

Phages show an elevated mutation rate and remarkably rapid evolution when attacked by the bacterial CRISPR/Cas system.
Abstract
Bacteria and bacteriophages arm themselves with various defensive and counterdefensive mechanisms to protect their own genome and degrade the other’s. CRISPR (clustered regularly interspaced short palindromic repeat)–Cas (CRISPR-associated) is an adaptive bacterial defense mechanism that recognizes short stretches of invading phage genome and destroys it by nuclease attack. Unexpectedly, we discovered that the CRISPR-Cas system might also accelerate phage evolution. When Escherichia coli bacteria containing CRISPR-Cas9 were infected with phage T4, its cytosine hydroxymethylated and glucosylated genome was cleaved poorly by Cas9 nuclease, but the continuing CRISPR-Cas9 pressure led to rapid evolution of mutants that accumulated even by the time a single plaque was formed. The mutation frequencies are, remarkably, approximately six orders of magnitude higher than the spontaneous mutation frequency in the absence of CRISPR pressure. Our findings lead to the hypothesis that the CRISPR-Cas might be a double-edged sword, providing survival advantages to both bacteria and phages, leading to their coevolution and abundance on Earth.
INTRODUCTION
Bacteriophages (phages) and bacteria are the most abundant organisms on Earth (1, 2). Phages infect bacteria and often kill them by using the cell as a factory to manufacture hundreds of new viruses and dissolving the cellular envelope to release the progeny. A single viral genome delivered by a single phage is sufficient to take control of the entire cell and divert the resources to assemble viruses (3). Bacteria have evolved strategies to defend themselves against this onslaught by phages, such as the production of restriction endonucleases that can digest the phage genome (4). Phages, in turn, have evolved counterdefenses such as modification of the genome, making it resistant to nucleases (3, 5). Although the molecular mechanisms of many of these innate defensive strategies are well understood, how the bacteria and phages, despite this perpetual “arms race,” have evolved to dominate Earth’s biomass remains poorly understood.
CRISPR (clustered regularly interspaced short palindromic repeat)–Cas (CRISPR-associated) is a remarkable adaptive defense system recently discovered in bacteria and archaea (6, 7). When a phage infects a bacterium, it incorporates short 20– to 40–base pair (bp) segments of phage genome (“spacers”) into a CRISPR array present in the bacterial genome. In the surviving bacteria, these spacers are expressed as CRISPR RNAs (crRNAs) and provide a surveillance mechanism for the descendant cells (6, 7). When the cells are infected by the same phage, the crRNAs guide the CRISPR-Cas system to the respective spacer sequence in the phage genome (protospacer) and cleave it (7). The bacterial genome is protected because the spacers in its CRISPR array lack additional recognition elements such as the PAM (protospacer adjacent motif) sequence. The cleaved phage genome is cannibalized, potentially to acquire additional spacers, and is no longer able to support a productive phage infection.
The type II CRISPR-Cas9 from Streptococcus pyogenes is the simplest and the best-studied bacterial adaptive immune system (6). It consists of three basic components—crRNA derived from the spacer sequences incorporated into the CRISPR array, tracrRNA (trans-activating crRNA) that is common to all spacers, and Cas9 nuclease—together assembling as a CRISPR-Cas9 complex. Guided by spacer-specific crRNA, the complex recognizes a three-nucleotide 5′-NGG-3′ PAM sequence plus the upstream complementary protospacer sequence in phage genome and makes a double-stranded DNA break in the protospacer sequence. The disrupted genome may be further degraded by nonspecific nucleases in the cell, resulting in the inactivation of phage genome and loss of plaque-forming ability.
The CRISPR-Cas9 system has been extensively exploited for targeted editing of mammalian genomes and to generate genetically modified cell lines and organisms (8). However, relatively little attention has been given to understand the basic biology of CRISPR-Cas and its role in host-virus relationships. The CRISPR-containing bacteria have the capability to essentially wipe out the susceptible phages, as documented by several studies (9). Rare CRISPR-escape mutant (CEM) phages would no doubt survive, but the bacteria can acquire additional spacer(s) from the resistant phage and become rapidly immune, gaining an upper hand in this arms race (9). This would not only deplete phage populations but also affect bacterial evolution because horizontal gene transfer, a key driver of bacterial evolution, is largely dependent on productive phage infections (10). Hence, robust levels of phages must coexist for both the bacteria and the phages to thrive (10).
Several anti-CRISPR mechanisms have been recently discovered in phages and in lysogenic bacteria containing integrated prophage genomes (11–13). These provide counterdefenses for phage survival by interfering with various steps of the CRISPR-Cas pathways and limiting the effectiveness of the CRISPR-mediated genome disruption. However, their role in phage and/or bacterial evolution is unknown.
Here, on the basis of some unexpected findings, we propose that the CRISPR-Cas system might have evolved not only to protect the bacterial host from phage infection but also to potentially benefit the phage by allowing rapid evolution. We recently reported that the wild-type (WT) phage T4 genome modified by cytosine hydroxymethylation and glucosylation (ghmC-T4) is much less vulnerable to S. pyogenes CRISPR-Cas9 cleavage when compared to the T4(C) mutant phage containing the unmodified cytosine genome (14). In this system, the crRNAs that are complementary to the protospacers in the T4 genome adjacent to a PAM sequence, as well as the tracrRNA and Cas9 nuclease, are expressed constitutively from a plasmid. Hence, the susceptibility of the T4 genome to CRISPR-Cas9 attack and the postcleavage mechanisms that respond to a single double-stranded break introduced into the T4 genome could be examined. Surprisingly, our analyses reveal that the plaques generated from the WT ghmC-phage infections accumulated CRISPR-escape mutations at extraordinary rates. It was so rapid that about 5 to 10% of the first-generation plaques predominantly contained the CEM phages, and essentially, 100% of the plaques became CEMs by the third generation. These results suggest that the CRISPR-Cas not only protects bacteria against phages but also drives rapid phage evolution, which, in turn, is essential for bacterial evolution. This double-edged role of CRISPR-Cas, and possibly other bacterial/phage defensive mechanisms, might suggest that these systems could provide selective advantages to both bacteria and phages, not merely to one or the other, that are essential for coevolution and, ultimately, their dominance on the planet.
RESULTS
Partial resistance of ghmC-modified DNA to CRISPR-Cas9 drives the evolution of phage T4 genome
Recently, we screened 25 spacer sequences across the T4 genome for their ability to restrict the WT T4 or the T4(C) mutant phage infection of Escherichia coli (E. coli) bacteria containing the S. pyogenes type II CRISPR-Cas9 system (14). All the components of the system (crRNA, tracrRNA, and Cas9 nuclease) were constitutively expressed from a resident plasmid under the control of appropriate promoters (14). Although the Cas9 nuclease is not native to E. coli, it is one of the best-defined models to analyze how phages respond to CRISPR-Cas attacks by the bacteria. The WT phage infections that deliver ghmC-modified genome, not surprisingly, produced more plaques when compared to the T4(C) mutant phages that deliver the unmodified cytosine (C) genome (Fig. 1, B and C). We hypothesized that this difference was due to more frequent escape of the ghmC-containing genome from cleavage by the Cas9 nuclease. Strikingly, however, the differences in plating efficiencies between the WT and T4(C) mutant phages, within even the same gene, varied vastly, up to five to six orders of magnitude (Fig. 1, A to C). For instance, spacers 23-2 and 20-1070, both in the essential genes coding for the major capsid protein gp23 and the portal protein gp20, respectively, were highly restrictive (high-restriction spacers). The plating efficiency was ~10−6 to 10−7 for both the WT and T4(C) phages. On the other hand, two other spacers in the same genes, 23-1490 and 20-995, showed a high level of restriction for the T4(C) phage (~10−6) but poor restriction for the WT phage (~10−1; low-restriction spacers). This was intriguing because no obvious differences in the spacer sequences, such as the C or GC content that could affect Cas9 cleavage, or the location of the spacer in the coding versus noncoding strand, could explain this difference. Further investigation is necessary to determine the underlying mechanism.
Fig. 1. Evolution of phage T4 genome under CRISPR-Cas9 pressure.
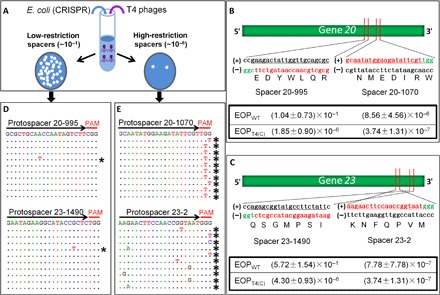
(A) Experimental scheme for testing the effect of CRISPR-Cas on phage T4 infection. Efficient cleavage by Cas9 nuclease at the protospacer sequence disrupts the phage genome, resulting in loss of plaque-forming ability (plate on the right; high-restriction spacer). Inefficient cleavage by Cas9 nuclease reduces plating efficiency (plate on the left; low-restriction spacer). (B and C) Plating efficiencies of high-restriction spacers 20-1070 and 23-2 and low-restriction spacers 20-995 and 23-1490. Shown are the locations of spacers on genes 20 and 23 and the nucleotide and amino acid sequences corresponding to the protospacer (red) and PAM (green) sequences. The sequences of the complementary strand are shown in black. Efficiency of plating (EOP) was determined, as described in Materials and Methods. The data shown are the average of three independent experiments ± SD. (D and E) Alignment of sequences corresponding to single plaques produced from infection of various spacer-expressing E. coli. The DNA from single plaques was amplified and sequenced, as described in Materials and Methods. The black arrows correspond to the spacer sequences, and the red lines correspond to the PAM sequences. The sequence at the top of each panel corresponds to the WT sequence. The dotted lines below correspond to the sequence obtained from each plaque. Only the mutated nucleotides are shown. The asterisks indicate the mutant sequences.
Sequencing of the CRISPR-resistant plaques (CRPs) from the high-restriction spacers showed that 100% of the plaques have mutations in the PAM or protospacer sequence (Fig. 1E). These represent rare preexisting mutations present in the phage stocks that prevented either binding of CRISPR-Cas to the PAM sequence or cleavage of the protospacer sequence by the Cas9 nuclease, thus escaping the CRISPR surveillance. On the other hand, sequencing of CRPs from the low-restriction spacers showed no mutations in PAM or protospacer sequences. This was consistent with our hypothesis that resistance here was not due to a mutation but due to escape of the WT ghmC-modified genome from Cas9-mediated gene disruption due to poor cleavage. Surprisingly, however, we found that, although most of the plaques have a WT protospacer sequence, ~1 in 10 to 20 (5 to 10%) have mutations in the protospacer or PAM sequence (Fig. 1D). This was observed in both genes 20 and 23 with the low-restriction spacers. This was completely unexpected because a mutation frequency of ~10−1 is too high to be due to preexisting mutations, which is expected and determined (Fig. 1, B and C) to be on the order of ~10−7 (15, 16). No such mutations were observed in the control plaques generated without the CRISPR-Cas9 pressure. Therefore, the high mutation rate of WT ghmC-modified T4 phage in CRISPR background must be due to rapid evolution and selection of mutants during active replication of phage genome in the infected cell under the pressure of CRISPR-Cas9.
A model for rapid evolution of phage T4 genome driven by CRISPR-Cas
We propose a model for the evolution of phage mutants under the pressure of CRISPR-Cas9 (Fig. 2). At a multiplicity of infection (MOI) of 0.001 (Fig. 1A), each plaque originates from infection of a single E. coli bacterium with a single WT phage. For a low-restriction spacer, about 10 to 20% of the genomes escape CRISPR-Cas9 cleavage (see Fig. 1, B and C) and enter phage replication cycle, triggering the production of new genomes. However, the constitutively expressed CRISPR-Cas components from the CRISPR plasmid can cleave the newly replicated genomes, albeit inefficiently (14), although continued production of CRISPR-Cas9 components would cease because early expression of T4 phage nucleases denA and denB degrade the CRISPR plasmid (17). The CRISPR-cleaved DNA would then initiate new replication events because phage T4 has no defined replication origin, and its replication is largely initiated by the recombination-dependent invasion of DNA ends into the actively replicating DNA (18–21). In addition, because T4 is a highly recombinogenic phage expressing potent recombination and repair enzymes (3), the ends could be repaired by a combination of mechanisms involving these enzymes, mechanisms that phage T4 uses for its own genome replication to generate a massively branched concatemeric DNA network. Consequently, the cleaved protospacer sequences might create hotspots for mutation as the T4-infected CRISPR–E. coli continually accumulates concatemeric DNA containing the repaired genomes of CRISPR-Cas cleaved DNA, which would then be encapsidated generating a burst of progeny (22, 23).
Fig. 2. A model for CRISPR-Cas9–driven evolution of phage T4 genome.
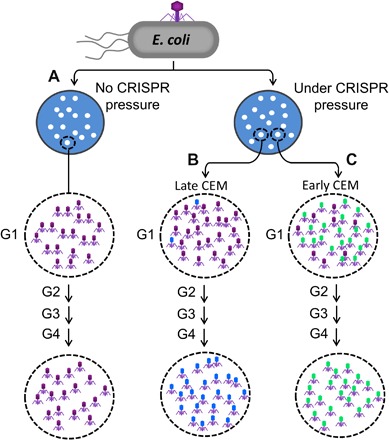
Schematic depicting the patterns of phage progeny in single plaques starting from a single phage infecting a single E. coli cell. Details of the model are described in Results.
A plaque represents a locus where a series of phage infection cycles productively lyse E. coli bacteria and concentrate ~107 progeny phages (Fig. 2). Spontaneous (random) mutants do exist in this population by classical error-prone replication mechanisms but at a very low frequency, roughly on the order of ~10−6 to 10−7 (15). Under CRISPR pressure (Fig. 2, B and C), however, if a CEM arose from repairing Cas9-cleaved ends, as described above, then that mutant phage will have greater fitness because it is no longer cleaved by Cas9 nuclease. Hence, it will produce a greater number of progeny viruses compared to the WT phage that has been partially restricted by Cas9. Thus, in subsequent generations, the fraction of CEM phages will raise markedly. Consequently, a plaque produced under CRISPR-Cas9 pressure would likely consist of a mixture of WT and CEM phages [generation 1 (G1) in Fig. 2, B and C], as opposed to a plaque produced without the CRISPR-Cas pressure (Fig. 2A). However, the fraction of CEM phages in a given G1 plaque would depend on the time at which the CEM arose. If it arose late after the initial infection (Fig. 2B), then the plaque will predominantly have WT phages and few CEMs. However, if it arose soon after the initial infection (Fig. 2C), then the CEM progeny will accumulate rapidly in the subsequent generations and predominate the population in the plaque. Consistent with this model, the CEMs were found at a remarkably high frequency among the first-generation (G1) plaques under CRISPR-Cas pressure, ~5% for the low-restriction gene 20 spacer 20-995 and ~10% for the low-restriction gene 23 spacer 23-1490 (Fig. 1D). In contrast, the CRISPR-escape mutation frequency for the high-restriction spacers in the same genes was ~10−6 to 10−7, similar to the expected spontaneous mutation frequency (Fig. 1, B and C).
Selection for CRISPR-driven mutations in the portal protein gene
We applied two tests to evaluate the above model. First, if the model is correct, then every WT phage infection, thus every plaque that arises as a result on CRISPR–E. coli containing a low-restriction spacer, should be on a trajectory to evolve into a CEM plaque. To test this prediction, each G1 plaque was transferred to a fresh CRISPR–E. coli lawn and allowed to form second-generation (G2) plaques (Fig. 3A). This was repeated up to five generations (G3 to G5). Individual plaques from G1 to G5 were picked, and the DNA flanking the protospacer/PAM sequence was sequenced (Fig. 3A). The data showed that the frequency of CEMs markedly increased from 10% in G1 plaques to 50% in G2 plaques and to 90 and 100% in G3 and G4 plaques, respectively. Furthermore, each plaque went through its own evolutionary trajectory both in time and sequence, selecting different CEMs, although some of the mutations were repeatedly selected. Furthermore, 100% of the CEMs are in the protospacer/PAM sequences. No CEMs were found in any of the control G1 to G5 plaques that were not under the CRISPR-Cas pressure.
Fig. 3. Selection of CEMs in the portal protein gene.
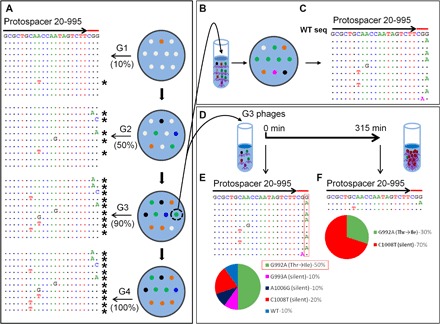
(A) Plaques produced from WT phage T4 infection of CRISPR–E. coli DH5α expressing spacer 20-995 (G1) were transferred to a fresh plate, and the process was repeated (G2 to G4). The DNA from single plaques was amplified and sequenced. Left: Alignment of sequences in the same manner, as described in legend to Fig. 1. (B) The mixture of phages from a G3 plaque was separated by serial dilution, and single plaques produced by individual variants were sequenced (C). “WT seq” indicates the wild-type sequence of protospacer. (D and E) To determine the relative fitness, we used the same phage mixture to infect E. coli DH5α expressing spacer 20-995 in a liquid culture. Single plaques obtained from the progeny produced after 315 min of infection were picked and sequenced (D). The percentages of each CEM in the starting sample (E) and after 315 min (F) of evolution are shown below as pie charts. The spacer and PAM sequences are marked with arrows and red lines, respectively. See Materials and Methods for details.
The second test was to capture an intermediate state of the evolutionary process. Our model predicts that, at an intermediate stage, a single plaque may contain more than one CEM phage plus the WT phages, but eventually, the most-fit mutant phage(s) under CRISPR-Cas9 pressure will predominate the population. To capture this state, we selected a G3 plaque that showed significant background in the sequencing chromatogram at certain positions of the PAM/protospacer sequence. This indicated the presence of a mixture of sequences. Individual phages present in this plaque were separated by serial dilution and plated on E. coli without the CRISPR-Cas pressure to ensure that no further evolution occurred (Fig. 3B). Of the 10 progeny phages sequenced, 1 had a WT sequence, 6 had mutations changing a G to an A of the two strictly required Gs of the 5′-NGG-3′ PAM recognition sequence, 2 had C-to-T mutations in the protospacer sequence, and 1 had an A-to-G mutation also in the protospacer sequence (Fig. 3, C and E). This pattern demonstrated independent evolution of different CEMs within the same plaque and near disappearance of the WT phage.
To determine the relative fitness of these CEMs, we then used this G3 phage mixture to infect the CRISPR–E. coli at a low MOI (0.001) and allowed it to grow for several hours (Fig. 3D). The progeny phages were plated, and single plaques were isolated and sequenced. The data showed that, although there were five different variants in the starting mixture of the G3 plaque (Fig. 3E), only two CEMs were recovered after several generations (Fig. 3F). Of these two, the C-to-T mutation in the protospacer sequence (silent mutation) predominated with 70% of phages in the progeny population, whereas the minor variant (30% of the progeny) had the G-to-A missense mutation (Thr to Ile) in the PAM sequence (Fig. 3F). The above sets of data confirm the basic predictions of our proposed CRISPR-driven evolution of the phage T4 genome (Fig. 2).
CRISPR-driven evolution of the major capsid protein gene
To test whether the CRISPR-driven evolution is applicable to any other (essential) gene in the phage T4 genome, we carried out the above analyses for another low-restriction spacer, 23-1490, which is part of the major capsid protein gene 23 (Fig. 1). The data demonstrated the same pattern (Fig. 4); the CEMs arose at a frequency of 10% among G1 plaques, which increased to 40% in G2 plaques and to 100% in G3 plaques. However, the pattern of the CEM phage selection in this protospacer region was different from that of the portal protein protospacer described above. This is expected because the mutations that can restore the major capsid function would be different from that of the portal protein. Here, only two types of CEMs were selected, a predominant G-to-T mutation (9 of 10 plaques) and a minor C-to-T mutation (1 of 10 plaques), both in the protospacer region. No mutations in the PAM sequence were recovered.
Fig. 4. Selection of CEMs in the major capsid protein gene.
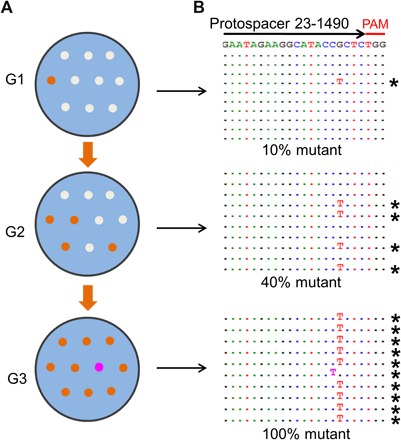
The experimental scheme (A) for analysis of CEM selection in the major capsid protein gene is the same as that used for the portal protein gene. (B) Sequences of CEMs. See Materials and Methods and legend to Fig. 3A for details.
CEMs exhibit dual phenotype
Forty CEMs were isolated from either the high-restriction spacers or the low-restriction spacers. Of these, 18 were unique variants, and the rest were repeat isolates of one of the variants (Figs. 5 and 6). All were single-point mutations, each retaining the reading frame as required to express the essential gene functions of the major capsid protein and the portal protein. Seven of the mutations were silent, and 11 involved amino acid changes. The four spacer regions contain 33 amino acid codons, which include a total of 276 possible single-point mutations while maintaining the reading frames of the essential genes 20 and 23 (fig. S1). Of these, 47 or 17% would be silent mutations, and the rest would be missense amino acid substitutions. However, because the percentage of recovered silent mutations was ~39% (7 of 18), more than twice that of what would be expected if the mutations were evenly distributed between the silent and missense mutations, it appears that the selection of CEMs is biased toward silent mutations. This might be because some (many) of the amino acid changes cost in fitness because these phage structural proteins are critical for head assembly (24, 25) and genome packaging (26). This was evident in at least one instance; when a cocktail of five CEM phages, as present in a single plaque, was used to infect E. coli, the C-to-T silent CEM at nucleotide 1008 of the gene 20 sequence was preferentially selected (Fig. 3, E and F). When this experiment was repeated slightly differently, by mixing the variants in equal proportions at the start (fig. S2, A and B), again this and another silent mutation were recovered at greater frequency, whereas the CEM with an amino acid change (Thr to Ile) became “extinct” after a few hours of growth (fig. S2C). However, as the following data show, selection of the CEMs was spacer-specific and exhibited different patterns, in part depending on the functional importance of the amino acid sequence encoded by the protospacer sequence.
Fig. 5. Characteristics of the CEMs obtained from gene 20 spacers.
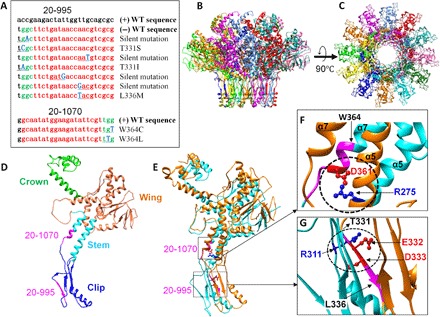
(A) List of CEM sequences obtained from gene 20 spacers. The spacer sequence and PAM are shown in red and green, respectively, and the mutated nucleotides are shown in blue (bold) and underlined. The WT sequences are shown at the top of each alignment. (B to G) Structural analysis of the CEMs of the portal protein gp20. Side (B) and top (C) views of the structure of the dodecameric gp20 portal assembly with each subunit shown in a different color. Single (D) and two (E) subunits of gp20 showing (i) the critical salt bridge between the D361 residue of one subunit and R275 residue of an adjacent subunit (circled) (F) and (ii) the functionally important residues of the clip domain, E332 and D333, of one subunit forming a salt bridge with R311 of an adjacent subunit (circled) (G). The regions corresponding to the protospacer and PAM sequences of spacers 20-1070 (D, E, and F) and 20-995 (D, E, and G) are shown in magenta. The positions of the mutated residues of the CEM phages corresponding to spacers 20-1070 (W364) (F) and 20-995 (T331 and L336) (G) are shown with arrows.
Fig. 6. Characteristics of the CEMs obtained from gene 23 spacers.
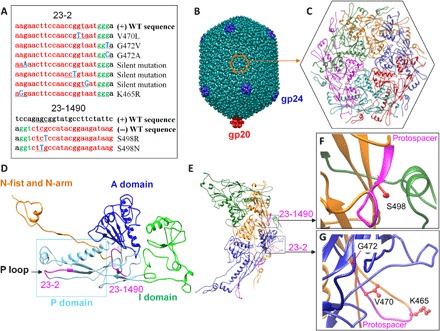
(A) List of CEM sequences obtained from gene 23 spacers. The spacer sequence and PAM are shown in red and green, respectively, and the mutated nucleotides are shown in blue (bold) and underlined. The WT sequences are shown at the top of each alignment. (B to G) Structural analysis of the CEMs of the major capsid protein gp23. (B) Side view of the phage T4 capsid. (C) Top view of the hexameric gp23 capsomer. Single (D) and three subunits of gp23 (E) involved in a network of intersubunit interactions. The regions corresponding to the protospacer and PAM sequences of spacers 23-1490 (D to F) and 23-2 (D, E, and G) are shown in magenta color. The side chains of the mutated residues of the CEM phages corresponding to spacers 23-1490 (S498) (F) and 23-2 (K465, V470, and G472) (G) are shown in red.
In the case of the low-restriction spacer 20-995, in addition to four silent CEMs, three mutants with amino acid changes were recovered (Fig. 5A). The amino acid sequence 331TEDYWLQR338 corresponding to the 20-995 protospacer encodes a β-strand in the “clip” domain of the portal protein (Fig. 5, B to E). It is part of the hydrophobic core of the domain and, in addition, contains two negatively charged residues, E332 and D333, that form a salt bridge with R311 of another β-strand of an adjacent subunit. Our mutational studies show that the salt bridge is critical for function. Consistent with the importance of this β-strand, the CEMs selected in this protospacer sequence fall at sites flanking the strand, the residues T331 (T331S and T331I) and L336 (L336M; Fig. 5G).
For the high-restriction spacer 20-1070, only two CEMs with changes at the same amino acid, W364C and W364L, were repeatedly selected (Figs. 1E and 5A). This is also consistent with our functional data in that the amino acid sequence encoded by this protospacer sequence (357GNMEDIRW364) is critical for head assembly and DNA packaging (Fig. 5, B to F) (27). The amino acid residues 361 to 374 form the helix α-7, which together with helix α-5 form the “stem” domain that lines the ~35 Å diameter central channel of the dodecameric portal vertex (27). The channel allows passage of DNA into the capsid during packaging and out of the capsid during infection. The D361 residue of the α-7 helix forms a critical intersubunit salt bridge with R275 residue of the α-5 helix of an adjacent subunit (Fig. 5, D to F). This interdigitation stabilizes the dodecameric portal structure and is essential for head assembly (27). Combinatorial mutagenesis of this region shows that no substitutions are tolerated at D361. The CEM selection confirmed this point because no mutations were recovered at or near D361. The CEM approach, however, seems more powerful because it allowed rapid scanning of seven amino acids spanning the protospacer sequence in one experiment and further identifying that the W364 residue that is upstream to D361 can be substituted without losing function.
Different CEMs were selected for the gene 23 spacers that also included both silent mutations and amino acid substitutions in the protospacer and PAM sequences (Fig. 6A). The low-restriction spacer 23-1490 encodes the amino acid sequence 497QSGMPSIL504 that links the axial domain (A domain) to the peripheral domain (P domain), whereas the high-restriction gene 23 spacer 23-2 comprises the amino acid sequence 465KNFQPVMG472 that is part of the P-loop sequence (Fig. 6, B to E) (28). The A domain through intersubunit interactions is responsible for assembling hexameric capsomers, whereas the P domain and the P-loop residues are important for interface interactions between capsomers (28). Consistent with our structural analyses (28), the recovered CEMs correspond to residues that do not appear to be involved in these interactions. For instance, the side chain of the S498 residue at which two CEMs were recovered (S498N and S498R) is fully exposed and not in proximity to any other side chains within a ~5 Å distance (Fig. 5F). The CEM G472V at the base of the P-loop resulted in a heat-sensitive phenotype probably because it affected the interaction of the P-loop with the “insertion” domain (I domain) linker of the adjacent subunit (Fig. 6, E and G, and fig. S3).
The above sets of data suggest that the selection of CEMs was driven not by whether the spacer is of low or high restriction type, or silent versus amino acid change, but rather by their ability to overcome two strong selection pressures: (i) resistance to Cas9 nuclease and (ii) retaining essential phage function. Although the sample size of the mutants analyzed here is small, it seems clear that the CRISPR-Cas selection approach can be used to generate pools of CEMs, the analysis of which may generate a detailed functional map and reveal the mechanistic requirements for a given phage function or for Cas9 cleavage. These are currently under investigation.
DISCUSSION
CRISPR-Cas is generally thought of as an adaptive immune system that has evolved to protect the bacterial host against phage infections, which are often lethal (7). An unexpected finding of this study is that the CRISPR-Cas might be a double-edged sword, not only a defensive mechanism against phages but also a potentially robust platform for phage evolution, which would ultimately benefit both the host and the virus.
The surprising observation was that mutations accumulated in the phage genome at an unusually high frequency and rapidity among the progeny produced from CRISPR-Cas9 E. coli infected with WT T4 phage containing the ghmC-modified genome. Virtually, each of these infections was found to be on an evolutionary trajectory to become CRISPR-resistant, with the mutations clustering exclusively in the protospacer and PAM sequences. These CEMs outcompeted the WT phage and predominated the population even among the first-generation plaques, about 5 to 10% of them, which increased to 40 to 50% in the second generation and nearly 100% in the third generation. These frequencies are striking, about six orders of magnitude greater than the spontaneous mutation frequency, which is on the order of ~10−7 (15, 16). All the CEMs exhibited dual phenotype, resistance to CRISPR-Cas9, and retention of the respective gene function. This seems to be a general pattern because it was observed with two essential phage structural genes, one coding for the major capsid protein gp23 and another for the portal vertex protein gp20.
The result that this high mutation frequency was observed with the low-restriction spacers (most of the spacers) suggests that the evolution of CEMs was linked to partial escape of the ghmC-modified phage genome from cleavage by Cas9 nuclease upon its first exposure to the CRISPR-Cas9 complex following delivery by phage injection. Otherwise, disruption of genome and loss of essential gene function would have destroyed the plaque-forming ability even if the cleaved ends were repaired, as was observed with a few high-restriction spacers or in infections by unmodified T4(C) mutant phage (14). Consistent with this reasoning, it has been well documented that the ghmC-modified genome is generally resistant to nucleases including the restriction endonucleases (3, 5, 14).
Escape from Cas9 cleavage means that phage genome replication would be initiated before the delivered genome is cleaved. Vigorous genome replication, a characteristic of phage life cycle spanning a mere 20 to 30 min, plus the continuing presence of Cas9 then drive evolution and selection of resistant mutations, as per the model described in Results (Fig. 2). This must be particularly robust in the case of phage T4, where replication is initiated largely by recombination events (19). The mechanisms are likely complex and not the main focus of this study, but the key implication of our findings is that the timing of CRISPR-Cas9 cleavage relative to the timing of the initiation of phage genome replication is critical for the evolution of the CEMs.
The time scales of CRISPR-Cas cleavage of phage genomes are unknown. A recent report (29) estimates that the association rate of CRISPR-Cas9 complex to a PAM site is ~40 ms if there were about five molecules of Cas9 per E. coli cell. Because the phage T4 genome contains 11,656 PAM sites, it would take about 6 min to scan the entire genome. The time taken might be even longer for the ghmC-modified T4 genome than for the unmodified C-genome, although the number of Cas9 molecules per cell is expected to be more than five. Therefore, it is safe to assume that it would take a few minutes for CRISPR-Cas9 to find a protospacer sequence in the ghmC-genome. By then, many, if not most, of the delivered T4 genomes would have initiated replication (21, 30). Consistent with this timeline, our data show that about 10 to 20% of ghmC-genomes survived Cas9 cleavage, and every one of these evolved into a CEM with varying fitness under the continuing pressure of CRISPR-Cas9. Because in nature, this would happen with spacers distributed throughout the phage genome, and in both strands of the genome, the CRISPR system can potentially drive large-scale evolution of phage genomes. Some of the mutant phages are expected to be more fit than the parental phage, whereas others, probably most as this study indicates, may not have a fitness advantage but would nevertheless remain in the population. Although the specific CRISPR-Cas9 system used here is not native to E. coli, this phenomenon might explain why numerous conservative substitutions in phage genes remain in the closely related phage families even though they may not confer any fitness gain (31, 32). At the same time, all the mutant phages by virtue of their resistance to CRISPR-Cas would be able to contribute to bacterial evolution by horizontal gene transfer and other mechanisms (10).
The timing of CRISPR-Cas cleavage, thus, might provide a critical window for fine-tuning the balance between defense against phages and evolution of phages and, in turn, bacteria. It could be accomplished by a variety of mechanisms, both phage-based such as the modification of genomes (14, 33–35), efficiency of initiation of genome replication (36), and inclusion of anti-CRISPR genes (12) and host-based such as the intrinsic catalytic rates of Cas9 cleavage and regulation of cleavage by accessory Cas proteins (37). All of these mechanisms have been described in the literature, and we predict that some of these slow down the rate of Cas9 cleavage, and the progeny phages thus produced likely contain a high frequency of mutations, as has been observed here. The CRISPR-Cas mechanism, thus, might be a part of the global evolutionary system that provides various degrees of advantages to both the bacteria and the phages.
In conclusion, our results suggest the possibility that the defensive and counterdefensive systems of the arms race between bacteria and phages such as the CRISPR-Cas may have been selected for the survival advantages that they provide to both the host and the virus, but not merely to one or the other, such that both the bacteria and the phages can coexist and coevolve, leading to their dominant presence on Earth.
MATERIALS AND METHODS
Plasmids, bacteria, and bacteriophage
The spacer plasmids, 20-995, 20-1070, 23-2, and 23-1490 were constructed in a previous study (14). WT T4 phage was propagated on E. coli P301 (sup0), as previously described (38–40). T4(C) is a mutant phage containing an amber mutation at amino acid 58 of gene 42 that codes for deoxycytidine monophosphate hydroxymethylase and an amber mutation at amino acid 124 of gene 56 that codes for deoxycytidine triphosphatase (3). The T4(C) mutant was propagated on E. coli B834 (hsdRB hsdMB met thi sup0) for only one generation to prevent accumulation of spontaneous revertants. The T4(C) phage stocks containing revertant phage at a frequency of <10−6 were used in all the experiments.
Plaque assays
The efficiency of individual spacers to restrict T4 phage infection was determined by plaque assay, as previously described (14). Briefly, the CRISPR-Cas plasmids with different spacers were transformed into E. coli DH5α [hsdR17(rK– mK+) sup2] individually. Up to ~107 plaque-forming units (PFU) of either WT T4 or T4(C) in 100 μl of Pi-Mg buffer (26 mM Na2HPO4, 22 mM KH2PO4, 70 mM NaCl, and 1 mM MgSO4) was added to 300 μl of E. coli (~108 cells/ml) containing the CRISPR-Cas plasmid. After a 7-min incubation at 37°C, 3 ml of 0.7% top agar with streptomycin (50 μg/ml) was added into each tube, mixed, and poured onto LB-streptomycin plates. The plates were then incubated at 37°C overnight. The EOP was calculated by dividing the PFU produced from infection of E. coli by the input PFU determined under permissive conditions.
DNA sequencing of single plaques
Single plaques were picked using a sterile Pasteur glass pipette and transferred into a 1.5-ml Eppedorf tube containing 200 μl of Pi-Mg buffer plus 2 μl of chloroform. After a 1-hour incubation at room temperature with mixing every few minutes, 4 μl of the sample was used as a template for polymerase chain reaction (PCR) using Phusion High-Fidelity PCR Master Mix (Thermo Fisher Scientific). Before starting PCR, the phage was denatured at 95°C for 10 min. Amplification was performed using appropriate primers flanking the protospacer sequence. The amplified DNA was purified by agarose gel electrophoresis using QIAquick Gel Extraction Kit (Qiagen) and sequenced (Retrogene).
Selection of CEMs
Three hundred microliters of E. coli DH5α containing the CRISPR-Cas plasmid (~108 cells/ml) was infected with WT T4 and mixed with 3 ml of 0.7% top agar with streptomycin (50 μg/ml) and poured onto an LB-streptomycin plate. After overnight incubation at 37°C, the plaques formed (G1) were picked by stabbing each plaque with a sterile toothpick and transferring them to another LB-streptomycin plate. The plaques formed (G2) are then subjected to the same process two to three more times (G3 to G5). Single plaques at each stage were sequenced, as described above, after amplification of the regions flanking the protospacer sequence using appropriate primers. For the 20-995 spacer, 1172-bp upstream and 226-bp downstream flanking regions were amplified; for 20-1070, 1247-bp upstream and 151-bp downstream flanking regions were amplified; for 23-1490, 227-bp upstream and 798-bp downstream flanking regions were amplified; and for 23-2, 129-bp upstream and 896-bp downstream flanking regions were amplified. E. coli DH5α without the CRISPR-Cas plasmid was used as a control.
Evolution of CEMs
Evolution of phages isolated from a single plaque was carried out, as shown in Fig. 3D. A single plaque was picked and transferred into a 1.5-ml Eppedoff tube containing 1 ml of Pi-Mg buffer plus 10 μl of chloroform. The phage titer was determined by plaque assay following serial dilutions. Four milliliters of log-phase E. coli DH5α cells (~2 × 108 cells/ml) containing the CRISPR-Cas plasmid was infected with phages at an MOI of 0.001 at 37°C. Three hundred fifteen minutes after infection, 400 μl of culture was collected and treated with a few drops of chloroform, and deoxyribonuclease I (7 μg/ml) and lysozyme (10 μg/ml) were then added to the sample and incubated at 37°C for 1 hour. The cell debris was removed by centrifugation of the suspension at 7000 rpm (4300g) for 10 min at 4°C. The supernatant was transferred into a new tube, and the phages were pelleted by centrifugation for 45 min at 15,000 rpm (21,130g) at 4°C. The pellet was resuspended in 200 μl of Pi-Mg buffer, serially diluted, and plated on LB plates. Ten single plaques were picked and sequenced, as described above.
Coculture of spacer 20-995 CEMs
An equal number of PFU of four T4 CEMs were mixed (fig. S2) and added to 1 ml of the log-phase E. coli DH5α cells (~2 × 108 cells/ml) at an MOI of 0.001 at 37°C. Three hundred fifteen minutes after infection, 100 μl of culture was collected and treated, as described above. Ten single plaques were picked and sequenced.
Plate spot test
Temperature sensitivity of each phage mutant was determined by plate spot test, as previously described (3). Briefly, 300 μl of E. coli DH5α (~108 cells/ml) was mixed with 3 ml of 0.7% top agar and poured onto an LB plate. About 1 μl of phage suspension (100 to 104 PFU) was applied on the top agar plate and left for 3 to 5 min at room temperature to let the drops dry. Three identical plates were prepared and incubated overnight at 42°, 37°, and 25°C, respectively.
Supplementary Material
Acknowledgments
We thank W.-c. Tang (Catholic University of America) for critical discussion of the manuscript and help with Fig. 5. Funding: This work was supported by the National Institute of Allergy and Infectious Diseases/NIH grants AI111538 and AI081726 to V.R. and in part by the NSF grant MCB-0923873 to V.R. and the Huazhong Agricultural University Research Foundation for Talented Scholars to P.T. Author contributions: P.T., X.W., and V.R. designed the experiments; P.T. and X.W. performed the experiments; and P.T. and V.R. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/2/eaar4134/DC1
fig. S1. List of all possible single mutations in each spacer.
fig. S2. Relative fitness of the CEMs that escaped Cas9 cleavage of the protospacer 20-995 of the portal protein gene.
fig. S3. Temperature sensitivity of the CEMs containing amino acid changes.
REFERENCES AND NOTES
- 1.Salmond G. P. C., Fineran P. C., A century of the phage: Past, present and future. Nat. Rev. Microbiol. 13, 777–786 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Hendrix R. W., Bacteriophage genomics. Curr. Opin. Microbiol. 6, 506–511 (2003). [DOI] [PubMed] [Google Scholar]
- 3.J. D. Karam, J. W. Drake, Molecular Biology of Bacteriophage T4 (American Society for Microbiology, 1994). [Google Scholar]
- 4.Seed K. D., Battling phages: How bacteria defend against viral attack. PLOS Pathog. 11, e1004847 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bryson A. L., Hwang Y., Sherrill-Mix S., Wu G. D., Lewis J. D., Black L., Clark T. A., Bushman F. D., Covalent modification of bacteriophage T4 DNA inhibits CRISPR-Cas9. MBio 6, e00648-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mohanraju P., Makarova K. S., Zetsche B., Zhang F., Koonin E. V., van der Oost J., Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science 353, aad5147 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Wiedenheft B., Sternberg S. H., Doudna J. A., RNA-guided genetic silencing systems in bacteria and archaea. Nature 482, 331–338 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Mali P., Esvelt K. M., Church G. M., Cas9 as a versatile tool for engineering biology. Nat. Methods 10, 957–963 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paez-Espino D., Sharon I., Morovic W., Stahl B., Thomas B. C., Barrangou R., Banfield J. F., CRISPR immunity drives rapid phage genome evolution in Streptococcus thermophilus. MBio 6, e00262-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koskella B., Brockhurst M. A., Bacteria–phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 38, 916–931 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pawluk A., Amrani N., Zhang Y., Garcia B., Hidalgo-Reyes Y., Lee J., Edraki A., Shah M., Sontheimer E. J., Maxwell K. L., Davidson A. R., Naturally occurring off-switches for CRISPR-Cas9. Cell 167, 1829–1838.e9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bondy-Denomy J., Pawluk A., Maxwell K. L., Davidson A. R., Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493, 429–432 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pawluk A., Staals R. H. J., Taylor C., Watson B. N. J., Saha S., Fineran P. C., Maxwell K. L., Davidson A. R., Inactivation of CRISPR-Cas systems by anti-CRISPR proteins in diverse bacterial species. Nat. Microbiol. 1, 16085 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Tao P., Wu X., Tang W.-C., Zhu J., Rao V., Engineering of bacteriophage T4 genome using CRISPR-Cas9. ACS Synth. Biol. 6, 1952–1961 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santos M. E., Drake J. W., Rates of spontaneous mutation in bacteriophage T4 are independent of host fidelity determinants. Genetics 138, 553–564 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drake J. W., A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. U.S.A. 88, 7160–7164 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carlson K., Ȗvervatin A., Bacteriophage T4 endonucleases II and IV, oppositely affected by dCMP hydroxymethylase activity, have different roles in the degradation and in the RNA polymerase-dependent replication of T4 cytosine-containing DNA. Genetics 114, 669–685 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mosig G., Recombination and recombination-dependent DNA replication in bacteriophage T4. Annu. Rev. Genet. 32, 379–413 (1998). [DOI] [PubMed] [Google Scholar]
- 19.Kreuzer K. N., Brister J. R., Initiation of bacteriophage T4 DNA replication and replication fork dynamics: A review in the Virology Journal series on bacteriophage T4 and its relatives. Virol. J. 7, 358 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.George J. W., Stohr B. A., Tomso D. J., Kreuzer K. N., The tight linkage between DNA replication and double-strand break repair in bacteriophage T4. Proc. Natl. Acad. Sci. U.S.A. 98, 8290–8297 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.A. W. Kozinski, Origins of T4 DNA replication, in Bacteriophage T4, C. K. Mathews, E. M. Kutter, G. Mosig, P. B. Berget, Eds. (America Society for Microbiology, 1983), pp. 111–119. [Google Scholar]
- 22.Black L. W., Rao V. B., Structure, assembly, and DNA packaging of the bacteriophage T4 head. Adv. Virus Res. 82, 119–153 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rao V. B., Feiss M., The bacteriophage DNA packaging motor. Annu. Rev. Genet. 42, 647–681 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Rao V. B., Black L. W., Structure and assembly of bacteriophage T4 head. Virol. J. 7, 356 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tao P., Mahalingam M., Marasa B. S., Zhang Z., Chopra A. K., Rao V. B., In vitro and in vivo delivery of genes and proteins using the bacteriophage T4 DNA packaging machine. Proc. Natl. Acad. Sci. U.S.A. 110, 5846–5851 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun S., Kondabagil K., Draper B., Alam T. I., Bowman V. D., Zhang Z., Hegde S., Fokine A., Rossmann M. G., Rao V. B., The structure of the phage T4 DNA packaging motor suggests a mechanism dependent on electrostatic forces. Cell 135, 1251–1262 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Sun L., Zhang X., Gao S., Rao P. A., Padilla-Sanchez V., Chen Z., Sun S., Xiang Y., Subramaniam S., Rao V. B., Rossmann M. G., Cryo-EM structure of the bacteriophage T4 portal protein assembly at near-atomic resolution. Nat. Commun. 6, 7548 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z., Sun L., Zhang Z., Fokine A., Padilla-Sanchez V., Hanein D., Jiang W., Rossmann M. G., Rao V. B., Cryo-EM structure of the bacteriophage T4 isometric head at 3.3-Å resolution and its relevance to the assembly of icosahedral viruses. Proc. Natl. Acad. Sci. U.S.A. 114, E8184–E8193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones D. L., Leroy P., Unoson C., Fange D., Ćurić V., Lawson M. J., Elf J., Kinetics of dCas9 target search in Escherichia coli. Science 357, 1420–1424 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.G. Mosig, Relationship of T4 DNA replication and recombination, in Bacteriophage T4, C. K. Mathews, E. M. Kutter, G. Mosig, P. B. Berget, Eds. (America Society for Microbiology, 1983), pp. 120–130. [Google Scholar]
- 31.Petrov V. M., Ratnayaka S., Nolan J. M., Miller E. S., Karam J. D., Genomes of the T4-related bacteriophages as windows on microbial genome evolution. Virol. J. 7, 292 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nolan J. M., Petrov V., Bertrand C., Krisch H. M., Karam J. D., Genetic diversity among five T4-like bacteriophages. Virol. J. 3, 30 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiro R., Shitrit D., Qimron U., Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol. 11, 42–44 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lemay M.-L., Tremblay D. M., Moineau S., Genome engineering of virulent lactococcal phages using CRISPR-Cas9. ACS Synth. Biol. 6, 1351–1358 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Martel B., Moineau S., CRISPR-Cas: An efficient tool for genome engineering of virulent bacteriophages. Nucleic Acids Res. 42, 9504–9513 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Molineux I. J., Panja D., Popping the cork: Mechanisms of phage genome ejection. Nat. Rev. Microbiol. 11, 194–204 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Wright A. V., Sternberg S. H., Taylor D. W., Staahl B. T., Bardales J. A., Kornfeld J. E., Doudna J. A., Rational design of a split-Cas9 enzyme complex. Proc. Natl. Acad. Sci. U.S.A. 112, 2984–2989 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tao P., Li Q., Shivachandra S. B., Rao V. B., Bacteriophage T4 as a nanoparticle platform to display and deliver pathogen antigens: Construction of an effective anthrax vaccine. Methods Mol. Biol. 1581, 255–267 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tao P., Mahalingam M., Rao V. B., Highly effective soluble and bacteriophage T4 nanoparticle plague vaccines against Yersinia pestis. Methods Mol. Biol. 1403, 499–518 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tao P., Mahalingam M., Kirtley M. L., van Lier C. J., Sha J., Yeager L. A., Chopra A. K., Rao V. B., Mutated and bacteriophage T4 nanoparticle arrayed F1-V immunogens from Yersinia pestis as next generation plague vaccines. PLOS Pathog. 9, e1003495 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/2/eaar4134/DC1
fig. S1. List of all possible single mutations in each spacer.
fig. S2. Relative fitness of the CEMs that escaped Cas9 cleavage of the protospacer 20-995 of the portal protein gene.
fig. S3. Temperature sensitivity of the CEMs containing amino acid changes.