

Abstract
The physical basis for enzymatic rate accelerations is a subject of great fundamental interest and of direct relevance to areas that include the de novo design of green catalysts and the pursuit of new drug regimens. Extensive investigations of C-H activating systems have provided considerable insight into the relationship between an enzyme’s overall structure and the catalytic chemistry at its active site. This Perspective highlights recent experimental data for two members of distinct, yet iconic C-H activation enzyme classes, lipoxygenases and prokaryotic alcohol dehydrogenases. The data necessitate a reformulation of the dominant textbook definition of biological catalysis. A multidimensional model emerges that incorporates a range of protein motions that can be parsed into a combination of global stochastic conformational thermal fluctuations and local donor-acceptor distance sampling. These motions are needed to achieve a high degree of precision with regard to internuclear distances, geometries, and charges within the active site. The available model also suggests a physical framework for understanding the empirical enthalpic barrier in enzyme-catalyzed processes. We conclude by addressing the often conflicting interface between computational and experimental chemists, emphasizing the need for computation to predict experimental results in advance of their measurement.
Graphical Abstract
INTRODUCTION
This laboratory has been deeply engrossed with studies of enzyme catalysis over a period of almost 50 years. Early investigations were primarily carried out and interpreted within an “accepted framework of enhanced transition state stabilization as the origin of catalysis”, that arises from a multitude of factors that include proximity,1 covalent catalysis,2 and acid, base and cofactor catalysis.3,4 Many large pharmaceutical companies, as well as smaller companies, invested decades of resources to apply knowledge of enzymatic transition state structure toward the development of new drugs. While a few important success stories emerged,5–7 in general the rational design of new drugs fell out of favor, with new drug discovery focused on an initial screening of compound libraries.8–11 At the same time, knowledge of enzymatic transition state structure held out the promise for the design and application of either small biomimetic systems or de novo designed protein catalysts that would be capable of reproducing the huge rate accelerations that characterize enzymes. Once again, moderate success ensued, with rate accelerations in the range to 103–109-fold,12–17 far short of established rate accelerations for enzymes of up to 1026-fold.18,19
If one is lucky as an experimental scientist, junctures occur where observations are made that cannot be fit into existing paradigms. This has occurred several times in our laboratory, with one of the more provocative turn of events occurring during a routine application of physical organic probes to the hydride transfer reaction catalyzed by yeast alcohol dehydrogenase. Seemingly inexplicable at first, the combined use of structure reactivity correlations and kinetic isotope effects led to conflicting conclusions regarding the degree to which the transition state structure resembled the reactant or the product.20 While an ill-conceived effort to reconcile these differences generated the (short-lived) proposal of free radical intermediates for what is a well-established hydride transfer reaction,21 these studies were the first step on the road to demonstrating the prevalent role for quantum hydrogen tunneling in enzyme reactions and its dependence on protein motions (dynamics). Changes in scientific direction are almost always a collective activity and this was certainly true for the demonstration that nuclear tunneling will often predominate for C-H activation at or near room temperature.22–24 In the course of repeated observations (by us and others) of a role for such behavior, new views of enzyme catalysis began to emerge. At the same time, progress in computing power made the application of QM/MM methods increasingly accessible, with an alternate emerging belief that computation alone might be able to solve the question of the source of enzyme catalytic power.25–28
This perspective has two primary goals: The first is to highlight experimental data, some of it quite new, that have forced us to extend our views of enzyme catalysis. Since such a large body of data has accrued over time, only a small subset will be described in detail and then related to emerging models for enzyme catalysis.29–34 The second is to address differences that arise from computational vs. experimental approaches to understanding catalysis, in particular the relevance of computation conclusions to emerging experimental observations as well as the need for computation to provide predictive and testable models of the origins of enzyme catalysis. To address this important and perplexing problem, a primary response from this laboratory has been to strengthen the experimental evidence, with the expectation that data speak for themselves. However, there have been repeated challenges that the outpouring of experimental findings is either irrelevant or wrong and, further, that a failure to respond to the computational results implies that functional inferences from experimental data are not representative of how enzymes work. We address this issue at the end of the current Perspective.
SOYBEAN LIPOXYGENASE AS A MODEL FOR THE CONTRIBUTION OF TUNNELING AND PROTEIN MOTIONS TO C-H ACTIVATION IN ENZYMES
Development of an analytical rate model that accommodates tunneling and incorporates protein motions
Soybean lipoxygenase (SLO) presents an exemplary experimental prototype for exploring the properties of room temperature hydrogen tunneling in biological systems and their relevance to a general theory for the origins of enzymatic rate accelerations. As has become clear from the behavior of SLO, together with that of a family of homologous prokaryotic alcohol dehydrogenases discussed below, a full understanding of enzyme catalysis requires an interrogation of protein structure and, importantly, dynamical properties that can extend far beyond the active site. Although much of enzymology, and de novo protein design, has been primarily focused on the properties of the enzyme active site, the role of the surrounding protein scaffold is increasingly recognized as a major, co-evolving component in the achievement of enormous rate accelerations.35–38
The first and rate limiting chemical step of the SLO reaction, under the condition of ambient O2, involves a net hydrogen atom transfer from the C-11 of the substrate to an active site ferric-hydroxide (formalized as a proton-coupled electron transfer, PCET) process, with transfer of a proton to the iron-bound hydroxide and an electron to the ferric center (Scheme 1).39,40 The initial observation of an enormous hydrogen/deuterium kinetic isotope effect (KIE ~ 80) in the reaction of the wild-type (WT) enzyme23,41 was met with considerable skepticism, stimulating extensive discussion in an effort to establish a theoretical underpinning. In subsequent years, through the use of site specific mutagenesis, trends in the temperature dependence of the KIE have become a major focus of study,42–44 leading ultimately to the currently robust model of “deep-tunneling” under the warm conditions of biology (see below). In the course of this ca. 30-year endeavor, SLO has been central to the development of robust models for enzyme catalyzed tunneling and PCET reactions in general.
Scheme 1.

Reaction mechanism for the peroxidation of long chain polyunsaturated fatty acids by soybean lipoxygenase.40 The first and rate limiting step is the hydrogen atom transfer from substrate to ferric hydroxide cofactor by a PCET process.
The theoretical basis of the tunneling mechanism in SLO has undergone a process of refinement, with the emergence of new data and accompanying adjustments of theory leading to a current consensus regarding the key features of the reaction. Models from this and other laboratories, have emphasized a need for a multidimensional landscape that separates the primary tunneling coordinate from the coordinates that describe the role of the surrounding protein matrix.42,45–47 In a generic model for enzymatic C-H activation, observed rate constants can be formalized in the context of two terms, Eq. 1:
| Eq. 1 |
where Fconf is the fraction of total enzyme that can achieve a subset of catalytically active enzyme-substrate (E•S) substates, multiplied by a rate constant that describes the subsequent H-tunneling process, ktun. The Fconf term is conceptually related to the earlier formulation of near attack conformations (NACs).29 According to the above formalism of Eq.1, kobs is a product of (i) the probability that stochastic sampling among a large number of conformational protein substates will achieve the “active” pre-tunneling E•S complexes (Fconf) and (ii) the barriers that control the tunneling probability within these pre-formed states (ktun). The explicit parameters within ktun have been discussed in numerous published papers and reviews.36,42,45–50
With a focus on the thermally activated reaction barrier that is determined by the protein environment, expressions for ktun can be derived that are related to Marcus formulations for electron tunneling, with the important addition of a term that describes the dependence of the rate of H-tunneling on a thermally averaged donor-acceptor distance (DAD) sampling coordinate, Eq. 2:50
| Eq. 2 |
According to Eq. 2, the is associated with a variety of factors: the electronic coupling between the donor and acceptor, the environmental reorganization energy (λ) and the reaction free energy (ΔG°), the proton vibrational energy levels, and the temperature independent wave function overlap between H-donor and acceptor.36,42,45–50 The P(R) term in Eq. 2 represents a stochastic sampling of a range of DADs, with the dominant tunneling distances representing a trade-off between repulsive interactions as the reacting atoms get close and the increased tunneling efficiencies at reduced DADs.42,48 The greater importance of DAD sampling in nuclear vs. electron tunneling is a direct result of the ca. 2000 greater mass for protium.22 The P(R) in Eq. 2 is similar to DAD sampling terms originally defined by Kuznetsov and Ulstrup45 and refined by Knapp et al.,42 and can be fully described by a force constant for the DAD sampling mode, together with an equilibrium DAD distance, R0. As will be discussed more fully below, R0 is not the initial DAD in resting enzyme, but is achieved via transient sampling within the conformational landscape that constitutes Fconf. In native enzymes, the expectation is that R0 will be significantly shorter than the inter-nuclear distance found in the dominant, thermodynamically most stable E•S structures.49 The size of the observed KIE is determined by both the isotopic sensitivities of hydrogenic wave function overlap and the accompanying DAD sampling contained within ktun.42,45
A unique and particularly insightful property of P(R) emerges when rates for protio- vs. isotopically-labelled substrates are compared over a range of temperatures, yielding the temperature dependence of the KIE, ΔEa or Ea(D)-Ea(H).42,48,49 This simplified parameter is much more straightforward in its interpretation than kobs, due to a cancellation of the majority of (isotopically insensitive) terms. A side by side analysis of the magnitude of the KIE and its ΔEa for mutants of SLO in relation to WT is typically carried out within the theoretical framework of Eq. 2. For the single site mutants most extensively characterized (L546A, L754A, I553X, see Figure 1), the value of the KIE is seen to remain largely unaltered, albeit with varied catalytic efficiencies (Table 1). The signature observation for these SLO variants is the increased temperature dependence of the KIEs compared to the nearly temperature-independence of KIEs in native WT SLO (ΔEa = 0.9 ± 0.2 kcal/mol).42 The parameters R0 and the relative frequencies of the DAD sampling describe this phenomenon quite well. In native SLO, local distance sampling is considered relatively unimportant, due to a precisely aligned geometry of H-donor and acceptor, R0 = 2.85Å, scripted by an active site environment comprised of conserved hydrophobic side chains.49,51 As will be discussed below, this type of “tight” active site geometry is highly dependent on more global protein motions that arise from an equilibrating conformational landscape.
Figure 1.

Structure of SLO and its active site. The two domains are color coded: wheat, N-terminal domain only present in human and plants, and light blue, C-terminal catalytic domain. The reactive portion of the substrate linoleic acid, pale green, has been modeled into the high resolution X-ray structure of SLO (3PZW; 1.4Å); the reactive carbon, C11, is colored black. The side chains discussed in the text are shown as pale yellow sticks. The ferric hydroxide cofactor is displayed as orange and red spheres, respectively. Side chains that ligate the catalytic cofactor are also shown.
Table 1.
Kinetic parameters of SLO and mutants in 0.1 M borate (pH 9) at 30°C
| Enzyme | kcat (s−1) | Ea(H)f (kcal/mol) | Dkcatg | ΔEah (kcal/mol) | R0i | Ωi |
|---|---|---|---|---|---|---|
| WTa | 297 (12) | 2.1 (0.2) | 81 (5) | 0.9 (0.2) | 2.85 | 319.9 |
| I553Vb | 91 (5) | 2.4 (0.5) | 77 (6) | 2.6 (0.5) | 2.96 | 271.0 |
| I553Lb | 273 (10) | 0.4 (0.7) | 81 (3) | 3.4 (0.6) | 3.073 | 243.2 |
| I553Aa | 280 (10) | 1.9 (0.2) | 93 (4) | 4.0 (0.3) | 3.043 | 251.5 |
| I553Gb | 58 (4) | 0.03 (0.04) | 178 (16) | 5.3 (0.7) | 3.317 | 217.9 |
| L546Aa | 4.8 (0.6) | 4.1 (0.4) | 93 (9) | 1.9 (0.6) | -- | -- |
| L754Aa | 0.31 (0.02) | 4.1 (0.3) | 112 (11) | 2.0 (0.5) | -- | -- |
| L546A/L754A (DM)c | 0.025 (0.01) | 9.9 (0.2) | 692 (43) | 0.3 (0.7) | See Figure 2 | |
| L546A/I553Ad | 2.21 (0.09) | 3.8 (0.4) | 128 (3) | 2.8 (0.4) | -- | -- |
| L754A/I553Ad | 0.56 (0.03) | 6.9 (0.2) | 85 (7) | 3.9 (0.5) | -- | -- |
| Y317Le | 267 (8) | 1.0 (0.1) | 68 (2) | 2.3 (0.2) | -- | -- |
The precise active site alignment in WT SLO produces a fairly stiff force constant for the DAD sampling mode, Ω, = 320 cm−1 using a mass of 14 amu for this mode.51 When a packing defect is introduced to the active site, via reduction in bulk of one of the key hydrophobic side chains, two things happen simultaneously: first, the H-donor and acceptor move apart, and second, the increase in the size of the active site cavity decreases the force constant for any subsequent DAD sampling. The latter facilitates an increased participation of a DAD sampling mode and a concomitant ability to recover the H-donor and acceptor distances characteristic of H-tunneling (2.7 Å).49–52 The combination of these features is the origin of the unaltered KIEs in almost every instance. What is remarkable is how readily a vibronically (electron/proton) nonadiabatic treatment of reactant and product proton wave function overlap is able to reproduce the patterns in both the KIEs and their temperature-dependence. Significantly, from the magnitude of Ω for the distance sampling coordinate treated anharmonically (Table 1), we estimate the DAD sampling motion to be fast (nanosecond to picosecond) and relatively local; additionally, all of the protein motions represented in Eq. 1 can be ascribed to stochastic sampling processes that occur on a time scale that is slower and distinct from the virtually instantaneous primary tunneling coordinate.
Extended experimental support of the derived analytical expressions for deep tunneling in SLO
Despite the enormous advances in our ability to demonstrate and describe deep tunneling during enzymatic C-H activation, there has been, until fairly recently, a lack of universal agreement regarding the physical origins of the data collected for SLO. In an effort to “push the envelope” of our understanding, a new variant of SLO was sought that would test the limits of the theory represented by Eqs. 1 and 2. In this context, a mutant form of enzyme was sought, with the goal of examining how far the theory would hold under the condition of an extreme perturbation. A double mutant (DM) of SLO was prepared in which the two key hydrophobic residues that normally confine the positioning of the reactive C-11 of the linoleic acid (LA) substrate were reduced in size: L546A/L754A. An X-ray structure of this variant shows a significantly enlarged active site cavity together with an unaltered backbone conformation and almost identical side chain conformers.53 The kinetic parameters for DM indicated a large, 104-fold, decrease in overall rate of catalysis and a substantially elevated Ea value. The most compelling feature of L546A/L754A emerged when the deuterium KIEs of the double mutant was measured with two independent experimental techniques, leading to a value of 537 ± 55 via pre-steady-state single turnover anaerobic measurement at 35°C53 and an average KIEs value of 661 ± 27 via steady-state measurement at six temperature between 5°C to 30°C.51 Though it was originally thought that a KIE of 80 was outside the bounds of possibility, the new value of 661± 27 appears to set a new standard for the largest room temperature value in condensed phase.
Fitting of this KIE value using the analytical nonadiabatic PCET expression in Eq. 2 provides a ready explanation for the observed behavior. The enormous and nearly temperature independent KIE of L546/L754A results from an elongation of the equilibrium donor acceptor distance for the active substates that occurs concomitant with an unaltered or even increased DAD sampling frequency (Figure 2). This is in contrast to all the active site single mutants (I553X, L754A, L754A)42,44 and alternative double mutants (L546A/I553A, L754A/I553A) that have been characterized,54,55 where mutation increases the temperature dependence of the KIE, corresponding to decreases in Ω (Table 1) and a resulting ability to recover a DAD that resembles WT.
Figure 2.

Vibronic nonadiabatic tunneling model fits of the experimental KIE data for DM-SLO. The experimental data for the temperature dependence of the KIE (black circles with error bars) are shown in b), together with the theoretical curves for different sets of the equilibrium DAD, R0, and DAD sampling frequency, Ω (colored lines). The colors of the lines represent the RMSD (upper bar) calculated as the differences between the theoretically predicted and experimentally measured KIEs for the six experimentally measured temperatures for a given R0 and Ω. The mass of the DAD sampling mode was chosen to be 14 amu as determined in independent molecular dynamics (MD) simulations of WT SLO. The panel a) shows the data for RMSD<150 (colored dots using same color scheme for the RMSD as in b), in relation to all possible fits (grey background) with 500 cm−1 as the upper limit for Ω.51 In c) the Dkcat value at 30 °C is plotted vs the Ea value for WT (black),42 I553X series (green),44 L546A and L754A (blue),42 other double mutant L546A/I553A (orange),55 and double mutant L546A/L754A (red).51 The vertical dashed line represents an extrapolation of the temperature-dependence of WT (Ea = 0.9 kcal/mol). In d) the different patterns that connect the changes in R0 and Ω are shown. A close distance between the H-donor (D) and H-acceptor (A) is expected from the properties of WT SLO. Below and pointing left, the initial increase in R0 (and accompanying decreases in Ω) can be compensated by DAD distance sampling, allowing recovery to WT-like behavior. Below and pointing right, only R0 gets elongated while Ω remains the same or even increases, as in the case of DM-SLO. The tightness of the spring represents the frequency of DAD sampling.
The outlier behavior of L546A/L754A is extremely fortuitous, providing a set of experimental parameters that strongly support the assumptions and predictions of the presented vibronically non-adiabatic model. The behavior of the DM-SLO is also of considerable structural interest, raising the question of the origin of the induced rigidity at the reactive carbon of substrate in relation to the ferric-hydroxide center. Studies under investigation suggest that the rate impairment may reflect an inability of the substrate to position itself properly in relation to catalytically-relevant protein motions.56
Insights into a key role for global conformational sampling, Fconf, during catalysis
One of the initial and foremost indications of the necessity for global conformational sampling in SLO came from the fitting of experimentally available parameters to the ktun term itself; as summarized in Table 1, the DAD distance in WT enzyme is 2.85 Å, prior to any DAD sampling to reach the dominant tunneling distance of 2.7 Å, ca. 0.3 to 0.5 Å shorter than an anticipated van der Waals interactions between the hydrogen donor carbon of substrate and the oxygen acceptor of the reaction center, Scheme 1. Given the deeply buried nature of the active site of SLO, it remained possible that an inherent property of this enzyme was an ability to “electrostatically compress” the DAD within the dominant ground state E-S complexes. While X-ray crystallography would be the “go to” method to resolve such an issue, decades of effort have failed to produce diffractable crystals for SLO in complex with either LA or a substrate analog.57 Fortunately, high-precision electron–nuclear double resonance (ENDOR) spectroscopy has now shown that the WT form of SLO adopts a ground state substrate complex with a DAD approximating van der Waals expectations;57 this finding provides strong support for the central role of transient conformational sampling among multiple protein substates as a means to reach the short DAD distances that are a prerequisite for efficient hydrogen tunneling.
ENDOR, typically regarded as “EPR-detected NMR”, has emerged as a central tool for determining the coordination environment at active sites of metallo-enzyme reaction intermediates.58,59 However, it has also shown promise to examine the active site architecture of substrate-enzyme complexes when the substrate is not coordinated to the metal.60 In the case of SLO, the canonical catalytic metal center (iron) was replaced with a S=5/2 Mn2+ probe surrogate to access suitable electronic spin properties; with this high spin Mn2+ ion, whose spin density was seen to behave effectively as a point dipole, the ENDOR responses at the outer transitions, ms = −5/2 to −3/2, provide frequency shifts that are nearly twice as large as that from the ms = +1/2 to −1/2 spin manifold. This property makes the ENDOR analysis of the E•S complex in SLO feasible out to (at least) 6 Å from the metal center.58,59
The intrinsic hyperfine interaction between the metal and carbon nucleus is dependent not only on internuclear distance (r), but is also sensitive to the orientation of the nuclear spin relative to the principal ZFS axis from the meal. By referencing the orientation of this spin axis with respect to the orientation of the metal bound water protons in the active site (through a combination of deuterium exchange and 1H ENDOR), the positions of specific 13C-labelled positions of substrate (either C10 or C11) could be determined in three dimensional space within the SLO active site. The ground state DAD (between the reactive C11 and the metal-bound oxygen) could, then, be calculated using the law of cosines (cf. Figure 3). Two dominant ground state conformations are indicated in SLO, with the closer of the observable Mn-C11 couplings approximating van der Waals contact for the DAD, Req. Similar analysis of an alternate Mn-C11 interaction places C11 at an even longer distance from the metal bound oxygen. The use of a Mn2+-substituted enzyme for such studies is supported by the high structural similarity between native enzyme and Mn-SLO. Importantly, these results validate that tunneling ready distances are not the result of an unusual shortening of the DAD in the ground state structure of the SLO E•S complex.56
Figure 3.
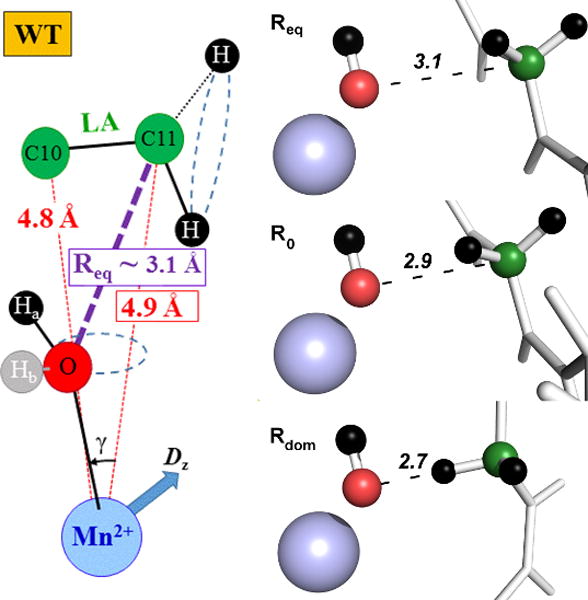
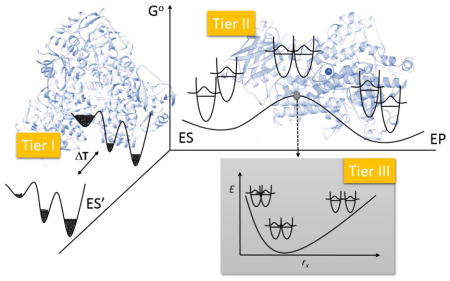
Model for active-site ground-state structure of WT SLO ES complex. a) The distances represented by the red dotted lines are ENDOR derived distances; the Mn-O distance is obtained from the X-ray crystal structure, (2.23 Å). The angle, γ = 26°, was calculated from MD simulations using ENDOR derived distances as restraints. The position of 1Hb was not well established and it is arbitrarily pictured (in grey), for completeness. The purple dashed line represents ground state equilibrium DAD distance (Req) calculated trigonometrically. The uncertainty of the C11-O distances was estimated as ± 0.2 Å. Dashed blue circles suggest the oxygen nucleus lies out of the x-z plane of the ZFS axes.57 b) A physical picture for the tiers of distances relevant for bringing the DAD from Req to Rdom, where 2.7Å represents the dominant distance for efficient hydrogen transfer by tunneling. The numbers in italics refer to the DAD in Angstroms. A physical picture of protein motions linked to the conversion of Req to Rdom can be found in Figure 13.
From the detailed kinetic parameters in Table 1, together with these critical ENDOR measurements, a physical picture for C-H activation begins to emerge, in which multiple tiers of distances progressively bring the donor and acceptor closer to a geometry that is sufficiently compacted to enable efficient wave function overlap (Figure 3). The first tier of distance, designated Req, represents the dominant enzyme-substrate ground state complex, which is typically characterized by van der Waals interactions of 3.2 to 3.5Å and, most generally, is observed by X-ray crystallography. The second tier of distance, R0, arises from the time-dependent, thermal sampling of a large family of higher energy protein substates (i.e. the protein’s conformational landscape), to achieve a subset of configurations that are optimal for catalysis. The transition from Req to R0 is expected to take place over a hierarchy of time scales and may involve large and distal parts of the protein; as noted, it is also considered one of the most challenging processes to link experimentally to the chemical bond breaking/making steps of catalysis. The last distance tier represents the tunneling distance, Rdom, at which the DAD hydrogenic wave function overlap becomes dominant. The transition from R0 to Rdom represents the fine tuning of active site geometries through local DAD sampling modes contained within the formalism of ktun, Eq. 2.
One additional feature relevant to the key contribution of Fconf to the rate expression in Eq. 2 is that computations of rate constants for H-tunneling (rather than isotopic rate ratios) uniformly lead to explicit values for ktun that are many orders of magnitude higher than observed rate constants.49 Agreement between measured and computed rate constants becomes possible upon inclusion of Fconf, a key term that reflects the very small but finite probability that motions within a protein conformational landscape will transiently access the subset of protein configurations that are competent to proceed along the reaction coordinate to product.
Biophysical probes that address the role of Fconf in C-H activation
Through the above described relationship between experimentally determined temperature dependences of KIEs and the theoretical modeling of a DAD distance sampling term in ktun, a transparent understanding of the role of local protein sampling in the catalysis of C-H activation has emerged. By contrast, obtaining compelling experimental evidence for the accompanying role of the fluctuating global protein conformational landscape in catalysis has presented a more formidable challenge.
A major direction within this laboratory has been first, to establish the impact of protein perturbations on a primary chemical step in each enzyme system (as illustrated in Table 1, via the targeting of bulky hydrophobic side chains in SLO) and second, to examine if there is a demonstrable impact of these mutations on select biophysical tools that have the potential to sense subtle changes in the protein conformational landscape. While the available tool box for such biophysical studies is large and growing, it is important to “find a good match” between the system under study and the appropriate biophysical protocol(s). In the case of SLO, its large size and dependence on an active site paramagnetic metal ion introduce significant challenges to potential NMR studies, while the site-selective analysis of the intrinsic protein fluorescence is precluded by the very large number of Trp residues distributed throughout the protein. Despite these limitations for SLO, quite a few tools have emerged as suitable and informative that include phylogenetic comparisons, high pressure kinetic analyses, room temperature X-ray structural analyses and hydrogen deuterium exchange.
Lipoxygenases are found to be widely distributed among plants, fungi, animals and more recently bacteria.61 These enzymes all display a highly conserved α-helical catalytic domain structure at their C-termini, coupled to low primary sequence homology.61,62 Of note, plant lipoxygenases such as SLO are ca. 20% larger than other members of this family, with this increase in size mapping to the addition of several new loops on the protein surface.62,63 Our recent investigation of a bacterial lipoxygenase from cynobaterial Nostoc sp (NspLOX) and its comparison to SLO emphasizes these differences.64 As anticipated, while the catalytic domains of NspLOX and SLO appear similar, there is only a 22% sequence identity and 40% similarity within the core structure. Their most striking differences can be seen from an overlay of the two structures, Figure 4, which highlights the enlarged loops in selected regions of the SLO surface. The impact of the loss of the surface loops in NspLOX is manifested by a modest rate reduction (37s−1 vs 300 s−1 for SLO at 30°C) and a very significantly elevated Arrhenius energy of activation with H-LA (Ea(H) = 12.4 ± 0.3 kcal/mol) that is accompanied by a similarly elevated Arrhenius prefactor (AH = (2.7± 1.5) × 1010 s−1).64 Notably, lipoxygenases from other sources lacking the surface loops in SLO - human reticulocyte 15-lipoxygnease (15-hLOX)65 and fungi (manganese lipoxygenase, Mn-LOX) 66-present considerably higher enthalpic barriers for C-H activation (ca. 8–11 kcal/mol) compared to SLO-1 (2 kcal/mol, Table 1). While loops have been increasingly recognized as playing important roles in many facets of protein function, these have been primarily focused on steps that control substrate binding and/or product release.67,68 In the case of lipoxygenase, the observed variability in loop structure and activation energies suggest a distinctly different function, in which flexible loops act to facilitate a transmission of thermal energy from the solvent to the enzyme active site. Even though the three lipoxygenase examples represent a small subset from the LOX family and could be a consequence of multiple features, the trends are remarkably similar to prior results reported for a family of prokaryotic alcohol dehydrogenases. In the earlier comparisons of enzymes from thermophilic, mesophilic and psychrophilic sources, perturbations away from optimal catalysis, via changes in temperature and/or use of site specific mutagenesis regularly produced a concomitant elevation of both the energies of activation and Arrhenius prefactors, ascribed to an induced perturbation of the conformational landscape that is dependent on distinct protein-solvent interfaces (pursued in further detail within the section on prokaryotic alcohol dehydrogenases below). 31,69–78
Figure 4.

Structural overlay of SLO (gray) and NspLOX (wheat).64 The SLO structure is from the high resolution X-ray model (1.4 Å) and the NspLOX model was generated from threading (Phyre) to a coral 8R-lipoxygenase. The loops unique to each structure are color coded: forest, SLO and navy blue, NspLOX.
As a first step toward a clear experimental distinction between catalytically relevant global vs. local motions in SLO, we initiated a study of the combined impact of pressure and temperature on the full set of kinetic parameters.79 The pressure dependence of activity with H-LA and D-LA was measured between 1 and 1034 bar at five temperature (bracketing 15°C to 35°C) using WT SLO and two single mutants, I553V and L546A. While the mutations of choice at position 553 were I553G and I553A, the significant reduction in the size of the side chain at this position led to rapid protein denaturation with elevated pressure and a compromise was reached with I553V that showed pressure stability similar to WT. Additionally, L754A and L546A/L754A were investigated, but with a focus on the H-LA substrate alone, since their excessively slow turnover rates with D-LA were incompatible with the available instrumentation. Prior kinetic and/or structural analyses of each of the single site mutations have shown interior packing defects that both increase DADs and active site flexibility.42,44
The observed impact of high pressure on rates and KIEs can be differentiated from the impact of elevated pressure on the enthalpic barriers for catalysis.79 With regard to rate and KIE, I553V and L546A display pressure-induced trends in kcat similar to WT: in these cases, the rate constants for H-LA and D-LA both increase with pressure (ca. 1–2 fold), leading to an almost constant Dkcat. The relatively small increases in rate constant are attributed to a small protein compression that becomes somewhat more pronounced for variants that contain L754A. Although these properties could arise from small changes in DADs, this is unlikely to be the only impact, since any significant decrease in DAD coordinate is also expected to significantly decrease the magnitude of the KIE. An accompanying explanation is that the observed effects of high pressure on rate are due to an aggregate of small changes in orientation and distance for protein side chains within the active site.
The more compelling feature of SLO that emerges from these studies is the high sensitivity of the Ea to elevated pressure for WT, I553V and L546A, in marked contrast to unaltered KIEs and their temperature dependence as reflected in the ΔEa values. The magnitude of Ea is expected to be influenced by many factors that appear in ktun (reorganization energy λ, reaction driving force ΔG°, and the DAD sampling frequency), together with the more global conformational landscape represented by Fconf. However, the finding that pressure effects on KIEs and ΔEa in WT and two other single mutants are minimal implies that local changes are likely small, and that the elevated Ea values with increasing pressure are primarily reflective of perturbations to a more global conformational landscape that is linked to changes at the protein/solvent interface.80 This fortuitous ability to distinguish functionally-linked global from local motions in SLO by means of high pressure may be due to the protein’s densely packed hydrophobic core and buried active site that are in communication with less structured loops on the protein surface (see below).
Surprisingly, the pressure effect on the conformational landscape trends become less pronounced or even eliminated for the mutants that contain L754A.51,79 The Ea value of L754A shows a very small increase under high pressure, while the DM-SLO displays an unaltered EaH value of ca. ~8 kcal/mol under all conditions (Figure 5). These trends in EaH value imply a pressure-resistant and more rigid region of the global conformational landscape compared to WT, L546A and I553V. We note that L546 and L754 are highly conserved throughout the very large lipoxygenase family62 and the proximity and kinetic properties of these two side chains with regard to the reactive carbon of LA has highlighted their importance in positioning the substrate for reaction. However, the distinctly different response of L546A and L754A to the compressive effects of high pressure suggests that they may play different roles in catalysis. With this distinction in mind, we proceeded towards a spatially resolved analysis of the global protein motions in SLO that could be linked to functionally relevant sites of mutation.
Figure 5.

The effect of elevated pressure on the empirical energy of activation (EaH) of kcat-H for WT, I553V, L546A, L754A, and L546A/L754A.79
Hydrogen deuterium exchange implicates an anisotropic network of thermal activation for C-H cleavage in SLO
Our initial quest was centered on detecting perturbations to conformational motions that arise from the site-specific substitution of active site residues. Two thermally averaged techniques were exploited to spatially resolve potential dynamical protein networks: X-ray crystallography using qFit analysis and hydrogen deuterium exchange mass spectrometry (HDXMS).81 We first carried out qFit analysis on the WT SLO crystal structure recorded at room temperature. qFit is a multi-conformer measure of constrained rotamer occupancies within an X-ray derived protein model; thus, it reveals otherwise hidden alternate conformations of the side chain and/or backbone.82,83 qFit analysis of the high resolution (1.7 Å) structure of WT SLO resulted in numerous alternate conformers including several active site side chains (e.g. L546, I553, and L754; see Figure 6). Many of these active site residues with alternate conformers were also determined in structures solved at cryogenic temperatures.44,53 From these structures, no significant temperature transition was isolated for residues with alternate side chains in SLO, as opposed to recent examples from other systems.84 Further, qFit analysis was extended to the active site mutant, I553G, whose kinetic parameters show an impact on the thermal barriers for active site catalysis (Table 1); in this structure, no significant deviations in side chain conformations could be discerned from WT. In fact, the only notable change in the electron density of the entire molecule was the loss of the side chain bulk at position I553 which caused the active site cavity to nearly double in volume. Thus, room temperature X-ray crystallography and qFit have not thus far provided a molecular link between networks of side chains and catalytic processes in SLO.
Figure 6.

Room temperature X-ray structure of SLO showing active site residues that occupy alternative side chain conformations.81 The catalytic iron is represented by the dark gray sphere; carbons for all metal ligand side chains are shown in pale yellow. Side chains with alternate conformers are colored in salmon; L546 lies behind I553.
With the current inability to uncover a structural network for catalytically relevant thermal motions from these X-ray tools, we turned to HDXMS.81 Time, temperature and mutation dependent HDXMS experiments were conducted for SLO under solution conditions in which EX-2 exchange behavior was both anticipated and demonstrated from the isotopic distribution of the mass spectrum envelope. The HDX process involves chemical exchange of peptidyl backbone amide hydrogens for deuterons when the target protein is incubated in D2O; subsequent proteolysis under lowered pH/pD to minimize back exchange generates hundreds of SLO peptides, enabling resolution of the exchange process in a spatially defined manner.85–87 Within the EX-2 regime, measured rate constants (kHDX) can be represented as the product of the intrinsic chemical exchange rate (kint) and the equilibrium constant for the interconversion between closed (solvent unexchangable) and open (solvent exchangeable) protein states (Kop = kopen/kclose). In this manner, EX-2 exchange provides a temperature dependent examination of backbone fluctuations that may be thermodynamically inaccessible to X-ray structural analysis.85–87 According to the EX-2 formulation, the major determinant for differences in observed HDX rate constants arises from the ability of the protein to transiently sample protein conformations that allow access of solvent for subsequent isotopic exchange. There are two distinctions between this process and catalysis: the first is their time constants, in which HDX experiments are generally interrogated on much longer times scales (seconds to hours) than catalysis (milliseconds); and the second is the degree of protein excursions which are expected to be more extensive in the case of HDX. Despite these distinctions, there is the underlying assumption that regional differences in protein flexibility that emerge from HDX studies will, in selected instances, be correlated with catalysis-dependent motions.
As in previous applications of HDXMS to enzymes, SLO samples in the absence of substrate were chosen for investigation, since binding of substrate was expected to lead to protection and a diminished sensitivity to HDX;88–90 For the WT SLO protein, proteolysis leads to 300–500 overlapping peptides. To reduce the complexity of data analyses as a function of both time and temperature, a family of 46 non-overlapping peptides, representing 89% of the total sequence within SLO’s catalytic core, were chosen as the primary focus. The temperature dependent analysis was further simplified via the introduction of pattern recognition, leading to the definition of four primary classes of HDXMS behavior. Pattern recognition was an essential development as not all of the peptides undergo an exchange process (or are too fast) within the dynamic range under the employed conditions, precluding quantitative analysis of the entire SLO sequence. Mapping these four defined classes onto the catalytic domain of SLO highlights the non-uniform effect of temperature on HDXMS. The more flexible regions of the protein, which exhibit rapid, temperature independent, exchange behavior, are concentrated along the outskirts and solvated part of the molecule. Solvent inaccessible peptides are primarily restricted to the buried, core peptides of the protein. The remaining central swath, approx. 45% of the SLO catalytic domain, defines the family of peptides that display significant and measurable temperature dependent exchange behavior in a manner that highlights the anisotropic response of peptidyl flexibility to temperature.81
While it is very difficult (and perhaps not possible) to deduce the relevance of observations of protein motions/flexibility to catalytic bond cleavage by examining a single protein variant (i.e. WT), the introduction of site-specific mutagenesis allows us to pinpoint which regions of protein flexibility are specifically altered by key mutants with defined impacts on the rate limiting C-H bond cleavage. Our first analysis of structure-function relationships via HDXMS in SLO involved a comparison of the series of mutations at position 553 at a single temperature (30 °C); as discussed above, these were previously shown to give rise to trends in the temperature dependence of the KIE that are attributed to increased active site flexibility. Analysis of the behavior of the full suite of 46 peptides in response to the bulk of the side chain at position 553 led to the identification of a very small set (414–423, 541–554, and 555–565) of peptides with this property. These peptides, which are within the active site region and close to the site of mutation, show a clear pattern in which the extent of exchange correlates (Figure 7) with previously measured values for ΔEa = Ea(D)-Ea(H). Although the active site of SLO is sequestered from solvent, there was the possibility that increased solvent accessibility could be the source of the observed HDX behavior with I553X. However, a high resolution, room temperature X-ray analysis of I553G, while producing the largest increase in active site volume, failed to indicate evidence for the presence of any additional water molecules. While such a result could be due to the presence of undetectable, disordered water, we note the ability to visualize additional bound water molecules in a range of other active site SLO mutants. 53,56
Figure 7.

Three active site peptides in SLO (yellow) exhibit mutation-induced increases in the percent deuterium exchange at 4 h.81 In (a), two key active site residues are shown: L546 (red) and I553 (blue). In (b), the percent HDX at 4h and 30˚C for the representative peptide, 555–565, is plotted as a correlation to the kinetically determined ΔEa (see Table 1).
The trends represented in Figure 7 thus provide biophysical evidence for the earlier kinetic models that had invoked increases in DAD sampling following side chain substitutions at I553.81 We next proceeded to extend the analysis of the weighted average rate constants characterizing the HDX of WT as a function of temperature (Figure 8) to the mutants, L546A and I553G. Once again, a thorough examination of the behavior of all peptides yielded only two representatives that display statistically different behavior from WT. These two peptides map to a completely different, solvent exposed region of SLO from those that are seen to undergo increased extent of exchange at 30 °C, Figure 8. The remarkable aspect of this new analysis was the emergence of a correlation between the enthalpy of activation describing active site C-H activation and that describing the rate of HDX within a single solvent exposed loop, 15–30 Å from the catalytic metal, the first of its kind. This mirroring of enthalpic barriers for HDX and catalysis further suggested a network of structural communication between the surface and active site that is strongly supported from the results of site specific mutagenesis studies (Y317L) at a strategically placed tyrosine residue buried between the loop and the substrate binding site (Table 1). These key observations have many implications for the evolution of optimal enzyme catalysis, highlighting how a small region of SLO may influence the thermal activation of catalysis via the thermally induced fluctuation of a single remote loop that resides at the protein/water interface.81
Figure 8.

Correlated activation energies for HDXMS (317–334) and kcat in SLO.81 a) The Arrhenius-like plots of the weighted average exchange rates constants, ln(kHDX(avg)), for peptide 317–334, are compared for WT, I553G and L546A. b) The correlation between trend in Ea[kHDX(avg)] and Ea[kcat] (Table 1). c): Peptide 317–334, which exhibits mutant variable Ea[kHDX(avg)], is colored orange. The residues predicted to be involved in the pathway of communication from this loop to the active site are shown in green spheres; L546 and I553 side chains are also included in this pathway and colored red and blue, respectively. The catalytic iron is shown as a dark gray sphere. A hydrogen bonded interaction between S749 and Y317 is colored in salmon.
GENERALIZATIONS BEYOND SLO: C-H ACTIVATION WITHIN A CONSERVED FAMILY OF PROKARYOTIC ALCOHOL DEHYDROGENASES
An ongoing tenet of enzymology is the search for general physical principles that will underlie all of catalysis. At this juncture, the reader may begin to question the extent to which the observations and concepts that have emerged for SLO are specific to this enzyme class. The data from this laboratory show that this is certainly not the case, with similar patterns of behavior seen for proteins that are characterized by completely different tertiary and quaternary structures and perform distinctive chemical reactions, dependent on a range of alternate cofactors. In this section, we summarize decades of work on a family of prokaryotic alcohol dehydrogenases characterized from thermophilic (ht-ADH), mesophilic (ms-ADH) and psychrophilic origins (ps-ADH),72 highlighting features that are similar to SLO and include (i) a role for global conformational excursions in the achievement of active site compaction, (ii) the contribution of both local DAD distance sampling modes and more global conformational sampling to reach catalytically competent active sites, and (iii) the importance of spatially resolved, dynamical networks that link solvent/protein interfaces to each domain within the active site.
Exploiting the temperature break in behavior for ht-ADH
Unlike the classical monomer structure of SLO, these ADHs are homo-tetramers (monomer size of 37 kDa for ht-ADH), with one Zn2+-containing active site and one structural Zn2+ in each subunit.91 Additionally, each subunit is comprised of multiple domains that bind the nicotinamide cofactor and substrate, respectively, with the active site zinc ion residing between these two domains. The reaction catalyzed by these proteins, illustrated in Scheme 2, is the transfer of a hydride ion between substrate and cofactor with concomitant proton removal or addition to substrate depending on the direction of the reaction. Studies of a related ADH from yeast in fact presented some of the initial compelling data for room temperature hydrogen tunneling in enzyme reactions and, further, showed the important role of active site hydrophobic side chains in facilitating the close DAD approach needed for optimal hydrogenic wave function overlap during catalysis.92
Scheme 2.

Generic reaction mechanism for the reversible oxidation of a primary alcohol by Zn(II)-dependent alcohol dehydrogenases. Proton transfer to a general base (B) accompanies the hydride transfer to NAD+.
However, the prokaryotic family of ADHs that functions in different temperature niches has provided the additional advantage of being sensitive to perturbations in temperature as well as presenting the ability to perform comparative biochemical analyses. The most studied member of the prokaryotic ADH family is the thermophilic enzyme, ht-ADH. This isozyme has been shown to undergo a break in kinetic behavior at 30 °C, in which the enthalpic barrier for catalysis and the temperature dependence of the KIE go in the same direction (Figure 9a).69 Specifically, ht-ADH shows a smaller Ea at the elevated temperatures of its function and this is accompanied by a temperature independent KIE; a decrease in temperature below a breakpoint of ca. 30 °C displays an alternate pattern in which the value of Ea rises and the KIE becomes temperature dependent. On the premise that smaller Ea values correspond to more flexible barriers for protein conformational sampling, this represents one of the earliest demonstrations of a correlation between global protein flexibility and active site compaction. Subsequent HDXMS measurements on ht-ADH complement these kinetic findings beautifully, showing a similar temperature break (at ca. 30 °C) in the weighted average rate constants for HDX,71 Figure 9b and c. Of considerable interest and importance regarding the anisotropic dynamical behavior of proteins, the five peptides that indicate a break in the rate constants for HDXMS are localized at the substrate binding domain of protein in a manner that radiates out from the active site toward two distinct protein/solvent interfaces (Figure 9c and Figure 11). Analogous to the studies of SLO, these HDX studies were carried out on enzyme free of cofactor and substrate, to increase the sensitivity of measurements to intrinsic protein flexibility, while the kinetics of substrate oxidation were accompanied by careful controls to show that hydrogen transfer is rate limiting under the conditions of the measurements. The aggregate data for ht-ADH support a model in which, above 30 °C, the protein is intrinsically flexible and capable of efficient conformational sampling to achieve compacted, catalytically activated active site geometries; this abrogates the need for significant DAD sampling, in analogy to the behavior for native SLO. Below 30 °C (the non-physiological condition), new protein conformations are introduced that are accompanied by active site configurations that are either less catalytically efficient (or inactive); this feature can be overcome to some extent by enhanced DAD sampling that increases the probability of effective wave function overlap.31
Figure 9.

Functionally relevant temperature dependent breaks in behavior for ht-ADH. In (a), illustration of the kinetic break at 30 ˚C for both the kcat and the KIE (i.e. Dkcat). The KIE is almost temperature independent above 30 ˚C (red) and transitions to a more temperature dependent KIE behavior below 30 ˚C (blue) with a concomitant increase in Ea for kcat.69 In (b), HDXMS analysis revealed 5 peptides (1–4, 7) that show an analogous temperature dependent transition in k[HX(WA)].71 In (c), these five peptides are numbered, mapped onto the ht-ADH structure and colored as red, orange, and magenta. The active-site Zn2+ is shown as a yellow sphere, adjacent to peptide 2, and the cofactor NAD+ is represented as yellow sticks.
Figure 11.

Defined interfaces for transfer of thermal energy from solvated subunit-subunit interfaces to the active site in ht-ADH.78
In another analogy to studies of SLO, site directed mutagenesis of hydrophobic active site residues has extended our understanding of the physical features that accompany the break at 30 °C and, by extension, the molecular basis for dynamically achieved active site compaction. Focusing on V260 that resides behind the nicotinamide ring within the cofactor binding domain (Figure 10a), it was shown that a step-wise reduction in size of this side chain leads to a systematic increase in the temperature dependence of the KIE that is reflective of the increased requirement for DAD sampling under physiological conditions.74 However, combining the same series of mutants at V260 with a reduction in temperature reverses the trend, enhancing the kinetic break at 30 °C to produce an Arrhenius prefactor of 1024 s−1 for the most impaired variant (Figure 10b).73 The enormous inflation of AH beyond a semiclassical limit of 1013 s−1 is attributed to a reversible trapping of ht-ADH into inactive enthalpic wells and the accompanying requirement for additional thermal excitation to reach the catalytically relevant protein substates. At the same time, the magnitude of ΔEa is found to decrease in value relative to WT, indicating that the combined impairments of low temperature and active site mutation result in an active site that is no longer capable of recovery to the most tunneling-efficient DADs. This is reminiscent of the active site rigidification seen with the double mutation, L546A/L754A in SLO, though in the case of ht-ADH the KIE has not undergone the huge increase characteristic of DM-SLO. This distinction raises the issue of the origin of the large differences in the absolute magnitude of KIEs in hydride vs. hydrogen atom transfer reactions. While our understanding of SLO has benefited tremendously from the development of a vibronically nonadiabatic analytical analysis of hydrogen tunneling, an analogous and predictive analytical formulation is still lacking for adiabatic processes that include the hydride transfer reaction described herein as well as hydride transfers in general.93–95 Despite this limitation, it is remarkable that similar trends in behavior are observed in both systems, highlighting the need for multidimensional formalisms that separate the coordinate controlling the hydrogenic wave function overlap from the coordinates that describe protein motions.
Figure 10.
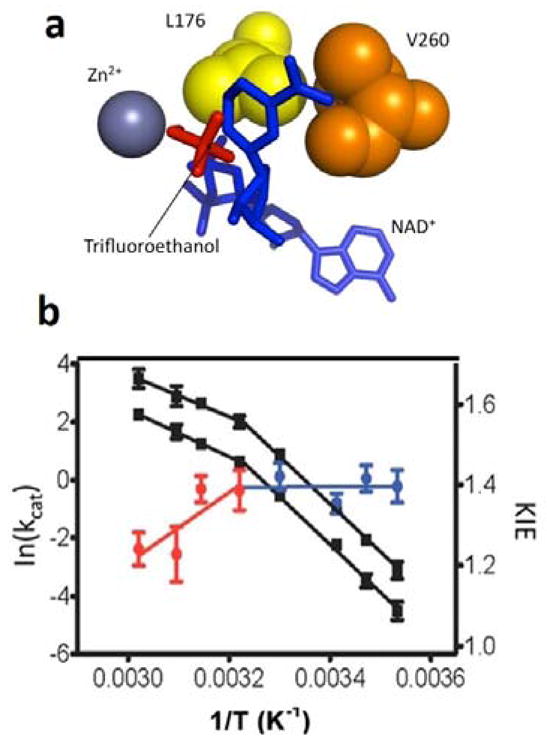
Impact of mutation at V260 in ht-ADH.74 Panel (a) displays a focused view of active-site hydrophobic side chains (Leu176 and Val260) that sit behind the nicotinamide ring of cofactor. Upon mutation of V260 to alanine (b), the break in kinetic at 30 °C in the WT ht-ADH is enhanced and the active site has been rigidified, as illustrated by the reversal of the temperature dependence of the KIE, above and below 30°C from that of WT (Figure 9a).
Fluorescence probes of ht-ADH extend our understanding of long range dynamical communication between a protein/solvent interface and bound substrate
While HDXMS experiments of ht-ADH, Figure 9, have defined a spatially resolved region at the substrate binding pocket that undergoes the same temperature dependent change in properties as the kinetic parameters, fluorescence studies have allowed considerable refinement and extension of this phenomenon. Structural comparisons between the psychrophilic and thermophilic prokaryotic ADHs indicate a ca. 60% sequence identity and virtually identical three dimensional structures.89 With regard to quaternary structure, the four subunits of these proteins are arranged via the dimerization of a pair of dimers. Consequently, two distinctive interfaces are formed, referred herein as either homologous (Interface I) or heterologous (Interface II), Figure 11. During the course of our search for structural differences that might explain the respective adaptation of ht-ADH and ps-ADH to different temperature niches, we identified a side chain in ht-ADH at the Interface I that undergoes pi stacking with itself, Y25-Y25. Of great interest, this side chain not only resides in the region of ht-ADH that shows a temperature break in HDX behavior, Figure 9c, but has also undergone mutation to Ala in the ps-ADH isozyme. Reasoning that a subunit interaction at a dimer interface could provide a means of communication from the protein surface to the active site, we prepared the Y25A variant of ht-ADH and characterized its kinetic and fluorescent properties.
The first key observation was an abolishment of the kinetic break observed in the native enzyme, Figure 9a.75 Of considerable benefit with regard to experimental design, ht-ADH contains only three Trp residues, facilitating the preparation of single Trp variants, Figure 12a. Using either the active site W87in near the substrate binding pocket or W165in near the surface as a control, ns to ps fluorescence lifetime measurements show a break in behavior for W87in at 30 °C; this behavior is absent for W165in77 and significantly is lost in the Y25A variant (Figure 12c, d).78 Additionally, by exacerbating the temperature transition through incorporation of V260A (see Figure 10 and accompanying text), Stokes shifts of the active site W87in also reveal two distinct conformations for ht-ADH above and below 30 °C (Figure 12b).78 These important findings show that both ns-ps time dependent fluorescent measurements and thermally averaged HDX experiments with ht-ADH mirror the kinetic behaviors of the C-H activation step, supporting a range of temporally and spatially distinctive motions in the tuning of active site reactivity.
Figure 12.

Key results from the time-resolved fluorescence studies on ht-ADH. For these studies, ht-ADH constructs were designed with one or two of the three native tryptophans substituted for phenylalanine side chains.77,78 Panel (a) shows the spatial location of these Trp residues (green sticks) with respect to the active site V260 (red) and Zn2+ (dark gray sphere). The NAD+ cofactor is represented in cyan sticks. Y25 at the subunit “Interface I” is shown for reference. Panel (b) presents the temperature dependent Stokes shifts (ps-ns timescale) that reveal two time constants, consistent with two conformations, for W87 fluorescence when the active site valine was mutated to alanine (V260A). Panels (c) and (d) show the temperature dependence of the fluorescence lifetime decay components for W87 (within the substrate binding domain) in WT and Y25A, correspondingly. A noticeable break at 30 °C is seen in the sub-nanosecond life time (black) in WT, but not for Y25A. Blue and red lines represent two longer, temperature independent components.
Temperature jump fluorescence and FRET identify a second long range communication pathway within the cofactor binding domain of ht-ADH
As discussed above, the time constants captured by fluorescent emission/ Stokes shifts measurements and HDX are operational at the extreme ends of the full time scale expected to describe protein motions that contribute to catalysis. Given the important role anticipated for microsecond motions in conformational sampling, we next turned our attention to the temperature-jump perturbations, first by examining the intrinsic Trp fluorescence of ht-ADH78 and later via FRET experiments.96 In the initial pump-probe exploration of Trp fluorescence, a short nanosecond pulse from an infrared laser was applied to a flowing ht-ADH sample (to avoid protein denaturation) generating a ca. 7 °C jump within the volume of the incident laser pulse. Electronic excitation of tryptophan was achieved with an additional laser pulse centered at 295 nm and resulted in time dependent emission intensities monitored near the peak emission line of tryptophan (335 nm). These experiments were pursued using a construct W87F, such that measured spectra reflect a combination of emission from W165 and W49. However, given the demonstrated insensitivity of W165 fluorescence to changes in either temperature or site specific mutagenesis, it was possible to attribute the observed behaviors to W49.78 This side chain is located within the cofactor binding domain and close to Interface II where it undergoes pi-stacking with F272 on the adjacent subunit (cf. Figure 11).
As anticipated, there was an observable decrease in the fluorescence emission from W49 upon the increase in temperature. Subsequently, however, there was also an unexpected and rapid recovery of fluorescence back to the initial intensity. Since this recovery process occurs on a time scale much faster than the overall millisecond cooling of the protein, an intra-protein heat transfer from the solvent accessible dimer Interface II to the interior of the protein was proposed.78 The behavior of W87 has been recently extended by examining the rate and temperature dependence of the transfer of W49 fluorescence emission to the nicotinamide ring of bound NADH in the presence of isobutyramide as a substrate analog. The FRET process is found to involve two microsecond relaxations, the faster of which essentially disappears below 30°C, to produce a break at the same temperature observed in the kcat for ht-ADH.96 Additionally, the energy of activation for the fast FRET rate constant above 30 °C and kcat are almost identical, providing a real-time link between a microsecond component of conformational sampling and active site tunneling. These fluorescence studies of W49 provide strong support for a second network that begins at Interface II where W89 resides, and interacts with the bound cofactor binding site over a distance of ≥ 8Å. Thus, two networks have been detected in the oligomeric, multi-domain ht-ADH, leading to the proposal of a role for separate solvent exposed dimer interfaces in modulating the positioning of substrate and cofactor in relation to one another.
DIFFERENT PROTEINS, DIFFERENT NETWORKS, BUT SIMILAR PHENOMENA
Herein we present a pictorial representation of the complexity and multidimensionality of hydrogen transfer reactions that have emerged from studies of SLO and ht-ADH (Figure 13). The combined experimental and theoretical work posits that stochastic conformational sampling is required to reduce the average DAD within thermodynamically stable, ground state configurations defined by van der Waals interactions to precisely positioned active sites with optimized geometries and electrostatics conducive to efficient wave function overlap. This protein conformational coordinate is orthogonal (not directly linked) to that for the reaction coordinate along which C-H cleavage is catalyzed (Figure 13a).36,51,73 Figure 13b illustrates an equilibration among the sets of inactive (or poorly active) and active enzyme-substrate substates, associated with Fconf in Eq. 1, while the Figure 13c–e illustrates the coordinates contained within modified Marcus models for hydrogen transfer, associated with ktun in Eq. 1.36 Both Fconf and ktun are temperature dependent, whereas only ktun is sensitive to isotopic substitution of the substrate, i.e., the impact of isotope substitution in the substrate(s) on the global conformational sampling of the protein-substrate system is expected to be small or negligible. For the reaction coordinate of hydrogen transfer, heavy atom preorganization first brings the system to the tunneling-ready state (middle graphs in Figure 13c and 13d), in which the hydrogenic potential surfaces are transiently isoenergetic, a prerequisite for tunneling between reactant and product in accord with the Franck-Condon principle. Further heavy-atom reorganization breaks the transient degeneracy and traps the hydrogen in the product well (bottom graphs in Figure 13c and 13d). The rate of reaching the crossing point depends on the reorganization energy (λ) and driving force (ΔG°), and is largely isotope independent. The wave function overlap itself decreases exponentially with increasing mass, a consequence of the much smaller overlap region for deuterium compared to the protium. The wave function overlap can be enhanced by the DAD sampling along the DAD coordinate once the tunneling ready state (R0) is reached (Figure 13e), as associated with P(R) in Eq 2. The DAD sampling is small or negligible in native forms of SLO and ht-ADH due to the highly precise alignment/compaction at the active site, but can become quite significant when a packing defect is introduced within/near the active site (often through site directed mutagenesis). Note that the tunneling of the particles is essentially instantaneous in the context of these other heavy-atom motions.
Figure 13.

A general multidimensional model for hydrogen transfer in SLO and ht-ADH.31,36,51 (a) Schematic representation of a free-energy landscape for the enzyme-substrate complex, that takes into account both the reaction coordinate for H-transfer and a conformational landscape coordinate; only some fraction of the E-S complex (labelled active) is competent to carry out catalysis. (b) Cartoon model of a stochastic conformational landscape. The conformational sampling landscape of enzyme is represented by a solid black line, with increasing energy along the vertical axis. For the example, only three enzyme substates are shown and colored in green, blue and red. The substate colored in red is designated as the catalytically active configuration, and the other two states are either inactive or poorly active. The ΔT is to represent the impact of temperature on the conformational coordinate. Panels (c)-(e) show the three slices of the energy surface along the reaction coordinate defined by ktun (Eq 2). Panel (c) represents the motions of the heavy atoms of protein that optimize ΔG° and λ for hydrogenic wave function overlap between the reactant (blue) and product (red) potential wells. Panel (d) illustrates the hydrogen wave functional overlap that occurs upon reaching transient degeneracy between the reactant and product wells. Panel (e) presents the effective potential surface along the DAD coordinate at the tunneling ready state and illustrates the additional role of DAD sampling on the efficiency of wave function overlap.
While SLO and ht-ADH both catalyze C-H activation based reactions, they differ greatly in their protein structures and chemical transformations, with SLO catalyzing PCET on a single substrate within the conserved catalytic core of a monomeric protein and ht-ADH catalyzing a substrate/cofactor hydride transfer reaction within a multi-domain, tetrameric protein structure. The fact that detailed studies of these highly disparate enzyme systems lead to similar conclusions regarding the temperature dependence of KIEs, the impact of the conformational landscape on Ea and the link of long range protein motions to C-H activation argues well for the generality of the described phenomena. Studies in progress on yet a different type of protein fold and chemical reaction that of the methyl transfer catalyzed by methyltransferase in which quantum tunneling effects are anticipated to be minimal, suggest that the conclusions highlighted in this Perspective will be generally applicable to a very broad array of enzymatic transformations.97–100
How is the reader to relate models for catalysis as shown in Figure 13 to original hypotheses developed by early pioneers in enzymology such as Pauling and Jencks.1,101,102 The proposal by Pauling of “enhanced transition state binding” is now understood to be a definition of catalysis and not a description of the underlying physical properties that determine catalysis.101 Jencks greatly advanced the conceptual basis for catalysis from a number of directions.102 A particularly important insight was his analysis of the reduction in the entropic barrier for enzyme reactions via the placement of a large number of covalently-linked, protein-derived functional groups in close proximity to bound substrate.103 This is an essential feature of biological catalysis that reduces the effective molecularity of a potentially many-component activated complex, to processes with rate constants of either s−1 (kcat) or M−1, s−1 (kcat/Km). The presence of multiple functional groups is also the origin of the electrostatic stabilization in models of catalysis that compare pre-organized enzyme active sites to a reference water reaction.104,105 The findings discussed herein build upon and extend these earlier models, to emphasize the link of protein conformational sampling to the generation of highly precise active site alignments; it is likely that this feature plays a central role in providing very high enzyme rate accelerations that, as noted earlier, can approach ca. 1026-fold. Further, in the case of models of hydrogen tunneling, as originally described for electron tunneling,106 the reaction barrier for the primary coordinate of hydrogen transfer can be ascribed completely to thermally activated motions within the protein itself.
COMPUTATION VS. EXPERIMENT
A key difference between these two endeavors is speed, with the ability of computers to deliver results in under a year once suitable programs are available. In contrast, well executed experiments often require many years first, for method development and later, for data collection with appropriate controls. This differential is reflected in the explosion of computational solutions to questions in enzymology. This has been especially true in the application of QM/MM methods that are still being validated with regard to the choice of basis sets, functionals, and the size of the QM region used in the calculation.99,107–109 Perhaps most critically, the output from a computation will be critically dependent on the model/algorithm employed and does not necessarily prove how enzymes work. In this section of the Perspective, we summarize our views on nine central issues.
Issue #1: Model selection
Model selection will be a primary determinant of conclusions arising from the computations. One of the theoretical models25,28,110–112 focuses on the comparison of a chosen reaction in solution to the same reaction on the enzyme. The ground state of each system under interrogation is defined as a preorganized cavity that is solvated either by water or by the active site of the protein. The substrate is then placed into this cavity and the rates and energetics for the solution vs enzymatic reactions computed. As concluded therein, the resulting barrier comes from the electrostatic environment that is enhanced within the enzyme active site as a result of the presence of well-placed and multiple active site residues, with less impact from the huge region of protein that extends beyond. In general, these computations are carried out until a match is made between theory and experiments. Little effort has been made thus far to predict a rate/free energy barrier as well as the impact of specific protein side chains on the former in advance of experimental measurements. The conclusion - that all of catalysis is electrostatic, and that protein motions are irrelevant - is not surprising and, in fact, the only possible conclusion in the context of this model. We note that with the exception of predictions from the developed vibronically non-adiabatic tunneling models,42,45,49,113,114 none of the findings described in this Perspective could have been anticipated in advance of experimental measurement.
Issue #2: The role of protein dynamics in active site preorganization
By preorganizing the reaction cavity in either water or on the enzyme, the role of the protein in facilitating the preorganization process is ignored. As discussed extensively in the experimental sections above, protein preorganization is one of the keys to successful catalysis and can be viewed as part of the evolutionary strategies that has led to the enormous rate accelerations characteristic of enzyme reactions. The preorganization event itself is not static and must arise from protein motions that are related to protein flexibility and are commonly referred to as “dynamics” by chemists working at the interface of biology. Further, the catalytically relevant substates reached as a function of such dynamical sampling are distinct from initial, ground state E•S structures, and may be extremely difficult to detect via computational approaches that include MD sampling. We note that this use of the term dynamics is distinctive from its usage in chemical kinetics where it is reserved for the efficiency of barrier crossing. Different fields – different usage of the same word.115
Issue # 3: Stochastic sampling vs. “direct coupling”
The time scales of protein motions and whether these are stochastic or directly coupled has been an issue of considerable discussion in the literature.32,116–118 This point becomes moot in vibronically nonadiabatic models for H-tunneling, since the reaction coordinate for H-tunneling is instantaneous and the reaction barrier is determined by slower protein motions. These protein motions can be local or distal and occur over a variety of time scales, with a subset of protein states entering into productive reaction. In any case, the motions described in all of our studies are considered to be stochastic, and this laboratory has not yet observed experimental features that require non-Boltzmann protein sampling. In this aspect, it appears that some of the models derived from computational analyses25,26,112 and experimental findings from this laboratory are in reasonable agreement.
Issue #4: Earlier use of the terms passive vs. active dynamics
Related to Issue #3, this laboratory’s earlier use of the terms active and passive dynamics was intended to distinguish the environmental reorganization parameter originally formulated by Marcus for electron transfer theory from an additional donor-acceptor distance sampling that comes into play for hydrogen tunneling. This definition of active dynamics was never intended to imply non-Boltzmann behavior and, in any case, the descriptor active vs. passive dynamics was replaced in this laboratory after 2002, 15 years ago.42
Issue # 5: Distinction between adiabatic and non-adiabatic treatments
The distinction between adiabatic and non-adiabatic treatments of chemical coordinates frequently enters into discussions of enzyme catalysis.49,113,114 While many reactions can be described as adiabatic, using this approach often leads to hydrogen tunneling being introduced as a correction to transition state theory.119,120 In many instances, computed rates, energy barriers and isotope effects can be made to match experimental measurements via adiabatic modeling. This can be satisfying, but as noted above, is not a proof of mechanism nor does it necessarily provide any new insights into the origins of catalysis. One important exception to the success of adiabatic analyses has been the temperature dependence of kinetic isotope effects, which as discussed here and in many other reviews, is most generally temperature-independent for native enzymes, becoming increasingly temperature-dependent following perturbation of the system, e.g., via site specific mutagenesis. This temperature independence of the KIE has been seen primarily in enzyme reactions and across a very broad range of different C-H activating systems.42,69,94,95,121–127 Successful modeling of temperature independent KIEs within an adiabatic QM/MM context has been achieved in a very few cases, via either the introduction of opposing trends of temperature on parameters such as the reaction driving force and position of the transitions state or the selective use of one among many QM functionals.128–130 As yet, this approach provides no predictive power and is difficult to extend to physical interpretations of, for example, the impact of site specific mutagenesis of hydrophobic side chains on enzyme function. In contrast, vibronically non-adiabatic theory, that allows a separation of the primary tunneling event from protein motions, shows a robust ability to both model and predict observed patterns in the data as a function of both temperature and the alteration of protein side chains. The emerging physical model shows that tight packing at the active site is required for temperature-independent KIEs and any perturbation/deviation from this property can lead to temperature-dependent KIEs.
Issue # 6: Experimental evidence for the link of conformational dynamics to catalysis
As noted earlier in this Perspective, while the formal analytical treatment of Eq. 2 may not pertain to adiabatic H-transfer processes, the results from combined kinetic characterizations and biophysical probes indicate identical trends independent of the nature of the H-transfer (H:−, H+, H−). The argument has been made that there is no compelling evidence in support of the link of protein conformational landscapes to active site chemistry.112 In fact, this statement is unfortunately an example of “false truth”, as there are quite a few lines of evidence that support an interplay between global conformational landscapes and catalytic efficiency, that go beyond protein motions related to substrate binding and product release steps.
To summarize briefly the experimental results described in some detail in this Perspective: Extensive characterization of hydrogen tunneling in a thermophillic alcohol dehydrogenase has shown a rate determining hydride transfer step, making it possible to perform detailed analyses of the temperature dependence of both rates and KIEs. The trends are remarkable, showing a marked increase in the enthalpy of activation below 30 °C that occurs concomitant with an increase in the temperature dependence of the KIE. These trends are exacerbated by site specific mutagenesis that reduces the volume of selectively placed hydrophobic side chains within the interior of the protein. The original interpretation, which remains undisputed, is a perturbation of the protein conformational landscape at low temperature that is easily accessed under optimal catalytic conditions but becomes trapped into low energy states when protein is impaired. Both the enthalpy and entropy of activation increase at low temperature (an example of enthalpy/entropy compensation) and this phenomenon can be understood by the introduction of deep valleys in the protein landscape that require an input of heat and an increase in disorder/entropy to sample catalytically productive protein substates.31,69–78
The recent verification of vibronically non-adiabatic models as a means of understanding the behavior of lipoxygenase has uncovered a surprising and informative feature.51 As shown in Eq. 2 above, the formalization reached, in which the rate expression for tunneling itself is multiplied by the probability of reaching the catalytically viable protein substates, turns out to be a critical feature. Using the concepts of preorganization and reorganization, we can assign the Fconf term to a dynamical sampling of the heavy atoms of the protein, while the additional motions are discussed within ktun and are comprised of both the Marcus environmental barrier, and the DAD sampling term. Most studies from this laboratory have been aimed at understanding the origin of KIEs, their temperature dependencies and trends with mutation; however, it is also possible to calculate absolute rates. When such rates are calculated for ktun, they are routinely found to be much faster than the observed rate and only approach the experimental rate when multiplied by a prefactor, Fconf.49 The new experimental HDXMS data for soybean lipoxygenase are of considerable significance in this regard, showing for the first time that thermal activation at a remote protein loop mirrors trends in the thermal activation driving hydrogen tunneling at the enzyme active, 15 to 30 Å away.81
While the described physical model was developed in the context of C-H activation, comparable properties are likely for all classes of enzyme reaction. Within this model, the barrier crossing for reaction will be virtually instantaneous (greater or equal to femtoseconds),36 once an enzyme has achieved, via conformational sampling, the set of transient active site configurations capable of very high catalytic turnover.
Issue #7: The temperature dependence of KIEs
The statement that catalysis is enhanced by rigidly configured active sites is formally correct and is corroborated experimentally by the observation of temperature independent KIEs in optimized enzyme systems. The ironic and initially non-intuitive aspect of active site rigidity is its link to global protein flexibility that facilitates the transient sampling of the substates that achieve catalysis. In the examples above, perturbations lead to enhanced enthalpies of activation for the chemical step and, most generally, increased temperature dependences for the KIEs. These properties indicate that perturbed conditions can enhance global rigidity that, concomitantly, leads to greater local motions within the active site. The converse is also true: optimal catalytic systems will show the property of a spatially resolved protein flexibility that is linked to the achievement of active site rigidity.
Issue #8: Quantum mechanical tunneling occurs as a primary process not as a “correction factor”
With the strong and growing evidence for the participation of “dynamical” protein landscapes in enzyme catalysis, we return to the issue of the contribution of tunneling as a catalytic advantage in enzyme reactions and its link to reduced barrier width. It is often argued, in the context of adiabatic treatments of the reaction barrier, that treat hydrogen tunneling fundamentally as a correction, that reduction in the barrier width will decrease not enhance the contribution of tunneling.112 This has been presented visually with diagrams that show a reduction in barrier height accompanying a decrease in barrier width, thereby increasing the fraction of reaction that occurs by an over rather than a through-the-barrier process (Figure 14). While the foregoing logic may be of value for systems where H-transfer occurs between hetero-atoms via the intermediacy of a strong and symmetrical H-bond,131–134 the logic can and will break down for systems in which a C-H bond is cleaved. In systems characterized by high bond dissociation energies, reducing the distance between an H-donor and acceptor can bring down the overall height of the barrier, but generally not enough to cause a change from through-the-barrier to over-the-barrier behavior. As summarized in this Perspective for SLO, alterations that change the DAD are found to alter the rate of reaction, the KIE and their temperature changes, but do not lead to any over-the-top barrier crossing. In this instance the only recourse for H-transfer is from a through-the-barrier process that involves facilitated hydrogenic wave function overlap. It has recently been suggested that the hydride transfer catalyzed by a dehydrogenase (yeast alcohol dehydrogenase) may occur by the creation of a compacted internuclear DAD that leads to an over-the-barrier reaction, for which the KIE arises solely from zero point energy difference between H and D.135 However, this type of model for hydrogen transfer from carbon has no underlying theoretical basis that can easily explain either the magnitude of single KIEs or the wide range of observed magnitudes and temperature dependencies for experimental KIEs. (Note the trends seen for ht-ADH as a function of temperature and mutation, Table 1 and Figures 9a and 10b.) According to the data discussed herein and their resulting theoretical interpretations, reactions in which the primary chemical event is a transfer of hydrogen from carbon occur via facilitated wave function overlap, analogous the movement of electrons which is never described as a partitioning between over and through the barrier processes.
Figure 14.

Potential energy as a function of donor acceptor distance. For hydrogen transfer, this has been proposed to lead from transfer through the barrier (a) to an over-the-barrier process (b).112 As discussed in the text, while this scenario may, in certain instances, explain proton transfer between heteroatoms, it does not offer a theoretically-based explanation for the range of KIEs and their temperature dependencies observed in enzymatic C-H cleavage reactions.
Issue # 9: Generalizations to non-tunneling reactions
More than ca. one third of biologically relevant chemical transformations require C-H activation, implicating a broad importance for the features of enzyme catalysis discussed within this Perspective. However, in an effort to move beyond extensive investigations of enzymatic C-H activation to other classes of enzyme reaction, we introduced a study of methyltransferases into this laboratory several years ago. Initial studies were focused on a human catechol O-methyltransferase (COMT)97–99 and have more recently been extended to a glycine N-methyltransferase.100 Analogous to C-H activation, the experimental studies have involved a combination of kinetic and KIE measurements that are found to be altered as a function of site specific mutagenesis. Whereas a number of scientists refer to our studies of methyl transfer reactions as tunneling reactions (perhaps because this laboratory has focused heavily on characterizing C-H activation reactions), in fact we chose methyl transfer as an alternative to reactions involving H-tunneling, since quantum tunneling effects are expected to be either absent or to play only a small role (amu of 12 for the methyl group vs. amu’s of 1, 2 and 3 for protium and its isotopes). The results from our interrogations of methyltransferases show trends in catalytic efficiency that can be directly related to trends in DADs, once again highlighting the role of compaction in achieving optimal catalysis.97–100 The further link to protein dynamical sampling is only now beginning to unfold and will be the subject of further publications.
SUMMARY AND SCOPE
We wish to stress that a major objective of the extensive experimental undertakings summarized within this Perspective has been to move beyond any generic description of catalysis, such as is invoked in “transition state stabilization”136–138 as well as “all” electrostatic models,28,110,112,139 and to refocus attention on achieving a detailed set of physical properties that take into account the co-evolved nature of enzyme active sites and their surrounding scaffolds. We posit that it is the elaboration of these principles that may ultimately allow us to design de novo catalysts that achieve the enormous rate accelerations documented for the full range of biological catalysts. As noted by many investigators, a temporal hierarchy of conformational fluctuations is expected to contribute to enzyme catalysis.35,76–78,96,140,141 In the context of C-H activation, the primary chemical coordinate occurs via hydrogenic wave function overlap and this can be viewed as an essentially instantaneous process. The observed rates, thus, depend critically on the slower, heavy atom motions that lead to the transient precise alignment of reactants and side chains, essential for high catalytic turnover rates. The major challenge has been to obtain a spatial and temporal description of these heavy atom motions and, as a result, an understanding of their relationship to the catalytic event.
Many important implications have emerged from the described experimental observations for H transfer by quantum tunneling and these are summarized below:
The necessity to bring the donor and acceptor in close proximity within families of protein substates that are distinct from the dominant ES ground state complex;
The engagement of protein motions, via tiers of increasingly close donor acceptor distances (i.e., a transient generation of close active site packing interactions that is central to enzyme catalytic efficiency);
The participation of large, hydrophobic residues within the interior of proteins, that can act in conjunction with conformational sampling to align the DAD as well as other active site interactions;
The involvement of discrete, evolved protein networks that communicate between the solvent/protein interface and the active site.
In short, decades of rigorous experimental examination of C-H activating enzymes has established that spatially resolved regions of protein dynamical sampling are required to achieve highly precise alignment/compaction at the active site that, in turn, accounts for the enormous rate accelerations that can arise in protein-based catalysts.
One additional insight emerging from the most recent studies of SLO is a molecular framework for the interpretation of the enthalpic barrier for catalytic PCET. Consistent with earlier interpretations from Tolman and Truhlar,142,143 our results support an empirical temperature dependence of enzyme catalyzed reactions that contains a contribution from stochastic conformational sampling (i.e. the conformational landscape, ΔHconf). In addition, because of the tunneling nature of SLO and ht-ADH, the enthalpic barrier will also consist of an intrinsic contributions from the PCET reaction (namely the Marcus parameters), so that the Ea(H) can be defined as:81
| Eq. (3) |
where ΔH°conf represents the thermodynamic contribution from the conformational landscape and ΔH°ΔG° and ΔHǂλ represent the temperature dependent portion of the Marcus terms, ΔG° (thermodynamic) and λ (kinetic). The observation of long-range impacts from distal mutations in both ht-ADH and SLO suggests that, in the context of networks of conformational communication, the contribution of λout (where λ = λin + λout) may play a dominant role in the temperature dependence of kinetics in natively evolved biological catalysts.
To conclude, we wish to emphasize that while the presence of conformational networks in proteins may seem increasingly obvious based on the accelerating amount of research in this area,35,144–150 the specific networks and their communication with the active site will be system dependent. Based on our currently limited focus, it is difficult and perhaps impossible to predict a dynamics network from mere inspection of each protein fold, and we suggest that a development of predictive algorithms may present an exciting new opportunity for computational chemists. Just as the Protein Structure Initiative was important for the categorization of the range of possible enzyme folds and their relationship to catalysis, we propose that the time has come for the creation of a Functionally Related Dynamics Initiative (FReDI), in which the application of a family of biophysical probes will produce a parallel categorization of the relationship of dynamical network(s) to catalytic efficiency. Progress in this area will naturally lead from the question of “Where are the catalytically relevant networks?” to the interrogation of the molecular details of how such networks may facilitate the relay of thermal activation from a protein surface to the active site.151 A combination of such activities has the potential to generate a new era of de novo protein-based catalyst design.
Acknowledgments
This work was supported by grants from the National Institute of Health (GM118117-01 to J.P.K. and GM113432 (F32) to A.R.O.). We thank Professor Dan Herschlag, Stanford University, for his critical and insightful comments.
Footnotes
Notes: The authors declare no competing financial interest.
References
- 1.Jencks WP. Binding energy, specificity, and enzymic catalysis: the circe effect. 1975. [DOI] [PubMed] [Google Scholar]
- 2.Zhang XY, Houk KN. Acc Chem Res. 2005;38:379–385. doi: 10.1021/ar040257s. [DOI] [PubMed] [Google Scholar]
- 3.Walsh C. Enzymatic reaction mechanisms. W. H. Freeman; San Francisco: 1979. [Google Scholar]
- 4.Frey PA, Hegeman AD. Enzymatic reaction mechanisms. Oxford University Press; Oxford ; New York: 2007. [Google Scholar]
- 5.De Clercq E. Rev Med Virol. 2009;19:287–299. doi: 10.1002/rmv.624. [DOI] [PubMed] [Google Scholar]
- 6.Schramm VL. ACS Chem Biol. 2013;8:71–81. doi: 10.1021/cb300631k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Itzstein M. Nat Rev Drug Discov. 2007;6:967–974. doi: 10.1038/nrd2400. [DOI] [PubMed] [Google Scholar]
- 8.Walters WP, Namchuk M. Nat Rev Drug Discov. 2003;2:259–266. doi: 10.1038/nrd1063. [DOI] [PubMed] [Google Scholar]
- 9.Klebe G. Drug Discovery Today. 2006;11:580–594. doi: 10.1016/j.drudis.2006.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Y, Shoichet BK. Nat Chem Biol. 2009;5:358–364. doi: 10.1038/nchembio.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li JW, Vederas JC. Science. 2009;325:161–165. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 12.Schultz PG, Lerner RA. Science. 1995;269:1835–1842. doi: 10.1126/science.7569920. [DOI] [PubMed] [Google Scholar]
- 13.Schultz PG, Yin J, Lerner RA. Angew Chem Int Ed. 2002;41:4427. doi: 10.1002/1521-3773(20021202)41:23<4427::AID-ANIE4427>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 14.Hilvert D. Annu Rev Biochem. 2013;82:447–470. doi: 10.1146/annurev-biochem-072611-101825. [DOI] [PubMed] [Google Scholar]
- 15.Burton AJ, Thomson AR, Dawson WM, Brady RL, Woolfson DN. Nat Chem. 2016;8:837–844. doi: 10.1038/nchem.2555. [DOI] [PubMed] [Google Scholar]
- 16.Obexer R, Godina A, Garrabou X, Mittl PRE, Baker D, Griffiths AD, Hilvert D. Nat Chem. 2017;9:50–56. doi: 10.1038/nchem.2596. [DOI] [PubMed] [Google Scholar]
- 17.Kries H, Blomberg R, Hilvert D. Curr Opin Chem Biol. 2013;17:221–228. doi: 10.1016/j.cbpa.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 18.Edwards DR, Lohman DC, Wolfenden R. J Am Chem Soc. 2012;134:525–531. doi: 10.1021/ja208827q. [DOI] [PubMed] [Google Scholar]
- 19.Radzicka A, Wolfenden R. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- 20.Welsh KM, Creighton DJ, Klinman JP. Biochemistry. 1980;19:2005–2016. doi: 10.1021/bi00551a001. [DOI] [PubMed] [Google Scholar]
- 21.Fisher HF, Conn EE, Vennesland B, Westheimer FH. J Biol Chem. 1953;202:687–697. [PubMed] [Google Scholar]
- 22.Cha Y, Murray CJ, Klinman JP. Science. 1989;243:1325–1330. doi: 10.1126/science.2646716. [DOI] [PubMed] [Google Scholar]
- 23.Jonsson T, Glickman MH, Sun SJ, Klinman JP. J Am Chem Soc. 1996;118:10319–10320. [Google Scholar]
- 24.Allemann RK, Scrutton NS. RSC Biomolecular sciences v 18. Royal Society of Chemistry; Cambridge: 2009. p. 410. [Google Scholar]
- 25.Olsson MHM, Parson WW, Warshel A. Chem Rev. 2006;106:1737–1756. doi: 10.1021/cr040427e. [DOI] [PubMed] [Google Scholar]
- 26.McGeagh JD, Ranaghan KE, Mulholland AJ. Biochim Biophys Acta. 2011;1814:1077–1092. doi: 10.1016/j.bbapap.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 27.van der Kamp MW, Mulholland AJ. Biochemistry. 2013;52:2708–2728. doi: 10.1021/bi400215w. [DOI] [PubMed] [Google Scholar]
- 28.Frushicheva MP, Mills MJL, Schopf P, Singh MK, Prasad RB, Warshel A. Curr Opin Chem Biol. 2014;21:56–62. doi: 10.1016/j.cbpa.2014.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bruice TC. Acc Chem Res. 2002;35:139–148. doi: 10.1021/ar0001665. [DOI] [PubMed] [Google Scholar]
- 30.Benkovic SJ, Hammes-Schiffer S. Science. 2003;301:1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- 31.Nagel ZD, Klinman JP. Nat Chem Biol. 2009;5:543–550. doi: 10.1038/nchembio.204. [DOI] [PubMed] [Google Scholar]
- 32.Schwartz SD, Schramm VL. Nat Chem Biol. 2009;5:552–559. doi: 10.1038/nchembio.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glowacki DR, Harvey JN, Mulholland AJ. Nat Chem. 2012;4:169–176. doi: 10.1038/nchem.1244. [DOI] [PubMed] [Google Scholar]
- 34.Hay S, Scrutton NS. Nat Chem. 2012;4:161–168. doi: 10.1038/nchem.1223. [DOI] [PubMed] [Google Scholar]
- 35.Henzler-Wildman K, Kern D. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 36.Klinman JP, Kohen A. Annu Rev Biochem. 2013;82:471–496. doi: 10.1146/annurev-biochem-051710-133623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanoian P, Liu CT, Hammes-Schiffer S, Benkovic S. Acc Chem Res. 2015;48:482–489. doi: 10.1021/ar500390e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hekstra DR, White KI, Socolich MA, Henning RW, Srajer V, Ranganathan R. Nature. 2016;540:400. doi: 10.1038/nature20571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glickman MH, Klinman JP. Biochemistry. 1995;34:14077–14092. doi: 10.1021/bi00043a013. [DOI] [PubMed] [Google Scholar]
- 40.Glickman MH, Klinman JP. Biochemistry. 1996;35:12882–12892. doi: 10.1021/bi960985q. [DOI] [PubMed] [Google Scholar]
- 41.Glickman MH, Wiseman JS, Klinman JP. J Am Chem Soc. 1994;116:793–794. [Google Scholar]
- 42.Knapp MJ, Rickert K, Klinman JP. J Am Chem Soc. 2002;124:3865–3874. doi: 10.1021/ja012205t. [DOI] [PubMed] [Google Scholar]
- 43.Meyer MP, Klinman JP. Chem Phys. 2005;319:283–296. doi: 10.1016/j.chemphys.2005.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meyer MP, Tomchick DR, Klinman JP. Proc Natl Acad Sci USA. 2008;105:1146–1151. doi: 10.1073/pnas.0710643105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuznetsov AM, Ulstrup J. Can J Chem. 1999;77:1085–1096. [Google Scholar]
- 46.Hatcher E, Soudackov AV, Hammes-Schiffer S. J Am Chem Soc. 2004;126:5763–5775. doi: 10.1021/ja039606o. [DOI] [PubMed] [Google Scholar]
- 47.Hatcher E, Soudackov AV, Hammes-Schiffer S. J Am Chem Soc. 2007;129:187–196. doi: 10.1021/ja0667211. [DOI] [PubMed] [Google Scholar]
- 48.Edwards SJ, Soudackov AV, Hammes-Schiffer S. J Phys Chem B. 2010;114:6653–6660. doi: 10.1021/jp100133p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soudackov AV, Hammes-Schiffer S. Faraday Discuss. 2016;195:171–189. doi: 10.1039/c6fd00122j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hammes-Schiffer S. Acc Chem Res. 2006;39:93–100. doi: 10.1021/ar040199a. [DOI] [PubMed] [Google Scholar]
- 51.Hu S, Soudackov AV, Hammes-Schiffer S, Klinman JP. Acs Catalysis. 2017;7:3569–3574. doi: 10.1021/acscatal.7b00688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pang JY, Hay S, Scrutton NS, Sutcliffe MJ. J Am Chem Soc. 2008;130:7092–7097. doi: 10.1021/ja800471f. [DOI] [PubMed] [Google Scholar]
- 53.Hu S, Sharma SC, Scouras AD, Soudackov AV, Carr CAM, Hammes-Schiffer S, Alber T, Klinman JP. J Am Chem Soc. 2014;136:8157–8160. doi: 10.1021/ja502726s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sharma SC, Klinman JP. J Am Chem Soc. 2008;130:17632–17633. doi: 10.1021/ja806354w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sharma SC, Klinman JP. Biochemistry. 2015;54:5447–5456. doi: 10.1021/acs.biochem.5b00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu S, Offenbacher AR, Poss EM, Gee CL, Wilcoxen J, Carr CAM, Scouras AD, Prigozhin DM, Britt RD, Alber T, Fraser JS, Kuriyan J, Klinman JP. In preparation. [Google Scholar]
- 57.Horitani M, Offenbacher AR, Carr CAM, Yu T, Hoeke V, Cutsail GE, Hammes-Schiffer S, Klinman JP, Hoffman BM. J Am Chem Soc. 2017;139:1984–1997. doi: 10.1021/jacs.6b11856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoffman BM. Acc Chem Res. 2003;36:522–529. doi: 10.1021/ar0202565. [DOI] [PubMed] [Google Scholar]
- 59.Hoffman BM. Proc Natl Acad Sci USA. 2003;100:3575–3578. doi: 10.1073/pnas.0636464100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang TC, Wolfe MD, Neibergall MB, Mekmouche Y, Lipscomb JD, Hoffman BM. J Am Chem Soc. 2003;125:7056–7066. doi: 10.1021/ja0214126. [DOI] [PubMed] [Google Scholar]
- 61.Ivanov I, Heydeck D, Hofheinz K, Roffeis J, O’Donnell VB, Kuhn H, Walther M. Arch Biochem Biophys. 2010;503:161–174. doi: 10.1016/j.abb.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 62.Newcomer ME, Brash AR. Protein Sci. 2015;24:298–309. doi: 10.1002/pro.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Prigge ST, Boyington JC, Gaffney BJ, Amzel LM. Proteins. 1996;24:275–291. doi: 10.1002/(SICI)1097-0134(199603)24:3<275::AID-PROT1>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 64.Carr CAM, Klinman JP. Biochemistry. 2014;53:2212–2214. doi: 10.1021/bi500070q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Segraves EN, Holman TR. Biochemistry. 2003;42:5236–5243. doi: 10.1021/bi0273462. [DOI] [PubMed] [Google Scholar]
- 66.Su C, Oliw EH. J Biol Chem. 1998;273:13072–13079. doi: 10.1074/jbc.273.21.13072. [DOI] [PubMed] [Google Scholar]
- 67.Nestl BM, Hauer B. ACS Catal. 2014;4:3201–3211. [Google Scholar]
- 68.Papaleo E, Saladino G, Lambrughi M, Lindorff-Larsen K, Gervasio FL, Nussinov R. Chem Rev. 2016;116:6391–6423. doi: 10.1021/acs.chemrev.5b00623. [DOI] [PubMed] [Google Scholar]
- 69.Kohen A, Cannio R, Bartolucci S, Klinman JP. Nature. 1999;399:496–499. doi: 10.1038/20981. [DOI] [PubMed] [Google Scholar]
- 70.Kohen A, Klinman JP. J Am Chem Soc. 2000;122:10738–10739. [Google Scholar]
- 71.Liang ZX, Lee T, Resing KA, Ahn NG, Klinman JP. Proc Natl Acad Sci USA. 2004;101:9556–9561. doi: 10.1073/pnas.0403337101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nagel ZD, Klinman JP. Chem Rev. 2006;106:3095–3118. doi: 10.1021/cr050301x. [DOI] [PubMed] [Google Scholar]
- 73.Nagel ZD, Dong M, Bahnson BJ, Klinman JP. Proc Natl Acad Sci USA. 2011;108:10520–10525. doi: 10.1073/pnas.1104989108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nagel ZD, Meadows CW, Dong M, Bahnson BJ, Klinman JP. Biochemistry. 2012;51:4147–4156. doi: 10.1021/bi3001352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nagel ZD, Cun SJ, Klinman JP. J Biol Chem. 2013;288:14087–14097. doi: 10.1074/jbc.M113.453951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meadows CW, Ou R, Klinman JP. J Phys Chem B. 2014;118:6049–6061. doi: 10.1021/jp500825x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Meadows CW, Tsang JE, Klinman JP. J Am Chem Soc. 2014;136:14821–14833. doi: 10.1021/ja506667k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Meadows CW, Balakrishnan G, Kier BL, Spiro TG, Kinman JP. J Am Chem Soc. 2015;137:10060–10063. doi: 10.1021/jacs.5b04413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hu S, Cattin-Ortola J, Munos JW, Klinman JP. Angew Chem Int Ed. 2016;55:9361–9364. doi: 10.1002/anie.201603592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Boonyaratanakornkit BB, Park CB, Clark DS. Biochim Biophys Acta. 2002;1595:235–249. doi: 10.1016/s0167-4838(01)00347-8. [DOI] [PubMed] [Google Scholar]
- 81.Offenbacher AR, Hu S, Poss EM, Carr CAM, Scouras AD, Prigozhin DM, Iavarone AT, Palla A, Alber T, Fraser JS, Klinman JP. ACS Cent Sci. 2017;3:570–579. doi: 10.1021/acscentsci.7b00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T. Proc Natl Acad Sci USA. 2011;108:16247–16252. doi: 10.1073/pnas.1111325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Keedy DA, Fraser JS, van den Bedem H. PLoS Comput Biol. 2015;11 doi: 10.1371/journal.pcbi.1004507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, Alber T. Nature. 2009;462:669. doi: 10.1038/nature08615. U149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hoofnagle AN, Resing KA, Ahn NG. Annu Rev Biophys Biomol Struct. 2003;32:1–25. doi: 10.1146/annurev.biophys.32.110601.142417. [DOI] [PubMed] [Google Scholar]
- 86.Englander SW. J Am Soc Mass Spectrom. 2006;17:1481–1489. doi: 10.1016/j.jasms.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wales TE, Engen JR. Mass Spectrom Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 88.Liang ZX, Tsigos I, Bouriotis V, Klinman JP. J Am Chem Soc. 2004;126:9500–9501. doi: 10.1021/ja047087z. [DOI] [PubMed] [Google Scholar]
- 89.Liang ZX, Tsigos I, Lee T, Bouriotis V, Resing KA, Ahn NG, Klinman JP. Biochemistry. 2004;43:14676–14683. doi: 10.1021/bi049004x. [DOI] [PubMed] [Google Scholar]
- 90.Kim HS, Damo SM, Lee SY, Wemmer D, Klinman JP. Biochemistry. 2005;44:11428–11439. doi: 10.1021/bi050630j. [DOI] [PubMed] [Google Scholar]
- 91.Karlsson A, El-Ahmad M, Johansson K, Shafqat J, Jornvall H, Eklund H, Ramaswamy S. Chem Biol Interact. 2003;143:239–245. doi: 10.1016/s0009-2797(02)00222-3. [DOI] [PubMed] [Google Scholar]
- 92.Bahnson BJ, Colby TD, Chin JK, Goldstein BM, Klinman JP. Proc Natl Acad Sci USA. 1997;94:12797–12802. doi: 10.1073/pnas.94.24.12797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Seymour SL, Klinman JP. Biochemistry. 2002;41:8747–8758. doi: 10.1021/bi020054g. [DOI] [PubMed] [Google Scholar]
- 94.Sikorski RS, Wang L, Markham KA, Rajagopalan PTR, Benkovic SJ, Kohen A. J Am Chem Soc. 2004;126:4778–4779. doi: 10.1021/ja031683w. [DOI] [PubMed] [Google Scholar]
- 95.Wang Z, Kohen A. J Am Chem Soc. 2010;132:9820–9825. doi: 10.1021/ja103010b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vaughn MB, Zhang J, Spiro TG, Dyer RB, Klinman JP. In preparation. [Google Scholar]
- 97.Zhang JY, Klinman JP. J Am Chem Soc. 2011;133:17134–17137. doi: 10.1021/ja207467d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang J, Kulik HJ, Martinez TJ, Klinman JP. Proc Natl Acad Sci USA. 2015;112:7954–7959. doi: 10.1073/pnas.1506792112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kulik HJ, Zhang J, Klinman JP, Martinez TJ. J Phys Chem B. 2016;120:11381–11394. doi: 10.1021/acs.jpcb.6b07814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang J, Klinman JP. J Am Chem Soc. 2016;138:9158–9165. doi: 10.1021/jacs.6b03462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pauling L. Nature. 1948;161:707–709. doi: 10.1038/161707a0. [DOI] [PubMed] [Google Scholar]
- 102.Jencks WP. Catalysis in chemistry and enzymology. McGraw-Hill; New York: 1969. [Google Scholar]
- 103.Page MI, Jencks WP. Proc Natl Acad Sci USA. 1971;68:1678. doi: 10.1073/pnas.68.8.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wolfenden R. Science. 1983;222:1087–1093. doi: 10.1126/science.6359416. [DOI] [PubMed] [Google Scholar]
- 105.Warshel A. J Biol Chem. 1998;273:27035–27038. doi: 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- 106.Marcus RA. Annu Rev Phys Chem. 1964;15:155. [Google Scholar]
- 107.Friesner RA, Guallar V. Annu Rev Phys Chem. 2005;56:389–427. doi: 10.1146/annurev.physchem.55.091602.094410. [DOI] [PubMed] [Google Scholar]
- 108.Lin H, Truhlar DG. Theor Chem Acc. 2007;117:185–199. [Google Scholar]
- 109.Senn HM, Thiel W. Angew Chem Int Ed. 2009;48:1198–1229. doi: 10.1002/anie.200802019. [DOI] [PubMed] [Google Scholar]
- 110.Kamerlin SCL, Warshel A. Faraday Discuss. 2010;145:71–106. doi: 10.1039/B907354J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kamerlin SCL, Warshel A. Proteins. 2010;78:1339–1375. doi: 10.1002/prot.22654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Warshel A, Bora RP. J Chem Phys. 2016;144 doi: 10.1063/1.4947037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hammes-Schiffer S, Soudackov AV. J Phys Chem B. 2008;112:14108–14123. doi: 10.1021/jp805876e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hammes-Schiffer S, Stuchebrukhov AA. Chem Rev. 2010;110:6939–6960. doi: 10.1021/cr1001436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kohen A. Acc Chem Res. 2015;48:466–473. doi: 10.1021/ar500322s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang Z, Antoniou D, Schwartz SD, Schramm VL. Biochemistry. 2016;55:157–166. doi: 10.1021/acs.biochem.5b01241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zoi I, Suarez J, Antoniou D, Cameron SA, Schramm VL, Schwartz SD. J Am Chem Soc. 2016;138:3403–3409. doi: 10.1021/jacs.5b12551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Harijan RK, Zoi I, Antoniou D, Schwartz SD, Schramm VL. Proc Natl Acad Sci USA. 2017;114:6456–6461. doi: 10.1073/pnas.1704786114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Garrett BC, Truhlar DG. J Phys Chem. 1979;83:200–203. [Google Scholar]
- 120.Bell RP. The tunnel effect in chemistry. Chapman and Hall; London ; New York: 1980. [Google Scholar]
- 121.Klinman JP. Quantum Tunnelling in Enzyme-Catalysed Reactions. The Royal Society of Chemistry; 2009. pp. 132–160. [Google Scholar]
- 122.Basran J, Sutcliffe MJ, Scrutton NS. Biochemistry. 1999;38:3218–3222. doi: 10.1021/bi982719d. [DOI] [PubMed] [Google Scholar]
- 123.Harris RJ, Meskys R, Sutcliffe MJ, Scrutton NS. Biochemistry. 2000;39:1189–1198. doi: 10.1021/bi991941v. [DOI] [PubMed] [Google Scholar]
- 124.Abad JL, Camps F, Fabrias G. Angew Chem Int Ed. 2000;39:3279. doi: 10.1002/1521-3773(20000915)39:18<3279::aid-anie3279>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 125.Basran J, Patel S, Sutcliffe MJ, Scrutton NS. J Biol Chem. 2001;276:6234–6242. doi: 10.1074/jbc.M008141200. [DOI] [PubMed] [Google Scholar]
- 126.Francisco WA, Knapp MJ, Blackburn NJ, Klinman JP. J Am Chem Soc. 2002;124:8194–8195. doi: 10.1021/ja025758s. [DOI] [PubMed] [Google Scholar]
- 127.Fan F, Gadda G. J Am Chem Soc. 2005;127:2067–2074. doi: 10.1021/ja044541q. [DOI] [PubMed] [Google Scholar]
- 128.Pu JZ, Ma SH, Gao JL, Truhlar DG. J Phys Chem B. 2005;109:8551–8556. doi: 10.1021/jp051184c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pang JY, Pu JZ, Gao JL, Truhlar DG, Allemann RK. J Am Chem Soc. 2006;128:8015–8023. doi: 10.1021/ja061585l. [DOI] [PubMed] [Google Scholar]
- 130.Swiderek K, Arafet K, Kohen A, Moliner V. Journal of Chemical Theory and Computation. 2017;13:1375–1388. doi: 10.1021/acs.jctc.6b01032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cleland WW, Frey PA, Gerlt JA. J Biol Chem. 1998;273:25529–25532. doi: 10.1074/jbc.273.40.25529. [DOI] [PubMed] [Google Scholar]
- 132.Wang L, Fried SD, Boxer SG, Markland TE. Proc Natl Acad Sci USA. 2014;111:18454–18459. doi: 10.1073/pnas.1417923111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Oltrogge LM, Boxer SG. ACS Cent Sci. 2015;1:148–156. doi: 10.1021/acscentsci.5b00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Salna B, Benabbas A, Sage JT, van Thor J, Champion PM. Nat Chem. 2016;8:874–880. doi: 10.1038/nchem.2527. [DOI] [PubMed] [Google Scholar]
- 135.Dzierlenga MW, Antoniou D, Schwartz SD. J Phys Chem Lett. 2015;6:1177–1181. doi: 10.1021/acs.jpclett.5b00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wolfenden R. Nature. 1969;223:704–705. doi: 10.1038/223704a0. [DOI] [PubMed] [Google Scholar]
- 137.Lienhard GE. Science. 1973;180:149–154. doi: 10.1126/science.180.4082.149. [DOI] [PubMed] [Google Scholar]
- 138.Schramm VL. Annu Rev Biochem. 2011;80:703–732. doi: 10.1146/annurev-biochem-061809-100742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Warshel A, Sharma PK, Kato M, Xiang Y, Liu HB, Olsson MHM. Chem Rev. 2006;106:3210–3235. doi: 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- 140.Yang H, Luo GB, Karnchanaphanurach P, Louie TM, Rech I, Cova S, Xun LY, Xie XS. Science. 2003;302:262–266. doi: 10.1126/science.1086911. [DOI] [PubMed] [Google Scholar]
- 141.Klinman JP. Acc Chem Res. 2015;48:449–456. doi: 10.1021/ar5003347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tolman RC. J Am Chem Soc. 1920;42:2506–2528. [Google Scholar]
- 143.Truhlar DG. J Chem Ed. 1978;55:309. [Google Scholar]
- 144.Emrick MA, Lee T, Starkey PJ, Mumby MC, Resing KA, Ahn NG. Proc Natl Acad Sci USA. 2006;103:18101–18106. doi: 10.1073/pnas.0608849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.McLaughlin RN, Poelwijk FJ, Raman A, Gosal WS, Ranganathan R. Nature. 2012;491:138–U163. doi: 10.1038/nature11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bhabha G, Biel JT, Fraser JS. Acc Chem Res. 2015;48:423–430. doi: 10.1021/ar5003158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Keedy DA, Kenner LR, Warkentin M, Woldeyes RA, Hopkins JB, Thompson MC, Brewster AS, Van Benschoten AH, Baxter EL, Uervirojnangkoorn M, McPhillips SE, Song JH, Alonso-Mori R, Holton JM, Weis WI, Brunger AT, Soltis SM, Lemke H, Gonzalez A, Sauter NK, Cohen AE, van den Bedem H, Thorne RE, Fraser JS. eLife. 2015;4 doi: 10.7554/eLife.07574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Singh P, Francis K, Kohen A. ACS Catal. 2015;5:3067–3073. doi: 10.1021/acscatal.5b00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Campbell E, Kaltenbach M, Correy GJ, Carr PD, Porebski BT, Livingstone EK, Afriat-Jurnou L, Buckle AM, Weik M, Hollfelder F, Tokuriki N, Jackson CJ. Nat Chem Biol. 2016;12:944. doi: 10.1038/nchembio.2175. [DOI] [PubMed] [Google Scholar]
- 150.Doshi U, Holliday MJ, Eisenmesser EZ, Hamelberg D. Proc Natl Acad Sci USA. 2016;113:4735–4740. doi: 10.1073/pnas.1523573113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Salna B, Benabbas A, Russo D, Champion PM. J Phys Chem B. 2017;121:6869–6881. doi: 10.1021/acs.jpcb.7b05570. [DOI] [PubMed] [Google Scholar]