201504594. [DOI] [PubMed] [Google Scholar]
- 15.Luo GZ, Blanco MA, Greer EL, He C, Shi Y. Nat Rev Mol Cell Biol. 2015;16:705–710. doi: 10.1038/nrm4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo GZ, He C. Nat Struct Mol Biol. 2017;24:503–506. doi: 10.1038/nsmb.3412. [DOI] [PubMed] [Google Scholar]
- 17.Shin JH, Xu L, Wang D. Transcription. 2016;7:57–62. doi: 10.1080/21541264.2016.1168506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shin JH, Xu L, Wang D. Cell Biosci. 2017;7:9. doi: 10.1186/s13578-016-0133-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu L, Wang W, Chong J, Shin JH, Xu J, Wang D. Crit Rev Biochem Mol Biol. 2015;50:503–519. doi: 10.3109/10409238.2015.1087960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fousteri M, Mullenders LH. Cell Res. 2008;18:73–84. doi: 10.1038/cr.2008.6. [DOI] [PubMed] [Google Scholar]
- 21.Hanawalt PC, Spivak G. Nat Rev Mol Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 22.van Luenen HG, Farris C, Jan S, Genest PA, Tripathi P, Velds A, Kerkhoven RM, Nieuwland M, Haydock A, Ramasamy G, Vainio S, Heidebrecht T, Perrakis A, Pagie L, van Steensel B, Myler PJ, Borst P. Cell. 2012;150:909–921. doi: 10.1016/j.cell.2012.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson MD, Harreman M, Svejstrup JQ. Biochim Biophys Acta, Gene Regul Mech. 2013;1829:151–157. doi: 10.1016/j.bbagrm.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 24.Lagerwerf S, Vrouwe MG, Overmeer RM, Fousteri MI, Mullenders LH. DNA Repair. 2011;10:743–750. doi: 10.1016/j.dnarep.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 25.Kellinger MW, Song CX, Chong J, Lu XY, He C, Wang D. Nat Struct Mol Biol. 2012;19:831–833. doi: 10.1038/nsmb.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Zhou Y, Xu L, Xiao R, Lu X, Chen L, Chong J, Li H, He C, Fu XD, Wang D. Nature. 2015;523:621–625. doi: 10.1038/nature14482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kireeva ML, Hancock B, Cremona GH, Walter W, Studitsky VM, Kashlev M. Mol Cell. 2005;18:97–108. doi: 10.1016/j.molcel.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 28.Charlet-Berguerand N, Feuerhahn S, Kong SE, Ziserman H, Conaway JW, Conaway R, Egly JM. EMBO J. 2006;25:5481–5491. doi: 10.1038/sj.emboj.7601403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belotserkovskii BP, Mirkin SM, Hanawalt PC. Chem Rev. 2013;113:8620–8637. doi: 10.1021/cr400078y. [DOI] [PubMed] [Google Scholar]
- 30.Wang D, Bushnell DA, Westover KD, Kaplan CD, Kornberg RD. Cell. 2006;127:941–954. doi: 10.1016/j.cell.2006.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roost C, Lynch SR, Batista PJ, Qu K, Chang HY, Kool ET. J Am Chem Soc. 2015;137:2107–2115. doi: 10.1021/ja513080v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugimoto K, Takeda S, Hirochika H. Plant J. 2003;36:550–564. doi: 10.1046/j.1365-313x.2003.01899.x. [DOI] [PubMed] [Google Scholar]
- 33.Karrer KM, VanNuland TA. Nucleic Acids Res. 2002;30:1364–1370. doi: 10.1093/nar/30.6.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang D, Bushnell DA, Huang X, Westover KD, Levitt M, Kornberg RD. Science. 2009;324:1203–1206. doi: 10.1126/science.1168729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kellinger MW, Ulrich S, Chong J, Kool ET, Wang D. J Am Chem Soc. 2012;134:8231–8240. doi: 10.1021/ja302077d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu L, Plouffe SW, Chong J, Wengel J, Wang D. Angew Chem, Int Ed. 2013;52:12341–12345. doi: 10.1002/anie.201307661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. Acta Crystallogr, Sect D: Biol Crystallogr. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Collaborative Computational Project, Number 4. Acta Crystallogr, Sect D: Biol Crystallogr. 1994;50:760–763. [Google Scholar]
- 39.Strong M, Sawaya MR, Wang S, Phillips M, Cascio D, Eisenberg D. Proc Natl Acad Sci U S A. 2006;103:8060–8065. doi: 10.1073/pnas.0602606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Emsley P, Cowtan K. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 41.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. Acta Crystallogr, Sect D: Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 42.Sternglanz H, Bugg CE. Science. 1973;182:833–834. doi: 10.1126/science.182.4114.833. [DOI] [PubMed] [Google Scholar]
- 43.Engel JD, von Hippel PH. Biochemistry. 1974;13:4143–4158. doi: 10.1021/bi00717a013. [DOI] [PubMed] [Google Scholar]
- 44.Engel JD, von Hippel PH. J Biol Chem. 1978;253:927–934. [PubMed] [Google Scholar]
- 45.Siwek W, Czapinska H, Bochtler M, Bujnicki JM, Skowronek K. Nucleic Acids Res. 2012;40:7563–7572. doi: 10.1093/nar/gks428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.