

Abstract
Appropriate level of protein phosphorylation on tyrosine is essential for cells to react to extracellular stimuli and keep cellular homeostasis. Faulty operation of signal pathways mediated by protein tyrosine phosphorylation causes numerous human diseases, which presents enormous opportunities for therapeutic interventions. While the importance of protein tyrosine kinases in orchestrating the tyrosine phosphorylation networks and in target-based drug discovery has long been recognized, the significance of protein tyrosine phosphatases (PTPs) in cellular signaling and disease biology has historically been underappreciated, due to a large extent an erroneous assumption that they are largely constitutive and housekeeping enzymes. Here, we provide a comprehensive examination on a number of regulatory mechanisms including redox modulation, allosteric regulation, and protein oligomerization, in controlling PTP activity. These regulatory mechanisms are integral to the myriad PTP-mediated biochemical events and reinforce the concept that PTPs are indispensable and specific modulators of cellular signaling. We also discuss how disruption of these PTP regulatory mechanisms can cause human diseases and how these diverse regulatory mechanisms can be exploited for novel therapeutic development.
Graphical Abstract
1. Introduction
The phosphorylation of protein represents a major post-translational modification that serves an essential role in controlling protein function.1 Latest estimations based on phosphoproteomic analyses suggest that under the appropriate conditions the bulk of cellular proteins are phosphorylated at one or more amino acid residues.2 The level of protein phosphorylation is determined by the opposing actions of protein kinases and protein phosphatases, which catalyze the addition or removal of the phosphate to or from proteins. Variation in the extent of protein phosphorylation brings about changes in protein activity, protein localization, protein–protein interaction, and protein stability, enabling cells to respond precisely to alterations in their environment and control almost all cellular processes. Not surprisingly, aberrant protein phosphorylation is known to disrupt key cellular pathways and is a well-established feature of many human diseases. In the human genome there are greater than 500 protein kinases, of which approximately 90 are Tyr specific and the remaining kinases prefer either Ser or Thr residues.3 All protein kinases share a conserved three-dimensional structure for the kinase domain and catalyze the phosphoryl transfer reaction with the same chemical mechanism. Interestingly, nature has evolved multiple strategies for protein dephosphorylation, utilizing distinct families of protein phosphatases that employ different catalytic mechanisms.
The protein tyrosine phosphatase (PTP) superfamily has 103 enzymes4 that catalyze substrate dephosphorylation through a covalent enzyme intermediate, involving a thiophosphate linkage from the active site cysteine residue.5 Strikingly, the human genome encodes a relatively small number of catalytic subunits for the classical Ser/Thr protein phosphatases (15 for the PPP family and 16 for the PPM family), which hydrolyze phospho-substrates through a metal-dependent mechanism.6–7 The relatively smaller number of Ser/Thr catalytic subunits combined with their broad in vitro activity has led to the misconception that protein phosphatases lack substrate specificity. We now know that the PPP family of Ser/Thr protein phosphatases function as obligatory oligomers inside the cell by associating with one or two regulatory subunits that modulate their substrate specificity, intracellular localization and overall activity of the holoenzymes.8 For example, PP1 typically functions as heterodimeric holoenzymes of one catalytic subunit and one regulatory subunit. With three different PP1 catalytic subunits and more than 200 PP1 regulatory subunits already reported, there exists a diverse array of functionally distinct PP1 holoenzymes inside the cell.9 Similarly, PP2A usually works as heterotrimeric holoenzymes containing a structural scaffold A subunit, a regulatory B subunit, and a catalytic C subunit. Potentially, more than 70 heterotrimeric PP2A complexes can be generated by combinatorial association of the two A subunits, dozens of B subunits and two C subunits.10 Unlike the PPP phosphatases, the PPM family members are generally believed to operate as monomers. Although PPP and PPM phosphatases share no sequence homology, they exhibit a similar three-dimensional structure in the phosphatase domain and utilize an analogous catalytic mechanism with a dinuclear (Fe3+ and Zn2+) metal-activated nucleophilic water molecule for phosphate monoester hydrolysis.11 Recent studies indicate that there are more than 40 novel protein phosphatases having the haloacid dehalogenase (HAD) fold.12 Notable members of the HAD family protein phosphatases include Fcp and Scp, which remove phosphates from the C-terminal domain of RNA polymerase II,13–14 and the Eyes absent (Eya) family of transcriptional co-activators that are vital for cell proliferation and organ formation.15–17 Unlike other classes of protein phosphatases, the HAD phosphatase catalyzed dephosphorylation reaction proceeds through an aspartyl phosphoenzyme intermediate.18
As is apparent from the above discussion, the overall number of protein phosphatases in the human genome is comparable to that of protein kinases. Given the dynamic and reversible nature of protein phosphorylation, this may be a necessary prerequisite for the cell to fine-tune and limit the extent and duration of signaling events through the opposing activity of specific protein kinases and phosphatases. However, while the role of protein kinases in controlling protein phosphorylation has long been recognized, the importance of protein phosphatases in signaling has historically been underappreciated, due to a large extent an erroneous assumption that the corresponding protein phosphatase activity needed to modulate the protein phosphorylation status may largely be constitutive.6,19 This is particularly disappointing despite ample evidence documenting that protein phosphatases in their own right are specific and crucial regulators of signal transduction and function as essential partners with protein kinases to orchestrate cellular responses to a wide range of biological stimuli.20–21 For example, a large-scale RNA interference study22 identified a significant number of survival kinases, as expected (11% of the total), but also exposed a positive role for protein phosphatases in cell survival, with 32% of the total promoting survival. An additional 5% of the total were identified as ‘death phosphatases’ for their apparent roles as tumor suppressors. This study provides a powerful demonstration that regulation of protein phosphatase activity is a key point of control in maintaining cellular homeostasis. As an effort to counter the misguided belief that protein phosphatases are constitutive and housekeeping enzymes, we will focus this review on a number of regulatory mechanisms identified for the PTPs in controlling the pleiotropic roles that these enzymes play in cell signaling. We will also discuss how disruption of these PTP regulatory mechanisms causes human diseases and how these diverse regulatory mechanisms can be exploited for novel therapeutic development.
2. Protein Tyrosine Phosphatases
2.1. Classification of PTP Family Members
Encoded by 103 genes in humans, the PTPs are composed of a large family of phosphatases with their structural diversity and functional complexity rival those of protein tyrosine kinases (PTKs).4 Together with the PTKs, the PTPs regulate a plethora of cellular activities, including proliferation and differentiation, survival, migration, metabolism, and the immune response.21,23 Not surprisingly, perturbed activities of either PTKs or PTPs often results in abnormal protein tyrosine phosphorylation, which has been associated with the etiology of various human diseases, including cancer, diabetes, and autoimmune dysfunctions.24–26 Accordingly, cellular processes coerced by dysregulated protein tyrosine phosphorylation present unique opportunities for targeted therapeutic modality.27–28 Drug discovery and development endeavors on the PTKs have already produced over two-dozen small molecule inhibitors and numerous antibodies that are used in the clinic today,29 further substantiating the concept that dysfunctional signaling events regulated by protein tyrosine phosphorylation can be manipulated for novel medicine. Naturally, there is considerable interest in selectively modulating disease biology, through targeting of the PTPs, for novel therapeutic applications.30–33
The main structural feature that characterizes the PTPs is the active site sequence (I/V)HCXAGXGR(S/T), also named the P-loop, housed within a conserved catalytic domain.5 Amino acid sequence examinations and substrate specificity studies suggest that the PTPs may be classified into: 1) the classical pTyr specific PTPs, 2) the dual specificity phosphatases (DSPs), 3) the Cdc25 phosphatases, and 4) the low molecular weight (LMW) PTPs (Figure 1). The pTyr specific PTP subfamily comprises 38 members, which may be further categorized into intracellular (17) and receptor-like (21) PTP enzymes (Figure 1). The intracellular PTPs, represented by PTP1B and SHP2, harbor a PTP domain and a variety of amino or carboxyl terminal extensions with either targeting or regulatory properties.34–36 The receptor-like PTPs, typified by CD4537 and PTPα,38 normally possess an extracellular ligand-binding segment, followed by a transmembrane region, and one or two cytoplasmic PTP domains. The DSPs consist of 61 members with diverse substrate specificity. Many of the dual specificity phosphatases are capable of dephosphorylating pSer/pThr as well as pTyr residues.39 Interestingly, PTEN and Myotubularins are also able to remove 3 position phosphate from phosphatidylinositol 3, 4, 5-triphosphate (PIP3) and phosphatidylinositol 3, 4-bisphosphate (PIP2), respectively.40–41 Laforin, another member of the DSP subfamily, is a glycogen phosphatase.42 The Cdc25 phosphatases (Cdc25A, B, and C) are responsible for taking off the inhibitory phosphates from Thr14 and Tyr15 in cyclin-dependent kinases, which activates the kinases and drives cell cycle progression.43–44 With the exception of PTP signature motif, the Cdc25 phosphatases have no significant amino acid sequence similarity with the classical PTPs, DSPs, or LMW-PTPs. The PTP signature motif in Cdc25 is flanked by two CH2 domains (~25 residues) and the X-ray crystal structures of the Cdc25 phosphatase domain resembles rhodanese fold.45 The LMW-PTP, encoded by Acp1, is an 18 kDa cytosolic enzyme found ubiquitously in both prokaryotic and eukaryotic organisms.46 There is little sequence homology between LMW-PTP and other PTPs beyond the active site signature motif. Interestingly, the active site sequence in LMW-PTP is positioned near the N-terminus, whereas it is localized near the C-terminal end of catalytic domains in all other PTPs and DSPs.
Figure 1.

Classification and schematic representation of the PTP superfamily.
2.2. Key Structural Features of PTPs
Despite variation in primary structure and difference in substrate specificity, structural studies reveal that the catalytic domains in pTyr specific PTPs and DSPs are evolutionarily related and share a similar tertiary fold consisting of several α-helices decorated on both sides of a central β-sheet (Figure 2A).47–49 Moreover, major structural features, such as the active site P-loop and the general acid/base containing WPD loop, that are important for substrate binding and catalysis are conserved among all members of the PTP superfamily.50–52 The PTP active site is situated within a ~ 9 Å deep cleft for pTyr specific PTPs and a ~ 6 Å deep cleft for the DSPs (Figure 2B). The P-loop sequence (I/V)HCXAGXGR(S/T) sits at the bottom of the active site crevice. The inward orientation of the P-loop amides creates a positive electrostatic potential to accommodate the negatively charged phosphoryl group in the substrate. The bound phosphoryl moiety is further stabilized by a α-helix dipole, positioned NH2-terminal to the P-loop, and the active site Arg residue (Figure 2A). The PTP active site is formed by several surface loops, which contain residues important for substrate recognition and catalytic turnover. One of the surface loops (referred as the WPD loop in the pTyr specific PTPs, see Figure 2) holds an essential Asp residue (Asp181 in PTP1B), which is required for general acid/base catalysis.53 This Asp is usually positioned 30–40 residues upstream of the PTP signature motif (I/V)HCXAGXGR(S/T) in both pTyr specific and dual specificity phosphatases. One key structural feature for the classical pTyr specific PTPs is that substrate binding stimulates a pronounced conformational change of the WPD loop, which brings Asp181 into close proximity to the phenolic oxygen of the pTyr substrate in order to render general acid catalysis.48,54 A second loop (α1–β1 loop), called the pTyr recognition loop, contains residues that engage in specific interactions with the peptide backbone and pTyr in the substrate.54–56 Together with the P-loop, WPD-loop, and α5–α6 loop (also called Q-loop, Figure 2), the α1–β1 pTyr recognition loop surrounds the active site and defines a 9 Å deep pTyr-binding pocket (Figure 2B), which may provide the structural basis for the classical PTP’s specificity for pTyr.54,57 DSPs lack the corresponding α1–β1 loop, resulting in an approximately 3 Å shallower active site, which may explain why DSPs can dephosphorylate both pTyr and pSer/pThr.49,58
Figure 2.

Key structure features of PTPs depicted on the crystal structure of PTP1B. (A) Overall structure of PTP1B catalytic domain in ribbon mode. All α-helix and β-sheet are labeled, and the four loop fragments (i.e. P-loop, WPD-loop, Q-loop and pTyr-loop) constituting active site pocket are highlighted. Essential residues for catalysis are shown in stick and labeled. (B) The representation of active site pocket on the surface of PTP1B. The surface contributed by the P-loop, WPD-loop, Q-loop and pTyr-recognition loop are colored in red, green, magenta and blue.
2.3. PTP Catalytic Mechanism
Extensive mechanistic and biochemical investigations have demonstrated that all PTPs employ the same mechanism for catalysis.5 The PTP catalyzed substrate dephosphorylation proceeds via a two-step chemical mechanism (Figure 3) involving a covalent thiophosphoryl enzyme intermediate (E-P), which results from the nucleophilic attach on the phosphorus atom in the substrate by the side chain of the active site Cys residue (Cys215 in PTP1B).59–61 In the first chemical step, the formation of the phosphoenzyme intermediate E-P is aided by the general acid (Asp181 in PTP1B) to defuse the build-up of a negative charge on the ester oxygen of the leaving group.53,62 In the second chemical step, E-P hydrolysis is initiated by the general base Asp181 mediated nucleophilic attack by a water molecule, regenerating the free enzyme and yielding inorganic phosphate. The EP formation and hydrolysis steps are further catalyzed by the PTPs through preferentially stabilization of the transition states with the guanidinium group of active site Arg (Arg221 in PTP1B).63–65 Finally, kinetic and structural studies suggest that the conserved Gln in the Q-loop (Gln262 in PTP1B) is important for the precise positioning of both the general base Asp181 and the water nucleophile which are essential for optimal E-P hydrolysis.66–67 The overall catalytic mechanism of substrate dephosphorylation is shared by the PTPs, DSPs and the more distantly-related Cdc25 phosphatases and LMW-PTP.5,68
Figure 3.

The catalytic mechanism for PTP-mediated dephosphorylation reaction. The gray dash lines indicate H-bonds which position the phosphate group and nucleophilic water, the blue dash lines show broken/formed bond in the transition state, and the blue solid line represent newly formed bond.
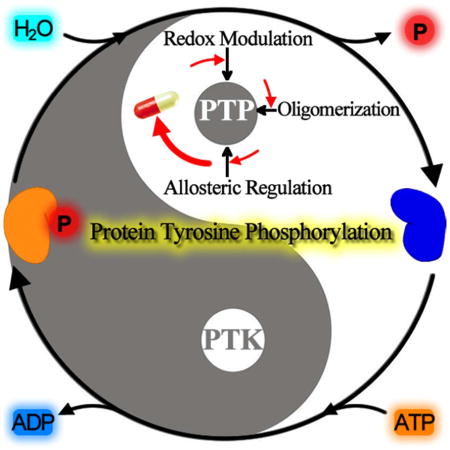
3. PTP Regulatory Mechanisms
To ensure proper level of protein tyrosine phosphorylation for cellular signaling, PTP activity is carefully organized through a number of regulatory mechanisms, including differential gene expression, confined subcellular localization, ligand binding, limited proteolysis, and post-translational modifications.69 PTPs can also be regulated through alterations in subcellular location, as exemplified by the observation that Cdc25C phosphatase can move between the cytoplasm and the nucleus, which appears to be an important regulatory mechanism.70 Here, we focus on additional regulatory mechanisms employed by the cell to modulate the intrinsic activity of PTPs. These include control of PTP activity by reversible active site Cys oxidation, allosteric mechanisms, and protein oligomerization. We will also discuss how inappropriate operation of these regulatory mechanisms may lead to pathological states and how they may be exploited for therapeutic purposes.
3.1. Redox Regulation
An increasingly appreciated mechanism of PTP regulation is reversible oxidation of the active site Cys through stimulus-mediated production of reactive oxygen species (ROS). Although initially regarded as toxic byproducts, controlled ROS production such as hydrogen peroxide (H2O2) is widely accepted as an important regulatory mechanism for cellular signaling processes.71–72 It has been demonstrated that engagement of cell surface receptors by ligands as diverse as insulin,73 EGF,74 PDGF75 or TGF-β76 induces rapid production of intracellular H2O2. Production of H2O2 requires NADPH oxidases in a regulated manner to govern cellular responses to a wide range of physiological stimuli (Figure 4).77 Importantly, it has been shown that intracellular H2O2 production is also required for growth factor-induced activation of receptor PTKs and tyrosine phosphorylation dependent cellular signaling. Evidence suggest that H2O2 can influence tyrosine phosphorylation-mediated cellular processes through transient and reversible oxidation of the PTP active site Cys (Figure 4).78–82 Indeed, reversible oxidation of the active site Cys thiol group by reactive oxygen species has been established as a viable mechanism of PTP regulation in a number of physiological contexts.83–86 Both cytosolic and receptor-like PTPs are known to subject to ROS mediated inactivation.
Figure 4.

Redox regulation of PTP activity by ROS. Intracellular ROS, including H2O2, O2·−, •OH and peroxidized lipids, converts PTP catalytic Cys to sulfenic acid and inactivates the PTPs. The sulfenic acid form could be further irreversibly oxidized to the sulfinic or sulfonic acid forms, or reversibly converted to a cyclic sulfenyl amide or intramolecular disulfide form.
As discussed above, an important feature for the PTP reaction is the involvement of the active site Cys for nucleophilic catalysis (Figure 3). Given the positively charged electrostatic potential in the PTP active site pocket, the pKa of the active site sulfhydryl group is ~5,87 which is substantially lower than a normal pKa for a typical Cys side chain in proteins (~8.5). Consequently, the sulfhydryl group within the PTP active site Cys must exist as a thiolate anion at physiological conditions. This not only increases the active site thiolate’s nucleophilicity but also makes it more susceptible to oxidation. Indeed, biochemical experiments show that the catalytic Cys can be converted to sulfenic acid (Cys-SOH) in the presence of H2O2, which effectively quenches the PTP reaction since the oxidized Cys-SOH is not capable of performing nucleophilic catalysis in PTP reaction (Figure 4).78,80,88 This provides a mechanistic basis for why H2O2 production promotes cell surface receptor mediated tyrosine phosphorylation. The resulting sulfenic acid could be rapidly reduced back to thiolate through reaction with small molecule cellular thiols89 or the thioredoxin system90, leading to full restoration of PTP activity. Accordingly, oxidation of the PTP active site Cys by H2O2 is transient and reversible and constitutes a dynamic regulatory mechanism of PTP activity.
For some time now, H2O2 has been viewed as the major ROS mediating reversible sulfhydryl oxidation within the cell. To that end, most investigations on PTP redox mechanisms have been carried out with H2O2. However it is important to recognize that, although H2O2 is able to oxidize the PTPs, it is not very reactive in chemical sense.91 Indeed, rate constants observed for H2O2-mediated PTP inactivation fall in the range of 10–20 M−1s−1,78,92–93 suggesting that PTP inactivation will be a rather slow process (t1/2 > 10 h) at the physiological concentrations of H2O2 (0.1–1 μM) thought to be existent upon growth factor stimulation.94–95 Strikingly, ROS-induced inactivation of PTP typically happens rapidly (2–5 min) inside the cell, which overlaps well with the temporary rise in H2O2 concentration upon exposure to growth factors.73,75,79,83 The disagreement between the kinetics for robust PTP inactivation observed during early cellular signaling events and the seemingly sluggish in vitro reactivity of H2O2 toward the PTPs may be reconciled if one assumes that H2O2 can undergo either spontaneous or enzymatic conversion to more reactive ROS capable of mediating rapid intracellular PTP inactivation (Figure 4).94–95 One recent study shows that the biological buffer bicarbonate/CO2 can potentiate H2O2’s ability to inactivate PTPs.96 The proposed mechanism involves oxidation of the active site Cys by peroxymonocarbonate produced by the reaction of H2O2 with HCO3/CO2 (Figure 4). Peroxymonophosphate (2−O3POOH) was also found an exceptional strong inactivator of PTP1B with a kinact/KI of 65,553 M−1 s−1, which is approximately 7,000 times more potent than H2O2.97 Similar to H2O2, PTP1B inactivation by peroxymonophosphate is also active site directed, and can be easily reversed by incubating the modified PTP1B with dithiothreitol. Another study suggests that hydroxyl radical, likely produced from Fenton reaction (H2O2 reduction mediated by Fe(II) or Cu(I)), may also serve as a more physiologically relevant ROS for effecting PTP inactivation (Figure 4).93 Indeed, hydroxyl radical was shown to inactivate PTPs through active site directed oxidation of the essential Cys to sulfenic acid, which can be reduced by small molecule thiol compounds. Importantly, it was shown that hydroxyl radical is kinetically more efficient than H2O2 to carry out oxidative inactivation of the PTPs. Thus, the second-order rate constants measured for the hydroxyl radical-induced PTP inactivation are at a minimum 2–3 orders of magnitude higher than those caused by H2O2 under the same reaction conditions. Hence at a physiologically relevant concentration of 1 μM H2O2, the half-life for the hydroxyl radical-induced PTP inactivation will be closer in time scale typically expected for cellular signaling, which takes place within 2–5 min upon growth factor stimulation. Other reactive oxygen species including superoxide anion (O2·−)80 and peroxidized lipids98 have also been implicated as physiologically relevant oxidizing agents in PTP regulation (Figure 4). Collectively, these studies establish redox regulation of PTP activity as a new layer of control over protein tyrosine phosphorylation mediated cellular events. From a kinetic perspective, either peroxymonophosphate or hydroxyl radical could serve as an endogenous hydrogen peroxide-derived redox regulator of cellular PTP activity under physiological or pathophysiological conditions. Further experiments are needed to conclusively identify the ROS responsible for the redox control of PTP activity in vivo.
3.2. Allosteric Mechanisms
3.2.1. MKP3 activation
A number of allosteric regulatory mechanisms have been reported for the control of PTP substrate specificity and phosphatase activity. A good case in point is how mitogen-activated protein kinase phosphatases (MKPs), which make a distinct sub-family of DSPs, recognize their substrates, the mitogen-activated protein (MAP) kinases. The MAP kinases are key players in relaying extracellular signals from cell membrane to the nucleus.99–101 There exist three main MAP kinase cascades: the ERK1/2 pathway which answers to stimuli that provoke cell proliferation and differentiation, as well as oncogenic transformation; the c-Jun N-terminal protein kinase (JNK) pathway; and the p38 pathway, both of which are stimulated in response to environmental stresses and cytokine mediated inflammations. While activation of protein kinases generally necessitates phosphorylation of a single amino acid within a structurally conserved activation loop,102 the MAP kinases are activated by phosphorylation of both the Thr and Tyr residues in the TxY motif (where x is Glu in ERK, Pro in JNK, and Gly in p38) within the activation loop.103–105 The MKPs are responsible for the inactivation of MAP kinases by removing the phosphate from both the pTyr and pThr in the MAP kinase activation loop. Earlier studies indicate that the MKPs exhibit distinct in vivo preferences for various MAP kinases as substrates.106 The best studied member of the MKP family is MKP3 (also known as DUSP6), which is known for its absolute substrate specificity for ERK1/2 over all other MAP kinases.107–108 Interestingly, MKP3 displays extremely low phosphatase activity towards small aryl phosphate substrates (e.g., kcat/Km =2.0 M−1s−1 for p-nitrophenyl phosphate and pTyr)109 and the bisphosphorylated peptide derived from the ERK2 activation loop (DHTGFLpTEpYVATR, kcat/Km =5.0 M−1s−1)110. Moreover, with the bisphosphorylated ERK2 activation loop-derived peptide as a substrate, MKP3 can only hydrolyze pTyr but not pThr. On the contrary, kinetic studies with the bisphosphorylated ERK2, the physiological substrate for MKP3, revealed that the kcat/Km value for the MKP3-catalyzed dephosphorylation of ERK2/pTpY (3.8 × 106 M−1s−1) is more than 106-fold higher than those measured for the hydrolysis of pNPP or the ERK2-derived bisphosphorylated peptide.111 Moreover, MKP3 can efficiently dephosphorylate both the pTyr and pThr in the activation loop of ERK2. In fact, the MKP3 catalyzed ERK2/pTpY dephosphorylation proceeds via an ordered, distributive mechanism in which MKP3 first associates with the bisphosphorylated ERK2/pTpY, removes the phosphate from the pTyr, and releases the monophosphorylated ERK2/pT, which then binds to a second MKP3 molecule for dephosphorylation, yielding the completely dephosphorylated ERK2. Given the vast difference in intrinsic reactivity (105-fold) between pTyr and pThr, it is quite noteworthy that MKP3 can promote pThr and pTyr hydrolysis with the same efficiency.
A series of biochemical and structural experiments were carried out to delineate the biochemical basis for the extraordinary substrate specificity of MKP3 for pERK2. The fact that small molecule and peptide substrates are weakly dephosphorylated suggests that substantial engagement of specific interactions between MKP3 and its physiological substrate pERK2 must occur. Like other members of the MKP subfamily, the MKP3 structure is comprised of an N-terminal regulatory segment harboring a kinase interaction motif (KIM) and a highly conserved C-terminal phosphatase domain.106 The crystal structure of the phosphatase domain of MKP3 revealed a distorted active site pocket that is incompatible for catalysis.58,112. Unlike other members of the PTP family, residues Cys293 and Arg299 within MKP3 active site are misaligned and the general acid Asp262 is found away from the catalytic site (Figure 5A). A critical clue came from the finding that ERK2 binding to MKP3 leads to a strong increase in MKP3 activity towards pNPP.113 Importantly, although the N-terminal domain of MKP3 is not needed for the MKP3-catalyzed pNPP reaction,110 it is required for ERK2 binding.114 To furnish biochemical understanding of the molecular basis for the ERK2 binding-induced MKP3 activation, detailed kinetic analyses of the MKP3-catalyzed aryl phosphate hydrolysis were conducted in the presence and absence of ERK2.109,115 Without ERK2, the hydrolysis of aryl phosphate substrates catalyzed by MKP3 shows little dependence on pH, viscosity, and leaving group properties. In the presence of ERK2, MKP3 exhibits increased phosphatase activity and higher affinity for oxyanions. Furthermore, general acid/base catalysis is restored for the MKP3 catalyzed reaction in the presence of ERK2. Taken together, the results indicate that, in the absence of ERK2, the rate-limiting step for the MKP3 catalyzed reaction probably corresponds to a substrate ERK2-induced conformational (allosteric) change in MKP3 that involves active site reorganization and closure of the general acid loop. ERK2 binding to MKP3 most likely serve to facilitate repositioning of key active site residues and accelerate WPD loop closure in MKP3 such that one of the chemical steps becomes rate-limiting. Binding of a nonphysiological substrate to MKP3 does not generate enough energy to trigger such a conformational reorganization within MKP3 for efficient catalysis. This rationalizes why MKP3 is unable to dephosphorylate pThr and displays weak activity toward the phosphopeptide derived from the ERK2 activation loop.
Figure 5.

Allosteric activation of MKP3 by MKP3-ERK2 interaction. (A) Distorted MKP3 active site in the X-ray crystal structure of the MKP3 phosphatase domain. (B) Key interaction elements presented in a structural model of MKP3-ERK2 complex. (C) Crystal structure revealed that a peptide from KIM sequence of MKP3 binds at the common docking site of ERK2, which consists of an acidic patch and a hydrophobic groove.
The studies described above indicate that conformational rearrangement within the MKP3 active site, induced by specific interactions between MKP3 and ERK2, is obligatory for MKP3 to attain full activity against pERK2. To define the structural basis for ERK2 recognition by MKP3 and ERK2 binding-induced MKP3 activation, systematic mutational and deletion analyses combined with X-ray crystallography and hydrogen-deuterium exchange mass spectrometry studies were executed. Results from mutational and biochemical experiments suggest that ERK2 recognition and ERK2-induced MKP3 activation involve multiple regions of MKP3.116 As expected, MKP3’s kinase interaction motif (KIM; residues 64RRLQKGNLPVR74) is a major contributor to high affinity binding to ERK2. In addition to KIM, a unique sequence motif in the middle of MKP3 also plays a role in binding ERK2. However, these two structural elements in MKP3 are not indispensable for the ERK2-induced MKP3 activation. A third binding site for ERK2 is found within the C-terminus of MKP3 (residues 348–381). Although C-terminus deletion or mutation of the putative ERK2 specific docking motif 364FTTP367 decrease MKP3’s affinity for ERK2 by only a modest amount, this C-terminal region is absolutely required for ERK2 binding-induced MKP3 activation. Similar analyses revealed that MKP3 activation by ERK2 also engages two distinct binding sites in ERK2.117 One site encompasses the so called common docking (CD) site (Figure 5B) in MAP kinases, which is comprised of several negatively charged residues (e.g., Asp316 and Asp319 in ERK2) that may interact with the positively charged residues in KIM through electrostatic interactions.118–120 It was shown that the common docking site, which is comprised of ERK2 residues Glu-79, Tyr-126, Arg-133, Asp-160, Tyr-314, Asp-316, and Asp-319, plays an important role in high affinity binding to MKP3 but is not essential for ERK2-provoked MKP3 activation. It turns out that ERK2-mediated activation of MKP3 requires ERK2 residues Tyr-111, Thr-116, Leu-119, Lys-149, Arg-189, Trp-190, Glu-218, Arg-223, Lys-229, and His-230 in the kinase substrate-binding (SB) region, which is distal to the ERK2 common docking (CD) site (Figure 5B).117
The three-dimensional structure of ERK2 in complex with the KIM sequence from MKP3 revealed that the common docking site (or the KIM docking site), situated in a non-catalytic segment opposite to the kinase active site pocket, is formed by a highly acidic patch and a hydrophobic groove, which are ideally positioned to interact with both the basic and Phi(A)-X-Phi(B) residues in the KIM motif (Figure 5C).121 Hydrogen-deuterium exchange mass spectrometry experiments showed that the KIM sequence in MKP3 and an MKP3-specific peptide fragment (101NSSDWNE107) associate with ERK2’s common docking site defined by amino acid residues in L16, L5, β7–β8, and αd-L8-αe, localized opposite to the kinase active site (Figure 5B). On top of this “tethering” effect, additional binding interactions between the 364FTTP367 motif in MKP3 and the ERK2 substrate-binding pocket, created by residues in the kinase activation lip, the P+1 site (β9-αf loop), L13 (αf–αg loop), and the MAP kinase insert (L14-α1L14-α2L14), are crucial for the ERK2-induced allosteric activation of MKP3 and formation of a productive enzyme-substrate complex by which the MKP3 active site is correctly juxtaposed to catalyze ERK2/pTpY dephosphorylation (Figure 5B).122 Collectively, the results demonstrate that specific interactions between the two proteins reside in structural elements that are remote from the active site of MKP3 and the ERK2 activation loop, through a bipartite recognition process. Accordingly, MKP3’s intrinsic substrate specificity is coupled to the ability of the ERK2 substrate to bring about productive reorientation of the MKP3 active site, which affords a powerful strategy to ensure high fidelity in down-regulating ERK2 activity. More recent crystal structure and NMR studies provide additional evidence on how residues outside the canonical KIM motif in MKPs interact with specific MAP kinases and contribute further to MAP kinase selectivity and signaling pathway fidelity.123–124
3.2.2 SHP2 molecular switch
The Src homology 2 (SH2)-domain containing PTP-2 (SHP2), encoded by the Ptpn11 gene, functions as a positive mediator in signal transduction.125–127 Results from biochemical and genetic studies reveal that SHP2 operates upstream of Ras, which functions as an essential signaling node that mediates growth factor-induced cell growth, migration, and survival.128–132 SHP2 is a ubiquitously expressed cytoplasmic PTP composed of two tandem SH2 domains at its NH2-terminus (N-SH2 and C-SH2), a central PTP catalytic domain, and a C-terminal tail (Figure 6A). Numerous studies show that the phosphatase activity of SHP2 is necessary for full activation of the Ras- ERK1/2 pathway.125,133 Interestingly, SHP2 is an allosteric enzyme modulated by an elegant ‘molecular switch’ regulatory mechanism (Figure 6A).35,134 SHP2 is auto-inhibited in the basal state by an intramolecular interaction between its N-SH2 domain and the PTP domain, which blocks the phosphatase active site for substrate binding.125,135 Upon growth factor receptor activation, the SHP2 N-SH2 domain binds specific pTyr motifs in receptor-PTKs, or more commonly scaffold proteins, which localize SHP2 to the vicinity of its substrates and simultaneously weakens the N-SH2/PTP domain interaction, enabling the PTP domain to bind and dephosphorylate SHP2 substrates.136–137
Figure 6.

Allosteric regulation of SHP2. (A) The schematic representation of SHP2 structure and allosteric regulation. (B) The comparison of N-SH2 domain conformation at I (gray) and A (cyan) state. (C) The pY peptide binding surface in N-SH2 domain at I and A state. BG- and EF-loop are depicted in purple and yellow, respectively. (D) The N-SH2/PTP interaction surface in the N-SH2 domain at the I and A state.
Structural analyses suggest that although the N-SH2 domain’s pTyr peptide-recognition site is distinct and remote from the binding interface between the N-SH2 and PTP domain, the two binding sites are linked via an allosteric switch associated with the two different N-SH2 domain conformations: an active (A) pTyr-containing peptide-bound state (Figure 6B&C, cyan) and an inactive (I) PTP domain bound state (Figure 6B&C, gray). In comparison with the crystal structures of the isolated N-SH2 domain, both with and without a bound pTyr peptide,138–139 the N-SH2 residues that interact with the SHP2 PTP domain (namely amino acids within β sheet βD’, βE, and βF, as well as helix αB) have experienced a concerted conformational movement of approximately 2 Å (Figure 6B). It appears that molecular motion of the αB helix in the PTP-binding site of N-SH2 can trigger a conformational change in the EF loop, which together with the adjacent BG loop makes the SH2 domain phosphopeptide binding site for residues C-terminal to the pTyr residue (Figure 6C). Consequently, the two N-SHP2 domain conformations have mutually exclusive properties. The I N-SHP2 domain conformation displays surface complementarity with the PTP catalytic site, but its phosphopeptide-binding site is distorted due to the shift of the EF loop towards the BG loop (Figure 6C). Conversely, in the phosphopeptide binding competent state (A) the PTP domain recognition surface (constituted by residues in βD’, βE, βF, and αB) is disrupted. This surface protrudes to and conflicts with PTP active site pocket at (A) state, thereby abolishing the N-SH2/PTP intramolecular interaction (Figure 6D). The allosteric conformation transition of the N-SH2 domain from the I to A state is regulated by pTyr-containing peptides derived from growth factor receptors or adapter proteins that bind to the N-SH2 domain. Thus binding of phosphopeptides to the N-SH2 domain promotes intramolecular “dissociation” of the N-SH2 domain from the PTP domain, shifting the SHP2 equilibrium to an open conformation in which the steric block of the phosphatase active site is relieved and SHP2 becomes catalytically active.
The importance of SHP2 in cell signaling is underlined by mutations in Ptpn11, which are linked to numerous human diseases. For example, germline mutations in Ptpn11 are associated with 50% Noonan syndrome (NS), an autosomal dominant developmental disorder manifested with features such as short stature, facial dysmorphia, and cardiac defects.140–141 Germline mutations in Ptpn11 are also found in 90% of LEOPARD syndrome (LS) cases.142 Both LS and NS are linked to heightened risk of malignancy.141,143–144 In addition, somatic mutations in Ptpn11 have been detected in patients with juvenile myelomonocytic leukemia (35%), acute myeloid leukemia (4%), myelodysplastic syndrome (10%), and acute lymphoid leukemia (7%).145–149 In addition to pediatric leukemia, Ptpn11 somatic mutations are also detected in adult acute myeloid leukemia (6%) and in solid tumors ranging from lung adenocarcinoma, melanoma, neuroblastoma, hepatocellular carcinoma and colon cancer.150–151 SHP2 has also been implicated in gastric carcinoma inflicted by Helicobacter pylori, which contains a virulence factor CagA, which, upon tyrosine phosphorylation by host Src kinases, can provoke SHP2 activation.152 Finally, due to the universal requirement of SHP2 in multiple receptor tyrosine kinase-mediated signal pathways, blocking SHP2 phosphatase activity may serve as a novel approach for malignancies caused by aberrant activation of receptor tyrosine kinases, some of which respond poorly to tyrosine kinase targeted monotherapy.153 Together, the existing biochemical and genetic evidence strongly identify SHP2 as the first bona fide oncoprotein in the PTP superfamily. There is increasing interest in therapeutic targeting of SHP2 for novel anti-cancer agents.32
Most of the NS or neoplasia-associated Ptpn11 mutations alter residues clustered at the N-SH2 and PTP domain binding interface (Figure 7A).150,154 These substitutions were thought to alter the allosteric mechanism of SHP2 to favor the activated state, resulting in gain-of-function properties.155–159 Indeed, studies have shown that these Ptpn11 mutations induce leukemia through cell-autonomous mechanisms that depend on SHP2 phosphatase activity.160–163 In contrast, LS associated Ptpn11 mutations have been found exclusively in the PTP phosphatase domain (Figure 7B). Recent experiments indicated that LS Ptpn11 mutations manifest in significantly reduced SHP2 phosphatase activity and are therefore regarded as loss-of-function genetic defects.159,164–166 These biochemical and genetic observations created a conundrum: how can Ptpn11 mutations with opposite effects on SHP2 phosphatase activity elicit similar overlapping phenotypes? The answer to this question came from the realization that disease-causing Ptpn11 mutations modify not only the phosphatase activity of SHP2 but also its molecular switch regulatory mechanism that could dictate phenotypic outcome.167–168
Figure 7.

Disease associated SHP2 mutations. (A) NS/cancer-associated SHP2 mutations mainly reside at the interface of N-SH2 and PTP domains. (B) LS-associated SHP2 mutations only appear within the PTP domain.
Detailed kinetic studies167–168 show that the full-length SHP2 phosphatase activity is less than 5% of the that of its catalytic domain, which is consistent with the finding from SHP2 crystal structures that SHP2 exists in an autoinhibited, closed conformation.35 In line with the expectation that gain-of-function SHP2 mutants exist in an open, constitutively active state, full-length D61Y and E76K proteins have kinetic parameters indistinguishable to those of the catalytic domain of SHP2. By contrast, the phosphatase domains of LS SHP2 mutants display kcat values that are 10- to 930-fold lower than that of the wild-type SHP2 counterpart, suggesting that LS mutants may be catalytically compromised. Importantly, the turnover numbers for full-length LS mutants are still 2 to 26-fold lower than those of their corresponding phosphatase domain alone.167–168 These results suggest that, similar to the wild-type SHP2, full-length LS mutants are also in a closed, autoinhibited conformation. Given the diverse nature of the SHP2 LS mutations, three-dimensional structures for all major LS mutants (Y279C, A461T, G464A, T468M, Q506P, and Q510E) were determined in an effort to fully delineate the molecular underpinnings for the disease.167–169 These crystal structures reveal why LS-causing SHP2 mutants have reduced phosphatase activity (Figure 8). The structural observations indicate that catalytic impairments induced by the LS mutations range from decreased substrate-binding to misalignment of active site residues and diminished transition state stabilization. Interestingly, the structures also show that, like the wild-type enzyme, all LS mutants are found in an auto-inhibited, closed conformation, in agreement with the prediction based on results from the kinetic experiments. Importantly, although the LS mutants adopt a closed conformation, these structures also disclose that the LS mutations weaken the autoinhibitory structure between the interfaces of the N-SH2 and PTP domains. Additional biophysical and computational analyses suggest that in comparison to native SHP2, the LS mutants display a greater tendency to switch from an autoinhibited closed conformation to a catalytically competent open state.167–168
Figure 8.

LS SHP2 mutations reduce SHP2 phosphatase activity by disturbing different step(s) in the catalytic process. In this figure, SHP2 wild-type (gray) and mutant (green) were superimposed onto PTP1B (cyan) structure representing transition state 1 or 2 to show mutation-induced disturbance at each specific step. Residue numbers are shown in blue for PTP1B and black for SHP2. Red dash lines represent mutation induced steric conflicts.
Based on thermodynamic principles and the predicted negative cooperativity between the two ligand binding sites (for pTyr peptide and the PTP domain) in the N-SHP2 domain, it follows that diminished interaction between the N-SH2 domain and the PTP domain must necessarily lead to increased association between the N-SH2 domain and its pTyr peptide ligands. Indeed, LS mutants exhibit greater affinity for upstream pTyr peptide motifs and are preferentially (as compared to native SHP2) activated by growth factor receptors or scaffolding proteins (Yu et al., 2013; 2014). Moreover, SHP2-associated LS mutants indeed display enhanced binding affinity for Grb2-associated binder-1 (Gab1)170 and stay longer on the scaffolding protein.167–168 Via its N-SH2 domain association with the pTyr-sequence motif in Gab1, SHP2 is directed to its physiological substrate(s) to drive Ras-ERK1/2 pathway activation.171–172 Accordingly, the LS mutants are more efficient in dephosphorylating Paxillin/pY118,167–168 a physiological substrate of SHP2 required for EGF-stimulated ERK1/2 activation.173 SHP2/Gab1 binding and SHP2-mediated dephosphorylation of Paxillin/pY118 are two recognized signaling events shown to be important for EGF-induced ERK1/2 phosphorylation.171,173 Given the LS mutants’ higher affinity for Gab1 and increased Paxillin/pY118 dephosphorylation by the LS SHP2 mutants, LS mutations are expected to engender a gain-of-function impact on ERK1/2 activation. Indeed, compared to cells expressing native SHP2, the ERK1/2 activity is more elevated and sustained after EGF stimulation in cells expressing the LS mutants.167–168 Furthermore, the biophysical outcome of the weakened association between the N-SH2 domain and the PTP phosphatase domain in the LS mutants is the augmented ability of the N-SH2 domain to bind the pTyr-peptide motifs in their physiological adaptor proteins under much lower concentrations of stimuli, thus effectively reducing the threshold for growth factor induced Ras/ERK1/2 pathway activation. As expected, the LS-associated SHP2 mutants induced ERK1/2 activation require considerably lower EGF concentration, which could confer significant developmental advantage under growth factor limiting conditions.167–168 Additional studies in Drosophila reveal that ubiquitous expression of SHP2/Y279C and SHP2/T468M, two of the most frequent LS-causing SHP2 alleles, leads to gain-of-function phenotypes due to heightened EGF mediated Ras-ERK1/2 pathway activation.174 These gain-of-function phenotypes observed for the LS mutants are similar to those observed with transgenic flies harboring NS-associated SHP2 mutants.158 Notably, experiments with SHP2/Y279C or SHP2/R468M transgenic fly suggest that LS mutant’s residual phosphatase activity is required for the gain-of-function developmental phenotypes.174 Expression of the most common LS-associated SHP2 mutants in embryos of zebrafish results in hyperactivation of ERK1/2 signaling, which leads to defective early heart development.175 Collectively, these structural and functional investigations provide strong evidence that catalytically deficient SHP2 mutants could still display gain-of-function phenotypes since they could preferentially engage upstream activator proteins with extended resident times, thus enabling prolonged substrate dephosphorylation by the mutant phosphatases and sustained activation of the Ras-ERK1/2 pathway.
3.3 Protein Oligomerization
3.3.1. Dimerization of transmembrane receptor-like PTPs
Protein dimerization forms the molecular basis for ligand-dependent catalytic activation of many receptor PTKs.176 In addition to receptor PTKs, an emerging mechanism for controlling the activity of intracellular protein kinases is to switch between inactive and active states by allosteric coupling with kinase domain surface residues involved in homo- or hetero-dimerization.177 Upon realization that resistance to Raf kinase-targeted therapeutics often stems from dimerization-dependent mechanisms,178 there is huge interest in gaining a better understanding of dimerization-induced allostery in order to develop improved therapeutic strategies.177 In the case of PTPs, protein oligomerization also represents an important regulatory mechanism, which was first recognized for transmembrane receptor-like PTPs. In addition to a ligand binding N-terminal domain situated outside the cell and a transmembrane segment, the majority of receptor PTPs also contains two cytoplasmic PTP domains. The PTP domain closer to the plasma membrane, named D1, harbors most, if not all, of the catalytic activity, while the plasma membrane distal second PTP domain, D2, is predominantly inactive but appears to be important for the function of the receptor PTP as a whole.179–180 In an analogous manner to receptor PTKs, the phosphatase activity of receptor PTPs can also be regulated by ligand-induced dimerization. A model linking ligand induced dimerization and receptor PTP phosphatase activity was suggested based on the three-dimensional structure of the membrane-proximal D1 domain of PTPα.181 In the X-ray crystal structure, the plasma membrane-proximal PTPα D1 domain forms a symmetrical dimer, in which a structurally restricted N-terminal helix-turn-helix ‘wedge’ from one monomer was found inserted into the active site pocket of the second (Figure 9). This observation let to the proposal that the phosphatase activity of receptor PTPs is inhibited in the dimeric state due to reciprocal occlusion of the respective active site pockets.181 Interestingly, the X-ray crystal structures of the tandem D1 and D2 domain constructs of LAR182 and CD45183 showed no evidence of dimerization even though the wedge motif was also present in these structures.182–183 Moreover, the D1 and D2 domains were positioned in such a way that both of their active site pockets were accessible. Indeed, the steric hindrance manifested by the D2 domain precludes the wedge-mediated D1 domain dimerization. Nonetheless, these structural observations may not accurately reflect the situation inside the cell since the relative D1 and D2 orientation and the accessibility of D1 domain active site to the helix-turn-helix wedge motif could be influenced by conformational changes in other parts of the molecule upon binding of a ligand to the extracellular fragment of the receptor PTP. In fact, biochemical and cell-based analyses of PTPα revealed that multiple structural elements in the D2 domain, the extracellular segment, and the transmembrane region make contribution to dimer formation inside the cell.184–185
Figure 9.

Dimeric structure of PTPα D1 domain. The N-terminal helix-turn-helix ‘wedge’ and catalytic Cys are highlighted in green and red respectively.
Using reconstitution of TCR signaling186 or rescue of Src family kinase activity187 as readouts, it was shown that forced dimerization of mutant receptor PTPs results presents loss of function phenotypes. Moreover, alteration of residues in the wedge motif attenuated dimerization-induced inhibition, indicating that this motif is important for the inhibitory effect of receptor PTP dimerization.187–188 Results from measurements using fluorescence resonance energy transfer (FRET) microscopy and cross-linking of the receptor inside the cell suggest that dimerization of PTPα is controlled not only by the wedge motif in D1 but also by structural features in the transmembrane and ectodomains.184–185 Furthermore, the D2 domain is also shown to play a role in regulating receptor PTP dimerization.184,189 Also, there is ample data supporting that dimerization of receptor PTPs occur inside cells and that receptor PTP dimerization inhibits the phosphatase activity.190–193 A clear example of receptor PTP regulation by a physiological extracellular molecule is furnished by the observation pleiotrophin inhibits the intrinsic phosphatase activity of its receptor, PTPbeta/zeta.194 Taken together, it appears that dimerization represents an important regulatory mechanism for receptor-like PTPs. Further studies will shed more light on the potential functional relevance of receptor PTP dimerization and of D1-wedge interactions.
3.3.2. Oligomerization of Intracellular PTPs
Recent studies suggest that protein oligomerization may also represent an emerging mechanism for the regulation of intracellular PTPs. The dual specificity phosphatase VH1 (Vaccinia virus H1 gene product) can attenuate host cell antiviral response by catalyzing STAT1 dephosphorylation. The VH1 crystal structure shows a novel dimeric arrangement (Figure 10A), with the two active sites positioned ~39 Å from each other.195 It appears that VH1 dimer formation involves a domain swap of the N-terminal helix and that specific recognition of STAT1 by VH1 requires the dimeric structure, which prevents STAT1 nuclear translocation, thereby blocking INFγ-mediated antiviral responses.196 The Vaccinia H1-related (VHR) phosphatase is a member of the DSP family shown to be important for the progression cell-cycle and tumorigenesis.197 Functional analysis of the “variable insert” unique to VHR by incorporating para-benzoylphenylalanine, a photo-cross-linkable amino acid, led to the discovery that VHR can dimerize inside cells.198 Further experiments indicate that VHR dimerization can lead to occlusion of its active site, blocking substrate accessibility. Evidence suggests that the glycogen phosphatase laforin can also form a dimer (Figure 10B), although the biological relevance of laforin dimerization is still the subject of active debates.199–202 The three-dimensional structure, along with complementary biophysical data, suggests that laforin can exist as an antiparallel dimer, which is mediated by residues in its phosphatase domain.203 Importantly, it was shown that the Lafora disease associated mutation F321S can interrupt dimerization of laforin and reduces its phosphatase activity toward glycogen. Myotubularin related protein 2 (MTMR2), a phosphoinositide lipid phosphatase, can also form as a homodimer (Figure 10C), which is important for its function inside the cell.204 Interestingly, while the laforin phosphatase domain contributes to dimer formation, the dimer interface for MTMR2 and VH1 phosphatases is formed via ancillary C-terminal coiled-coil domain and a swapped N-terminal α helix, respectively.196,204
Figure 10.

Dimeric structures of (A) VH1, (B) laforin, and (C) MTMR2. In the MTMR2 dimer presentation, the coiled-coil domain is not observed in the crystal structure and delineated as a cylinder to show its relative location.
The importance of protein homodimerization in regulating PTP activity was further emphasized by the recent findings that homodimerization of PTEN is important for its phosphatase activity toward the physiological substrate phosphatidylinositol 3, 4, 5-triphosphate.205 As a major tumor repressor, malfunction PTEN has been implicated in the pathogenesis of both hereditary and sporadic cancers.206 Strikingly, several cancer-associated catalytically impaired PTEN mutants were shown to form heterodimers with native PTEN and hamper its catalytic activity in a dominant-negative fashion.205 As a consequence, cells and tissues expressing heterozygous PTEN(C124S/+) and PTEN(G129E/+) mutants display increased sensitivity to PI3K/Akt pathway activation in comparison to native and PTEN(+/−) heterozygous counterpart. In addition, catalytically inactive PTEN mutant knock-in mice are found to be more prone to tumor formation and exhibit phenotypes reminiscent of complete loss of PTEN. Further structural and biophysical studies reveal that PTEN can form homodimers in solution, which is stabilized by the C-terminal tail, and that C-terminal tail phosphorylation interferes with this stabilization.207 These results provide new insight into the cellular regulatory mechanism of PTEN phosphatase activity.
Finally, PRL (Phosphatase of Regenerating Liver) phosphatases provide another striking example where protein oligomerization may play a crucial role in regulating PTP function. PRLs constitute a novel class of C-terminal prenylated phosphatases within the PTP superfamily, with three closely related members (PRL1, 2 and 3, also referred to PTP4A1, PTP4A2, and PTP4A3).208–211 PRL1 was initially discovered as an immediate early gene induced upon partial hepatectomy.212 PRL2 and PRL3 were subsequently discovered by their sequence similarity to PRL1. Unlike many other PTPs that antagonize the action of protein kinases, the PRLs rank among the most oncogenic PTPs and function to promote activation of both ERK1/2213–217 and Akt218–221, two of the key pathways that are abnormally up-regulated in cancer. PRLs are overexpressed in many cancer cell lines, and those with high level PRL expressions show increased proliferation, migration and anchorage-independent cell growth.212–213,222–225 Moreover, cells overexpressing PRLs form tumors in mice with high metastatic potential,215,226–228 whereas reduction in PRL expression decreases cell proliferation and migration as well as tumorigenesis in vivo.215,217,229–233 Importantly, PRL level is elevated in various human cancers where PRL overexpression is strongly correlated with late stage metastasis as well as poor clinical outcomes.224,230,234–241 Recent functional studies using engineered genetic mouse models indicate that deletion of PRL2 or PRL1 causes placental insufficiency, hematopoietic stem cell renewal, and spermatogenesis due to increased PTEN level and decreased Akt activity.220–221,242–243 The ability of PRLs to activate Akt and down-regulating the tumor repressor PTEN provides a biochemical basis for their oncogenic potential. Taken together, the current data implicate PRLs as novel molecular markers and therapeutic targets for metastatic cancers. Consequently, PRLs have garnered considerable interest for drug discovery.211
Early structural studies uncovered that PRL1 crystallizes as a homotrimer (Figure 11).92,244 The PRL1 homotrimer features a three-fold rotational symmetry with the C-terminal tail of each monomer facing the same side of the trimer (Figure 11A). Since the C-terminal tail of PRL contains a prenylation motif, it is most likely that the side of the trimer facing the plasma membrane is made up by the C-residues. Accordingly, the active site of PRL1 is pointed away from the trimer interface and opposite to the C-terminal plasma membrane binding region (Figure 11A&B). PRL1 trimer formation is mediated by both polar and hydrophobic interactions. The sum of buried accessible surface area at the dimer interface is 1130–1150 Å2 for each monomer, which account for ~29% of the whole surface area of each PRL1 monomer.92 In the PRL1 homotrimer structure, helix α5 of one PRL1 monomer (amino acids 124–134) makes contacts with helix α1, the β1 – β2 hairpin, and the α3 – β5 loop from another monomer (Figure 11C). Remarkably, Arg138 in helix α5 inserts into the hydrophobic pocket formed by Lys39 and Tyr40 in helix α1, making a specific salt bridge with Glu36. On the other hand, Pro96 from the α3 – β5 loop protrudes into a hydrophobic pocket bordered by residues Leu146 in helix α6, Val130 and Tyr126 in helix α5 (Figure 11D). Additional interactions include six hydrogen bonds made by residues Asp128, Gln131, Arg134, and Gln135 with main chain amides or carbonyls in the α3 – β5 loop, the β1 – β2 hairpin, and the side chain of Arg18. Since residues (Arg18, Glu36, Pro96, Met124, Tyr126, Asp128, Val130, Arg138, and Leu146) observed at the dimer interface are conserved among all PRLs, trimer formation is likely a general property for all PRL phosphatases.
Figure 11.

Trimeric arrangement of PRL1 in the crystal structure. (A) Overall structure of the PRL1 trimer in two orientations. In each monomer, the secondary structures are labeled; the P-loop and C-terminal tail are highlighted in red and blue, respectively. (B) Surface representation of the PRL1 trimer. Two dimer interfaces in monomer b are highlighted in purple (ba-interface) and yellow (bc-interface), and all residues involved in these interfaces in monomer b are listed accordingly. The close-up view of key interactions are respectively shown in (C) and (D) for ba- and bc-interface.
Subsequent biochemical experiments established that all PRLs have the ability to form trimers in solution and inside the cells.92,214,216,244 The PRLs are known to be prenylated at CaaX motif in their C-termini,222,245–246 which serves to attach them to plasma membrane. This anchoring effect is expected to increase the local PRL concentration in cell membrane and significantly enhance the propensity of PRL trimerization. Thus PRL1 homotrimers observed in the crystal structures may have direct relevance to intact PRL1 inside the cell since its C-terminal prenylation motif sits on the membrane-facing surface.92 Consequently, PRL1 trimerization may operate as a novel regulatory mechanism for PRL function. Indeed, it has been shown that trimer formation is required for PRL-mediated cell proliferation and motility because disruption of PRL trimer formation by mutations of residues in the dimer interface results in a loss-of-function phenotype.214,247 Overexpression of PRLs, as observed in numerous cancer cell lines, is expected to escalate trimer formation. Thus it is possible that the increased proliferation and metastasis observed in cancer cells with elevated PRL expression is the consequence of increased PRL trimerization.
4. Therapeutic targeting of PTP regulatory mechanisms
4.1. Targeting the oxidized active site Cys
Selective oxidation of the active site thiolate in PTPs by ROS initially produces the sulfenic acid form (Cys-SOH), but high concentrations of oxidant can cause irreversible generation of sulfinic (Cys-SO2H) or sulfonic acid (Cys-SO3H) derivatives (Figure 4). To prevent irreversible oxidation, the sulfenic acid product can form covalent adduct with residues in close proximity of the catalytic Cys. For example, it has been shown that sulfenic acid is capable of reacting with a backbone amide to yield a cyclic sulfenyl amide88,248–249 or with an adjacent thiol to form an intramolecular disulfide92,250 (Figure 4). These secondary reaction adducts can be reversed by reduction, and serve as stabilizing mechanisms to shield the active site Cys from further oxidation. Interestingly, the ability of the reversibly oxidized active site thiol group to transform into a cyclic sulfenyl-amide intermediate presents an exciting opportunity to target the PTPs. Structural studies reveal that this sulfenyl-amide formation induces a large conformational change in the active site of PTP1B which transiently blocks substrate binding and catalysis.88,249 There is considerable interest in targeting PTP1B for diabetes and obesity treatment, given that fact that PTP1B deficient mice are healthy, exhibit increased insulin sensitivity, and do not develop obesity when they are fed with high-fat diet.251–252 To demonstrate whether stabilization of the oxidized form could be a viable approach for therapeutic targeting of the PTPs, single-chain variable antibody fragments that specifically recognizes the cyclic sulfenamide form of PTP1B were generated against the active-site double mutant PTP1B-C215A/S216A, which adopts a conformation similar to the oxidized PTP1B.253–254 When expressed in mammalian cells, clone scFv45 immunoprecipitated oxidized PTP1B after H2O2 or insulin stimulation and colocalized with the oxidized PTP1B. Importantly, expression of scFv45, but not a non-targeting intrabody, promotes insulin-induced tyrosine phosphorylation of the insulin receptor, its substrate IRS1, and downstream Akt phosphorylation.253 These results provide the needed proof of principle that strategies to stabilize the oxidized, inactive PTP1B could present a new therapeutic approach to target PTP1B. To that end, a number of small molecule compounds, including sulfone-stabilized carbanions and 1,3-diketone derivatives (Figure 12), have been identified for covalent capture of the cyclic sulfenamide intermediate in oxidized PTP1B.255–256 These sulfone carbon acids could be used as building blocks for the construction of therapeutic agents that inhibit PTP1B phosphatase activity via transient trapping of the cyclic sulfenamide produced during growth factor signaling. In addition, the dimedone scaffold (Figure 12) when incorporated into an appropriate binding module has also been demonstrated to exhibit selectivity for the sulfenic acid generated in PTP active site.257 More recently, a novel immunochemical approach consisting of an antibody designed to recognize the conserved sequence of the PTP active site (VHCDMDSAG) harboring the catalytic cysteine modified with dimedone has been developed to directly profile oxidized PTPs.258 It is anticipated that future work in this area will lead to novel therapeutic molecules that can covalently capture the oxidized forms of PTPs generated inside the cell under disease conditions.
Figure 12.

Structures of small molecule PTP inhibitors discussed in this review, which target the regulatory mechanisms unique to each PTP.
4.2. Targeting allosteric activation of MKP3
In order to develop an improved understanding of the roles of MKPs in health and diseases, potent and selective small molecule probes will be invaluable. Unfortunately the shallower active sites for the DSPs have evaded many traditional high throughput screening and medicinal chemistry efforts. A zebrafish based chemical screen led to the identification of a small molecule inhibitor for MKP3, named (E)-2-benzylidene-3-(cyclohexylamino)-2,3-dihydro-1H-inden-1-one (BCI) (Figure 12), which blocks MKP3 catalyzed dephosphorylation of a small aryl phosphate substrate in the presence of ERK1/2 and enhances the expression of fibroblast growth factor target genes in zebrafish embryos.259 Further structure and activity study and molecular modeling suggest that the BCI series of molecules inhibit MKP3 by binding the crevice located between helix α7 and the general acid loop in MKP3, preventing the re-positioning of Asp262 to act as a general acid during catalysis.260 Treatment of patient-derived pre-B acute lymphoblastic leukemia cells with BCI resulted in ERK1/2 hyperphosphorylation and induced cell death, suggesting potential for targeting MKP3 in ALL indications.261 These MKP3 inhibitors can be useful in helping reveal the role of MKP3 in normal physiology and in diseases.
4.3. Targeting the SHP2 molecular switch mechanism
The above discussion on SHP2 highlights the importance of the allosteric mechanism, namely the intramolecular switch between its N-SH2 domain and the PTP domain, not only in controlling SHP2-mediated signaling under normal physiological conditions but also in pathogenic settings where the molecular switching mechanism is perturbed. Recent progress in targeting SHP2 for drug discovery suggests that the unique allosteric mechanism may also be exploited to develop compounds that selectively inhibit SHP2 relative to other PTPs for therapeutic purposes.262–263 Utilizing the full-length SHP2 construct and screening for compounds that can block SHP2 activation by an N-SH2 domain binding pTyr peptide, the Novartis team identified several small molecule compounds that stabilize the auto-inhibited SHP2 conformation.263 Medicinal chemistry optimization afforded SHP099 (Figure 12)), which has a Kd of 70 nM for SHP2 and exhibits no significant activity against other PTPs. The three-dimensional structure of SHP2 complexed with SHP099 shows that the inhibitor binding to a tunnel-like region at the intersection of the C-SH2, N-SH2, and PTP domains (Figure 13A). In essence, SHP099 acts like “molecular glue” to lock SHP2 in the inactive conformation and prevents its enzymatic activation. In receptor PTK-driven cancer cells SHP099 was shown to suppress Ras–ERK1/2 signaling and attenuate cell proliferation.262 SHP099 was also efficacious in mouse tumor xenograft models.262 These results with the allosteric inhibitor SHP099 together with similar results generated with active site directed SHP2 inhibitors264–267 provide the proof-of-concept that pharmacological modulation of SHP2 activity represents a promising therapeutic intervention for cancer treatment.
Figure 13.

The binding modes for two allosteric PTP inhibitors. (A) SHP099 binds at the inter-domain interfaces of SHP2 to stabilize the autoinhibited conformation. (B) Analog 3 binds at the trimer interfaces of PRL1 to prevent trimer formation.
4.4. Targeting receptor PTP dimerization
The potential impacts of receptor PTP dimerization on its phosphatase activity raise the prospect of developing small molecules that can either stabilize or disrupt the dimeric structure.268 The recent finding that a monoclonal antibody targeting the extracellular regions of CD148/DEP-1 can block endothelial cell proliferation and angiogenesis serves as an example for this approach.269 Based upon the dimerization model of regulation, several ‘wedge domain’ peptides have been identified as inhibitors of receptor PTPs.270 A more recent case is provided by targeting PTPσ, a receptor for chondroitin sulphate proteoglycans, with a cell-penetrating wedge peptide that can bind PTPσ and lessen the chondroitin sulphate proteoglycans-induced inhibition. Administration of the PTPσ cell-penetrating wedge peptide mimetic inhibits PTPσ promoted innervation and functional restoration in mice following spinal cord injury.271–272 Treatment with the PTPσ cell-penetrating wedge peptide mimetic also promotes cardiac innervation in a mouse model of myocardial infarction.273 Finally, in silico screening for compounds predicted to bind the interface between CD45’s D1 and D2 domains led to compound 211 (Figure 12).274 Compound 211 increased Tyr394 phosphorylation in Lck, a CD45 substrate in Jurkat, but not CD45-null (J.45) cells, and blocked early and proximal TCR signaling.274 Compound 211 also dose-dependently reduced inflammation in the delayed-type hypersensitivity mouse model.274
4.5. Targeting PRL3 trimerization
The PRL phosphatases have recently emerged as compelling molecular targets for cancer drug discovery. However, the fairly flat PRL active site pocket and its structural similarity to other PTPs pose significant challenge for PRL inhibitor development. Indeed, active site directed PRL inhibitors disclosed from the literature are neither sufficiently potent nor selective, and thus are not appropriate for in vivo functional study and drug development.211 Recent reports suggest that intracellular PTPs such as PRLs, can be targeted with antibodies.275–277 Given the functional requirement of PRL trimerization, pharmacologic disruption of PRL trimerization represents an innovative approach for the treatment of human cancers with elevated PRL expression. By targeting the unique, noncatalytic trimerization interfaces that are not related to any other PTP family members, such PRL detrimerizers would be highly specific to the PRL. Starting from a stepwise computer-based virtual screening approach, Cmpd-43 (Figure 12) and several of its analogs were identified that are capable of preventing PRL trimerization.247 Biochemical and structural studies establish that Cmpd-43 can effectively block PRL1 trimerization. Interestingly, PRL1 crystallized as a monomer in the presence of Analog-3 (Figure 12), a close derivative of Cmpd 43.247 The crystal structure of PRL1-Analog-3 shows that the compound makes direct contact with residues Tyr14, Met124, Phe132, Asp128, Thr13, Tyr14, Lys15, Asn16, and Arg159 in the trimer interface of PRL1 and obstructs its trimerization (Figure 13B). Cmpd-43 can also specifically attenuate the activation of both ERK1/2 and Akt and inhibit PRL-induced cell proliferation and migration. Importantly, Cmpd-43 exhibits excellent anti-cancer efficacy both in tumor cell lines and in a melanoma xenograft mouse model.247 This study provides pharmacological validation that trimerization is important for PRL1 function and that targeting PRL trimerization is a viable approach for therapeutic development.
5. Concluding Remarks
We have made great strides in delineating the biological functions of a number of PTP family members.20,24,278 These PTPs have been demonstrated to control a wide range of physiological processes, including cell proliferation, migration, metabolism, survival and immune response. As might be expected for enzymes that participate in such an important capacity in cellular signaling, PTP activity must be tightly regulated. That expectation is further reinforced by the examples presented here showing how various regulatory mechanisms, such as oxidation of active site Cys, allostery and oligomerization, are utilized to control PTP activity. Moreover, a growing body of work also implicates perturbation of PTP regulatory mechanisms in the pathogenesis of human diseases, ranging from metabolic disorders to cancer. Structure and function investigation has yielded critical insights into the regulatory mechanisms that control the PTP activity. Promising case studies covered in this review indicate that targeting PTP regulatory mechanisms could provide a new way forward to developing a broad array of PTP modulating agents, one that exploits the remarkable diversity of PTP regulatory elements while avoiding the challenges of pursuing the PTP active site.30–33
It is important to point out that comparing to PTKs, the study of PTPs is still in its early stages as the majority of the PTP superfamily remain largely uncharacterized. It is anticipated that application of state-of-the-art CRISPR/Cas9 gene editing techniques279 and novel genetically modified animal models280–281 will continue to provide new functional perspectives for the remaining PTP family members and reveal new linkage of PTP dysfuctions to the causes of human diseases. As more potent and specific PTP inhibitors become available,31,282 these small molecule chemical probes are expected to play increasingly important role in interrogating the biological functions of PTPs in both normal physiological processes and pathological conditions. Further understanding of the biochemical underpinnings of PTP regulation shall continue to offer new perspectives on the importance of these enzymes in both health and disease. Continued and persistent efforts to exploit PTP regulatory mechanisms will bring a new dimension to the armory of PTP-based drug discovery, ultimately delivering novel medicines for various human diseases.
Acknowledgments
Studies in our laboratory have been supported by National Institutes of Health Grants RO1 CA69202 and RO1 CA207288.
Abbreviations
- PTK
protein tyrosine kinase
- PTP
protein tyrosine phosphatase
- DSP
dual specificity phosphatase
- pTyr
phosphotyrosine
- MAP
mitogen-activated protein
- MKPs
mitogen-activated protein kinase phosphatases
- PRL
phosphatase of regenerating liver
- SHP2
The Src homology 2 (SH2)-domain containing protein tyrosine phosphatase-2
- ROS
Reactive oxygen species
Biographies
Zhong-Hong Yu is an Assistant Research Scientist in the Department of Medicinal Chemistry and Molecular Pharmacology at Purdue University. He received his bachelor’s degree in Chemistry in 2001 and his Ph.D. degree in Chemical Biology in 2007 from Nankai University in China. His training and research during the Ph.D. program were in the area of computer-assisted drug design. He started his postdoctoral research in 2008 at Indiana University School of Medicine with Professor Zhong-Yin Zhang and moved to Purdue University in 2016. Dr. Yu’s current research focuses on the discovery of small molecular inhibitors for specific PTPs by integrated computational and experimental approaches, including high throughput virtual screening, molecular docking, molecular dynamics simulation, enzyme kinetics, mutagenesis, and X-ray crystallography. He also utilizes these techniques to investigate the structure and function of PTPs implicated as therapeutic targets and understand the molecular basis of how these PTPs cause disease.
Zhong-Yin Zhang is Distinguished Professor of Medicinal Chemistry, the Robert C. and Charlotte P. Anderson Chair in Pharmacology, and Head of the Department of Medicinal Chemistry and Molecular Pharmacology at Purdue University. He obtained his undergraduate degree in Chemistry from Nankai University and his Ph.D. in Chemistry from Purdue University. After postdoctoral training at the Upjohn Company and the University of Michigan, he started his independent career the Albert Einstein College of Medicine in 1994 and rose through the ranks to become full professor in 2002. Before returning to Purdue in 2016, he was Robert A. Harris Professor and Chairman of the Department of Biochemistry and Molecular Biology at Indiana University School of Medicine from 2005–2015. Professor Zhang’s research spans both chemistry and biology with a specific focus on the structure and function of protein tyrosine phosphatases (PTPs), PTP-mediated signaling mechanisms, mechanistic enzymology, and development of PTP inhibitors as tool compounds to interrogate PTP biology and as novel therapeutics for the treatment of human diseases.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Cohen P. The regulation of protein function by multisite phosphorylation - a 25 year update. Trends Biochem Sci. 2000;25:596–601. doi: 10.1016/s0968-0004(00)01712-6. [DOI] [PubMed] [Google Scholar]
- 2.Sharma K, D’Souza RCJ, Tyanova S, Schaab C, Wisniewski JR, Cox J, Mann M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014;8:1583–1594. doi: 10.1016/j.celrep.2014.07.036. [DOI] [PubMed] [Google Scholar]
- 3.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 4.Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 5.Zhang ZY. Chemical and mechanistic approaches to the study of protein tyrosine phosphatases. Acc Chem Res. 2003;36:385–392. doi: 10.1021/ar020122r. [DOI] [PubMed] [Google Scholar]
- 6.Brautigan DL. Protein Ser/Thr phosphatases - the ugly ducklings of cell signalling. FEBS J. 2013;280:324–345. doi: 10.1111/j.1742-4658.2012.08609.x. [DOI] [PubMed] [Google Scholar]
- 7.Shi YG. Serine/threonine phosphatases: mechanism through structure. Cell. 2009;139:468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 8.Virshup DM, Shenolikar S. From promiscuity to precision: protein phosphatases get a makeover. Mol Cell. 2009;33:537–545. doi: 10.1016/j.molcel.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 9.Bollen M, Peti W, Ragusa MJ, Beullens M. The extended PP1 toolkit: designed to create specificity. Trends Biochem Sci. 2010;35:450–458. doi: 10.1016/j.tibs.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wurzenberger C, Gerlich DW. Phosphatases: providing safe passage through mitotic exit. Nat Rev Mol Cell Biol. 2011;12:469–482. doi: 10.1038/nrm3149. [DOI] [PubMed] [Google Scholar]
- 11.Barford D, Das AK, Egloff MP. The structure and mechanism of protein phosphatases: Insights into catalysis and regulation. Annu Rev Biophys Biomol Struct. 1998;27:133–164. doi: 10.1146/annurev.biophys.27.1.133. [DOI] [PubMed] [Google Scholar]
- 12.Seifried A, Schultz J, Gohla A. Human HAD phosphatases: structure, mechanism, and roles in health and disease. FEBS J. 2013;280:549–571. doi: 10.1111/j.1742-4658.2012.08633.x. [DOI] [PubMed] [Google Scholar]
- 13.Archambault J, Pan GH, Dahmus GK, Cartier M, Marshall N, Zhang S, Dahmus ME, Greenblatt J. FCP1: the RAP74-interacting subunit of a human protein phosphatase that dephosphorylates the carboxyl-terminal domain of RNA polymerase IIO. J Biol Chem. 1998;273:27593–27601. doi: 10.1074/jbc.273.42.27593. [DOI] [PubMed] [Google Scholar]
- 14.Yeo M, Lin PS, Dahmus ME, Gill GN. A novel RNA polymerase II C-terminal domain phosphatase that preferentially dephosphorylates serine 5. J Biol Chem. 2003;278:26078–26085. doi: 10.1074/jbc.M301791200. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, Nigam SK, Aggarwal AK, Maas R, Rose DW, et al. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature. 2003;426:247–254. doi: 10.1038/nature02083. [DOI] [PubMed] [Google Scholar]
- 16.Rayapureddi JP, Kattamuri C, Steinmetz BD, Frankfort BJ, Ostrin EJ, Mardon G, Hegde RS. Eyes absent represents a class of protein tyrosine phosphatases. Nature. 2003;426:295–298. doi: 10.1038/nature02093. [DOI] [PubMed] [Google Scholar]
- 17.Tootle TL, Silver SJ, Davies EL, Newman V, Latek RR, Mills IA, Selengut JD, Parlikar BEW, Rebay I. The transcription factor Eyes absent is a protein tyrosine phosphatase. Nature. 2003;426:299–302. doi: 10.1038/nature02097. [DOI] [PubMed] [Google Scholar]
- 18.Zhang MM, Liu J, Kim Y, Dixon JE, Pfaff SL, Gill GN, Noel JP, Zhang Y. Structural and functional analysis of the phosphoryl transfer reaction mediated by the human small C-terminal domain phosphatase, Scp1. Protein Sci. 2010;19:974–986. doi: 10.1002/pro.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trinkle-Mulcahy L, Lamond AI. Mitotic phosphatases: no longer silent partners. Curr Opin Cell Biol. 2006;18:623–631. doi: 10.1016/j.ceb.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 20.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 21.Tonks NK. Protein tyrosine phosphatases - from housekeeping enzymes to master regulators of signal transduction. FEBS J. 2013;280:346–378. doi: 10.1111/febs.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacKeigan JP, Murphy LO, Blenis J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat Cell Biol. 2005;7:591–600. doi: 10.1038/ncb1258. [DOI] [PubMed] [Google Scholar]
- 23.Hunter T. Tyrosine-phosphorylation: thirty years and counting. Curr Opin Cell Biol. 2009;21:140–146. doi: 10.1016/j.ceb.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Julien SG, Dube N, Hardy S, Tremblay ML. Inside the human cancer tyrosine phosphatome. Nat Rev Cancer. 2011;11:35–49. doi: 10.1038/nrc2980. [DOI] [PubMed] [Google Scholar]
- 25.Cohen P, Alessi DR. Kinase drug discovery - What’s next in the field? ACS Chem Biol. 2013;8:96–104. doi: 10.1021/cb300610s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He RJ, Yu ZH, Zhang RY, Zhang ZY. Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol Sin. 2014;35:1227–1246. doi: 10.1038/aps.2014.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. New Engl J Med. 2005;353:172–187. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- 28.Ventura JJ, Nebreda AR. Protein kinases and phosphatases as therapeutic targets in cancer. Clin Transl Oncol. 2006;8:153–160. doi: 10.1007/s12094-006-0005-0. [DOI] [PubMed] [Google Scholar]
- 29.Wu P, Nielsen TE, Clausen MH. Small-molecule kinase inhibitors: an analysis of FDA- approved drugs. Drug Discov Today. 2016;21:5–10. doi: 10.1016/j.drudis.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 30.Vintonyak VV, Antonchick AP, Rauh D, Waldmann H. The therapeutic potential of phosphatase inhibitors. Curr Opin Chem Biol. 2009;13:272–283. doi: 10.1016/j.cbpa.2009.03.021. [DOI] [PubMed] [Google Scholar]
- 31.He RJ, Zeng LF, He YT, Zhang S, Zhang ZY. Small molecule tools for functional interrogation of protein tyrosine phosphatases. FEBS J. 2013;280:731–750. doi: 10.1111/j.1742-4658.2012.08718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Butterworth S, Overduin M, Barr AJ. Targeting protein tyrosine phosphatase SHP2 for therapeutic intervention. Future Med Chem. 2014;6:1423–1437. doi: 10.4155/fmc.14.88. [DOI] [PubMed] [Google Scholar]
- 33.Lazo JS, Sharlow ER. Drugging undruggable molecular cancer targets. Annu Rev Pharmacol Toxicol. 2016;56:23–40. doi: 10.1146/annurev-pharmtox-010715-103440. [DOI] [PubMed] [Google Scholar]
- 34.Frangioni JV, Beahm PH, Shifrin V, Jost CA, Neel BG. The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino-acid C-terminal sequence. Cell. 1992;68:545–560. doi: 10.1016/0092-8674(92)90190-n. [DOI] [PubMed] [Google Scholar]
- 35.Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998;92:441–450. doi: 10.1016/s0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 36.Haj FG, Verveer PJ, Squire A, Neel BG, Bastiaens PIH. Imaging sites of receptor dephosphorylation by PTP1B on the surface of the endoplasmic reticulum. Science. 2002;295:1708–1711. doi: 10.1126/science.1067566. [DOI] [PubMed] [Google Scholar]
- 37.Trowbridge IS, Thomas ML. CD45 - an emerging role as a protein-tyrosine-phosphatase required for lymphocyte-activation and development. Annu Rev Immunol. 1994;12:85–116. doi: 10.1146/annurev.iy.12.040194.000505. [DOI] [PubMed] [Google Scholar]
- 38.Pallen CJ. Protein tyrosine phosphatase alpha (PTP alpha): A src family kinase activator and mediator of multiple biological effects. Curr Top Med Chem. 2003;3:821–835. doi: 10.2174/1568026033452320. [DOI] [PubMed] [Google Scholar]
- 39.Guan KL, Broyles SS, Dixon JE. A Tyr/Ser protein phosphatase encoded by vaccinia virus. Nature. 1991;350:359–362. doi: 10.1038/350359a0. [DOI] [PubMed] [Google Scholar]
- 40.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1: dephosphorylates the lipid second messenger, phosphatidylinositol 3:4:5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 41.Taylor GS, Maehama T, Dixon JE. Myotubularin, a protein tyrosine phosphatase mutated in myotubular myopathy, dephosphorylates the lipid second messenger, phosphatidylinositol 3-phosphate. Proc Natl Acad Sci U S A. 2000;97:8910–8915. doi: 10.1073/pnas.160255697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tagliabracci VS, Turnbull J, Wang W, Girard JM, Zhao X, Skurat AV, Delgado-Escueta AV, Minassian BA, DePaoli-Roach AA, Roach PJ. Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc Natl Acad Sci U S A. 2007;104:19262–19266. doi: 10.1073/pnas.0707952104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dunphy WG, Kumagai A. The CDC25 protein contains an intrinsic phosphatase activity. Cell. 1991;67:189–196. doi: 10.1016/0092-8674(91)90582-j. [DOI] [PubMed] [Google Scholar]
- 44.Millar JBA, Russell P. The CDC25 M-phase inducer - an unconventional protein phosphatase. Cell. 1992;68:407–410. doi: 10.1016/0092-8674(92)90177-e. [DOI] [PubMed] [Google Scholar]
- 45.Fauman EB, Cogswell JP, Lovejoy B, Rocque WJ, Holmes W, Montana VG, Piwnica-Worms H, Rink MJ, Saper MA. Crystal structure of the catalytic domain of the human cell cycle control phosphatase, Cdc25A. Cell. 1998;93:617–625. doi: 10.1016/s0092-8674(00)81190-3. [DOI] [PubMed] [Google Scholar]
- 46.Maccari R, Ottana R. Low molecular weight phosphotyrosine protein phosphatases as emerging targets for the design of novel therapeutic agents. J Med Chem. 2012;55:2–22. doi: 10.1021/jm200607g. [DOI] [PubMed] [Google Scholar]
- 47.Barford D, Flint AJ, Tonks NK. Crystal structure of human protein tyrosine phosphatase 1B. Science. 1994;263:1397–1404. [PubMed] [Google Scholar]
- 48.Stuckey JA, Schubert HL, Fauman EB, Zhang ZY, Dixon JE, Saper MA. Crystal structure of Yersinia protein tyrosine phosphatase at 2. 5-angstrom and the complex with tungstate. Nature. 1994;370:571–575. doi: 10.1038/370571a0. [DOI] [PubMed] [Google Scholar]
- 49.Yuvaniyama J, Denu JM, Dixon JE, Saper MA. Crystal structure of the dual specificity protein phosphatase VHR. Science. 1996;272:1328–1331. doi: 10.1126/science.272.5266.1328. [DOI] [PubMed] [Google Scholar]
- 50.Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, Jansen PG, Andersen HS, Tonks NK, Moller NPH. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol Cell Biol. 2001;21:7117–7136. doi: 10.1128/MCB.21.21.7117-7136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang ZY. Protein tyrosine phosphatases: Structure and function, substrate specificity, and inhibitor development. Annu Rev Pharmacol Toxicol. 2002;42:209–234. doi: 10.1146/annurev.pharmtox.42.083001.144616. [DOI] [PubMed] [Google Scholar]
- 52.Barr AJ, Ugochukwu E, Lee WH, King ONF, Filippakopoulos P, Alfano I, Savitsky P, Burgess-Brown NA, Muller S, Knapp S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell. 2009;136:352–363. doi: 10.1016/j.cell.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang ZY, Wang YA, Dixon JE. Dissecting the catalytic mechanism of protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 1994;91:1624–1627. doi: 10.1073/pnas.91.5.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jia ZC, Barford D, Flint AJ, Tonks NK. Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science. 1995;268:1754–1758. doi: 10.1126/science.7540771. [DOI] [PubMed] [Google Scholar]
- 55.Sarmiento M, Zhao Y, Gordon SJ, Zhang ZY. Molecular basis for substrate specificity of protein-tyrosine phosphatase 1B. J Biol Chem. 1998;273:26368–26374. doi: 10.1074/jbc.273.41.26368. [DOI] [PubMed] [Google Scholar]
- 56.Sarmiento M, Puius YA, Vetter SW, Keng YF, Wu L, Zhao Y, Lawrence DS, Almo SC, Zhang ZY. Structural basis of plasticity in protein tyrosine phosphatase 1B substrate recognition. Biochemistry. 2000;39:8171–8179. doi: 10.1021/bi000319w. [DOI] [PubMed] [Google Scholar]
- 57.Dunn D, Chen L, Lawrence DS, Zhang ZY. The active site specificity of the Yersinia protein-tyrosine phosphatase. J Biol Chem. 1996;271:168–173. doi: 10.1074/jbc.271.1.168. [DOI] [PubMed] [Google Scholar]
- 58.Stewart AE, Dowd S, Keyse SM, McDonald NQ. Crystal structure of the MAPK phosphatase Pyst1 catalytic domain and implications for regulated activation. Nat Struct Biol. 1999;6:174–181. doi: 10.1038/5861. [DOI] [PubMed] [Google Scholar]
- 59.Guan KL, Dixon JE. Evidence for protein tyrosine phosphatase catalysis proceeding via a Cysteine-phosphate intermediate. J Biol Chem. 1991;266:17026–17030. [PubMed] [Google Scholar]
- 60.Cho HJ, Krishnaraj R, Kitas E, Bannwarth W, Walsh CT, Anderson KS. Isolation and structural elucidation of a novel phosphocysteine intermediate in the LAR protein tyrosine phosphatase enzymatic pathway. J Am Chem Soc. 1992;114:7296–7298. [Google Scholar]
- 61.Denu JM, Lohse DL, Vijayalakshmi J, Saper MA, Dixon JE. Visualization of intermediate and transition-state structures in protein-tyrosine phosphatase catalysis. Proc Natl Acad Sci U S A. 1996;93:2493–2498. doi: 10.1073/pnas.93.6.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu L, Zhang ZY. Probing the function of Asp128 in the low molecular weight protein-tyrosine phosphatase-catalyzed reaction. A pre-steady-state and steady-state kinetic investigation. Biochemistry. 1996;35:5426–5434. doi: 10.1021/bi952885a. [DOI] [PubMed] [Google Scholar]
- 63.Zhang ZY, Wang Y, Wu L, Fauman EB, Stuckey JA, Schubert HL, Saper MA, Dixon JE. The Cys(X)5Arg catalytic motif in phosphoester hydrolysis. Biochemistry. 1994;33:15266–15270. doi: 10.1021/bi00255a007. [DOI] [PubMed] [Google Scholar]
- 64.Hengge AC, Sowa GA, Wu L, Zhang ZY. Nature of the transition-state of the protein- tyrosine phosphatase-catalyzed reaction. Biochemistry. 1995;34:13982–13987. doi: 10.1021/bi00043a003. [DOI] [PubMed] [Google Scholar]
- 65.Hoff RH, Wu L, Zhou B, Zhang ZY, Hengge AC. Does positive charge at the active sites of phosphatases cause a change in mechanism? The effect of the conserved arginine on the transition state for phosphoryl transfer in the protein-tyrosine phosphatase fromYersinia. J Am Chem Soc. 1999;121:9514–9521. [Google Scholar]
- 66.Zhao Y, Wu L, Noh SJ, Guan KL, Zhang ZY. Altering the nucleophile specificity of a protein-tyrosine phosphatase-catalyzed reaction - Probing the function of the invariant glutamine residues. J Biol Chem. 1998;273:5484–5492. doi: 10.1074/jbc.273.10.5484. [DOI] [PubMed] [Google Scholar]
- 67.Pannifer ADB, Flint AJ, Tonks NK, Barford D. Visualization of the cysteinyl-phosphate intermediate of a protein-tyrosine phosphatase by X-ray crystallography. J Biol Chem. 1998;273:10454–10462. doi: 10.1074/jbc.273.17.10454. [DOI] [PubMed] [Google Scholar]
- 68.Jackson MD, Denu JM. Molecular reactions of protein phosphatases - Insights from structure and chemistry. Chem Rev. 2001;101:2313–2340. doi: 10.1021/cr000247e. [DOI] [PubMed] [Google Scholar]
- 69.den Hertog J, Oestman A, Bohmer FD. Protein tyrosine phosphatases: regulatory mechanisms. FEBS J. 2008;275:831–847. doi: 10.1111/j.1742-4658.2008.06247.x. [DOI] [PubMed] [Google Scholar]
- 70.Graves PR, Lovly CM, Uy GL, Piwnica-Worms H. Localization of human Cdc25C is regulated both by nuclear export and 14–3-3 protein binding. Oncogene. 2001;20:1839–1851. doi: 10.1038/sj.onc.1204259. [DOI] [PubMed] [Google Scholar]
- 71.Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rhee SG. H2O2: a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 73.Mahadev K, Zilbering A, Zhu L, Goldstein BJ. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1B in vivo and enhances the early insulin action cascade. J Biol Chem. 2001;276:21938–21942. doi: 10.1074/jbc.C100109200. [DOI] [PubMed] [Google Scholar]
- 74.Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide - Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- 75.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth-factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 76.Thannickal VJ, Day RM, Klinz SG, Bastien MC, Larios JM, Fanburg BL. Ras-dependent and -independent regulation of reactive oxygen species by mitogenic growth factors and TGF-beta1. FASEB J. 2000;14:1741–1748. doi: 10.1096/fj.99-0878com. [DOI] [PubMed] [Google Scholar]
- 77.Lambeth JD. Nox enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 78.Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: Evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 79.Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 80.Barrett WC, DeGnore JP, Keng YF, Zhang ZY, Yim MB, Chock PB. Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatase 1B. J Biol Chem. 1999;274:34543–34546. doi: 10.1074/jbc.274.49.34543. [DOI] [PubMed] [Google Scholar]
- 81.Xu D, Rovira II, Finkel T. Oxidants painting the cysteine chapel: Redox regulation of PTPs. Dev Cell. 2002;2:251–252. doi: 10.1016/s1534-5807(02)00132-6. [DOI] [PubMed] [Google Scholar]
- 82.Paulsen CE, Carroll KS. Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem Biol. 2010;5:47–62. doi: 10.1021/cb900258z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 84.Meng TC, Buckley DA, Galic S, Tiganis T, Tonks NK. Regulation of insulin signaling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. J Biol Chem. 2004;279:37716–37725. doi: 10.1074/jbc.M404606200. [DOI] [PubMed] [Google Scholar]
- 85.Loh K, Deng HY, Fukushima A, Cai XC, Boivin B, Galic S, Bruce C, Shields BJ, Skiba B, Ooms LM, et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009;10:260–272. doi: 10.1016/j.cmet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Frijhoff J, Dagnell M, Godfrey R, Ostman A. Regulation of protein tyrosine phosphatase oxidation in cell adhesion and migration. Antioxid Redox Signal. 2014;20:1994–2010. doi: 10.1089/ars.2013.5643. [DOI] [PubMed] [Google Scholar]
- 87.Zhang ZY, Dixon JE. Active-site labeling of the yersinia protein-tyrosine-phosphatase - the determination of the pk(a) of the active-site cysteine and the function of the conserved histidine-402. Biochemistry. 1993;32:9340–9345. doi: 10.1021/bi00087a012. [DOI] [PubMed] [Google Scholar]
- 88.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 89.Claiborne A, Yeh JI, Mallett TC, Luba J, Crane EJ, Charrier V, Parsonage D. Protein-sulfenic acids: Diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry. 1999;38:15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- 90.Schwertassek U, Haque A, Krishnan N, Greiner R, Weingarten L, Dick TP, Tonks NK. Reactivation of oxidized PTP1B and PTEN by thioredoxin 1. FEBS J. 2014;281:3545–3558. doi: 10.1111/febs.12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Halliwell B, Clement MV, Long LH. Hydrogen peroxide in the human body. FEBS Lett. 2000;486:10–13. doi: 10.1016/s0014-5793(00)02197-9. [DOI] [PubMed] [Google Scholar]
- 92.Sun JP, Wang WQ, Yang H, Liu SJ, Liang F, Fedorov AA, Almo SC, Zhang ZY. Structure and biochemical properties of PRL-1: a phosphatase implicated in cell growth, differentiation, and tumor invasion. Biochemistry. 2005;44:12009–12021. doi: 10.1021/bi0509191. [DOI] [PubMed] [Google Scholar]
- 93.Meng FG, Zhang ZY. Redox regulation of protein tyrosine phosphatase activity by hydroxyl radical. Biochim Biophys Acta. 2013;1834:464–469. doi: 10.1016/j.bbapap.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stone JR, Yang SP. Hydrogen peroxide: A signaling messenger. Antioxid Redox Signal. 2006;8:243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 95.Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 96.Zhou HY, Singh H, Parsons ZD, Lewis SM, Bhattacharya S, Seiner DR, LaButti JN, Reilly TJ, Tanner JJ, Gates KS. The biological buffer bicarbonate/CO2 potentiates H2O2-mediated inactivation of protein tyrosine phosphatases. J Am Chem Soc. 2011;133:15803–15805. doi: 10.1021/ja2077137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.LaButti JN, Gates KS. Biologically relevant chemical properties of peroxymonophosphate (=O(3)POOH) Bioorg Med Chem Lett. 2009;19:218–221. doi: 10.1016/j.bmcl.2008.10.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ostman A, Frijhoff J, Sandin A, Bohmer FD. Regulation of protein tyrosine phosphatases by reversible oxidation. J Biochem. 2011;150:345–356. doi: 10.1093/jb/mvr104. [DOI] [PubMed] [Google Scholar]
- 99.Chang LF, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 100.Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: A 10-year update. Physiol Rev. 2012;92:689–737. doi: 10.1152/physrev.00028.2011. [DOI] [PubMed] [Google Scholar]
- 101.Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- 102.Johnson LN, Noble MEM, Owen DJ. Active and inactive protein kinases: Structural basis for regulation. Cell. 1996;85:149–158. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- 103.Ahn NG, Seger R, Bratlien RL, Diltz CD, Tonks NK, Krebs EG. Multiple components in an epidermal growth factor-stimulated protein kinase cascade - In vitro activation of a myelin basic-protein microtubule-associated protein-2 kinase. J Biol Chem. 1991;266:4220–4227. [PubMed] [Google Scholar]
- 104.Payne DM, Rossomando AJ, Martino P, Erickson AK, Her JH, Shabanowitz J, Hunt DF, Weber MJ, Sturgill TW. Identification of the regulatory phosphorylation sites in PP42/mitogen-activated protein-kinase (MAP kinase) EMBO J. 1991;10:885–892. doi: 10.1002/j.1460-2075.1991.tb08021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Canagarajah BJ, Khokhlatchev A, Cobb MH, Goldsmith EJ. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell. 1997;90:859–869. doi: 10.1016/s0092-8674(00)80351-7. [DOI] [PubMed] [Google Scholar]
- 106.Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs): Shaping the outcome of MAP kinase signalling. FEBS J. 2013;280:489–504. doi: 10.1111/j.1742-4658.2012.08716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Muda M, Theodosiou A, Rodrigues N, Boschert U, Camps M, Gillieron C, Davies K, Ashworth A, Arkinstall S. The dual specificity phosphatases M3/6 and MKP-3 are highly selective for inactivation of distinct mitogen-activated protein kinases. J Biol Chem. 1996;271:27205–27208. doi: 10.1074/jbc.271.44.27205. [DOI] [PubMed] [Google Scholar]
- 108.Groom LA, Sneddon AA, Alessi DR, Dowd S, Keyse SM. Differential regulation of the MAP, SAP and RK/p38 kinases by Pyst1: a novel cytosolic dual-specificity phosphatase. EMBO J. 1996;15:3621–3632. [PMC free article] [PubMed] [Google Scholar]
- 109.Zhou B, Zhang ZY. Mechanism of mitogen-activated protein kinase phosphatase-3 activation by ERK2. J Biol Chem. 1999;274:35526–35534. doi: 10.1074/jbc.274.50.35526. [DOI] [PubMed] [Google Scholar]
- 110.Wiland AM, Denu JM, Mourey RJ, Dixon JE. Purification and kinetic characterization of the mitogen-activated protein kinase phosphatase rVH6. J Biol Chem. 1996;271:33486–33492. doi: 10.1074/jbc.271.52.33486. [DOI] [PubMed] [Google Scholar]
- 111.Zhao Y, Zhang ZY. The mechanism of dephosphorylation of extracellular signal-regulated kinase 2 by mitogen-activated protein kinase phosphatase 3. J Biol Chem. 2001;276:32382–32391. doi: 10.1074/jbc.M103369200. [DOI] [PubMed] [Google Scholar]
- 112.Camps M, Nichols A, Arkinstall S. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J. 2000;14:6–16. [PubMed] [Google Scholar]
- 113.Camps M, Nichols A, Gillieron C, Antonsson B, Muda M, Chabert C, Boschert U, Arkinstall S. Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science. 1998;280:1262–1265. doi: 10.1126/science.280.5367.1262. [DOI] [PubMed] [Google Scholar]
- 114.Muda M, Theodosiou A, Gillieron C, Smith A, Chabert C, Camps M, Boschert U, Rodrigues N, Davies K, Ashworth A, et al. The mitogen-activated protein kinase phosphatase-3 N-terminal noncatalytic region is responsible for tight substrate binding and enzymatic specificity. J Biol Chem. 1998;273:9323–9329. doi: 10.1074/jbc.273.15.9323. [DOI] [PubMed] [Google Scholar]
- 115.Fjeld CC, Rice AE, Kim Y, Gee KR, Denu JM. Mechanistic basis for catalytic activation of mitogen-activated protein kinase phosphatase 3 by extracellular signal-regulated kinase. J Biol Chem. 2000;275:6749–6757. doi: 10.1074/jbc.275.10.6749. [DOI] [PubMed] [Google Scholar]
- 116.Zhou B, Wu L, Shen K, Zhang JL, Lawrence DS, Zhang ZY. Multiple regions of MAP kinase phosphatase 3 are involved in its recognition and activation by ERK2. J Biol Chem. 2001;276:6506–6515. doi: 10.1074/jbc.M009753200. [DOI] [PubMed] [Google Scholar]
- 117.Zhang JL, Zhou B, Zheng CF, Zhang ZY. A bipartite mechanism for ERK2 recognition by its cognate regulators and substrates. J Biol Chem. 2003;278:29901–29912. doi: 10.1074/jbc.M303909200. [DOI] [PubMed] [Google Scholar]
- 118.Tanoue T, Adachi M, Moriguchi T, Nishida E. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat Cell Biol. 2000;2:110–116. doi: 10.1038/35000065. [DOI] [PubMed] [Google Scholar]
- 119.Tanoue T, Maeda R, Adachi M, Nishida E. Identification of a docking groove on ERK and p38 MAP kinases that regulates the specificity of docking interactions. EMBO J. 2001;20:466–479. doi: 10.1093/emboj/20.3.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bardwell AJ, Abdollahi M, Bardwell L. Docking sites on mitogen-activated protein kinase (MAPK) kinases, MAR phosphatases and the Elk-1 transcription factor compete for MAPK binding and are crucial for enzymic activity. Biochem J. 2003;370:1077–1085. doi: 10.1042/BJ20021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu SJ, Sun JP, Zhou B, Zhang ZY. Structural basis of docking interactions between ERK2 and MAP kinase phosphatase 3. Proc Natl Acad Sci U S A. 2006;103:5326–5331. doi: 10.1073/pnas.0510506103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhou B, Zhang JL, Liu SJ, Reddy S, Wang F, Zhang ZY. Mapping ERK2-MKP3 binding interfaces by hydrogen/deuterium exchange mass spectrometry. J Biol Chem. 2006;281:38834–38844. doi: 10.1074/jbc.M608916200. [DOI] [PubMed] [Google Scholar]
- 123.Kumar GS, Zettl H, Page R, Peti W. Structural basis for the regulation of the mitogen-activated protein (MAP) kinase p38 alpha by the dual specificity phosphatase 16 MAP kinase binding domain in solution. J Biol Chem. 2013;288:28347–28356. doi: 10.1074/jbc.M113.499178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Peti W, Page R. Molecular basis of MAP kinase regulation. Protein Sci. 2013;22:1698–1710. doi: 10.1002/pro.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Neel BG, Gu HH, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28:284–293. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 126.Chan RJ, Feng GS. PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood. 2007;109:862–867. doi: 10.1182/blood-2006-07-028829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Grossmann KS, Rosario M, Birchmeier C, Birchmeier W. The tyrosine phosphatase Shp2 in development and cancer. Adv Cancer Res. 2010;106:53–89. doi: 10.1016/S0065-230X(10)06002-1. [DOI] [PubMed] [Google Scholar]
- 128.Herbst R, Carroll PM, Allard JD, Schilling J, Raabe T, Simon MA. Daughter of sevenless is a substrate of the phosphotyrosine phosphatase corkscrew and functions during sevenless signaling. Cell. 1996;85:899–909. doi: 10.1016/s0092-8674(00)81273-8. [DOI] [PubMed] [Google Scholar]
- 129.Malumbres M, Barbacid M. Timeline - RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 130.Noguchi T, Matozaki T, Horita K, Fujioka Y, Kasuga M. Role of SH-PTP2: a protein tyrosine phosphatase with Src homology-2 domains, in insulin-stimulated Ras activation. Mol Cell Biol. 1994;14:6674–6682. doi: 10.1128/mcb.14.10.6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Perkins LA, Larsen I, Perrimon N. Corkscrew encodes a putative protein tyrosine phosphatase that functions to transduce the terminal signal from the receptor tyrosine kinase torso. Cell. 1992;70:225–236. doi: 10.1016/0092-8674(92)90098-w. [DOI] [PubMed] [Google Scholar]
- 132.Gutch MJ, Flint AJ, Keller J, Tonks NK, Hengartner MO. The Caenorhabditis elegans SH2 domain-containing protein tyrosine phosphatase PTP-2 participates in signal transduction during oogenesis and vulval development. Genes Dev. 1998;12:571–585. doi: 10.1101/gad.12.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tiganis T, Bennett AM. Protein tyrosine phosphatase function: the substrate perspective. Biochem J. 2007;402:1–15. doi: 10.1042/BJ20061548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Barford D, Neel BG. Revealing mechanisms for SH2 domain mediated regulation of the protein tyrosine phosphatase SHP-2. Structure. 1998;6:249–254. doi: 10.1016/s0969-2126(98)00027-6. [DOI] [PubMed] [Google Scholar]
- 135.Pei DH, Wang J, Walsh CT. Differential functions of the two Src homology 2 domains in protein tyrosine phosphatase SH-PTP1. Proc Natl Acad Sci U S A. 1996;93:1141–1145. doi: 10.1073/pnas.93.3.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pluskey S, Wandless TJ, Walsh CT, Shoelson SE. Potent stimulation of SH-PTP2 phosphatase activity by simultaneous occupancy of both SH2 domains. J Biol Chem. 1995;270:2897–2900. doi: 10.1074/jbc.270.7.2897. [DOI] [PubMed] [Google Scholar]
- 137.Sugimoto S, Wandless TJ, Shoelson SE, Neel BG, Walsh CT. Activation of the SH2-containing protein-tyrosine-phosphatase, SH-PTP2: by phosphotyrosine-containing peptides derived from insulin-receptor substrate-1. J Biol Chem. 1994;269:13614–13622. [PubMed] [Google Scholar]
- 138.Lee CH, Kominos D, Jacques S, Margolis B, Schlessinger J, Shoelson SE, Kuriyan J. Crystal structures of peptide complexes of the amino-terminal SH2 domain of the SYP tyrosine phosphatase. Structure. 1994;2:423–438. doi: 10.1016/s0969-2126(00)00044-7. [DOI] [PubMed] [Google Scholar]
- 139.Eck MJ, Pluskey S, Trub T, Harrison SC, Shoelson SE. Spatial constraints on the recognition of phosphoproteins by the tandem SH2 domains of the phosphatase SH-PTP2. Nature. 1996;379:277–280. doi: 10.1038/379277a0. [DOI] [PubMed] [Google Scholar]
- 140.Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S, et al. Mutations in PTPN11: encoding the protein tyrosine phosphatase SHP-2: cause Noonan syndrome. Nat Genet. 2001;29:465–468. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 141.Tartaglia M, Gelb BD. Noonan syndrome and related disorders: Genetics and pathogenesis. Annu Rev Genom Hum Genet, Annual Reviews: Palo Alto. 2005;6:45–68. doi: 10.1146/annurev.genom.6.080604.162305. [DOI] [PubMed] [Google Scholar]
- 142.Digilio MC, Conti E, Sarkozy A, Mingarelli R, Dottorini T, Marino B, Pizzuti A, Dallapiccola B. Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am J Hum Genet. 2002;71:389–394. doi: 10.1086/341528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Merks JHM, Caron HN, Hennekam RCM. High incidence of malformation syndromes in a series of 1:073 children with cancer. Am J Med Genet A. 2005;134A:132–143. doi: 10.1002/ajmg.a.30603. [DOI] [PubMed] [Google Scholar]
- 144.Keren B, Hadchouel A, Saba S, Sznajer Y, Bonneau D, Leheup B, Boute O, Gaillard D, Lacombe D, Layet V, et al. PTPN11 mutations in patients with LEOPARD syndrome: a French multicentric experience. J Med Genet. 2004;41:117–119. doi: 10.1136/jmg.2004.021451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tartaglia M, Niemeyer CM, Fragale A, Song XL, Buechner J, Jung A, Hahlen K, Hasle H, Licht JD, Gelb BD. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–150. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 146.Tartaglia M, Martinelli S, Cazzaniga G, Cordeddu V, Iavarone I, Spinelli M, Palmi C, Carta C, Pession A, Arico M, et al. Genetic evidence for lineage-related and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia. Blood. 2004;104:307–313. doi: 10.1182/blood-2003-11-3876. [DOI] [PubMed] [Google Scholar]
- 147.Loh ML, Vattikuti S, Schubbert S, Reynolds MG, Carlson E, Lieuw KH, Cheng JW, Lee CM, Stokoe D, Bonifas JM, et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood. 2004;103:2325–2331. doi: 10.1182/blood-2003-09-3287. [DOI] [PubMed] [Google Scholar]
- 148.Loh ML, Reynolds MG, Vattikuti S, Gerbing RB, Alonzo TA, Carlson E, Cheng JW, Lee CM, Lange BJ, Meshinchi S. PTPN11 mutations in pediatric patients with acute myeloid leukemia: results from the Children’s Cancer Group. Leukemia. 2004;18:1831–1834. doi: 10.1038/sj.leu.2403492. [DOI] [PubMed] [Google Scholar]
- 149.Kratz CP, Niemeyer CM, Castleberry RP, Cetin M, Bergstrasser E, Emanuel PD, Hasle H, Kardos G, Klein C, Kojima S, et al. The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood. 2005;106:2183–2185. doi: 10.1182/blood-2005-02-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, Maris JM, Richardson A, Bardelli A, Sugarbaker DJ, et al. Activating mutations of the Noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004;64:8816–8820. doi: 10.1158/0008-5472.CAN-04-1923. [DOI] [PubMed] [Google Scholar]
- 151.Miyamoto D, Miyamoto M, Takahashi A, Yomogita Y, Higashi H, Kondo S, Hatakeyama M. Isolation of a distinct class of gain-of-function SHP-2 mutants with oncogenic RAS-like transforming activity from solid tumors. Oncogene. 2008;27:3508–3515. doi: 10.1038/sj.onc.1211019. [DOI] [PubMed] [Google Scholar]
- 152.Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer. 2004;4:688–694. doi: 10.1038/nrc1433. [DOI] [PubMed] [Google Scholar]
- 153.Stommel JM, Kimmelman AC, Ying HQ, Nabioullin R, Ponugoti AH, Wiedemeyer R, Stegh AH, Bradner JE, Ligon KL, Brennan C, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318:287–290. doi: 10.1126/science.1142946. [DOI] [PubMed] [Google Scholar]
- 154.Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008;27:179–192. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- 155.Fragale A, Tartaglia M, Wu J, Gelb BD. Noonan syndrome-associated SHP2/PTPN11 mutants cause EGF-dependent prolonged GAB1 binding and sustained ERK2/MAPK1 activation. Hum Mutat. 2004;23:267–277. doi: 10.1002/humu.20005. [DOI] [PubMed] [Google Scholar]
- 156.Keilhack H, David FS, McGregor M, Cantley LC, Neel BG. Diverse biochemical properties of Shp2 mutants. Implications for disease phenotypes. J Biol Chem. 2005;280:30984–30993. doi: 10.1074/jbc.M504699200. [DOI] [PubMed] [Google Scholar]
- 157.Martinelli S, Torreri P, Tinti M, Stella L, Bocchinfuso G, Flex E, Grottesi A, Ceccarini M, Palleschi A, Cesareni G, et al. Diverse driving forces underlie the invariant occurrence of the T42A, E139D, I282V and T468M SHP2 amino acid substitutions causing Noonan and LEOPARD syndromes. Hum Mol Genet. 2008;17:2018–2029. doi: 10.1093/hmg/ddn099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Oishi K, Gaengel K, Krishnamoorthy S, Kamiya K, Kim IK, Ying HW, Weber U, Perkins LA, Tartaglia M, Mlodzik M, et al. Transgenic Drosophila models of Noonan syndrome causing PTPN11 gain-of-function mutations. Hum Mol Genet. 2006;15:543–553. doi: 10.1093/hmg/ddi471. [DOI] [PubMed] [Google Scholar]
- 159.Tartaglia M, Martinelli S, Stella L, Bocchinfuso G, Flex E, Cordeddu V, Zampino G, van der Burgt I, Palleschi A, Petrucci TC, et al. Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am J Hum Genet. 2006;78:279–290. doi: 10.1086/499925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, Yang WT, Pao LI, Gilliland DG, Epstein JA, et al. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med. 2004;10:849–857. doi: 10.1038/nm1084. [DOI] [PubMed] [Google Scholar]
- 161.Chan G, Kalaitzidis D, Usenko T, Kutok JL, Yang WT, Mohi MG, Neel BG. Leukemogenic Ptpn11 causes fatal myeloproliferative disorder via cell-autonomous effects on multiple stages of hematopoiesis. Blood. 2009;113:4414–4424. doi: 10.1182/blood-2008-10-182626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mohi MG, Williams IR, Dearolf CR, Chan G, Kutok JL, Cohen S, Morgan K, Boulton C, Shigematsu H, Keilhack H, et al. Prognostic, therapeutic, and mechanistic implications of a mouse model of leukemia evoked by Shp2 (PTPN11) mutations. Cancer Cell. 2005;7:179–191. doi: 10.1016/j.ccr.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 163.Xu D, Liu X, Yu WM, Meyerson HJ, Guo CY, Gerson SL, Qu CK. Non-lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. J Exp Med. 2011;208:1977–1988. doi: 10.1084/jem.20110450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hanna N, Montagner A, Lee WH, Miteva M, Vidal M, Vidaud M, Parfait B, Raynal P. Reduced phosphatase activity of SHP-2 in LEOPARD syndrome: Consequences for PI3K binding on Gab1. FEBS Lett. 2006;580:2477–2482. doi: 10.1016/j.febslet.2006.03.088. [DOI] [PubMed] [Google Scholar]
- 165.Kontaridis MI, Swanson KD, David FS, Barford D, Neel BG. PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J Biol Chem. 2006;281:6785–6792. doi: 10.1074/jbc.M513068200. [DOI] [PubMed] [Google Scholar]
- 166.Marin TM, Keith K, Davies B, Conner DA, Guha P, Kalaitzidis D, Wu X, Lauriol J, Wang B, Bauer M, et al. Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. J Clin Invest. 2011;121:1026–1043. doi: 10.1172/JCI44972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yu ZH, Xu J, Walls CD, Chen L, Zhang S, Zhang RY, Wu L, Wang LN, Liu SJ, Zhang ZY. Structural and mechanistic insights into LEOPARD Syndrome-associated SHP2 mutations. J Biol Chem. 2013;288:10472–10482. doi: 10.1074/jbc.M113.450023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yu ZH, Zhang RY, Walls CD, Chen L, Zhang S, Wu L, Liu S, Zhang ZY. Molecular basis of gain-of-function LEOPARD syndrome-associated SHP2 mutations. Biochemistry. 2014;53:4136–4151. doi: 10.1021/bi5002695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Qiu W, Wang XN, Romanov V, Hutchinson A, Lin A, Ruzanov M, Battaile KP, Pai EF, Neel BG, Chirgadze NY. Structural insights into Noonan/LEOPARD syndrome-related mutants of protein-tyrosine phosphatase SHP2 (PTPN11) BMC Struct Biol. 2014;14:10. doi: 10.1186/1472-6807-14-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.HolgadoMadruga M, Emlet DR, Moscatello DK, Godwin AK, Wong AJ. A Grb2- associated docking protein in EGF- and insulin-receptor signalling. Nature. 1996;379:560–564. doi: 10.1038/379560a0. [DOI] [PubMed] [Google Scholar]
- 171.Cunnick JM, Dorsey JF, Munoz-Antonia T, Mei L, Wu J. Requirement of SHP2 binding to Grb2-associated binder-1 for mitogen-activated protein kinase activation in response to lysophosphatidic acid and epidermal growth factor. J Biol Chem. 2000;275:13842–13848. doi: 10.1074/jbc.275.18.13842. [DOI] [PubMed] [Google Scholar]
- 172.Cunnick JM, Mei L, Doupnik CA, Wu J. Phosphotyrosines 627 and 659 of Gab1 constitute a bisphosphoryl tyrosine-based activation motif (BTAM) conferring binding and activation of SHP2. J Biol Chem. 2001;276:24380–24387. doi: 10.1074/jbc.M010275200. [DOI] [PubMed] [Google Scholar]
- 173.Ren Y, Meng SS, Mei L, Zhao ZJ, Jove R, Wu J. Roles of Gab1 and SHP2 in paxillin tyrosine dephosphorylation and Src activation in response to epidermal growth factor. J Biol Chem. 2004;279:8497–8505. doi: 10.1074/jbc.M312575200. [DOI] [PubMed] [Google Scholar]
- 174.Oishi K, Zhang H, Gault WJ, Wang CJ, Tan CC, Kim IK, Ying HW, Rahman T, Pica N, Tartaglia M, et al. Phosphatase-defective LEOPARD syndrome mutations in PTPN11 gene have gain-of-function effects during Drosophila development. Hum Mol Genet. 2009;18:193–201. doi: 10.1093/hmg/ddn336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bonetti M, Overman JP, Tessadori F, Noel E, Bakkers J, den Hertog J. Noonan and LEOPARD syndrome Shp2 variants induce heart displacement defects in zebrafish. Development. 2014;141:1961–1970. doi: 10.1242/dev.106310. [DOI] [PubMed] [Google Scholar]
- 176.Weiss A, Schlessinger J. Switching signals on or off by receptor dimerization. Cell. 1998;94:277–280. doi: 10.1016/s0092-8674(00)81469-5. [DOI] [PubMed] [Google Scholar]
- 177.Lavoie H, Li JJ, Thevakumaran N, Therrien M, Sicheri F. Dimerization-induced allostery in protein kinase regulation. Trends Biochem Sci. 2014;39:475–486. doi: 10.1016/j.tibs.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 178.Poulikakos PI, Persaud Y, Janakiraman M, Kong XJ, Ng C, Moriceau G, Shi HB, Atefi M, Titz B, Gabay MT, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Felberg J, Johnson P. Characterization of recombinant CD45 cytoplasmic domain proteins - Evidence for intramolecular and intermolecular interactions. J Biol Chem. 1998;273:17839–17845. doi: 10.1074/jbc.273.28.17839. [DOI] [PubMed] [Google Scholar]
- 180.Streuli M, Krueger NX, Thai T, Tang M, Saito H. Distinct functional roles of the two intracellular phosphatase like domains of the receptor-linked protein tyrosine phosphatases LCA and LAR. EMBO J. 1990;9:2399–2407. doi: 10.1002/j.1460-2075.1990.tb07415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bilwes AM, denHertog J, Hunter T, Noel JP. Structural basis for inhibition of receptor protein-tyrosine phosphatase-alpha by dimerization. Nature. 1996;382:555–559. doi: 10.1038/382555a0. [DOI] [PubMed] [Google Scholar]
- 182.Nam HJ, Poy F, Krueger NX, Saito H, Frederick CA. Crystal structure of the tandem phosphatase domains of RPTP LAR. Cell. 1999;97:449–457. doi: 10.1016/s0092-8674(00)80755-2. [DOI] [PubMed] [Google Scholar]
- 183.Nam HJ, Poy F, Saito H, Frederick CA. Structural basis for the function and regulation of the receptor protein tyrosine phosphatase CD45. J Exp Med. 2005;201:441–452. doi: 10.1084/jem.20041890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Jiang GQ, Den Hertog J, Hunter T. Receptor-like protein tyrosine phosphatase alpha homodimerizes on the cell surface. Mol Cell Biol. 2000;20:5917–5929. doi: 10.1128/mcb.20.16.5917-5929.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tertoolen LGJ, Blanchetot C, Jiang GQ, Overvoorde J, Gadella TWJ, Hunter T, den Hertog J. Dimerization of receptor protein-tyrosine phosphatase alpha in living cells. BMC Cell Biol. 2001;2:8. doi: 10.1186/1471-2121-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Desai DM, Sap J, Schlessinger J, Weiss A. Ligand-mediated negative regulation of a chimeric transmembrane receptor tyrosine phosphatase. Cell. 1993;73:541–554. doi: 10.1016/0092-8674(93)90141-c. [DOI] [PubMed] [Google Scholar]
- 187.Jiang GL, den Hertog J, Su J, Noel J, Sap J, Hunter T. Dimerization inhibits the activity of receptor-like protein-tyrosine phosphatase-alpha. Nature. 1999;401:606–610. doi: 10.1038/44170. [DOI] [PubMed] [Google Scholar]
- 188.Majeti R, Bilwes AM, Noel JP, Hunter T, Weiss A. Dimerization-induced inhibition of receptor protein tyrosine phosphatase function through an inhibitory wedge. Science. 1998;279:88–91. doi: 10.1126/science.279.5347.88. [DOI] [PubMed] [Google Scholar]
- 189.Blanchetot C, Tertoolen LG, Overvoorde J, den Hertog J. Intra- and intermolecular interactions between intracellular domains of receptor protein-tyrosine phosphatases. J Biol Chem. 2002;277:47263–47269. doi: 10.1074/jbc.M205810200. [DOI] [PubMed] [Google Scholar]
- 190.Hower AE, Beltran PJ, Bixby JL. Dimerization of tyrosine phosphatase PTPRO decreases its activity and ability to inactivate TrkC. J Neurochem. 2009;110:1635–1647. doi: 10.1111/j.1471-4159.2009.06261.x. [DOI] [PubMed] [Google Scholar]
- 191.Lee S, Faux C, Nixon J, Alete D, Chilton J, Hawadle M, Stoker AW. Dimerization of protein tyrosine phosphatase sigma governs both ligand binding and isoform specificity. Mol Cell Biol. 2007;27:1795–1808. doi: 10.1128/MCB.00535-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Takeda A, Matsuda A, Paul RMJ, Yaseen NR. CD45-associated protein inhibits CD45 dimerization and up-regulates its protein tyrosine phosphatase activity. Blood. 2004;103:3440–3447. doi: 10.1182/blood-2003-06-2083. [DOI] [PubMed] [Google Scholar]
- 193.Toledano-Katchalski H, Tiran Z, Sines T, Shani G, Granot-Attas S, den Hertog J, Elson A. Dimerization in vivo and inhibition of the nonreceptor form of protein tyrosine phosphatase epsilon. Mol Cell Biol. 2003;23:5460–5471. doi: 10.1128/MCB.23.15.5460-5471.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Meng K, Rodriguez-Pena A, Dimitrov T, Chen W, Yamin M, Noda M, Deuel TF. Pleiotrophin signals increased tyrosine phosphorylation of beta-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase beta/zeta. Proc Natl Acad Sci U S A. 2000;97:2603–2608. doi: 10.1073/pnas.020487997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Koksal AC, Nardozzi JD, Cingolani G. Dimeric quaternary structure of the prototypical dual specificity phosphatase VH1. J Biol Chem. 2009;284:10129–10137. doi: 10.1074/jbc.M808362200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Koksal AC, Cingolani G. Dimerization of Vaccinia virus VH1 is essential for dephosphorylation of STAT1 at tyrosine 701. J Biol Chem. 2011;286:14373–14382. doi: 10.1074/jbc.M111.226357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Pavic K, Duan GY, Kohn M. VHR/DUSP3 phosphatase: structure, function and regulation. FEBS J. 2015;282:1871–1890. doi: 10.1111/febs.13263. [DOI] [PubMed] [Google Scholar]
- 198.Pavic K, Rios P, Dzeyk K, Koehler C, Lemke EA, Kohn M. Unnatural amino acid mutagenesis reveals dimerization as a negative regulatory mechanism of VHR’s phosphatase activity. ACS Chem Biol. 2014;9:1451–1459. doi: 10.1021/cb500240n. [DOI] [PubMed] [Google Scholar]
- 199.Dukhande VV, Rogers DM, Roma-Mateo C, Donderis J, Marina A, Taylor AO, Sanz P, Gentry MS. Laforin, a dual specificity phosphatase involved in Lafora disease, is present mainly as monomeric form with full phosphatase activity. PLoS One. 2011;6:e24040. doi: 10.1371/journal.pone.0024040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Liu Y, Wang Y, Wu C, Liu Y, Zheng P. Dimerization of Laforin is required for its optimal phosphatase activity, regulation of GSK3 beta phosphorylation, and Wnt signaling. J Biol Chem. 2006;281:34768–34774. doi: 10.1074/jbc.M607778200. [DOI] [PubMed] [Google Scholar]
- 201.Liu Y, Wang Y, Wu C, Liu Y, Zheng P. Deletions and missense mutations of EPM2A exacerbate unfolded protein response and apoptosis of neuronal cells induced by endoplasm reticulum stress. Hum Mol Genet. 2009;18:2622–2631. doi: 10.1093/hmg/ddp196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Sanchez-Martin P, Raththagala M, Bridges TM, Husodo S, Gentry MS, Sanz P, Roma-Mateo C. Dimerization of the glucan phosphatase Laforin requires the participation of cysteine 329. PLoS One. 2013;8:e69523. doi: 10.1371/journal.pone.0069523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Raththagala M, Brewer MK, Parker MW, Sherwood AR, Wong BK, Hsu S, Bridges TM, Paasch BC, Hellman LM, Husodo S, et al. Structural mechanism of Laforin function in glycogen dephosphorylation and Lafora disease. Mol Cell. 2015;57:261–272. doi: 10.1016/j.molcel.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Berger P, Schaffitzel C, Berger I, Ban N, Suter U. Membrane association of myotubularin-related protein 2 is mediated by a pleckstrin homology-GRAM domain and a coiled-coil dimerization module. Proc Natl Acad Sci U S A. 2003;100:12177–12182. doi: 10.1073/pnas.2132732100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Papa A, Wan LX, Bonora M, Salmena L, Song MS, Hobbs RM, Lunardi A, Webster K, Ng C, Newton RH, et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell. 2014;157:595–610. doi: 10.1016/j.cell.2014.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Worby CA, Dixon JE. PTEN. Annu Rev Biochem. 2014;83:641–669. doi: 10.1146/annurev-biochem-082411-113907. [DOI] [PubMed] [Google Scholar]
- 207.Heinrich F, Chakravarthy S, Nanda H, Papa A, Pandolfi PP, Ross AH, Harishchandra RK, Gericke A, Losche M. The PTEN tumor suppressor forms homodimers in solution. Structure. 2015;23:1952–1957. doi: 10.1016/j.str.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Bessette DC, Qiu DX, Pallen CJ. PRL PTPs: mediators and markers of cancer progression. Cancer Metastasis Rev. 2008;27:231–252. doi: 10.1007/s10555-008-9121-3. [DOI] [PubMed] [Google Scholar]
- 209.Stephens BJ, Han HY, Gokhale V, Von Hoff DD. PRL phosphatases as potential molecular targets in cancer. Mol Cancer Ther. 2005;4:1653–1661. doi: 10.1158/1535-7163.MCT-05-0248. [DOI] [PubMed] [Google Scholar]
- 210.Rios P, Li X, Kohn M. Molecular mechanisms of the PRL phosphatases. FEBS J. 2013;280:505–524. doi: 10.1111/j.1742-4658.2012.08565.x. [DOI] [PubMed] [Google Scholar]
- 211.Campbell AM, Zhang ZY. Phosphatase of regenerating liver: a novel target for cancer therapy. Expert Opin Ther Targets. 2014;18:555–569. doi: 10.1517/14728222.2014.892926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Diamond RH, Cressman DE, Laz TM, Abrams CS, Taub R. PRL-1: a unique nuclear protein tyrosine phosphatase, affects cell-growth. Mol Cell Biol. 1994;14:3752–3762. doi: 10.1128/mcb.14.6.3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Liang FB, Liang J, Wang WQ, Sun AP, Udho E, Zhang ZY. PRL3 promotes cell invasion and proliferation by down-regulation of Csk leading to Src activation. J Biol Chem. 2007;282:5413–5419. doi: 10.1074/jbc.M608940200. [DOI] [PubMed] [Google Scholar]
- 214.Sun JP, Luo Y, Yu X, Wang WQ, Zhou B, Liang FB, Zhang ZY. Phosphatase activity, trimerization, and the C-terminal polybasic region are all required for PRL1-mediated cell growth and migration. J Biol Chem. 2007;282:29043–29051. doi: 10.1074/jbc.M703537200. [DOI] [PubMed] [Google Scholar]
- 215.Hardy S, Wong NN, Muller WJ, Park M, Tremblay ML. Overexpression of the protein tyrosine phosphatase PRL-2 correlates with breast tumor formation and progression. Cancer Res. 2010;70:8959–8967. doi: 10.1158/0008-5472.CAN-10-2041. [DOI] [PubMed] [Google Scholar]
- 216.Bai YP, Luo Y, Liu SJ, Zhang LJ, Shen K, Dong YS, Walls CD, Quilliam LA, Wells CD, Cao YJ, et al. PRL-1 protein promotes ERK1/2 and RhoA protein activation through a non-canonical interaction with the Src homology 3 domain of p115 Rho GTPase-activating protein. J Biol Chem. 2011;286:42316–42324. doi: 10.1074/jbc.M111.286302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Wang Y, Lazo JS. Metastasis-associated phosphatase PRL-2 regulates tumor cell migration and invasion. Oncogene. 2012;31:818–827. doi: 10.1038/onc.2011.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Wang HH, Quah SY, Dong JM, Manser E, Tang JP, Zeng Q. PRL-3 down-regulates PTEN expression and signals through PI3K to promote epithelial-mesenchymal transition. Cancer Res. 2007;67:2922–2926. doi: 10.1158/0008-5472.CAN-06-3598. [DOI] [PubMed] [Google Scholar]
- 219.Stephens B, Han HY, Hostetter G, Demeure MJ, Von Hoff DD. Small interfering RNA-mediated knockdown of PRL phosphatases results in altered Akt phosphorylation and reduced clonogenicity of pancreatic cancer cells. Mol Cancer Ther. 2008;7:202–210. doi: 10.1158/1535-7163.MCT-07-0542. [DOI] [PubMed] [Google Scholar]
- 220.Dong YS, Zhang LJ, Zhang S, Bai YP, Chen HY, Sun XX, Yong WD, Li W, Colvin SC, Rhodes SJ, et al. Phosphatase of regenerating liver 2 (PRL2) is essential for placental development by down-regulating PTEN (Phosphatase and Tensin Homologue Deleted on Chromosome 10) and activating Akt protein. J Biol Chem. 2012;287:32172–32179. doi: 10.1074/jbc.M112.393462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Dong YS, Zhang LJ, Bai YP, Zhou HM, Campbell AM, Chen HY, Yong WD, Zhang WJ, Zeng Q, Shou WN, et al. Phosphatase of regenerating liver 2 (PRL2) deficiency impairs Kit signaling and spermatogenesis. J Biol Chem. 2014;289:3799–3810. doi: 10.1074/jbc.M113.512079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Cates CA, Michael RL, Stayrook KR, Harvey KA, Burke YD, Randall SK, Crowell PL, Crowell DN. Prenylation of oncogenic human PTP(CAAX) protein tyrosine phosphatases. Cancer Lett. 1996;110:49–55. doi: 10.1016/s0304-3835(96)04459-x. [DOI] [PubMed] [Google Scholar]
- 223.Wang J, Kirby CE, Herbst R. The tyrosine phosphatase PRL-1 localizes to the endoplasmic reticulum and the mitotic spindle and is required for normal mitosis. J Biol Chem. 2002;277:46659–46668. doi: 10.1074/jbc.M206407200. [DOI] [PubMed] [Google Scholar]
- 224.Wang Q, Holmes DIR, Powell SM, Lu QL, Waxman J. Analysis of stromal-epithelial interactions in prostate cancer identifies PTPCAAX2 as a potential oncogene. Cancer Lett. 2002;175:63–69. doi: 10.1016/s0304-3835(01)00703-0. [DOI] [PubMed] [Google Scholar]
- 225.Werner SR, Lee PA, DeCamp MW, Crowell DN, Randall SK, Crowell PL. Enhanced cell cycle progression and down regulation of p21(Cip1)/(Waf1) by PRL tyrosine phosphatases. Cancer Lett. 2003;202:201–211. doi: 10.1016/s0304-3835(03)00517-2. [DOI] [PubMed] [Google Scholar]
- 226.Guo K, Li J, Tang JP, Koh V, Gan BQ, Zeng Q. Catalytic domain of PRL-3 plays an essential role in tumor metastasis - Formation of PRL-3 tumors inside the blood vessels. Cancer Biol Ther. 2004;3:945–951. doi: 10.4161/cbt.3.10.1111. [DOI] [PubMed] [Google Scholar]
- 227.Wu XP, Zeng H, Zhang XM, Zhao Y, Sha HB, Ge XM, Zhang MY, Gao X, Xu Q. Phosphatase of regenerating liver-3 promotes motility and metastasis of mouse melanoma cells. Am J Pathol. 2004;164:2039–2054. doi: 10.1016/S0002-9440(10)63763-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Zeng Q, Dong JM, Guo K, Li J, Tan HX, Koh V, Pallen CJ, Manser E, Hong W. PRL-3 and PRL-1 promote cell migration, invasion, and metastasis. Cancer Res. 2003;63:2716–2722. [PubMed] [Google Scholar]
- 229.Achiwa H, Lazo JS. PRL-1 tyrosine phosphatase regulates c-Src levels, adherence, and invasion in human lung cancer cells. Cancer Res. 2007;67:643–650. doi: 10.1158/0008-5472.CAN-06-2436. [DOI] [PubMed] [Google Scholar]
- 230.Kato H, Semba S, Miskad UA, Seo Y, Kasuga M, Yokozaki H. High expression of PRL-3 promotes cancer cell motility and liver metastasis in human colorectal cancer: A predictive molecular marker of metachronous liver and lung metastases. Clin Cancer Res. 2004;10:7318–7328. doi: 10.1158/1078-0432.CCR-04-0485. [DOI] [PubMed] [Google Scholar]
- 231.Li ZR, Zhan WH, Wang Z, Zhu BH, He YL, Peng JS, Cai SR, Ma JP. Inhibition of PRL-3 gene expression in gastric cancer cell line SGC7901 via microRNA suppressed reduces peritoneal metastasis. Biochem Biophys Res Commun. 2006;348:229–237. doi: 10.1016/j.bbrc.2006.07.043. [DOI] [PubMed] [Google Scholar]
- 232.Polato F, Codegoni A, Fruscio R, Perego P, Mangioni C, Saha S, Bardelli A, Broggini M. PRL-3 phosphatase is implicated in ovarian cancer growth. Clin Cancer Res. 2005;11:6835–6839. doi: 10.1158/1078-0432.CCR-04-2357. [DOI] [PubMed] [Google Scholar]
- 233.Qian F, Li YP, Sheng X, Zhang ZC, Song R, Dong W, Cao SX, Hua ZC, Xu Q. PRL-3 siRNA inhibits the metastasis of B16-BL6 mouse melanoma cells in vitro and in vivo. Mol Med. 2007;13:151–159. doi: 10.2119/2006-00076.Qian. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Fagerli UM, Holt RU, Holien T, Vaatsveen TK, Zhan FH, Egeberg KW, Barlogie B, Waage A, Aarset H, Dai HY, et al. Overexpression and involvement in migration by the metastasis-associated phosphatase PRL-3 in human myeloma cells. Blood. 2008;111:806–815. doi: 10.1182/blood-2007-07-101139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Fang XY, Song R, Chen W, Yang YY, Gu YH, Shu YQ, Wu XD, Wu XF, Sun Y, Shen Y, et al. PRL-3 promotes the malignant progression of melanoma via triggering dephosphorylation and cytoplasmic localization of NHERF1. J Invest Dermatol. 2015;135:2273–2282. doi: 10.1038/jid.2015.154. [DOI] [PubMed] [Google Scholar]
- 236.Jin SW, Wang KM, Xu K, Xu JY, Sun J, Chu ZH, Lin DC, Koeffler PH, Wang J, Yin D. Oncogenic function and prognostic significance of protein tyrosine phosphatase PRL-1 in hepatocellular carcinoma. Oncotarget. 2014;5:3685–3696. doi: 10.18632/oncotarget.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Miskad UA, Semba S, Kato H, Yokozaki H. Expression of PRL-3 phosphatase in human gastric carcinomas: Close correlation with invasion and metastasis. Pathobiology. 2004;71:176–184. doi: 10.1159/000078671. [DOI] [PubMed] [Google Scholar]
- 238.Park JE, Yuen HF, Zhou JB, Al-aidaroos AQO, Guo K, Valk PJ, Zhang SD, Chng WJ, Hong CW, Mills K, et al. Oncogenic roles of PRL-3 in FLT3-ITD induced acute myeloid leukaemia. EMBO Mol Med. 2013;5:1351–1366. doi: 10.1002/emmm.201202183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Radke I, Gotte M, Kersting C, Mattsson B, Kiesel L, Wulfing P. Expression and prognostic impact of the protein tyrosine phosphatases PRL-1: PRL-2: and PRL-3 in breast cancer. Br J Cancer. 2006;95:347–354. doi: 10.1038/sj.bjc.6603261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Ren TT, Jiang BH, Xing XF, Dong B, Peng LR, Meng L, Xu HY, Shou CC. Prognostic significance of phosphatase of regenerating liver-3 expression in ovarian cancer. Pathol Oncol Res. 2009;15:555–560. doi: 10.1007/s12253-009-9153-1. [DOI] [PubMed] [Google Scholar]
- 241.Saha S, Bardelli A, Buckhaults P, Velculescu VE, Rago C, St Croix B, Romans KE, Choti MA, Lengauer C, Kinzler KW, et al. A phosphatase associated with metastasis of colorectal cancer. Science. 2001;294:1343–1346. doi: 10.1126/science.1065817. [DOI] [PubMed] [Google Scholar]
- 242.Bai YP, Zhou HM, Zhang LJ, Dong YS, Zeng Q, Shou WN, Zhang ZY. Role of phosphatase of regenerating liver 1 (PRL1) in spermatogenesis. Sci Rep. 2016;6:12. doi: 10.1038/srep34211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kobayashi M, Bai YP, Dong YS, Yu H, Chen SS, Gao R, Zhang LJ, Yoder MC, Kapur R, Zhang ZY, et al. PRL2/PTP4A2 phosphatase is important for hematopoietic stem cell self-renewal. Stem Cells. 2014;32:1956–1967. doi: 10.1002/stem.1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Jeong DG, Kim SJ, Kim JH, Son JH, Park MR, Lim SM, Yoon TS, Ryu SE. Trimeric structure of PRL-1 phosphatase reveals an active enzyme conformation and regulation mechanisms. J Mol Biol. 2005;345:401–413. doi: 10.1016/j.jmb.2004.10.061. [DOI] [PubMed] [Google Scholar]
- 245.Bardelli A, Saha S, Sager JA, Romans KE, Xin BZ, Markowitz SD, Lengauer C, Velculescu VE, Kinzler KW, Vogelstein B. PRL-3 expression in metastatic cancers. Clin Cancer Res. 2003;9:5607–5615. [PubMed] [Google Scholar]
- 246.Zeng Q, Si XN, Horstmann H, Xu Y, Hong WJ, Pallen CJ. Prenylation-dependent association of protein-tyrosine phosphatases PRL-1:-2: and-3 with the plasma membrane and the early endosome. J Biol Chem. 2000;275:21444–21452. doi: 10.1074/jbc.M000453200. [DOI] [PubMed] [Google Scholar]
- 247.Bai YP, Yu ZH, Liu SJ, Zhang LJ, Zhang RY, Zeng LF, Zhang S, Zhang ZY. Novel anticancer agents based on targeting the trimer interface of the PRL phosphatase. Cancer Res. 2016;76:4805–4815. doi: 10.1158/0008-5472.CAN-15-2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Sivaramakrishnan S, Keerthi K, Gates KS. A chemical model for redox regulation of protein tyrosine phosphatase 1B (PTP1B) activity. J Am Chem Soc. 2005;127:10830–10831. doi: 10.1021/ja052599e. [DOI] [PubMed] [Google Scholar]
- 249.van Montfort RLM, Congreve M, Tisi D, Carr R, Jhoti H. Oxidation state of the active- site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423:773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 250.Sohn J, Rudolph J. Catalytic and chemical competence of regulation of Cdc25 phosphatase by oxidation/reduction. Biochemistry. 2003;42:10060–10070. doi: 10.1021/bi0345081. [DOI] [PubMed] [Google Scholar]
- 251.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 252.Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, Moghal N, Lubkin M, Kim YB, Sharpe AH, et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol. 2000;20:5479–5489. doi: 10.1128/mcb.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Haque A, Andersen JN, Salmeen A, Barford D, Tonks NK. Conformation-sensing antibodies stabilize the oxidized form of PTP1B and inhibit its phosphatase activity. Cell. 2011;147:185–198. doi: 10.1016/j.cell.2011.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Haque A, Tonks NK. The use of phage display to generate conformation-sensor recombinant antibodies. Nat Protoc. 2012;7:2127–2143. doi: 10.1038/nprot.2012.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Parsons ZD, Ruddraraju KV, Santo N, Gates KS. Sulfone-stabilized carbanions for the reversible covalent capture of a posttranslationally-generated cysteine oxoform found in protein tyrosine phosphatase 1B (PTP1B) Biorg Med Chem. 2016;24:2631–2640. doi: 10.1016/j.bmc.2016.03.054. [DOI] [PubMed] [Google Scholar]
- 256.Ruddraraju KV, Parsons ZD, Llufrio EM, Frost NL, Gates KS. Reactions of 1:3-diketones with a dipeptide isothiazolidin-3-one: Toward agents that covalently capture oxidized protein tyrosine phosphatase 1B. J Org Chem. 2015;80:12015–12026. doi: 10.1021/acs.joc.5b01949. [DOI] [PubMed] [Google Scholar]
- 257.Leonard SE, Garcia FJ, Goodsell DS, Carroll KS. Redox-based probes for protein tyrosine phosphatases. Angew Chem Int Ed. 2011;50:4423–4427. doi: 10.1002/anie.201007871. [DOI] [PubMed] [Google Scholar]
- 258.Garcia FJ, Carroll KS. An immunochemical approach to detect oxidized protein tyrosine phosphatases using a selective C-nucleophile tag. Mol Biosyst. 2016;12:1790–1798. doi: 10.1039/c5mb00847f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Molina G, Vogt A, Bakan A, Dai WX, de Oliveira PQ, Znosko W, Smithgall TE, Bahar I, Lazo JS, Day BW, et al. Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nat Chem Biol. 2009;5:680–687. doi: 10.1038/nchembio.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Korotchenko VN, Saydmohammed M, Vollmer LL, Bakan A, Sheetz K, Debiec KT, Greene KA, Agliori CS, Bahar I, Day BW, et al. In vivo structure-activity relationship studies support allosteric targeting of a dual specificity phosphatase. ChemBioChem. 2014;15:1436–1445. doi: 10.1002/cbic.201402000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Shojaee S, Caeser R, Buchner M, Park E, Swaminathan S, Hurtz C, Geng HM, Chan LN, Klemm L, Hofmann WK, et al. Erk negative feedback control enables Pre-B cell transformation and represents a therapeutic target in acute lymphoblastic leukemia. Cancer Cell. 2015;28:114–128. doi: 10.1016/j.ccell.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Chen YNP, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, Antonakos B, Chen CHT, Chen ZL, Cooke VG, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. 2016;535:148–152. doi: 10.1038/nature18621. [DOI] [PubMed] [Google Scholar]
- 263.Fortanet JG, Chen CHT, Chen YNP, Chen ZL, Deng Z, Firestone B, Fekkes P, Fodor M, Fortin PD, Fridrich C, et al. Allosteric inhibition of SHP2: Identification of a potent, selective, and orally efficacious phosphatase inhibitor. J Med Chem. 2016;59:7773–7782. doi: 10.1021/acs.jmedchem.6b00680. [DOI] [PubMed] [Google Scholar]
- 264.Bunda S, Burrell K, Heir P, Zeng LF, Alamsahebpour A, Kano Y, Raught B, Zhang ZY, Zadeh G, Ohh M. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nature Commun. 2015;6:8859. doi: 10.1038/ncomms9859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Grosskopf S, Eckert C, Arkona C, Radetzki S, Bohm K, Heinemann U, Wolber G, von Kries JP, Birchmeier W, Rademann J. Selective inhibitors of the protein tyrosine phosphatase SHP2 block cellular motility and growth of cancer cells in vitro and in vivo. Chemmedchem. 2015;10:815–826. doi: 10.1002/cmdc.201500015. [DOI] [PubMed] [Google Scholar]
- 266.Zeng LF, Zhang RY, Yu ZH, Li SJ, Wu L, Gunawan AM, Lane BS, Mali RS, Li XJ, Chan RJ, et al. Therapeutic potential of targeting the oncogenic SHP2 phosphatase. J Med Chem. 2014;57:6594–6609. doi: 10.1021/jm5006176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Zhang RY, Yu ZH, Zeng LF, Zhang S, Bai YP, Miao JM, Chen L, Xie JW, Zhang ZY. SHP2 phosphatase as a novel therapeutic target for melanoma treatment. Oncotarget. 2016;7:73817–73829. doi: 10.18632/oncotarget.12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Bohmer F, Szedlacsek S, Tabernero L, Ostman A, den Hertog J. Protein tyrosine phosphatase structure-function relationships in regulation and pathogenesis. FEBS J. 2013;280:413–431. doi: 10.1111/j.1742-4658.2012.08655.x. [DOI] [PubMed] [Google Scholar]
- 269.Takahashi T, Takahashi K, Mernaugh RL, Tsuboi N, Liu H, Daniel TO. A monoclonal antibody against CD148: a receptor-like tyrosine phosphatase, inhibits endothelial-cell growth and angiogenesis. Blood. 2006;108:1234–1242. doi: 10.1182/blood-2005-10-4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Xie YM, Massa SM, Ensslen-Craig SE, Major DL, Yang T, Tisi MA, Derevyanny VD, Runge WO, Mehta BP, Moore LA, et al. Protein-tyrosine phosphatase (PTP) wedge domain peptides - A novel approach for inhibition of PTP function and augmentation of protein-tyrosine kinase function. J Biol Chem. 2006;281:16482–16492. doi: 10.1074/jbc.M603131200. [DOI] [PubMed] [Google Scholar]
- 271.Lang BT, Cregg JM, DePaul MA, Tran AP, Xu K, Dyck SM, Madalena KM, Brown BP, Weng YL, Li SX, et al. Modulation of the proteoglycan receptor PTP sigma promotes recovery after spinal cord injury. Nature. 2015;518:404–408. doi: 10.1038/nature13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Li H, Wong C, Li W, Ruven C, He LM, Wu XL, Lang BT, Silver J, Wu WT. Enhanced regeneration and functional recovery after spinal root avulsion by manipulation of the proteoglycan receptor PTP sigma. Sci Rep. 2015;5:14. doi: 10.1038/srep14923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Gardner RT, Wang L, Lang BT, Cregg JM, Dunbar CL, Woodward WR, Silver J, Ripplinger CM, Habecker BA. Targeting protein tyrosine phosphatase sigma after myocardial infarction restores cardiac sympathetic innervation and prevents arrhythmias. Nature Commun. 2015;6:6235. doi: 10.1038/ncomms7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Perron MD, Chowdhury S, Aubry I, Purisima E, Tremblay ML, Saragovi HU. Allosteric noncompetitive small molecule selective inhibitors of CD45 tyrosine phosphatase suppress T-cell receptor signals and inflammation in vivo. Mol Pharmacol. 2014;85:553–563. doi: 10.1124/mol.113.089847. [DOI] [PubMed] [Google Scholar]
- 275.Guo K, Li J, Tang JP, Tan CP, Hong CW, Al-Aidaroos AQ, Varghese L, Huang C, Zeng Q. Targeting intracellular oncoproteins with antibody therapy or vaccination. Sci Trans Med. 2011;3:99ra85. doi: 10.1126/scitranslmed.3002296. [DOI] [PubMed] [Google Scholar]
- 276.Guo K, Tang JP, Jie L, Al-Aidaroos AQ, Hong CW, Tan CP, Park JE, Varghese L, Feng Z, Zhou J, et al. Engineering the first chimeric antibody in targeting intracellular PRL-3 oncoprotein for cancer therapy in mice. Oncotarget. 2012;3:158–171. doi: 10.18632/oncotarget.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Thura M, Al-Aidaroos AQ, Yong WP, Kono K, Gupta A, Lin YB, Mimura K, Thiery JP, Goh BC, Tan P, et al. PRL3-zumab, a first-in-class humanized antibody for cancer therapy. JCI Insight. 2016;1:e87607. doi: 10.1172/jci.insight.87607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Hendriks W, Elson A, Harroch S, Pulido R, Stoker A, den Hertog J. Protein tyrosine phosphatases in health and disease. FEBS J. 2013;280:708–730. doi: 10.1111/febs.12000. [DOI] [PubMed] [Google Scholar]
- 279.Wright AV, Nunez JK, Doudna JA. Biology and applications of CRISPR systems: Harnessing Nature’s toolbox for genome engineering. Cell. 2016;164:29–44. doi: 10.1016/j.cell.2015.12.035. [DOI] [PubMed] [Google Scholar]
- 280.Sharpless NE, DePinho RA. Model organisms - The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov. 2006;5:741–754. doi: 10.1038/nrd2110. [DOI] [PubMed] [Google Scholar]
- 281.Frese KK, Tuveson DA. Maximizing mouse cancer models. Nat Rev Cancer. 2007;7:645–658. doi: 10.1038/nrc2192. [DOI] [PubMed] [Google Scholar]
- 282.Zhang ZY. Drugging the undruggable: Therapeutic potential of targeting protein tyrosine phosphatases. Acc Chem Res. 2017;50:122–129. doi: 10.1021/acs.accounts.6b00537. [DOI] [PMC free article] [PubMed] [Google Scholar]