

Abstract
During terminal differentiation, the global protein complement is remodeled, as epitomized by erythrocytes, whose cytosol is ~98% globin. The erythroid proteome undergoes a rapid transition at the reticulocyte stage; however, the mechanisms driving programmed elimination of preexisting cytosolic proteins are unclear. We found that a mutation in the murine Ube2o gene, which encodes a ubiquitin-conjugating enzyme induced during erythropoiesis, results in anemia. Proteomic analysis suggested that UBE2O is a broad-spectrum ubiquitinating enzyme that remodels the erythroid proteome. In particular, ribosome elimination, a hallmark of reticulocyte differentiation, was defective in Ube2o−/− mutants. UBE2O recognized ribosomal proteins and other substrates directly, targeting them to proteasomes for degradation. Thus, in reticulocytes, the induction of ubiquitinating factors may drive the transition from a complex to a simple proteome.
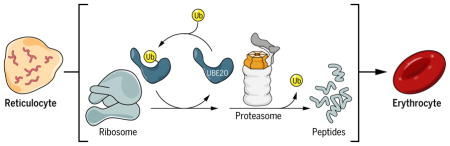
Graphical Abstract
Main Text
The cytosol of mature red blood cells (RBCs) is ~98% globin (1), constituting one of the simplest cellular proteomes known. Achieving this unique state may require programmed degradation of pre-existing cellular proteins in terminally differentiating erythroid precursors. Interest in this problem led to the discovery of the ubiquitin-proteasome system (UPS) in reticulocytes, where it is highly active (2, 3). The synthesis of certain components of the UPS, particularly ubiquitin-conjugating enzyme UBE2O (also called E2-230K), is highly induced in the erythroid lineage in parallel with globin, contemporaneously with the loss of many other UPS components (4). The gene encoding UBE2O appears to share a GATA1-dependent mechanism of transcriptional induction with globin (5), leading to UBE2O expression in reticulocytes as well as their erythroblast precursors (6–8). Thus, a program of ubiquitination that is specific to terminal differentiation might drive remodeling of the erythroid proteome. We assessed this model after identifying a mutation, hem9, in the gene encoding UBE2O.
The autosomal recessive hem9 mutation was identified in a systematic ethylnitrosourea screen for mouse mutants (9). Homozygous hem9 mutants have a form of anemia characterized by small cells (microcytosis), reduced concentrations of hemoglobin (hypochromia), and elevated RBC counts (erythrocytosis) (Table 1). Reciprocal transplantation studies demonstrated that these phenotypes are intrinsic to the hematopoietic system (fig. S1). Histological characterization of peripheral blood smears (fig. S1, A and B) confirmed the phenotype of hypochromic anemia.
Table 1. Ube2o−/− mice have hypochromic, microcytic anemia.
Erythropoietic parameters in Ube2ohem9 (Ube2o−/−) and genetic interaction between Ube2o−/− and the β-globin deficient Hbbth3 (th3) allele.
| Hbb | Ube2o | Spleen:BW (mg/g) | RBC (x106/μl) | HGB (g/dl) | MCV (fl) | MCH (pg) | RETIC ABS (x103/μl) | CHr (pg) |
|---|---|---|---|---|---|---|---|---|
| +/+ | +/+ | 3.9±0.2 | 8.50±0.27 | 13.2±0.8 | 58.9±1.7 | 15.6±0.5 | 2.2x102±0.2x102 | 15.4±0.6 |
| +/+ | −/− (hem9) | 3.8±0.2 | 11.59±0.39‡ | 11.6±0.6‡ | 43.3±1.3‡ | 10.0±0.2‡ | 4.4x102±1.2x102 | 12.0±0.3‡ |
| th3/+ | +/+ | 22.4±1.1‡ƒ | 6.86±0.41‡ƒ | 6.6±0.2‡ƒ | 48.5±1.1‡ƒ | 9.8±0.6‡ | 1.7x103±3.5x103‡ƒ | 12.9±0.3‡ƒ |
| th3/+ | −/− (hem9) | 9.8±4.0‡ƒ* | 11.53±0.86‡* | 8.3±0.8‡ƒ* | 34.0±1.1‡ƒ* | 7.1±0.4‡ƒ* | 1.2x103±2.2x103‡ƒ* | 9.7±0.2‡ƒ* |
Spleen:BW, spleen weight:body weight. RBC, red blood cells; HGB, hemoglobin; MCV, mean cell volume; MCH, mean corpuscular hemoglobin; RETIC ABS, absolute reticulocytes; CHr, mean reticulocyte hemoglobin content. All studies were performed in 8-week-old female littermates, with n=6 animals in each group. Significance was calculated by one-way ANOVA with Tukey’s multiple comparison test.
<0.05 vs WT;
<0.05 vs Hbb+/+ Ube2o−/−;
<0.05 vs Hbbth3/+ Ube2o+/+.
Values are presented ± s.d. (See also fig. S3F).
We mapped hem9 to a ~309-kb interval on chromosome 11. A single gene in this interval, Ube2o, was expressed in erythroid precursors far above its basal level in other cell types (4, 10, 11). Sequencing revealed a nonsense mutation predicted to truncate the C-terminal 168 amino acids. UBE2O protein was undetectable by immunoblot analysis of reticulocyte-rich blood from Ube2ohem9 animals (fig. S2C). To validate the mutation, we bred hem9 mutants to two Ube2o gene trap alleles. Compound heterozygotes lacked UBE2O protein and exhibited an RBC phenotype similar to that of hem9 homozygotes (fig. S2D). Thus, hem9 is a null allele of Ube2o (hereafter Ube2o−/−).
Ube2o−/− reticulocytes showed reductions in many ubiquitin conjugate species (Fig. 1A). Given that hundreds of ubiquitinating factors are expressed in mammals, it is unusual to find such a dominant role of a single component. Unassembled and misfolded globins are preferentially ubiquitinated, as seen in β-thalassemia, where unpaired α-globin precipitates cause cellular damage, ineffective erythropoiesis, hemolysis, and anemia (12). Purified UBE2O ubiquitinated purified α-globin, though some ubiquitination of endogenous α-globin persisted in the Ube2o−/− mutant (fig. S3, A and B).
Fig. 1. Ube2o−/− reticulocytes are deficient in eliminating ribosomal proteins.

(A) Proteins from reticulocyte lysates were resolved by SDS-PAGE and immunoblotted with an anti-ubiquitin antibody. Each sample is from a different mouse whose reticulocytes were induced by serial bleeding.
(B) Ubiquitination was reconstituted in Ube2o−/− reticulocyte lysates using recombinant UBE2O. Reactions were supplemented with biotin-tagged ubiquitin and ubiquitin activating enzyme (UBE1), and incubated for 45 min at 37°C. Samples were resolved by SDS-PAGE. Proteins were electroblotted and visualized using streptavidin-HRP. The catalytically inactive C1037A mutant (UBE2O-CA) was used as a control. UBE2O-CA did not have conjugating activity in lysates, as the only bands evident in this case were biotin-ubiquitin and autoubiquitinated E1. In the absence of lysate, UBE2O was autoubiquitinated, as previously reported (27). W, UBE2O-WT; C, UBE2O-CA.
(C) UBE2O ubiquitinates ribosomal proteins in reconstituted Ube2o−/− lysates. Ubiquitin conjugates (see Fig. 2A) purified by NeutrAvidin-biotin pulldown were digested on beads with trypsin. Peptides containing di-glycine modified lysines (ubiquitination sites) were identified and mapped by LC-MS/MS. All ribosomal protein ubiquitination events were unique to the wild-type UBE2O sample. Δ spectral counts denotes subtraction of all spectral counts collected in the UBE2O-CA sample from those of the UBE2O-WT reaction for a given di-glycine peptide over six replicate experiments. Ub sites, unique ubiquitination sites identified.
(D), Levels of ribosomal proteins from Ube2o−/− and wild-type reticulocytes assessed by immunoblotting. GAPDH and β-spectrin are loading controls. 100 μg of protein was loaded per lane.
(E) Volcano plot of quantitative proteomics analysis, representing the relation of the log10 of the p-value adjusted using Benjamini-Hochberg correction [-log (adj. p-value)] and the log2 of the fold change [log2 (Ube2o−/−/Ube2o+/+)]. Proteins significantly (adj. p-value < 0.05) upregulated or downregulated at least 25% in Ube2o−/− samples were displayed in red and blue respectively. Highly significant enrichment of ribosomes (adj. p-value=3.23 x 10−6) was found by KEGG pathway enrichment analysis of all proteins upregulated significantly and by more than 25% in Ube2o−/− reticulocytes. Brackets, number of proteins per group.
We tested whether UBE2O-dependent degradation of excess α-globin might contribute to the Ube2o−/− phenotype by crossing a deletion of the β-globin loci (Hbbth3 allele), which causes a β-thalassemia intermedia phenotype (13), with the Ube2o−/− line. However, UBE2O deficiency actually increased hemoglobin levels in the Hbbth3/+ mutant (Table 1). Mitigation of the anemia in the double mutant was accompanied by increased numbers of RBCs (Table 1), which were smaller in mean volume than controls and had diminished hemoglobin content.
UBE2O deficiency markedly reduced precipitated hemoglobin aggregates that are associated with β-globin deficiency, and accordingly the accumulation of insoluble α-globin, without affecting the steady-state α/β-globin ratio for soluble protein (fig. S4, A to D). Furthermore, double mutants showed reduced splenomegaly, a major consequence of β-thalassemia caused by increased RBC destruction and heightened splenic erythropoiesis. RBC lifespan was also increased in the double mutants, which is consistent with a reduction in harmful unpaired α-globin chains (fig. S4E).
In summary, although α-globin is a UBE2O substrate, defective ubiquitination and degradation of α-globin could not fully explain the Ube2o−/− phenotype; instead, the loss of UBE2O ameliorated phenotypes attributable to α-globin excess. The reduced globin levels of the mutant appear to reflect attenuated translation of α- and β-globin, as indicated by ribosome profiling (fig. S5, A and B). In the mutant, eukaryotic initiation factor 2α (eIF2α) phosphorylation was induced, which is suppressive of translation and may therefore account for the reduction of globin expression (fig. S5C).
Ribosomal proteins are abundantly ubiquitinated by UBE2O
To identify additional substrates of UBE2O, we added recombinant enzyme or a catalytically inactive mutant (UBE2O-C1037A; hereafter UBE2O-CA), at physiological levels, to reticulocyte lysates from null animals. Prior to the reaction, endogenous E2 activity was quenched chemically. Recombinant UBE2O formed ubiquitin conjugates robustly, while UBE2O-CA was inactive (Fig. 1B). Newly formed conjugates were identified by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis (Table S1). Although numerous targets were identified, including α- and β-globin (fig. S6A), the major class of targets was ribosomal proteins (RPs), comprising 87% of spectral counts of all ubiquitinated peptides that were specific to wild-type UBE2O, other than those from UBE2O itself (Fig. 1C).
The two most frequently identified ubiquitinated RPs were RPL29 and RPL35, which had been suggested to interact with UBE2O by high-throughput affinity-purification mass spectrometry (14). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis (15) of all 105 UBE2O targets (Table S1) showed a strong selection for ribosomal proteins (adjusted P value = 3.65x10−17); STRING analysis (16) also showed a definitive cluster of ribosomal proteins (fig. S6, B and C). When conjugate purification was repeated under denaturing conditions, RPs remained the major class of ubiquitination targets (fig. S6D and Table S1). Thus, the ubiquitination pattern does not reflect pre-existing UBE2O-independent ubiquitin modifications that cofractionated with UBE2O-dependent modifications simply because they were present on the same ribosome.
A hallmark of the transition from the reticulocyte to the erythrocyte stage of differentiation is the elimination of ribosomes (17). The associated mechanism is poorly understood (18, 19). We probed reticulocyte lysates from wild-type and Ube2o−/− mice with antibodies to seven RPs (Fig. 1D). In all cases, we observed elevated levels in the mutants, consistent with a broad defect in RP elimination in Ube2o−/− mice.
To search globally for proteins that may be targets of UBE2O, we compared the proteomes of wild-type and Ube2o−/− reticulocytes by tandem mass tagging (TMT) mass spectrometry (20) (fig. S7). Of 1235 proteins quantified (Table S2 and Fig. 1E), 183 were elevated in mutant reticulocytes. The results suggested that UBE2O, when induced to high levels, might execute a broad program of ubiquitination and degradation. KEGG pathway enrichment analysis yielded a highly significant enrichment of ribosomes (Fig. 1E). Ferritins (FTL1 and FTH1) were also elevated in Ube2o−/− reticulocytes (Fig. 1E). The increase in ferritin levels appears to reflect translational control, as ribosome profiling studies showed elevated levels of translating mRNA for Ftl1 and Fth1 in the mutant (fig. S8A). Ribosome pausing was prominent at iron-responsive (IRE) elements with the Ftl1 and Fth1 mRNAs in wild-type but not mutant reticulocytes (fig. S8B); this result suggested that Ube2o−/− reticulocytes are responding to increased cytosolic iron or heme levels and that reticulocyte iron deficiency is unlikely to be the cause of the anemia. Accordingly, the heme-regulated eIF2α kinase (HRI), a sensor of iron deficiency, was not induced in Ube2o−/− mutants, nor did a null mutation in the HRI-encoding gene modify the anemic phenotype (fig. S9)
Ube2o−/− mice are deficient in ribosome elimination
We next tested whether UBE2O governs the level of ribosomes or, more narrowly, that of free RPs, which are typically unstable (21). Sucrose gradient analysis showed that Ube2o−/− reticulocytes have elevated ribosome levels, particularly 80S monosomes (Fig. 2A and fig. S10).
Fig. 2. Ube2o−/− reticulocytes have elevated levels of ribosomes.

(A) Polysome profiles were generated by fractionating Ube2o−/− and WT reticulocyte lysates on 20–50% sucrose gradients. The X-axis represents position in the gradient, the Y-axis OD260. Left, freshly-isolated Ube2o−/− reticulocytes showed an 80S monosome peak that is elevated in comparison to WT. Right, as above but after 31 hours of ex vivo differentiation.
(B) Flow cytometry analysis after ex vivo differentiation. WT CD71+ reticulocytes showed progressive loss of both MitoTracker Deep Red and thiazole orange staining. Ube2o−/− CD71+ reticulocytes retained thiazole orange staining after 72 hours of ex vivo differentiation (upper panels), with mitochondrial elimination unaffected (lower panels).
When cultured ex vivo, reticulocytes mature into erythrocytes over 48 to 72 hours (19, 22), with both ribosomes and mitochondria being eliminated. Cultured reticulocytes were analyzed by flow cytometry using MitoTracker Deep Red, which stains viable mitochondria, and thiazole orange, which stains RNA; in reticulocytes, the most abundant RNA is ribosomal (rRNA) (Fig. 2B). After 72 hours of ex vivo differentiation, the median fluorescence intensity of thiazole orange staining in Ube2o−/− reticulocytes was higher than in the wild type by a factor of 28, whereas MitoTracker staining was higher in the mutant only by a factor of 1.6. The lack of significant UBE2O involvement in mitochondrial elimination is consistent with previous indications that maturation-associated mitophagy, an autophagic process, is ubiquitin-independent in reticulocytes (19, 23).
Polysome profiles from wild-type reticulocytes cultured ex vivo showed that ribosome elimination was nearly complete by 31 hours; by contrast, the 80S monosome peak remained prominent in Ube2o−/− reticulocytes (Fig. 2A), consistent with delayed turnover of multiple RPs during maturation (fig. S11). Thus, ribosome levels are elevated in Ube2o−/− reticulocytes because of a defect in ribosome elimination during terminal differentiation. Despite the persistence of this peak, few polysomes were evident in the gradient profile, perhaps because of low levels of mRNA or translational initiation factors at this stage of differentiation.
UBE2O is sufficient to drive ribosome elimination in non-erythroid cells
Having found that UBE2O is critical for ribosome elimination in reticulocytes, we tested whether it is sufficient for elimination in a non-erythroid cell, as might be expected if UBE2O recognized RPs directly. We integrated the full length UBE2O open reading frame into Flp-In T-REx 293 cells under a doxycycline-inducible promoter. The UBE2O-CA mutant, integrated at the same locus, was employed as a control (Fig. 3A and Fig S12, A and B). UBE2O was induced for 48 to 72 hours to recapitulate the period of terminal erythroid maturation (Fig. 3A). 293-E2O cells remained viable, with no indication of induced cell death (fig. S12C). The effect of UBE2O expression on the proteome was assessed by mass spectrometry, which quantified 7807 proteins (fig. S13, A and B, and Table S3). Upon UBE2O induction, we identified 685 proteins present at substantially lower levels (≤50%) than in UBE2O-CA by day 3 (Fig. 3B), with a clear preferential effect on basic proteins (Fig. 3D). Thus, ectopic UBE2O induction led to extensive proteome remodeling.
Fig. 3. UBE2O is sufficient to drive the elimination of ribosomes in HEK293 cells.

(A) UBE2O induction by doxycycline treatment in Flp-In T-REx 293 cells was assessed by immunoblotting, using antibodies to UBE2O. Full length human UBE2O-WT and UBE2O-CA genes were genomically integrated in an untagged form, and induced with doxycycline for 24 hr. 20 μg of cell lysate was loaded per lane. GAPDH is a loading control.
(B, C) Mass spectrometry (TMT) quantification of 7,808 proteins after UBE2O (WT or CA) induction for 0, 12, 24, 48 and 72 hours. All 685 proteins that were downregulated more than 50% after 72h of UBE2O-WT induction, in comparison to UBE2O-CA, are shown in (B). Coloring reflects Pearson correlation (r) to the median pattern from high (red) to low (black). The bar graph represents results from KEGG pathway enrichment analysis, which indicated highly significant enrichment (adj. p-value=2.77 x 10−6) of ribosomes, with 18 proteins downregulated (shown in C).
(D) Isoelectric point (pI) comparison of all quantified proteins (red), and all downregulated proteins (blue), based on data from panel B. pI values were obtained from the proteome pI database (46).
(E) After induction of UBE2O (WT or CA) for the indicated times (48 and 72 hours), ribosomal protein levels were analyzed by immunoblotting. GAPDH is a loading control. 20 μg of cell lysate was loaded per lane, and all samples were from the same experiment.
(F) Sucrose gradient analysis of cells overexpressing UBE2O-WT showed a reduced 80S monosome peak after 72 hours of doxycycline treatment.
Among proteins whose levels were reduced more than 50% upon UBE2O induction, KEGG pathway enrichment analysis identified a highly significant down-regulation of RPs (Fig. 3, B and C). This was confirmed by immunoblotting for RPL29, RPL35, and RPL23a (Fig. 3E). RPs were not degraded at comparable rates, inconsistent with what would be expected for an autophagocytic mechanism. Ribosome degradation might proceed in steps, with RPs showing the fastest degradation more likely to be direct substrates of UBE2O. We also observed significant enrichment in ribosome biogenesis and rRNA processing pathways, which are nonfunctional in reticulocytes, as they are enucleate (Fig. 3B and fig. S13, C to E). These effects of UBE2O are likely direct, because multiple ribosomal and nucleolar proteins were ubiquitinated by recombinant UBE2O when added to extracts of human embryonic kidney (HEK) 293 cells and analyzed by mass spectrometry (fig. S14, A to C, and Table S1).
After UBE2O induction, polysome profiles showed a strong reduction of the 80S monosome peak (Fig. 3F). Although reticulocytes cannot produce ribosomes de novo, a reduction of ribosome levels in 293-E2O cells could reflect either degradation of pre-existing ribosomes or interference with ribosome synthesis. To discriminate between these possibilities, we suppressed the transcription of new rRNA with a selective inhibitor of RNA polymerase I, CX-5461 (24, 25). Under these “chase” conditions, UBE2O drove the destabilization of RPs after 24–48 hours of inhibitor treatment, with no effect observed in the catalytic null mutant (fig. S15). Therefore, pre-existing ribosomal proteins can be eliminated by UBE2O.
In summary, the 293-E2O cell data recapitulate a critical aspect of normal erythroid differentiation, and confirm the inducible breakdown of ribosomes—complexes that are thought to be highly stable under basal conditions. Furthermore, UBE2O is sufficient to drive ribosome elimination in non-erythroid cells in the absence of reticulocyte-specific factors, which suggests that the control of erythroid ribosome turnover may be dependent principally on UBE2O itself.
UBE2O executes a broad program of ubiquitination
On the basis of proteomic data on reticulocytes and 293-E2O cells, we selected seven potential nonribosomal targets of UBE2O for confirmation by immunoblotting. We observed strongly elevated levels of all of these proteins in mutant reticulocytes, including a chaperone for α-globin AHSP (α-hemoglobin stabilizing protein) (Fig. 4A). AHSP was strongly stabilized in a time-course study using ex vivo cultured Ube2o−/− reticulocytes (fig. S16A). However, elevated AHSP levels do not explain the erythroid phenotype of Ube2o−/− mutants, because an Ahsp−/− mutation did not alleviate the Ube2o−/− phenotype (fig. S16, B and C). In summary, immunoblot analysis validated the proteomics data and thus suggested that an extensive set of RPs and non-RPs is eliminated from reticulocytes under the control of UBE2O.
Fig. 4. UBE2O recognizes substrates directly.

(A) Immunoblotting of Ube2o−/− and WT reticulocyte lysates showed an elevation of putative non-ribosomal UBE2O substrates. GAPDH and β-spectrin are loading controls. 100 μg of protein was loaded per lane.
(B) Schematic representation of domains within UBE2O. The T1 construct lacks conserved region 1 (CR1), whereas T2 lacks both CR1 and CR2. In the CA mutant, the active-site Cys1037 is substituted with Ala. Domain assignment is described in the Methods.
(C) In vitro ubiquitination assays were performed in a purified system with UBE1, HA-ubiquitin, UBE2O (WT or CA, W and C respectively) and the indicated candidate substrates for 4 hours at 37°C. Histone H2B is a model substrate of UBE2O (27). N-terminal truncations (T1 and T2) abrogate the ubiquitination of ribosomal proteins and of H2B, but do not prevent modification of AHSP. Samples were analyzed by immunoblotting with either anti-HA or anti-AHSP antibodies, after SDS-PAGE. The left and right panels represent different exposures from the same gel.
(D) Left, UBE2O can directly recognize and ubiquitinate putative non-ribosomal substrates DDX56, NOL12, NOP16, and PTRF, which are elevated in either Ube2o−/− reticulocytes or decreased in 293-E2O cells. Right, purified UBE2O can ubiquitinate individual ribosomal proteins outside of the 80S complex. Samples were analyzed by immunoblotting with anti-HA antibodies, after gradient SDS-PAGE. W, UBE2O-WT; C, UBE2O-CAThe apparent molecular masses of substrates (in kDa) were determined by SDS-PAGE followed by Coomassie staining (data not shown): calmodulin, 17; DDX56, 64; NOL12, 27; NOP16, 25; PTRF, 55–65; RPL35, 17; RPL36a, 16; RPL37, 15.
(E) CR1 and CR2 provide substrate-recognition with distinct specificities. The CR1 and CR2 fragments were expressed in E. coli in a biotin-tagged form, and loaded onto streptavidin resin, using amounts sufficient to saturate the resin’s binding capacity. Purified UBE2O substrates were mixed with resin at 20-fold excess in the presence of carrier. The resin was washed in 50 mM NaCl, 20 mM Tris-HCl (pH 7.4). Bound material was eluted and 25% of the eluate was resolved by SDS-PAGE followed by immunoblotting. The input represents 2.5% of the total sample; consequently CR1 and CR2 are not visualized in the input lanes, though present. Note, rows 6–8 are from the same experiment.
Our finding that UBE2O is sufficient to direct the degradation of its target proteins suggests that it may mediate direct recognition of these proteins. However, E3 rather than E2 enzymes typically mediate substrate recognition (26). The molecular mass of UBE2O is 143 kDa (Fig. 4B), whereas typical E2 enzymes are of 20–25 kDa. It therefore seemed likely that UBE2O may function as an E3 enzyme fused to an E2 (27–29). To test this model, we reconstituted the ubiquitination of candidate UBE2O substrates with purified components.
When partially purified ribosomes were tested by immunoblot analsis, we observed that ubiquitination of RPs was efficient relative to that of histone H2B, a model substrate of UBE2O (Fig. 4C). The ubiquitin acceptor proteins in this reaction were confirmed as RPs by LC-MS/MS-mediated ubiquitination site mapping (fig. S17 and Table S1). Although these proteins were added to the assay principally in the form of ribosome complexes, it is possible and perhaps likely that UBE2O prefers the free forms of these proteins as substrates. Indeed, when tested individually, recombinant RPL35, RPL36a, and RPL37 all proved to be UBE2O substrates (Fig. 4D). Other proteins predicted from proteomics data to be UBE2O substrates were all efficiently ubiquitinated, while calmodulin, a negative control, was not (Fig. 4D). UBE2O added multiple ubiquitin groups to these substrates, although almost exclusively in the form of multi-monoubiquitin modification (fig. S18). In summary, several in vivo targets of UBE2O were also modified by this enzyme in a purified system, which suggests that UBE2O, in addition to being an E2 enzyme, is also an E3–a specificity determining ubiquitination factor.
In addition to its E2 (or UBC) domain, UBE2O has four distinct evolutionarily conserved regions, CR1, CR2, CR3, and a predicted coiled-coil (CC) element (Fig. 4B). When truncated forms of UBE2O were assayed, the importance of the tested domains for conjugation rates varied depending on the substrate tested, consistent with CR1 and perhaps other conserved domains being substrate specificity elements (Fig. 4C). The hypothesis that CR1 and CR2 are substrate recognition domains was assessed more directly by testing them for interaction with UBE2O substrates in pull-down binding assays. Both CR1 and CR2 bound UBE2O substrates, although their binding specificity differed (Fig. 4E). CR1 and CR2 contain acidic patches, which may be involved in their binding to basic proteins. A confirmed acidic substrate, AHSP, did not show significant binding to CR1 or CR2 (Fig. 4E), nor was its ubiquitination dependent on these domains (Fig. 4C). These results indicate that UBE2O is capable of multiple modes of substrate recognition. In summary, UBE2O has the expected features of an E2–E3 hybrid enzyme in which ubiquitin is charged by the UBC domain of UBE2O, to be subsequently donated to proteins that are recognized in most cases by the CR1 and CR2 domains.
Ribosomal proteins are degraded by the proteasome
Previous studies have shown that regulated ribosome degradation occurs via autophagy in yeast (30–33). In reticulocytes, however, inhibitors of autophagy do not affect ribosome breakdown (19). Selective autophagy in reticulocytes is mediated by NIX, but this factor drives the elimination of mitochondria rather than ribosomes (19, 23, 34).
To test whether RPs are eliminated by the proteasome, we incubated reticulocytes with proteasome inhibitors PS-341 and epoxomicin. After ex vivo differentiation, treated wild-type reticulocytes retained thiazole orange staining, resembling Ube2o−/− reticulocytes that had not been exposed to proteasome inhibitor (Fig. 5A; see also fig. S19A). These data suggest that RPs are degraded by the proteasome. In fact, treated wild-type reticulocytes retained their thiazole orange staining more fully than did untreated Ube2o−/− reticulocytes (Fig. 5A), suggesting the existence of alternative pathways of ribosome elimination that are UBE2O-independent, but proteasome-dependent. In contrast to the proteasome, p97–which dissociates components of several multisubunit complexes in a ubiquitin-dependent manner (35)–had no detectable effect on ribosome elimination (fig. S20).
Fig. 5. The ubiquitin-proteasome system in late erythroid differentiation.

(A) WT and Ube2o−/− reticulocytes were differentiated ex vivo at 37°C for 48 hours with proteasome inhibitors (50nM epoxomicin and 50nM PS-341) or DMSO vehicle control, then analyzed by FACS.
(B) Reconstitution of ribosomal protein degradation in Ube2o−/− reticulocyte lysates. Ubiquitin levels were supplemented to prevent depletion of free ubiquitin upon proteasome inhibition. Proteasomes were inhibited by adding PS-341 (50 nM) and epoxomicin (50 nM) together. GAPDH is a loading control. 100 μg of protein was loaded per lane.
(C) WT reticulocytes were treated with proteasome inhibitors (50 nM epoxomicin and 50 nM PS-341) and differentiated ex vivo for 48 hours. Quantitative metabolomic profiling of treated and untreated WT reticulocytes showed a depletion of multiple free amino acids due to proteasome inhibition, with Lys and Arg most strongly affected. Significance was calculated by two-sample t-test. *p<0.05, **p<0.01, ***p<0.001.
(D) An ensemble of ubiquitin ligases and ubiquitin conjugating enzymes is induced in late erythroid differentiation. Stages are arranged in a temporal progression. Stage 1 includes proerythroblasts and early basophilic erythroblasts; stage 2, early and late basophilic erythroblasts; stage 3, polychromatophilic and orthochromatophilic erythroblasts; and stage 4, late orthochromatophilic erythroblasts and reticulocytes. The induction of globin mRNA is shown for comparison. Raw RNA-seq data are taken from (6).
Ubiquitin depletion is evident in reticulocytes treated with the highest concentrations of proteasome inhibitors (fig. S19B). To address this potential caveat, we reconstituted RP degradation in a cell-free reticulocyte lysate system, which permitted supplementation of free ubiquitin. The degradation of RPs was reconstituted by adding back UBE2O to null reticulocyte lysate. RPs were not destabilized by UBE2O-CA, or under conditions of proteasome inhibitor treatment, while maintaining free ubiquitin levels (Fig. 5B and fig. S19C). However, the stabilizing effect of the inhibitors, although apparently complete for RPL29 and RPS23, was partial in the case of RPL35 and RPL36A, as judged from the intensity of the major protein band (Fig. 5B and fig. S19C). For both proteins, a high-molecular weight species appeared upon proteasome inhibition, which likely represented ubiquitinated forms of RPL35 and RPL36A. To confirm that proteasome inhibitors completely blocked RP degradation, we treated the reaction with USP21 to deubiquitinate high-molecular weight forms of the protein. This treatment concentrated RPL35 into a single band that was constant in intensity over the time course of incubation (fig. S19D).
The importance of the proteasome in remodeling the proteome in reticulocytes was further tested by metabolomic analysis. Amino acids such as Lys and Arg, which are abundant in RPs, were indeed depleted by proteasome inhibitors (Fig. 5C). We therefore considered whether UBE2O might cooperate with other ubiquitinating factors that may be induced in late erythroid differentiation. Using RNA-sequencing data for erythroid cells taken from progressive stages of differentiation (6–8), we identified eight E2 and E3 enzymes that are induced in late erythroid cells in parallel with UBE2O (Fig. 5D). We hypothesize that these enzymes work in concert to globally remodel the erythroid proteome. Expression of the vast majority of ubiquitinating enzymes is extinguished during this period (fig. S21, A and B), so that the UPS is not simply amplified during erythroid differentiation, but instead deeply reconfigured. The unique transformation of the UPS in these cells may reflect a strategy to eliminate thousands of cellular proteins from reticulocytes, thus enabling extreme concentration of globin.
DISCUSSION
In many cases of terminal cellular differentiation, the proteome is remodeled on a global scale to generate a highly specific cellular phenotype. The extreme case is erythroid differentiation, where a single species, hemoglobin, is concentrated to ~98% of soluble protein (1). Accomplishing this rapid transformation of the cellular phenotype requires not only highly active protein synthesis, but also degradation of a vast set of pre-existing, otherwise relatively stable, soluble proteins. This degradation must also be selective, so as to spare properly assembled globin and other components of the mature RBC. Despite this active degradation, most components of the ubiquitin pathway are downregulated transcriptionally during erythroid differentiation. However, a small subset of ubiquitin conjugating enzymes and ubiquitin ligases is induced, among them UBE2O. We propose that UBE2O plays a major role in proteomic remodeling, with tens and perhaps hundreds of proteins being eliminated through its activity.
As an E2–E3 hybrid enzyme, UBE2O functions autonomously and can thus remain active well into the reticulocyte stage, promoting maturation at a time when most E2 enzymes have been lost. The ability of UBE2O to promote reticulocyte maturation also depends upon its broad substrate specificity; multiple substrate recognition domains enable the recognition of diverse substrates and may, through avidity, allow coordinate low-affinity substrate interactions to be productive of ubiquitination. In an accompanying paper, Yanagitani et al. report that UBE2O is a quality-control ubiquitin ligase (36). Because quality-control ligases are characteristically broad in their specificity, a ligase of this type may be pre-adapted to serve in global proteome remodeling when induced to high levels.
UBE2O prefers basic substrates, including RPs, and this specificity may underlie a defining feature of the reticulocyte to erythrocyte transition, the elimination of ribosomes (17). Although the ubiquitination of ribosomes and of free RPs has previously been observed (21, 37–39), our findings show that ubiquitination of RPs, in either their free or assembled state, can be used to promote the elimination of ribosomes from cells. Indeed, ribosome ubiquitination has previously been associated with the suppression of their turnover via autophagy (33).
Contrary to the longstanding paradigm that multiubiquitin chains are required to initiate degradation, UBE2O-dependent ubiquitination events efficiently targeted substrates to the proteasome through multi-monoubiquitination events. Indeed, recent work from several groups supports the sufficiency of multi-monoubiquitination for proteasomal degradation, depending on the substrate (40–42).
The robust proteasome-dependent protein turnover seen during the reticulocyte stage of differentiation not only serves to eliminate proteins other than globin from these cells, but may also boost globin synthesis by supplying amino acids for late-stage synthesis of globin. The loss of amino acid transporters during erythroid maturation (7, 43) may impose a strong link between proteins synthesis and degradation in these cells. Consistent with the proposed interconnection between proteasome function and translation, UBE2O-deficient mice exhibit hypochromic anemia, and ribosome occupancy of the globin genes–the major synthetic output–is decreased. Loss of UBE2O accordingly ameliorates phenotypes attributable to α-globin excess, as seen for a model of β-thalassemia. UBE2O is thus a potential therapeutic target, not only for mixed-lineage leukemia and other cancers, as recently proposed (44, 45), but also for β-thalassemia, one of the most prevalent inherited diseases worldwide.
Supplementary Material
Synopsis.
Introduction
The reticulocyte-red blood cell transition is a canonical example of terminal differentiation. The mature red blood cell has one of the simplest cellular proteomes known, with hemoglobin remarkably concentrated to ~98% of soluble protein. During reticulocyte maturation, the proteome is remodeled through the programmed elimination of most generic constituents of the cell, in parallel with abundant synthesis of cell-type-specific proteins, such as hemoglobin. The mechanisms that drive rapid turnover of soluble and normally stable proteins in terminally differentiating cells remain largely unclear.
Rationale
The ubiquitin-proteasome system (UPS) was discovered in reticulocytes, where it is highly active. However, its function in this developmental context has not been established. UBE2O is an E2 (ubiquitin-conjugating) enzyme that is co-induced with globin and expressed at elevated levels late in the erythroid lineage. We identified an anemic mouse line with a null mutation in Ube2o, and used multiplexed quantitative proteomics to identify candidate substrates of UBE2O in an unbiased and global manner. We found that the protein compositions of mutant and wild-type reticulocytes differed markedly, suggesting that UBE2O-dependent ubiquitination might target its substrates for degradation to effect remodeling of the proteome.
To test whether UBE2O was sufficient for proteome remodeling, we engineered a nonerythroid cell line to inducibly express UBE2O above its basal level. Upon induction, we observed the decline of hundreds of proteins from these cells, in many cases the same proteins as those eliminated from reticulocytes. Overexpression of an active-site mutant of UBE2O did not show these effects. Therefore, a major component of the specificity underlying differentiation-linked proteome remodeling appears to be carried by UBE2O itself. These results also indicate that UBE2O may function as a hybrid enzyme with both E2 and E3 (ubiquitin-ligating) activities. In support of this model, candidate substrates identified by proteomics were ubiquitinated by purified UBE2O without the assistance of additional specificity factors.
Results
The most prominent phenotypes of the Ube2o mutant are an anemia characterized by small cells with low hemoglobin content (microcytic hypochromic anemia), and a defect in the elimination of ribosomes, the latter being a key aspect of reticulocyte maturation. When we added recombinant UBE2O protein to reticulocyte lysates from the null mutant, ubiquitin was conjugated primarily to ribosomal proteins. Moreover, immunoblot analysis and quantitative proteomics revealed elevated levels of multiple ribosomal proteins in mutant reticulocytes. Sucrose gradient analysis indicated persistence of not only ribosomal proteins but of ribosomes themselves during ex vivo differentiation of mutant reticulocytes. Accordingly, ribosomes were eliminated upon induction of UBE2O in non-erythroid cells. The elimination of organelles from reticulocytes, as exemplified by that of mitochondria, was not affected in the Ube2o mutant, indicating the specificity of its effects on programmed protein turnover.
Free ribosomal proteins were ubiquitinated by purified UBE2O, which suggests that these proteins are true substrates of the enzyme. However, UBE2O substrates are diverse in nature and not limited to ribosomal proteins. Individual domains of UBE2O bound substrates with distinct specificities. Thus, the broad specificity of UBE2O reflects the presence of multiple substrate recognition domains within the enzyme.
Proteasome inhibitors blocked the degradation of UBE2O-dependent substrates in reticulocytes, although UBE2O does not form polyubiquitin chains. Rather, UBE2O adds single ubiquitin groups to substrates at multiple sites. Proteasome inhibitor treatment ex vivo led to depletion of the pools of many amino acids, suggesting that the flux of ubiquitinated substrates through the reticulocyte proteasome is sufficient to supply amino acids needed for late-stage translation of mRNA. In late erythropoiesis, several ubiquitin-conjugating enzymes and ligases are induced together with Ube2o while most components of the UPS disappear. We propose that the UPS is not simply amplified during erythroid maturation, but is instead broadly reconfigured to promote remodeling of the proteome.
Conclusion
A highly specialized UPS is expressed in the reticulocyte and is used to remodel the proteome of these cells on a global scale. UBE2O, a hybrid E2–E3 enzyme, functions as a major specificity factor in this process. In reticulocytes, and perhaps other differentiated cells such as in the lens, the induction of ubiquitinating factors may drive the transition from a complex to a simple proteome.
Caption
UBE2O drives remodeling of the proteome during erythroid differentiation.
The transformation of reticulocytes into erythrocytes involves the elimination of myriad proteins. UBE2O is an E2–E3 hybrid enzyme that directly recognizes and ubiquitinates proteins that are fated for elimination. The target protein is degraded by the proteasome; ubiquitin (Ub) is recycled. UBE2O substrates include ribosomal proteins, recognized in a free form or possibly within the ribosome complex.
Acknowledgments
Special thanks to S. Elsasser for bioinformatics analysis of Ube2o, and to B. Wadas for characterizing autophagy inhibitors. We thank M. Hegde for communicating results prior to publication. Supported by NIH grants 5R21HL116210 and 5R01HL125710, Biogen-Idec grant 6780680-01, and the Diamond Blackfan Anemia Foundation (D.F. and M.D.F.); NIH Medical Scientist Training Program grant T32GM007753 and NIH F30 grant HL124980 (A.T.N.); NIH K01 grant DK098285 (J.A.P.); and NIH grants U01 HD39372 and R01 CA115503 (M.J.J.). We thank R. King, C. Widmaier, S. de Poot, K. Y. Hung, and D. Brown for advice and assistance. Hem9 mice are available from M.J.J. under a material transfer agreement with Baylor College of Medicine. The data presented in this paper are tabulated in the main paper and the supplementary materials.
Footnotes
References and Notes
- 1.Roux-Dalvai F, et al. Extensive analysis of the cytoplasmic proteome of human erythrocytes using the peptide ligand library technology and advanced mass spectrometry. Mol Cell Proteomics. 2008;7:2254–2269. doi: 10.1074/mcp.M800037-MCP200. [DOI] [PubMed] [Google Scholar]
- 2.Hass AL. In: Red blood cell aging. Magnani M, De Flora A, editors. Plenum Press; New York and London: 1991. pp. 191–205. [Google Scholar]
- 3.Ciehanover A, Hod Y, Hershko A. A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem Biophys Res Commun. 1978;81:1100–1105. doi: 10.1016/0006-291x(78)91249-4. [DOI] [PubMed] [Google Scholar]
- 4.Wefes I, et al. Induction of ubiquitin-conjugating enzymes during terminal erythroid differentiation. Proc Natl Acad Sci U S A. 1995;92:4982–4986. doi: 10.1073/pnas.92.11.4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li L, et al. Ldb1-nucleated transcription complexes function as primary mediators of global erythroid gene activation. Blood. 2013;121:4575–4585. doi: 10.1182/blood-2013-01-479451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wong P, et al. Gene induction and repression during terminal erythropoiesis are mediated by distinct epigenetic changes. Blood. 2011;118:e128–138. doi: 10.1182/blood-2011-03-341404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.An X, et al. Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood. 2014;123:3466–3477. doi: 10.1182/blood-2014-01-548305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Madzo J, et al. Hydroxymethylation at gene regulatory regions directs stem/early progenitor cell commitment during erythropoiesis. Cell Rep. 2014;6:231–244. doi: 10.1016/j.celrep.2013.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kile BT, et al. Functional genetic analysis of mouse chromosome 11. Nature. 2003;425:81–86. doi: 10.1038/nature01865. [DOI] [PubMed] [Google Scholar]
- 10.Haldeman MT, Finley D, Pickart CM. Dynamics of ubiquitin conjugation during erythroid differentiation in vitro. J Biol Chem. 1995;270:9507–9516. doi: 10.1074/jbc.270.16.9507. [DOI] [PubMed] [Google Scholar]
- 11.Wu C, et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaeffer JR. ATP-dependent proteolysis of hemoglobin alpha chains in beta-thalassemic hemolysates is ubiquitin-dependent. J Biol Chem. 1988;263:13663–13669. [PubMed] [Google Scholar]
- 13.Yang B, et al. A mouse model for beta 0-thalassemia. Proc Natl Acad Sci U S A. 1995;92:11608–11612. doi: 10.1073/pnas.92.25.11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huttlin EL, et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell. 2015;162:425–440. doi: 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuleshov MV, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szklarczyk D, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glowacki ER, Millette RL. Polyribosomes and the Loss of Hemoglobin Synthesis in the Maturing Reticulocyte. J Mol Biol. 1965;11:116–127. doi: 10.1016/s0022-2836(65)80177-2. [DOI] [PubMed] [Google Scholar]
- 18.Zhang T, Shen S, Qu J, Ghaemmaghami S. Global Analysis of Cellular Protein Flux Quantifies the Selectivity of Basal Autophagy. Cell Rep. 2016;14:2426–2439. doi: 10.1016/j.celrep.2016.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sandoval H, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McAlister GC, et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Analytical chemistry. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sung MK, et al. A conserved quality-control pathway that mediates degradation of unassembled ribosomal proteins. Elife. 2016;5 doi: 10.7554/eLife.19105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geminard C, de Gassart A, Vidal M. Reticulocyte maturation: mitoptosis and exosome release. Biocell. 2002;26:205–215. [PubMed] [Google Scholar]
- 23.Schweers RL, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104:19500–19505. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drygin D, et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011;71:1418–1430. doi: 10.1158/0008-5472.CAN-10-1728. [DOI] [PubMed] [Google Scholar]
- 25.Bywater MJ, et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell. 2012;22:51–65. doi: 10.1016/j.ccr.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berndsen CE, Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol. 2014;21:301–307. doi: 10.1038/nsmb.2780. [DOI] [PubMed] [Google Scholar]
- 27.Berleth ES, Pickart CM. Mechanism of ubiquitin conjugating enzyme E2-230K: catalysis involving a thiol relay? Biochemistry. 1996;35:1664–1671. [Google Scholar]
- 28.Zhang X, et al. Fine-tuning BMP7 signalling in adipogenesis by UBE2O/E2-230K-mediated monoubiquitination of SMAD6. EMBO J. 2013;32:996–1007. doi: 10.1038/emboj.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mashtalir N, et al. Autodeubiquitination protects the tumor suppressor BAP1 from cytoplasmic sequestration mediated by the atypical ubiquitin ligase UBE2O. Mol Cell. 2014;54:392–406. doi: 10.1016/j.molcel.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 30.Kraft C, Deplazes A, Sohrmann M, Peter M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol. 2008;10:602–610. doi: 10.1038/ncb1723. [DOI] [PubMed] [Google Scholar]
- 31.Ossareh-Nazari B, et al. Cdc48 and Ufd3, new partners of the ubiquitin protease Ubp3, are required for ribophagy. EMBO Rep. 2010;11:548–554. doi: 10.1038/embor.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pestov DG, Shcherbik N. Rapid cytoplasmic turnover of yeast ribosomes in response to rapamycin inhibition of TOR. Mol Cell Biol. 2012;32:2135–2144. doi: 10.1128/MCB.06763-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ossareh-Nazari B, et al. Ubiquitylation by the Ltn1 E3 ligase protects 60S ribosomes from starvation-induced selective autophagy. J Cell Biol. 2014;204:909–917. doi: 10.1083/jcb.201308139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Novak I, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia D, Tang WK, Ye Y. Structure and function of the AAA+ ATPase p97/Cdc48p. Gene. 2016;583:64–77. doi: 10.1016/j.gene.2016.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yanagitani K, Juszkiewicz S, Hegde RS. UBE2O is a quality control factor for orphans of multiprotein complexes. Science. 2017;357:472–475. doi: 10.1126/science.aan0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spence J, et al. Cell cycle-regulated modification of the ribosome by a variant multiubiquitin chain. Cell. 2000;102:67–76. doi: 10.1016/s0092-8674(00)00011-8. [DOI] [PubMed] [Google Scholar]
- 38.Silva GM, Finley D, Vogel C. K63 polyubiquitination is a new modulator of the oxidative stress response. Nat Struct Mol Biol. 2015;22:116–123. doi: 10.1038/nsmb.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Higgins R, et al. The Unfolded Protein Response Triggers Site-Specific Regulatory Ubiquitylation of 40S Ribosomal Proteins. Mol Cell. 2015;59:35–49. doi: 10.1016/j.molcel.2015.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Braten O, et al. Numerous proteins with unique characteristics are degraded by the 26S proteasome following monoubiquitination. Proc Natl Acad Sci U S A. 2016;113:E4639–4647. doi: 10.1073/pnas.1608644113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shabek N, et al. The size of the proteasomal substrate determines whether its degradation will be mediated by mono- or polyubiquitylation. Mol Cell. 2012;48:87–97. doi: 10.1016/j.molcel.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 42.Dimova NV, et al. APC/C-mediated multiple monoubiquitylation provides an alternative degradation signal for cyclin B1. Nat Cell Biol. 2012;14:168–176. doi: 10.1038/ncb2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benderoff S, Blostein R, Johnstone RM. Changes in amino acid transport during red cell maturation. Membr Biochem. 1978;1:89–106. doi: 10.3109/09687687809064161. [DOI] [PubMed] [Google Scholar]
- 44.Liang K, et al. Therapeutic Targeting of MLL Degradation Pathways in MLL-Rearranged Leukemia. Cell. 2017;168:59–72 e13. doi: 10.1016/j.cell.2016.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vila IK, et al. A UBE2O-AMPKalpha2 Axis that Promotes Tumor Initiation and Progression Offers Opportunities for Therapy. Cancer Cell. 2017;31:208–224. doi: 10.1016/j.ccell.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kozlowski LP. Proteome-pI: proteome isoelectric point database. Nucleic Acids Res. 2017;45:D1112–D1116. doi: 10.1093/nar/gkw978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian M, Campagna DR, Woodward LS, Justice MJ, Fleming MD. hem6: an ENU-induced recessive hypochromic microcytic anemia mutation in the mouse. Blood. 2008;112:4308–4313. doi: 10.1182/blood-2007-09-111500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han AP, et al. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 2001;20:6909–6918. doi: 10.1093/emboj/20.23.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kihm AJ, et al. An abundant erythroid protein that stabilizes free alpha-haemoglobin. Nature. 2002;417:758–763. doi: 10.1038/nature00803. [DOI] [PubMed] [Google Scholar]
- 50.Ohgami RS, et al. nm1054: a spontaneous, recessive, hypochromic, microcytic anemia mutation in the mouse. Blood. 2005;106:3625–3631. doi: 10.1182/blood-2005-01-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Torrance JD, Bothwell TH. In: Methods in Hematology. Cook J, editor. Vol. 1. Churchill Livingstone Press; New York: 1980. chap. 104–109. [Google Scholar]
- 52.Shechter D, Dormann HL, Allis CD, Hake SB. Extraction, purification and analysis of histones. Nat Protoc. 2007;2:1445–1457. doi: 10.1038/nprot.2007.202. [DOI] [PubMed] [Google Scholar]
- 53.Rappsilber J, Ishihama Y, Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Analytical chemistry. 2003;75:663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- 54.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robinson JT, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paulo JA, O’Connell JD, Gaun A, Gygi SP. Proteome-wide quantitative multiplexed profiling of protein expression: carbon-source dependency in Saccharomyces cerevisiae. Mol Biol Cell. 2015;26:4063–4074. doi: 10.1091/mbc.E15-07-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Paulo JA, O’Connell JD, Gygi SP. A Triple Knockout (TKO) Proteomics Standard for Diagnosing Ion Interference in Isobaric Labeling Experiments. J Am Soc Mass Spectrom. 2016;27:1620–1625. doi: 10.1007/s13361-016-1434-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huttlin EL, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elias JE, Gygi SP. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol Biol. 2010;604:55–71. doi: 10.1007/978-1-60761-444-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 63.Paulo JA, et al. Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources. J Proteomics. 2016;148:85–93. doi: 10.1016/j.jprot.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tyanova S, et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 65.Chen EY, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vizcaino JA, et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44:D447–456. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sievers F, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Letunic I, Doerks T, Bork P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 2015;43:D257–260. doi: 10.1093/nar/gku949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jones DT, Cozzetto D. DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics. 2015;31:857–863. doi: 10.1093/bioinformatics/btu744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Belin S, et al. Purification of ribosomes from human cell lines. Curr Protoc Cell Biol. 2010;Chapter 3(Unit 3):40. doi: 10.1002/0471143030.cb0340s49. [DOI] [PubMed] [Google Scholar]
- 71.Ye Y, et al. Polyubiquitin binding and cross-reactivity in the USP domain deubiquitinase USP21. EMBO Rep. 2011;12:350–357. doi: 10.1038/embor.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Klemperer NS, Berleth ES, Pickart CM. A novel, arsenite-sensitive E2 of the ubiquitin pathway: purification and properties. Biochemistry. 1989;28:6035–6041. doi: 10.1021/bi00440a047. [DOI] [PubMed] [Google Scholar]
- 73.Beckett D, Kovaleva E, Schatz PJ. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 1999;8:921–929. doi: 10.1110/ps.8.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gell D, Kong Y, Eaton SA, Weiss MJ, Mackay JP. Biophysical characterization of the alpha-globin binding protein alpha-hemoglobin stabilizing protein. J Biol Chem. 2002;277:40602–40609. doi: 10.1074/jbc.M206084200. [DOI] [PubMed] [Google Scholar]
- 75.Paulo JA. Sample preparation for proteomic analysis using a GeLC-MS/MS strategy. J Biol Methods. 2016;3 doi: 10.14440/jbm.2016.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gallien S, et al. Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol Cell Proteomics. 2012;11:1709–1723. doi: 10.1074/mcp.O112.019802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.MacLean B, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lu L, Han AP, Chen JJ. Translation initiation control by heme-regulated eukaryotic initiation factor 2alpha kinase in erythroid cells under cytoplasmic stresses. Mol Cell Biol. 2001;21:7971–7980. doi: 10.1128/MCB.21.23.7971-7980.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.