

Summary
Sponges are benthic filter feeders that play pivotal roles in coupling benthic-pelagic processes in the oceans that involve transformation of dissolved and particulate organic carbon and nitrogen into biomass. While the contribution of sponge holobionts to the nitrogen cycle has been recognized in past years, their importance in the sulfur cycle, both oceanic and physiological, has only recently gained attention. Sponges in general, and Theonella swinhoei in particular, harbor a multitude of associated microorganisms that could affect sulfur cycling within the holobiont. We reconstructed the genome of a Chromatiales (class Gammaproteobacteria) bacterium from a metagenomic sequence dataset of a T. swinhoei-associated microbial community. This relatively abundant bacterium has the metabolic capability to oxidize sulfide yet displays reduced metabolic potential suggestive of its lifestyle as an obligatory symbiont. This bacterium was detected in multiple sponge orders, according to similarities in key genes such as 16S rRNA and polyketide synthase genes. Due to its sulfide oxidation metabolism and occurrence in many members of the Porifera phylum, we suggest naming the newly described taxon Candidatus Porisulfidus.
Introduction
Sulfur is assimilated into amino acids, vitamins, secondary metabolites, and sulfo-lipid compounds, making it an essential element for all living organisms. Beyond its importance as a building block, many bacteria and archaea use reduced sulfur compounds as electron sources to generate energy and assimilate inorganic carbon (Vogler et al., 1942). Photoautotroph sulfur oxidizing bacteria (SOB) oxidize hydrogen sulfide and other forms of reduced sulfur under anoxic conditions, such as those that occur in stratified lakes (Jorgensen et al., 1979). Their counterparts, the chemolithotrophic SOB, can be found in a multitude of environments, from oxic to anoxic, terrestrial to deep-ocean hydrothermal vents (extensively reviewed by Gohsh and Dam (2009)). In extreme environments, such as hydrothermal vents, sulfur-chemolitotrophs support communities of heterotrophic organisms by filling the role of primary producers (Stewart et al., 2005). In many of these cases SOB form intra- or extra-cellular symbioses with their heterotrophic hosts (Bright and Giere, 2005; Taylor and Glover, 2006; Nakagawa et al., 2014).
Sponges are heterotrophic filter-feeders, some hosting up to 5 × 1010 bacteria (mL sponge)-1. They inhabit marine and freshwater ecosystems (Taylor et al., 2007) and mostly feed by filtering up to 50,000 times their body volume per day (Weisz et al., 2008). Their evolutionary success may be ascribed to their complex association with diverse consortia of microorganisms (Schmitt et al., 2012), that provide them with various services, from chemical defense (Guo et al., 2011) to recycling metabolic waste (Mohamed et al., 2010). The role of sponge-associated microorganisms in biogeochemical cycles has been studied extensively over the past four decades (Maldonado et al., 2012; Webster and Thomas, 2016). However, most studies have focused on the carbon and nitrogen cycles. For example, it is now well established that ammonia excreted by the sponge cells serves as an energy and N source for its nitrifying symbionts (Southwell et al., 2008), and urea is used as a nitrogen source by sponge-associated bacteria (Su et al., 2013). Not only nitrification but also denitrification occurs within a sponge. The anoxic environment within the sponge body may provide the necessary conditions for bacteria and archaea responsible for such activities (Schläppy et al., 2010; Lavy et al., 2016).
While nitrogen metabolism in the sponge holobiont is widely studied, our knowledge of the sulfur cycle, and the role of sponge-associated bacteria in it, is very limited. The most thorough work on sulfur metabolism within sponges was conducted on the high microbial abundance (HMA) sponge Geodia barretti (Hoffmann et al., 2005). In that study, sulfate reduction rates were higher in areas close to filtration chambers compared to the sponge cortex. In accordance with the higher reduction rates, 5.3 × 109 cells (mL sponge)-1 of Deltaproteobacteria (of the Desulfoarculus/Desulfomonile/Syntrophus cluster) were found in the vicinity of filtration chambers using fluorescence in-situ hybridization (FISH) probes and none were found in the cortex.
Gene amplification studies have provided evidence of archaeal and bacterial sulfur reduction/oxidation processes in sponges. A recent work found that 87% of all amplified adenosine-5′-phosphosulfate reductase alpha subunit gene (aprA) sequences, which encodes a key enzyme in microbial sulfate reduction and sulfur oxidation, from G. barretti were affiliated with sulfur oxidizing Alphaproteobacteria and Gammaproteobacteria. (Jensen et al., 2016). The remaining 13% of sequences clustered with sulfate-reducing archaea of the phylum Euryarchaeota. Also, DGGE profiles of spatial samples taken at three locations within the sponge were indistinguishable, suggesting that the microbial community is similar in composition throughout the sponge body. Although these findings may seem to contradict those of Hoffmann et al. (2005), it should be noted that the two studies used different methods. In another study of the deep-sea sponge Polymastia cf. corticata (Meyer and Kuever, 2008), members of six identified sulfur-oxidizing and sulfate reducing lineages containing the aprA gene were identified. Among them were the gammaproteobacterial SOB and non-sponge-specific alphaproteobacterial SOB that were present in the entire sponge body. However, the putative sponge-specific alphaproteobacterial sulfur-oxidizers and archaeal sulfate-reducing strains were restricted to the inner tissue sections. The co- occurrence of SOB and sulfate reducing bacteria (SRB) in both studies suggests the two processes take place in the same sponge.
DGGE and 16S rRNA gene amplicon sequencing studies are used to assess the structure of microbial communities in the environment in general and sponges in particular. Several such studies have identified Chromatiales bacteria in several sponges. Kennedy et al. (2014) have found Chromatiales related sequences in four deep water sponges (Lissodendoryx diversichela, Poecillastra compressa, Inflatella pellicular, and Stelletta normans). Members of the Chroamtiales order were also detected in Hymeniacidon sinapium (Jeong et al., 2015), Rhabdastrella globostellata (Steinert et al., 2016), Axinella corrugate (White et al., 2012), and Cinachyrella australiensis (Cleary et al., 2013). In the latter, the 16S rRNA gene sequence clustered with sequences of other Nitrosococcus species. Chromatiales sequences were found in Aplysina fulva from Brazilian water (Hardoim et al., 2009) and in Sarcotragus spinosulus (Hardoim et al., 2012). Both studies suggested that these bacteria may carry out sulfide oxidation as other members of this order contains purple sulfur bacteria that are able to carry out anoxygenic photosynthesis using hydrogen sulfide as the electron donor. The sulfur-oxidizing bacteria of this order were found to be abundant in Amphimedon queenslandica and were further identified as members of the family Ectothiorhodospiraceae (Gauthier et al., 2016). The sequences clustered with other uncultivated clones, including some from a chemoautotrophic sulfur-oxidizing bacterium associated with the demosponge Haliclona cymaeformis detected by Tian et al. (2014). Using electron-microscopy, the authors found that the microbial cell lacks photosynthetic structures, but contain globules inside the cell membrane, hypothesized to be sulfur, as occurs in other purple-sulfur bacteria. While greatly improving our understanding of microbial community structure, amplicon studies are limited by the short reads length, thus can typically only identify microbes down to the family or genus level. Further, metabolic insights rely the assumption that the physiology of the organism with the most closely related 16S rRNA gene is similar to that of the organism of interest.
Imhoff and Trüper (1976) isolated four Gammaproteobacterial phototrophic SOB (Chromatium gracile, C. vinosum, C. minutissimum and Ectothiorhodospira mobilis) and two Alphaproteobacteria (Rhodopseudomonas sulfidophila and R. palustris) SOB from four sponge species. The authors concluded that a special micro-environment is formed, allowing suitable growth conditions for the anaerobic phototrophic bacteria in the sponge. However, it was only in 2014 that a genome of a sponge-associated SOB was published (Tian et al., 2014). The genomic bin Gspo, named Thioalkalivibrio sp. HK1 (order Chromatiales) in its NCBI GenBank entry, was obtained from a metagenome of the sponge Haliclona cymaeformis. HK1 displays genetic characteristics such as a lack of transposases and an abundance of ankyrin repeat protein domains, which are common in bacterial symbionts. The presence of soxABXYZ and dsrAB genes, as well as absence of soxCD suggest that this organism oxidizes sulfide or sulfite through the reverse sulfate reduction pathway (Friedrich et al., 2001), as do other Chromatiaceae. Interestingly, the bacterium was found to be a non- photosynthetic mixotroph. The addition of heterotrophic capability could be crucial in the oxidized environments that frequently occur in the sponge body throughout the day (Lavy et al., 2016). Heterotrophic metabolism could also support these bacteria if they are horizontally transferred between sponges via an oxidized seawater environment.
In the past three years, attention has focused on the role of sulfur metabolizing bacteria in sponges. Three new draft genomes of sponge-associated sulfur oxidizing bacteria were published. Gsub from Suberites sp. and SOB1 from Lophophysema eversa, both of the order Thiotrichales are closely related to Candidatus Vesicomyosocius okutanii and Candidatus Ruthia maganifica, endosymbionts of deep sea clams (Tian et al., 2016, 2017). These bacteria show the potential to oxidize sulfite to sulfate using the Sox operon. A third sulfur oxidizing sponge-associated Chromatiales named AqS1, closely related to HK1, was recently found in the sponge Amphimedon queenslandica (Gauthier et al., 2016). The genomic differences between AqS1 and HK1 suggest that the two bacteria form two distinct species and maybe even genera. Moreover, Gauthier et al. (2016) show that Thioalkalivibrio HK1 may in fact, not be a Thioalkalivibrio at all, and therefore will be named HK1 from now on.
The sponge Theonella swinhoei is common on Indo-Pacific coral reefs, including the Gulf of Aqaba, and Eilat northern Red Sea (Ilan et al., 2004). It harbors a dense consortium of photosynthetic and heterotrophic bacteria, with up to 1010 bacteria (mL sponge)-1 (Magnino et al., 1999). So far, nine bacterial phyla (Gemmatimonadetes, Chloriflexi, Cyanobacteria, Nitrospira, Proteobacteria, Spirochaetes, Verrucomicrobia, Actinobacteria, Acidobacteria) and four candidate phyla (Poribacteria, Tectomicrobia, OP10 and OS-K) were detected by culture-independent methods in T. swinhoei (Schmitt et al., 2012). This study found 16S rRNA gene sequences related to sulfate reducing Desulfobulbaceae and Desulfovibrio. However, no sulfur oxidizing microbes were identified. Members of three additional phyla (Bacteriodetes, Firmicutes, and Lentisphaerae) were recently detected in a culturing experiment (Lavy et al., 2014). In the latter experiment, two Desulfovibrio isolates were grown under microaerobic conditions. The two isolates (TSAR6 and TSAR16) are closely related to known SRB (964 bp 16S rRNA >97% similar, 100% coverage). Moreover, a recent study reported barite (BaSO4) formation, which is linked to sulfur oxidation, within T. swinhoei (Keren et al., 2017). Numerous secondary metabolites have been extracted from T. swinhoei, some are novel antitumor active polyketides, whereas others have antifungal or antibacterial activity (Bewley et al., 1996; Schmidt et al., 2000; Okada et al., 2002; Piel, 2004). Some of these compounds have been attributed to extracellular bacterial symbionts (Bewley et al., 1996; Piel, 2004, 2009; Wilson et al., 2014). Similar to other high microbial abundance (HMA) sponges, T. swinhoei has dense tissue and slow water flow through its body (Yahel et al., 2003; Weisz et al., 2008). A recent study found that microaerobic conditions occur temporally and spatially within the sponge over long periods of time (Lavy et al., 2016).
As in the case of many other sponge species, very little is known about the sulfur cycle in T. swinhoei and its symbionts. This could be because sulfur chemical species such as sulfide are not stable and therefore hard to detect in-situ. In order to overcome this barrier, we studied the sulfur biogeochemical cycle within the sponge through analysis of the microbial community. Using genome resolved metagenomics we revealed the presence of a SOB in the sponge. In this study, we present the genome of the discovered bacterium and discuss its role in the sulfur cycle in T. swinhoei and in other sponge species.
Results and discussion
General genomic characteristics
Sponges are considered as holobionts, as their microbial counterparts undertake important roles in their host’s survival. Studies in the past decade have shown that these symbionts provide numerous services for their host, from chemical protection (Wakimoto et al., 2014) to nitrogen cycling (Hoffmann et al., 2009). Genome resolved metagenomics, which provides an opportunity to investigate the vast microbial community of the coral reef sponge T. swinhoei, revealed a bacterium that seems to participate in the sponge’s sulfur metabolism by oxidizing sulfide and sulfite to sulfate.
The recovered T. swinhoei SOB (TsSOB) draft genome consists of 21 contigs and has a total size of 1.59 Mbp (Figure S1). The longest contig size is 444 Kbp and the draft genome N50 is 153 Kbp (Table 1). The genome has an average GC content of 59.4% and a coding density of 94%. TsSOB was found to be the 10th most abundant bacterium in T. swinhoei’s microbial community, based on rank abundance analysis using the rpS3 gene sequences (Figure S2). The genome has genes coding for 19 tRNA types, 17 tRNA synthetases, 51 ribosomal proteins, and a total of 1567 genes (Table S1). Completeness and possible contamination were evaluated by CheckM (Parks et al., 2015) to be 92.9% and 0%, respectively. The reads mapping to the contigs of TsSOB were also present in the metagenome sequenced from a second T. swinhoei sample (breadth 0.96, average coverage 7.3), and partially in the third sample (breadth 0.48, average coverage 1.7) but not in a seawater sample (breadth 0.01, average coverage 1.7).
Table 1.
Genome features.
| Feature | Value |
|---|---|
| Genome size (Mb) | 1.59 |
| Number of contigs | 21 |
| N50 (scaffolds) | 153662 |
| GC (%) | 59.48 |
| Predicted genes | 1567 |
| Total rRNA | 1 |
| 5S rRNA | 1 |
| 16S rRNA | 1 |
| 23S rRNA | 1 |
| tRNA | 42 |
| Bacterial single copy genes | 51 |
| Ribosomal proteins* | 51 |
| Coding density (%) | 94 |
| Completeness estimate (%) | 92.98 |
| Genome size estimate (Mb) | 1.72 |
List of ribosomal proteins, tRNA and tRNA synthetases that are present in the genome is given is Table S1.
A phylogenetic analysis based on the protein sequences of 16 concatenated ribosomal proteins reveals that TsSOB is a Gammaproteobacteria from the order Chromatiales (Figure 1 and Figure S3). TsSOB forms a long branching sister clade to Halothiobacillus neapolitanus, a non-photosynthetic, carbon fixing, sulfide oxidizing bacterium (Garrity et al., 2010). It is therefore suggested that TsSOB may be a member of Halothiobacillaceae family which currently has only one genus, Halothiobaillus, or even represents a new family. Members of the Halothiobacillaceae family are known to be aerobic, halophilic, and considered to play an important role in global carbon and sulfur cycles (Pokorna and Zabranska, 2015). They depend entirely on inorganic compounds (CO2 and reduced sulfur) for their carbon and energy needs (Garrity et al., 2010). The average nucleotide identity (ANI) between TsSOB and Halothiobacillus neapolitanus is 67.3%, suggesting that TsSOB is a distinct genus or family (Goris et al., 2007). The two other SOB sponge symbionts HK1 and AqS1, which are the symbionts of the sponges Haliclona cymaeformis and Amphimedon queenslandica, respectively, form a cluster close to Ectothiorhodospiraceae, another class of Chromatiales.
Figure 1. TsSOB is member of the Chromatiales order, most closely related to the sulfur-oxidizing Halothiobacillus neapolitanus.
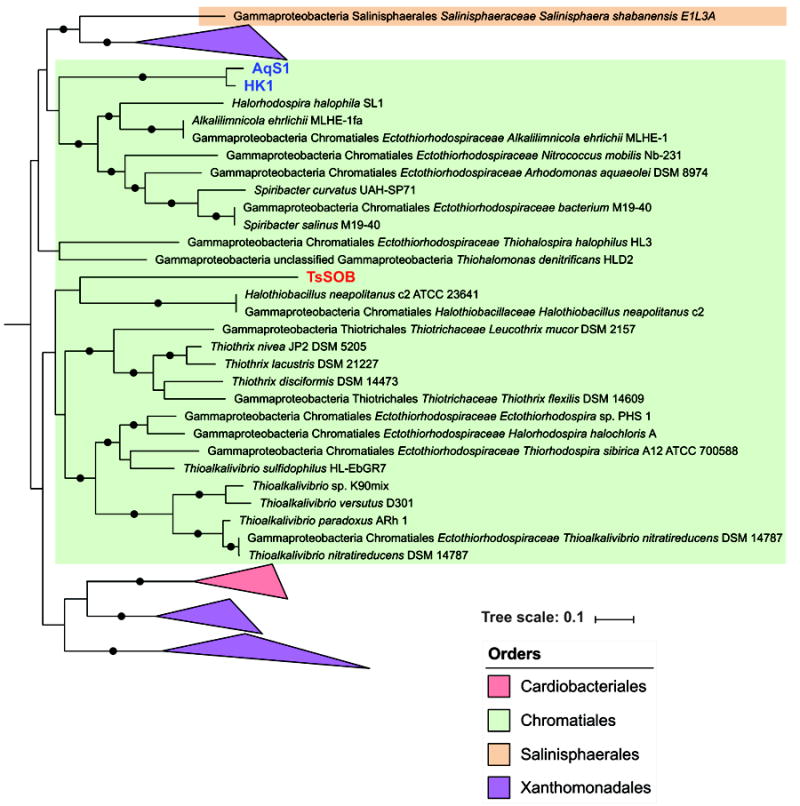
A subset of a Maximum-likelihood (ML) phylogenetic tree which includes taxons within the vicinity of TsSOB. The tree was calculated with RaXML with 100 bootstraps and based on sequences of 16 concatenated ribosomal proteins. Bootstrap values greater than 0.8 are shown as black dots on branches. Blue text indicates sulfur-oxidizing sponge symbionts reported in the literature, whereas TsSOB is marked in red.
Sulfide oxidation
The presence of dissimilatory (bi)sulfite reductase (Dsr) genes as well as sulfide dehydrogenase flavocytochrome-C (FCC) suggests that TsSOB is capable of oxidizing sulfide to be used as an electron source. The 13 Dsr genes are arranged in one operon that consists of dsrABEFHCMKLJOPN (Figure 2 and Figure S4). As expected, the additional DsrD, which is considered a marker for sulfate reduction (Sander et al., 2006), is not present. Sulfurtransferase tusE, which is a member of the dsrC/tusE and may function as a regulatory sulfur-related protein, is located immediately after the dsr operon (Venceslau et al., 2014). The two cytochrome subunits of FCC are found outside on another scaffold. The cytochrome complex allows TsSOB to oxidize sulfide to elemental sulfur, which can then be further oxidized by the proteins coded by dsr. Upstream to the dsr operon are the sulfite oxidation genes SoxABXYZ (Figure 2 and Figure S4). While dsr and FCC allow sulfide oxidation, the presence of sox genes suggests that thiosulfate oxidation is another possible process carried out by the bacterium (Figure 3). Previous studies showed that when soxCD are missing, as in the case of TsSOB, cytochrome c reduction rates decrease to 25% (Friedrich et al., 2001). In order to compensate for the loss of efficiency, sulfur oxidation may continue by the product of dsr genes, as in the case of Thioploca ingrica (Kojima et al., 2015). Sulfur oxidation is a trait not unique to T. swinhoei’s associated bacteria. Previous studies showed that this process may be mediated by symbionts of H. cymaeformis (HK1) (Tian et al., 2014), Suberites sp. (Gsub) (Tian et al., 2016) Lophophysema eversa (SOB1) (Tian et al., 2017) and A. queenslandica (AqS1) (Gauthier et al., 2016). AqS1 and Thialkalivibrio sp. HK1 both possess a polycistronic cassette of 12 dsr genes. FCC, which is involved in catalyzing the formation of sulfur globules and enables sulfide oxidation, was not detected. It is likely that both bacteria do have FCC as they are closely related to members of the Ectothiorhodospiraceae family which are capable of sulfide oxidation.
Figure 2. Schematic representation of sulfide oxidation operon structure found in the draft genome of TsSOB.
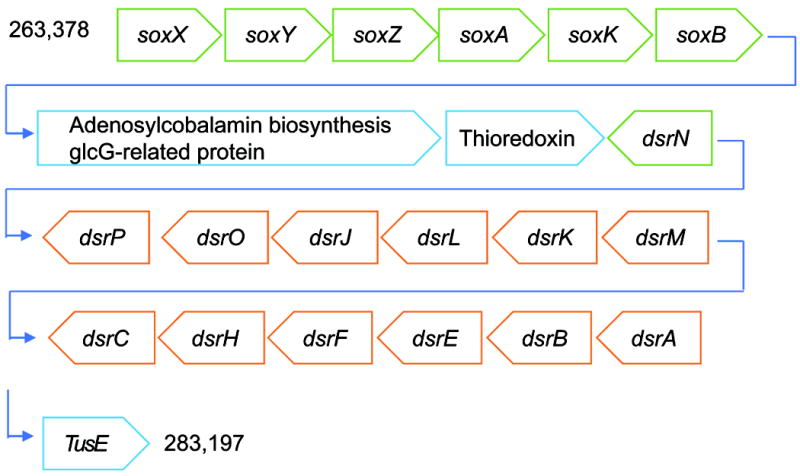
The operon is present in scaffold_2 of the draft genome. The soxXYZAKB genes are found on the forward strand while dsrABEFHCMKLJOP are on the complement strand. As expected, the regulatory gene dsrD, which is considered a marker for sulfate reduction, is missing from the operon. Both cytochrome subunits of sulfide dehydrogenase Flavocytochrome-C (FCC) are present in another scaffold. Size-accurate representation of the genes is given in Figure S4. Numbers at the beginning and end of the sequence indicate the location of the structure along the scaffold. Green and orange colors denote genes of the Sox and Dsr families, respectively.
Figure 3. Sulfide and thiosulfate oxidation pathways present in the genome.
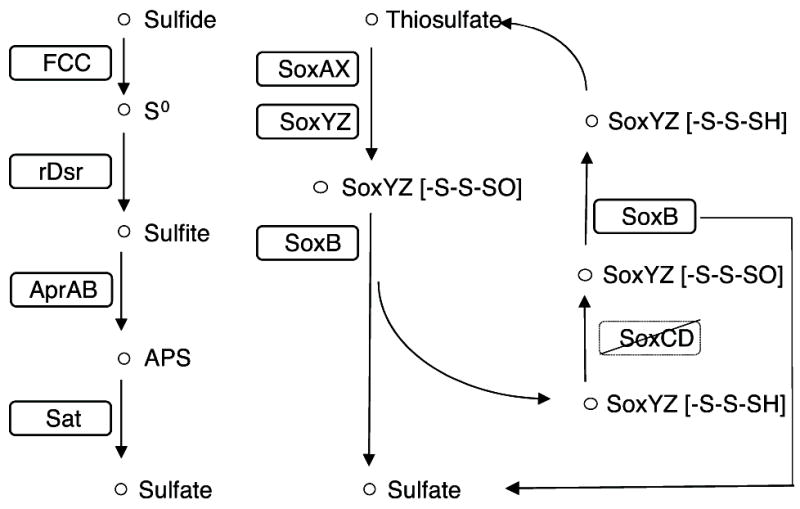
TsSOB can oxidize sulfide to sulfate through the reverse-Dsr pathway. Both subunits of Flavocytochrome-C (FCC), which are required in order to oxidize sulfide to sulfur intracellularly, are present in the genome. Thiosulfate is oxidized to sulfate through the Sox pathway. Solid outline - genes present in the draft genome. Crossed-out genes are missing from the draft genome.
In the case of sulfur oxidizers and reducers, the phylogeny of these traits could be traced according to the accumulation of changes in the sequence of the dsrAB genes that code for dissimilatory (bi)sulfite reductase subunits dsrA and dsrB (Muller et al., 2015). A Maximum Likelihood (ML) phylogenetic tree based on the dsrAB amino-acid sequences of TsSOB and other sulfur oxidizing bacteria agrees with the 16 concatenated ribosomal proteins tree, as TsSOB forms a clade distinct from HK1 and AqS1 (Figure 4). This evidence strengthens the finding that the sulfur oxidation property of TsSOB is not related to that of other sulfur oxidizing sponge symbionts such as HK1 and Gsub.
Figure 4. The dsrAB genes of TsSOB are distinct from those HK1 and AqS1 sponge symbionts.
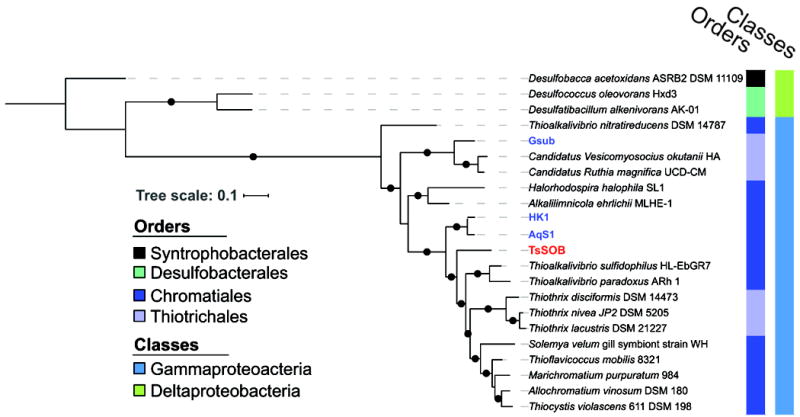
The Maximum-Likelihood phylogenetic tree of the amino-acid sequences of dsrAB depicts the evolutionary distance in dsrAB between TsSOB and other sulfur oxidizing sponge symbionts. The dsrAB sequences were identified with HMMs TIGR02064 and TIGR02066. No dsrAB sequence was found in SOB1 from Lophophysema eversa. Bootstrap values greater than 0.7 are markde as black dots on branches. . Blue text indicates sulfur-oxidizing sponge symbionts reported in the literature, whereas TsSOB is marked in red.
The Red Sea is an oligotrophic body of water, poor in labile sources of carbon, nitrogen and phosphate (Yahel et al., 2003; Batayneh et al., 2014). One significant source of sulfur are corals, which release dimethylsulfoniopropionate (DMSP) into the surrounding water (Hill et al., 1995). Being the only known external source for reduced sulfur in coral reefs, it is surprising that TsSOB lacks the genes required to catabolize DMSP and dimethylsulfide (DMS). While it is possible that genes coding for enzymes responsible for these processes are present in the missing parts of the genome, they are absent from other genomes of Halothiobacillus currently available in the Integrated Microbial Genomes (IMG) database (N=3). It is also possible that marine bacteria liberate sulfate from DMSP (Raina et al., 2010), and the sulfate is then reduced to sulfur by anaerobic Desulfovibrio, an SRB that was previously isolated from the same host sponge (Lavy et al., 2014). Sulfate concentrations in the Red Sea surface water are approximately 2.1 mM (El-Manharawy and Hafez, 2003). At a pumping rate of 6 ml min-1 (ml sponge)-1, which fits the pumping rates of HMA sponges in general (Weisz et al., 2008) and T. swinhoei in particular (Yahel et al., 2003), 18.8 mmol sulfate would pass through 1 ml of sponge tissue daily. This sulfate could be reduced by SRB in the sponge, becoming available for TsSOB. A similar mechanism was suggested for the sulfur cycle in the sponge G. barretti (Jensen et al., 2016).
Carbon, nitrogen and amino-acids metabolism
Metabolic prediction shows that TsSOB is an auxotroph for amino acids other than alanine, glycine, asparagine and glutamine. While genes involved in these pathways might be found in the missing parts of the genome, auxotrophy for alanine, histidine, phenylalanine, tyrosine and tryptophan is shared with the three other Halothiobacillus species in the IMG database. However, a search for amino-acids synthesis-related genes using Hidden Markov models (HMMs) suggests that all genes required for the synthesis of valine, leucine and isoleucine are present in the genome of TsSOB (Figure S5). While it has the potential for synthesizing branched amino acids, TsSOB also has genes encoding for branched amino-acid transporters (livFGHKM) in its genome. Interestingly, while it is capable of importing and synthesizing branched amino-acids, most of the genes associated with valine, leucine and isoleucine degradation are missing (Figure S6). The ability to synthesize branched amino-acids and the presence of their transporters may indicate that the TsSOB can export these amino-acids to be used by the host or other host-associated microbes.
The genome of TsSOB may also provide clues as to the sources of carbon and nitrogen that the bacterium requires for growth. The urtABCDE and ureDABCEFG genes that code for urea transport proteins and urease, respectively, were found in one operon (scaffold 6). Their occurrence suggests that urea, a valuable source of nitrogen and carbon, can be taken up and catabolized by the bacterium. Interestingly, the KEGG reference genome of its close relative, the free-living Halothiobacillus neapolitanus, does not have the genes coding for urease. Urea, along with creatinine were recently suggested to fuel the nitrogen cycle within sponges (Moitinho-Silva et al., 2017). Upon cleavage, urea breaks down to ammonia and CO2. Although ammonia oxidation-related genes are absent from its genome, two other genes that are related to ammonia utilization are present. The first is cytidine triphosphate (CTP) synthase which takes ammonia and Uridine-5’-triphosphate (UTP) and converts them to CTP. The second is glutamine-synthetase adenylyltransferase which regulates the activity of glutamine-synthetase. While glutamine-synthetase is missing from the genome, the presence of its regulator suggests that it may be present in the full genome.
TsSOB is probably capable of fixing carbon through the reductive citric acid cycle, as its genome shows most of genes required for this metabolic pathway (Figure S7). This trait is common for Halothiobacillus. By using urea, which is a metabolic waste product of the sponge, TsSOB gains CO2 and ammonia from its breakdown. Interestingly, AqS1 and HK1, which are the also sulfur oxidizing sponge symbionts, both lack genes encoding for ureases (Gauthier et al., 2016).
All members of the family Halothiobacillaceae are aerobic bacteria, often found in environments rich in hydrogen sulfide (Garrity et al., 2010). TsSOB may be an aerobe or microaeorbe. Sponges are aerobic animals but microaerobic conditions can occur within their bodies for prolonged periods of time (Hoffmann et al., 2007, 2008). A recent study reports consistent suboxic and anoxic conditions in T. swinhoei (Lavy et al., 2016). Moreover, ribosomal protein S3 sequences of Deltaproteobacteria and Clostridia, which are likely to be anaerobes, were abundant (x25 and x9 coverage, respectively) (Fig. S2). Previous studies have isolated anaerobic sulfate reducing Desulfovibrio from T. swinhoei (Lavy et al., 2014; Keren et al., 2015, 2016). A co-localization of sulfate reducing and sulfide oxidizing bacteria was recently found in the sponge G. barretti (Jensen et al., 2016). Similarly, alternating aeration conditions within the body of T. swinhoei, as was found by Lavy et. al (2016), could support both sulfate reduction by Desulfovibrio as well as sulfide oxidation by TsSOB.
Symbiont lifestyle
DNA repair-related enzymatic functions are known to deteriorate in host-associated bacteria, and therefore can be used to assess genome streamlining (Moran and Wernegreen, 2000). Out of 26 genes that are reported by Moran and Wernegreen (2000) to indicate genome streamlining, 10 are missing in TsSOB’s draft genome (alkA, mutH, mutT, nfo, phnW, phrB, recB, recC, recD and tag). Their absence could be attributed to the incompleteness of the genome, but it could also be that as a symbiont, TsSOB genome has lost these functions. According to checkM estimate of completeness (92%), the full genome of TsSOB is predicted to be 1.7 Mbp in length. For comparison, the genome size of its most closely related free-living bacterium Halothiobacillus neapolitanus c2 ATCC 23641 (GenBank accession NC_013422.1) is 2.58 Mbp. Other free living Chromatiales have genomes that are larger than TsSOB. For example, Thioalkalivibrio sp. K90mix isolated from sediment samples has a 2.74 Mbp genome (Muyzer, et al., 2011), and the genome of Thioalkalivibrio sulfidophilus HL-EbGr7, isolated from a bioreactor removing sulfide from gas, is 3.46 Mbp (Muyzer, et al., 2011). Sponge-associated sulfur oxidizing bacteria have estimated genome sizes in the range of 1.37 Mbp (Gsub 98% complete) to 3.46 Mbp (HK1, 99.1% complete).
While Halothiobacilli are motile via a single flagellum (Garrity et al., 2010), only flhF, a regulatory gene that affects the transcription of other flagellum biosynthesis genes, is present in the genome. The absence of genes might be related to the incomplete genome or indicate that the bacterium lacks a functional flagellum. It also possesses one copy of the gliding motility protein gldG and two twitching motility related proteins pilT. We calculated the iRep replication index to test whether TsSOB was actively replicating at the time of sampling. The index looks at the entire population, not at single cells, and its calculation assumes that more reads would be mapped to the origin of replication compared to the terminus in a replicating bacterium. The index is calculated as a ratio between the highest and lowest number of reads mapped along the genome at question, where a greater ratio value indicates initiation of more replication forks. Any value greater than 1 would mean that at least some part of the population is actively replicating. An iRep replication index of 1.23 was calculated for TsSOB (r2=0.99, %windows=100) suggesting it is actively replicating, and not merely being digested by the sponge.
TsSOB is a sponge-coral specific bacterium
The genome of TsSOB contains two polyketide synthase gene clusters supA and supB, which are common in sponge-specific bacteria (Figure S8 and Table S2). The supA cluster is composed of ketosynthase, acyltransferase, dehydratase, methyl transferases, enoyl reductases and ketoreductases modules, whereas supB contains only one phosphopantetheine attachment site. The supA amino-acid sequences are 73% and 62% similar to genes of bacteria from the sponges T. swinhoei and Aplysina aerophoba, respectively. The 76 amino-acid sequence of supB, was 56% and 39% similar to that of bacteria from A. aerophoba and T. swinhoei.
The presence of similar supA and supB genes in other sponge samples called for a broader search of TsSOB in microbial samples. Substantial effort has been made in recent years to sequence 16S rRNA tags from sponge-associated microbial communities from around the world. This resulted in a detailed description of microbial communities of many sponges (Thomas et al., 2016). Searching NCBI’s non-redundant nucleotide database for the 16S rRNA sequence of TsSOB shows that the bacteria with almost identical gene sequence are common in nine sponge and two coral orders (Figure 5, Table S3 and Figure S9). Sequences of 13 symbionts were ≥97% identical, suggesting they are of the same species and three are potentially from the same genus (>94% identity). While TsSOB-related sequences were found in 13 sponge species from 9 families, the next most common SOB, HK1, is found in 12 sponge species from 9 families. It should be noted that the available 16S rRNA sequence of HK1 was only 324 bp, and therefore some the matches with identity over 97% could be due to size bias. The similarity between AqS1 and HK1, as inferred from their ribosomal proteins and dsrAB sequences, as well as the comparison that was conducted by Gauthier et al. (2016), suggest that they belong to the same genus. The occurrence of HK1 in every sponge species in which AqS1 was found (except for Ancorina alata) further strengthens this finding.
Figure 5. Association and grouping of sponge symbionts to their HMA/LMA hosts.
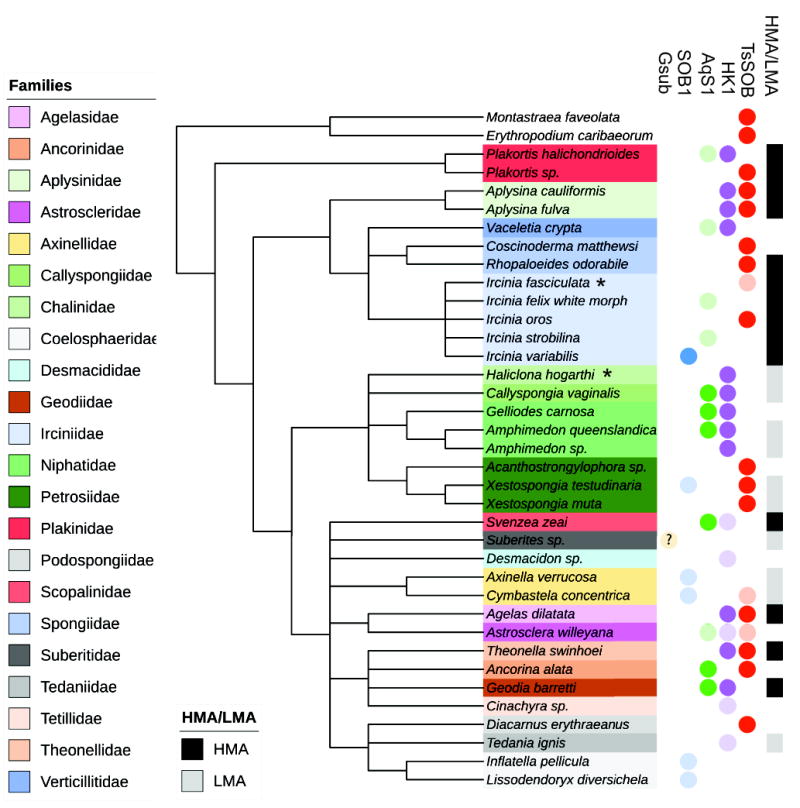
16S rRNA gene of TsSOB is present in multiple orders, and most often in HMA sponges. The 16S rRNA gene of other sulfide oxidizing sponge symbionts (SOB1, AqS1 and HK1, but not Gsub) were also in several sponges. The phylogenetic relations between sponges was adopted from Morrow and Cardenas (2015) and World Porifera Database (Van Soest et al., 2017). The 16S rRNA sequence of Gsub did not match any sponge associated bacterium in the database. The Gsub genome was originally reported from Suberites sp. and therefore it is marked in the tree with a question mark. Sponges were assigned as HMA or LMA according to the works of Gloeckner et al. (2014) and Moitinho-Silva et al. (2015). Lightly colored circles indicate matches that had less than 97% identity and therefor may represent another bacterial species. Two coral species, in which TsSOB was detected, are used as an outgroup. Asterisks denote species names which were updated since the sequences were submitted to NCBI database: Haliclona hogarti is now Haliclona tubifera, Ircinica fasciculata is now Ircinica variabilis, and Ancorinca alata is now Ecionemia alata. The full results of the NCBI nucleotide nr BLAST search are given in Table S3, and the phylogenetic relationship between the 16S rRNA sequences is presented in Figure S9.
TsSOB is mostly found in HMA sponges. Out of nine sponge families in which TsSOB was detected (> 97% identity of 16S rRNA), only members of Axinellidae and Petrosidae are low microbial abundance (LMA) sponges (Figure 4) (Gloeckner et al., 2014, 2014). The 16S rRNA sequence of TsSOB also matched sequences of bacteria derived from two corals, Montastaraea faveolata and Erythropodium caribaeorum, with 99% coverage and identity (Table S3). HK1 and AqS1 do not show a preference to either HMA or LMA sponges while the 16S rRNA of SOB1 is mostly found in LMA sponges. Although Gsub did not have any match to sponge-associated bacteria in NCBI’s nucleotide database, Tian et al. (2017) reported its isolation from a Suberites sp. which is a LMA sponge, and therefore it is considered as an LMA-associated bacterium. The occurrence of TsSOB primarily in HMA sponges may indicate that it relies on interactions with other symbionts of the sponges or corals, for its survival. Another possibility, which seems more likely, would be that this pattern is related to the temporal microaerobic and anaerobic conditions that may occur in the body of the slow water pumping HMA sponges (Weisz et al., 2008).
With a 19/20 matches for the 16S rRNA sequence of TsSOB matching sponge- and coral-associated symbionts in NCBI nucleotide database, TsSOB might be considered a sponge-coral-associated bacterium (Simister et al., 2012). This is further supported by a 16S rRNA Maximum-Likelihood phylogenetic tree that shows that the TsSOB related sequences are distinct from those of other SOB symbionts (Figure S9). The occurrence of the bacterium in both sponges and corals could suggest it has a pivotal role in sulfide and thiosulfate oxidation in the coral reef environment.
The occurrence of TsSOB in sponges from different orders and different locations around the globe, as well as it metabolic properties (i.e., ability to import and export amino acids and urea as well as ability for sulfide oxidation), may suggest that it is a true symbiont. The fact that the 16S rRNA sequence of TsSOB was not detected in seawater samples in NCBI’s nr database is similar to the case of Candidatus Poribacteria, which are readily found in sponges but are rarely detected in seawater (Taylor et al., 2012). This may indicate that the relationship between the bacterium and its host is an old one, potentially established by the last common ancestor of the nine sponge families, and that this bacterium is being vertically transferred (i.e., from parent to daughter). To date, no other Halothiobacillaceae is known to be sponge-associated.
Following the taxonomy guidelines for uncultivated prokaryotes (Konstantinidis et al., 2017), we suggest that TsSOB is a representative of a novel taxon, and propose naming it Candidatus Porisulfidus. The name indicates that this taxon mainly occurs in sponges (phylum Porifera), and is capable of sulfide oxidation.
Conclusions
Based on the metagenomic analysis of a sponge-associated microbial community, we identified a relatively abundant Chromatiales bacterium TsSOB with the capacity to oxidize sulfide and thiosulfate, for which we suggest the name “Candidatus Porisulfidus”. The bacterium, which is somewhat similar to Halothiobacillus, is predicted to use urea derived from its Theonella swinhoei sponge host, as a nitrogen and carbon source. The size of its genome, presence of urease genes, indication of active replication within the sponge and detection of its 16S rRNA sequence in several sponge orders but not seawater, suggest that TsSOB is a sponge-coral-associated, rather than a transient free-living bacterium. The only other organisms in the community that appear to be capable of sulfur compound transformations belong to the genus Desulfovibrio, the members of which can reduce sulfate. All of these bacteria may be responsible for sulfur cycling within T. swinhoei, regulating the amount of sulfate and sulfide available to the host and other symbionts residing in its mesohyl. Sulfate likely represents a significant resource for the sponge microbiome, given the concentration of sulfate in the seawater and the large volume of water that passes through the sponge on a daily basis. Its transformation to sulfide by Desulfovibrio and subsequently back to sulfate by TsSOB would form a complete cycle occurring within the holobiont.
Experimental Procedures
Sampling and DNA preparation
Three T. swinhoei specimens were collected at Eilat, Red Sea (29°29’57.63”N / 34°54’54.61”E) by SCUBA diving at 20 m depth, sealed in closed bags, and immediately processed on site at the Interuniversity Institute for Marine Sciences. All work henceforth was performed in a laminar flow hood under sterile conditions. Sponges were thoroughly rinsed in sterile calcium-magnesium-free artificial seawater (CMF-ASW) to remove transient bacteria and loosen cellular connections, and were cut to 1 cm layers. The outer-most layer of each sponge that contains cyanobacteria was removed, and cores were collected from each layer of each sponge into sterile tubes. Each sub-sample was homogenized in CMF_ASW and the resulting homogenate was repetitively agitated by stirring, to separate clumps of cells. Sponge cells were separated by passive settling and centrifugation at 100 g (Wilson et al., 2014). Unicellular cells were collected by centrifugation at 3000 g and discarding of the supernatant.
Five liters of seawater were collected on site in a pre-bleached container. The seawater was serially filtered through 11 μm, 1.2 μm and finally a 0.22 μm Sterivex filter within 1 hour of sampling. A lysis buffer was added to the 0.22 μm Sterivex filter prior to DNA extraction as according to a protocol published by Wright et al. (2009).
DNA was extracted by following the CTAB extraction protocol. Briefly, the bacterial pellet from sponge samples and the seawater sample lysis buffer were suspended in 0.8 ml CTAB buffer and incubated at 60°C for one hour. Resulting lysate was mixed with an equal amount of chloroform:isoamylalchohol 24:1 (v/v), followed by centrifugation (15 min, 4°C). The aquatic phase was transferred into a new tube and treated with 1 μl RNase for 30 min at 37°C. For DNA precipitation, sodium acetate (1:10 v/v) and isopropanol (1:1 v/v) were added and the sample was incubated overnight (-20°C). Following incubation, DNA was pelleted (15300 g, 15 min, 4°C), washed with cold ethanol and suspended in ddH2O.
A metagenomic library was prepared for each of the three sponges and the seawater sample at the Tauber Bioinformatics Research Center of Haifa University after validating DNA concentration and integrity by using Qubit (Thermo Fisher Scientific) and gel electrophoresis, respectively. The sample was sheared using Covaris E220 (settings: 40 seconds, 10% duty cycle, 200 cycles per burst, 175 peak incident power) and a library was prepared using NEB’s Ultra DNA Library Prep kit for Illumina with AmpureXP bead selection aimed to give fragments of 250 bp according to the manufacturer’s protocol. The library was sequenced at the Technion Institute using an Illumina Hiseq 2500, paired-end 150 base-pairs (bp) sequencing.
Sequence analysis, annotation and assembly
Illumina adapters were removed and raw sequences were quality-trimmed with Sickle (Joshi and Fass, 2011). Sequences of one sponge sample were then matched and assembled using IDBA-UD (Peng et al., 2012). Thereafter reads were mapped with bowtie2 (Langmead and Salzberg, 2012) to calculate reads coverage. Genes were predicted by Prodigal (Hyatt et al., 2010) and predicted protein sequences were annotated using usearch (Edgar, 2010) against KEGG, UniRef100 and UniProt databases.
Initial binning of scaffolds was done based on similarity in GC content and coverage of scaffolds as well as taxonomic identity of genes in each scaffold. The putative genomes were identified using the ggKbase binning tools (http://ggkbase.berkeley.edu). Bins were assessed partly based on the number of Bacterial Single Copy Genes (BSCG), and Ribosomal Proteins (RP) found in each bin. Each putative genome bin was downloaded along with other bins that share similar GC content and coverage. Scaffolds were split into 2500 bp chunks and the tetramer frequency of each segment was calculated. Scaffolds were clustered according to their tetramer frequency based on Emergent Self-Organizing Maps (ESOM) with Databionic ESOM Analyzer (Ultsch and Moerchen, 2005; Dick et al., 2009). Scaffolds that had less than 50% of their segments present inside an ESOM cluster, were omitted from the bin. The updated bins were loaded back into ggKbase for further inspection. Scaffolds present in the original bin that were < 2000 bp in length were retained so long as their gene taxonomy profile matched that of the rest of the bin. Scaffolding errors introduced by idba_ud were corrected using an in-house script as described by Brown et. al (2015), and fusion of overlapping scaffolds was done with Geneious version 8.0 (https://www.geneious.com, (Kearse et al., 2012)).
CheckM v1.0.7 (Parks et al., 2015) was used to verify the genome completeness and percentage of possible contamination, with the expected single copy gene sets defined based on the genomes of other Gammaproteobacteria. A phylogenetic tree was created using a standard set of 16 ribosomal proteins sequences (S2, S8, S10, S17, S19, L2, L3, L4, L5, L6, L14, L15, L16, L18, L22, L24) (Hug et al., 2013), with references from 52 other sulfur oxidizing bacteria. The amino acid sequences were obtained by predicted open reading frames (ORFs) for each genome using Prodigal v2.6.3, then searching these predictions for the 16 ribosomal proteins using an in-house script (Probst et al., 2017). Proteins were aligned with MUSCLE (Edgar, 2004) and non-informative ends were removed. The sequences were then concatenated, and columns with more than 95% gaps were stripped, resulting in a total of 2379 informative positions (i.e., positions along an alignment which at which at least 5% of the sequences have a non-gap information). The total number of informative position for each genome was verified, keeping genomes which spanned over 50% of the total length of the alignment. A Maximum-Likelihood tree was constructed with PhyML algorithm (v3.0) (Guindon et al., 2010) using LG+α+γ substitution model, and 20 substitution rate categories.
Functional profile of the genome was evaluated using ggKbase, KEGG KAAS (Moriya et al., 2007), the IMG system (Chen et al., 2017), and Hidden Markov Models for of shared KEGG orthologies (KOs) as follows. Predicted proteins of TsSOB were searched against a database of HMMs representing all the KOs (Kanehisa, 2000). The HMM database was compiled using the HMMER suite (Finn et al., 2015), based on assignment of proteins to KOs according to KEGG FTP Release 2015-06-22. Individual trusted thresholds were calculated by running HMM search of all the proteins with assigned KOs against the HMM database. Polyketide Synthase sequences were sought for by AntiSmash antibiotics & Secondary Metabolite Analysis Shell (Weber et al., 2015).
The taxonomy of sulfur oxidizing bacteria can be often assisted by observing the phylogeny of their dsrAB genes (Muller et al., 2015). The amino acid sequences of dsrA and dsrB were sought for in the 52 genomes that were used to construct the phylogenetic tree (Figure 1) using TIGRFAM HMMs for dsrA (TIGR02064) and dsrB (TIGR02066). Briefly, the HMMs were downloaded from TIGRFAM database and HMMER (v 3.0) (HMMER; Eddy, 1998) was used with default cutoffs to search for sequences within the protein sequences of the genomes. Amino acid sequences for dsrAB were found in 22 genomes, including TsSOB. The sequences were aligned and non-informative positions, as well non-informative ends were stripped, as described above. The 838 informative positions were aligned using phyml, and a ML tree with 100 bootstraps was constructed.
The 16S rRNA sequence of Thioalkalivibrio sp. HK1 was downloaded from JGI’s IMG web site (Chen et al., 2017), while the sequences of Gsub, AqS1, and SOB were downloaded from NCBI Genebank database. The sequences were used for a BLAST search against NCBI nucleotide nr database. The results were filtered so that the top 20 unique hits were retained. Hits were considered unique if they originate from different studies or if they are from the same study but from different host/isolation source. The full results of this search are given in Table S2. The sequences of those hits were downloaded and aligned with MUSCLE and positions with >95% gaps were removed (as was previously described). A Maximum-Likelihood phylogenetic tree was then calculated with PHYML using GTR model and estimated transition/transversion ratio, proportion of invariable sites and gamma-shape parameter. Invertebrates that were found to host any of the five bacteria were placed on a taxonomic dendrogram which relies on the latest Porifera taxonomy found in the World Porifera Database (Van Soest et al., 2017) and the taxonomy of demosponges published by Morrow and Cardenas (2015). Sponges were identified as either LMA or HMA based on the two most recent publications by Gloeckner et al. (2014) and Moitinho-Silva et al. (2017). Post processing of all phylogenetic trees was done in iTol (Letunic and Bork, 2016).
A rank abundance curve was calculated using sequencing coverage of scaffolds that encode ribosomal protein S3 (rpS3) as a proxy for genome abundance. All unique rpS3 sequences in the sample were aligned against 2887 known rpS3 sequences using MUSCLE. A Neighbor-Joining tree was then constructed to determine the identity of each rpS3 sequence to the class or phylum level.
The in-situ replication rate was inferred based on the sequencing coverage trend that results from bi-directional genome replication from a single origin of replication using iRep (Brown et al., 2016). The presence of TsSOB in the two un-binned sponge samples as well as in the seawater sample was determined by calculate_breadth.py, a python script which calculates the breadth and coverage of the genome according to the mapped reads from each of the metagenomic samples (Olm et al., 2017).
Supplementary Material
Originality-Significance Statement.
Sponges and their microbial symbionts are key members in marine benthic environments. There is great interest in the study of sponge-associated microbes in areas ranging from microbial ecology of host-symbiont interactions, to production of secondary metabolites for biomedical research. Yet, the fundamental metabolic interactions at the basis of symbiotic relationships are poorly understood. Here we investigated the potential role of a novel bacterial sponge-symbiont in sulfur metabolism. We show that this bacterium is common in sponge species from different orders. The work further elucidates an ecological foundation for the association of sponges and their microbial consortia.
Acknowledgments
Funding
The authors would like to acknowledge the Interuniversity Institute, Eilat and its staff for the ongoing support and use of their facilities. We thank Dr. Noa Sher for assisting with metagenomic library preparation and Dr. Itai Sharon for his input. The study has been partially supported by the following institutions and grants; Israel Science Foundation (grant number 957/14). A.L. was supported by an Eshkol scholarship from the Israeli Ministry of Science, Technology and Space. A.L was also funded by the Tel Aviv University Global Research & Training Fellowship (GRTF). R.K. was supported by the Fellowship of the Argentina PhD Honors Program of the Smolarz Family. Support was also provided by a grant from the US Department of Energy (grant number DOE-SC10010566) and NIEHS grant R42 ES004705-19.
Footnotes
Conflict of Interest
No conflict of interest is known to the authors
Nucleotide sequence accession number
TsSOB draft genome is publically available at Genbank, with the accession number: MRSX00000000. WGS raw sequences are available at NCBI’s Sequence Read Archive (SRA) under BioSample accession number SAMN06111390
References
- Batayneh A, Elawadi E, Zaman H, Al-Taani AA, Nazzal Y, Ghrefat H. Environmental assessment of the Gulf of Aqaba coastal surface waters, Saudi Arabia. J Coast Res. 2014;30:283–290. [Google Scholar]
- Bewley CA, Holland ND, Faulkner DJ. Two classes of metabolites from Theonella swinhoei are localized in distinct populations of bacterial symbionts. Experientia. 1996;52:716–722. doi: 10.1007/BF01925581. [DOI] [PubMed] [Google Scholar]
- Bright M, Giere O. Microbial symbiosis in Annelida. Symbiosis. 2005;38:1–45. [Google Scholar]
- Brown CT, Hug LA, Thomas BC, Sharon I, Castelle CJ, Singh A, et al. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature. 2015;523:208–211. doi: 10.1038/nature14486. [DOI] [PubMed] [Google Scholar]
- Chen I-MA, Markowitz VM, Chu K, Palaniappan K, Szeto E, Pillay M, et al. IMG/M: integrated genome and metagenome comparative data analysis system. Nucleic Acids Res. 2017;45:D507–D516. doi: 10.1093/nar/gkw929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary DFR, Becking LE, de Voogd NJ, Pires ACC, Polónia ARM, Egas C, Gomes NCM. Habitat- and host-related variation in sponge bacterial symbiont communities in Indonesian waters. FEMS Microbiol Ecol. 2013;85:465–482. doi: 10.1111/1574-6941.12135. [DOI] [PubMed] [Google Scholar]
- Dick GJ, Andersson AF, Baker BJ, Simmons SL, Thomas BC, Yelton AP, Banfield JF. Community-wide analysis of microbial genome sequence signatures. Genome Biol. 2009;10:R85. doi: 10.1186/gb-2009-10-8-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- El-Manharawy S, Hafez A. Study of seawater alkalization as a promising RO pretreatment method. Desalination. 2003;153:109–120. [Google Scholar]
- Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, et al. HMMER web server: 2015 update. Nucleic Acids Res. 2015;43:W30–W38. doi: 10.1093/nar/gkv397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich CG, Rother D, Bardischewsky F, Quentmeier A, Fischer J. Oxidation of reduced inorganic sulfur compounds by bacteria: Emergence of a common mechanism? Appl Environ Microbiol. 2001;67:2873–2882. doi: 10.1128/AEM.67.7.2873-2882.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrity G, Brenner DJ, Krieg NR, Staley JR, editors. Bergey’s manual of systematic bacteriology the Proteobacteria, Part B: The Gammaproteobacteria. Springer Verlag; 2010. [Google Scholar]
- Gauthier M-EA, Watson JR, Degnan SM. Draft genomes shed light on the dual bacterial symbiosis that dominates the microbiome of the coral reef sponge Amphimedon queenslandica. Front Mar Sci. 2016;3:196. [Google Scholar]
- Ghosh W, Dam B. Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. Fems Microbiol Rev. 2009;33:999–1043. doi: 10.1111/j.1574-6976.2009.00187.x. [DOI] [PubMed] [Google Scholar]
- Gloeckner V, Wehrl M, Moitinho-Silva L, Gernert C, Schupp P, Pawlik JR, et al. The HMA-LMA dichotomy revisited: an electron microscopical survey of 56 sponge species. Bio Bull. 2014;227:78–88. doi: 10.1086/BBLv227n1p78. [DOI] [PubMed] [Google Scholar]
- Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Guo XC, Zheng L, Zhou WH, Cui ZS, Han P, Tian L, Wang XR. A case study on chemical defense based on quorum sensing: antibacterial activity of sponge-associated bacterium Pseudoalteromonas sp NJ6-3-1 induced by quorum sensing mechanisms. Ann Microbiol. 2011;61:247–255. [Google Scholar]
- Hardoim CCP, Costa R, Araújo FV, Hajdu E, Peixoto R, Lins U, et al. Diversity of bacteria in the marine sponge Aplysina fulva in brazilian coastal waters. Appl Environ Microbiol. 2009;75:3331–3343. doi: 10.1128/AEM.02101-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardoim CCP, Esteves AIS, Pires FR, Gonçalves JMS, Cox CJ, Xavier JR, Costa R. Phylogenetically and spatially close marine sponges harbour divergent bacterial communities. PLOS ONE. 2012;7:e53029. doi: 10.1371/journal.pone.0053029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill RW, Dacey JWH, Krupp DA. Dimethylsulfoniopropionate in reef corals. Bull Mar Sci. 1995;57:489–494. [Google Scholar]
- Hoffmann F, Larsen O, Tore Rapp H, Osinga R. Oxygen dynamics in choanosomal sponge explants. Mar Biol Res. 2005;1:160–163. [Google Scholar]
- Hoffmann F, Radax R, Woebken D, Holtappels M, Lavik G, Rapp HT, et al. Complex nitrogen cycling in the sponge Geodia barretti. Environ Microbiol. 2009 doi: 10.1111/j.1462-2920.2009.01944.x. [DOI] [PubMed] [Google Scholar]
- Hoffmann F, Røy H, Bayer K, Hentschel U, Pfannkuchen M, Brümmer F, Beer D. Oxygen dynamics and transport in the Mediterranean sponge Aplysina aerophoba. Mar Biol. 2008;153:1257–1264. doi: 10.1007/s00227-008-0905-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann F, Sauter E, Sachs O, Roy H, Klages M. In: Oxygen distribution in Tentorium semisuberites and in its habitat in the Arctic deep sea. Custódio M, Lôbo-Hajdu G, Hajdu E, Muricy G, editors. Museu Nacional – Universidade Federal do Rio de Janeiro; 2007. pp. 379–382. [Google Scholar]
- Hug LA, Castelle CJ, Wrighton KC, Thomas BC, Sharon I, Frischkorn KR, et al. Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome. 2013;1:22. doi: 10.1186/2049-2618-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilan M, Gugel J, Van Soest RWM. Taxonomy, reproduction and ecology of new and known Red Sea sponges. Sarsia. 2004;89:388–410. [Google Scholar]
- Imhoff JF, Trüper HG. Marine sponges as habitats of anaerobic phototrophic bacteria. Microb Ecol. 1976;3:1–9. doi: 10.1007/BF02011449. [DOI] [PubMed] [Google Scholar]
- Jensen S, Fortunato SAV, Hoffmann F, Hoem S, Rapp HT, Øvreås L, Torsvik VL. The relative abundance and transcriptional activity of marine sponge-associated microorganisms emphasizing groups involved in sulfur cycle. Microb Ecol. 2016:1–9. doi: 10.1007/s00248-016-0836-3. [DOI] [PubMed] [Google Scholar]
- Jeong J-B, Kim K-H, Park J-S. Sponge-specific unknown bacterial groups detected in marine sponges collected from Korea through barcoded pyrosequencing. J Microbiol Biotechnol. 2015;25:1–10. doi: 10.4014/jmb.1406.06041. [DOI] [PubMed] [Google Scholar]
- Jorgensen BB, Kuenen JG, Cohen Y. Microbial transformations of sulfur compounds in a stratified lake (Solar Lake, Sinai)1. Limnol Oceanogr. 1979;24:799–822. [Google Scholar]
- Joshi NA, Fass JN. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33) [Software] 2011 Available https://github.com/najoshi/sickle.
- Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy J, Flemer B, Jackson SA, Morrissey JP, O’Gara F, Dobson AD. Evidence of a putative deep sea specific microbiome in marine sponges. PLoS ONE. 2014;9:e91092. doi: 10.1371/journal.pone.0091092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren R, Lavy A, Ilan M. Increasing the richness of culturable arsenic-tolerant bacteria from Theonella swinhoei by addition of sponge skeleton to the growth medium. Microb Ecol. 2016;71:873–886. doi: 10.1007/s00248-015-0726-0. [DOI] [PubMed] [Google Scholar]
- Keren R, Lavy A, Mayzel B, Ilan M. Culturable associated-bacteria of the sponge Theonella swinhoei show tolerance to high arsenic concentrations. Front Microbiol. 2015;6 doi: 10.3389/fmicb.2015.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren R, Mayzel B, Lavy A, Polishchuk I, Levy D, Fakra SC, et al. Sponge-associated bacteria mineralize arsenic and barium on intracellular vesicles. Nat Commun. 2017;8:14393. doi: 10.1038/ncomms14393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima H, Ogura Y, Yamamoto N, Togashi T, Mori H, Watanabe T, et al. Ecophysiology of Thioploca ingrica as revealed by the complete genome sequence supplemented with proteomic evidence. ISME J. 2015;9:1166–1176. doi: 10.1038/ismej.2014.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis KT, Rosselló-Móra R, Amann R. Uncultivated microbes in need of their own taxonomy. ISME J. 2017 doi: 10.1038/ismej.2017.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Meth. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavy A, Keren R, Haber M, Schwartz I, Ilan M. Implementing sponge physiological and genomic information to enhance the diversity of its culturable associated bacteria. FEMS Microbiol Ecol. 2014;87:16. doi: 10.1111/1574-6941.12240. [DOI] [PubMed] [Google Scholar]
- Lavy A, Keren R, Yahel G, Ilan M. Intermittent hypoxia and prolonged suboxia measured in situ in a marine sponge. Front Mar Sci. 2016;3 [Google Scholar]
- Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–W245. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnino G, Sara A, Lancioni T, Gaino E. Endobionts of the coral reef sponge Theonella swinhoei (Porifera, Demospongiae) Inverteb Biol. 1999;118:213–220. [Google Scholar]
- Maldonado M, Ribes M, Van Duyl FC. Nutrient fluxes through sponges: Biology, budgets, and ecological implications. In: Becerro MA, Uriz MJ, Maldonado M, Turon X, editors. Advances in sponge science: physiology, chemical and microbial diversity, biotechnology, advances in marine biology. Elsevier Academic Press Inc; San Diego: 2012. pp. 113–182. [DOI] [PubMed] [Google Scholar]
- Meyer B, Kuever J. Phylogenetic diversity and spatial distribution of the microbial community associated with the Caribbean deep-water sponge Polymastia cf. corticata by 16S rRNA, aprA, and amoA gene analysis. Microb Ecol. 2008;56:306–321. doi: 10.1007/s00248-007-9348-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed NM, Saito K, Tal Y, Hill RT. Diversity of aerobic and anaerobic ammonia-oxidizing bacteria in marine sponges. ISME J. 2010;4:38–48. doi: 10.1038/ismej.2009.84. [DOI] [PubMed] [Google Scholar]
- Moitinho-Silva L, Díez-Vives C, Batani G, Esteves AI, Jahn MT, Thomas T. Integrated metabolism in sponge microbe symbiosis revealed by genome-centered metatranscriptomics. ISME J. 2017;11:1651–1666. doi: 10.1038/ismej.2017.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moitinho-Silva L, Steinert G, Nielsen S, Hardoim CCP, Wu Y-C, McCormack GP, et al. Predicting the HMA-LMA status in marine sponges by machine learning. Front Microbiol. 2017;8 doi: 10.3389/fmicb.2017.00752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow C, Cárdenas P. Proposal for a revised classification of the Demospongiae (Porifera) Front Zool. 2015;12:7. doi: 10.1186/s12983-015-0099-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller AL, Kjeldsen KU, Rattei T, Pester M, Loy A. Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi)sulfite reductases. ISME J. 2015;9:1152–1165. doi: 10.1038/ismej.2014.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyzer G, Sorokin DY, Mavromatis K, Lapidus A, Clum A, Ivanova N, et al. Complete genome sequence of Thioalkalivibrio “sulfidophilus” HL-EbGr7. Stand Genomic Sci. 2011;4:23–35. doi: 10.4056/sigs.1483693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyzer G, Sorokin DY, Mavromatis K, Lapidus A, Foster B, Sun H, et al. Complete genome sequence of Thioalkalivibrio sp. K90mix. Stand Genomic Sci. 2011;5:341–355. doi: 10.4056/sigs.2315092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Shimamura S, Takaki Y, Suzuki Y, Murakami S, Watanabe T, et al. Allying with armored snails: the complete genome of gammaproteobacterial endosymbiont. ISME J. 2014;8:40–51. doi: 10.1038/ismej.2013.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Matsunaga S, van Soest RWM, Fusetani N. Nagahamide A, an antibacterial depsipeptide from the marine sponge Theonella swinhoei. Org Lett. 2002;4:3039–3042. doi: 10.1021/ol0262791. [DOI] [PubMed] [Google Scholar]
- Olm MR, Brown CT, Brooks B, Firek B, Baker R, Burstein D, et al. Identical bacterial populations colonize premature infant gut, skin, and oral microbiomes and exhibit different in situ growth rates. Genome Res. 2017;27:601–612. doi: 10.1101/gr.213256.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Leung HC, Yiu SM, Chin FY. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28:1420–8. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- Piel J. Metabolites from symbiotic bacteria. Nat Prod Rep. 2004;21:519. doi: 10.1039/b310175b. [DOI] [PubMed] [Google Scholar]
- Piel J. Metabolites from symbiotic bacteria. Nat Prod Rep. 2009;26:338–362. doi: 10.1039/b703499g. [DOI] [PubMed] [Google Scholar]
- Pokorna D, Zabranska J. Sulfur-oxidizing bacteria in environmental technology. Biotechnol Adv. 2015;33:1246–1259. doi: 10.1016/j.biotechadv.2015.02.007. [DOI] [PubMed] [Google Scholar]
- Probst AJ, Castelle CJ, Singh A, Brown CT, Anantharaman K, Sharon I, et al. Genomic resolution of a cold subsurface aquifer community provides metabolic insights for novel microbes adapted to high CO 2 concentrations: Genomic resolution of a high-CO2 subsurface community. Environ Microbiol. 2017;19:459–474. doi: 10.1111/1462-2920.13362. [DOI] [PubMed] [Google Scholar]
- Raina J-B, Dinsdale EA, Willis BL, Bourne DG. Do the organic sulfur compounds DMSP and DMS drive coral microbial associations? Trends Microbiol. 2010;18:101–108. doi: 10.1016/j.tim.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Sander J, Engels-Schwarzlose S, Dahl C. Importance of the DsrMKJOP complex for sulfur oxidation in Allochromatium vinosum and phylogenetic analysis of related complexes in other prokaryotes. Arch Microbiol. 2006;186:357–366. doi: 10.1007/s00203-006-0156-y. [DOI] [PubMed] [Google Scholar]
- Schläppy ML, Weber M, Mendola D, Hoffmann F, de Beer D. Heterogeneous oxygenation resulting from active and passive flow in two Mediterranean sponges, Dysida avara and Chondrosia reniformis. Limnol Ocean. 2010;55:1289–1300. [Google Scholar]
- Schmidt EW, Obraztsova AY, Davidson SK, Faulkner DJ, Haygood MG. Identification of the antifungal peptide-containing symbiont of the marine sponge Theonella swinhoei as a novel delta-proteobacterium, “Candidatus Entotheonella palauensis”. Mar Biol. 2000;136:969–977. [Google Scholar]
- Schmitt S, Tsai P, Bell J, Fromont J, Ilan M, Lindquist N, et al. Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. ISME J. 2012;6:564–576. doi: 10.1038/ismej.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simister RL, Deines P, Botte ES, Webster NS, Taylor MW. Sponge-specific clusters revisited: a comprehensive phylogeny of sponge-associated microorganisms. Env Microbiol. 2012;14:517–24. doi: 10.1111/j.1462-2920.2011.02664.x. [DOI] [PubMed] [Google Scholar]
- Southwell MW, Weisz JB, Martens CS, Lindquist N. In situ fluxes of dissolved inorganic nitrogen from the sponge community on Conch Reef, Key Largo, Florida. Limnol Oceanogr. 2008;53:986–996. [Google Scholar]
- Steinert G, Taylor MW, Deines P, Simister RL, Voogd NJ, de Hoggard M, Schupp PJ. In four shallow and mesophotic tropical reef sponges from Guam the microbial community largely depends on host identity. PeerJ. 2016;4:e1936. doi: 10.7717/peerj.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart FJ, Newton ILG, Cavanaugh CM. Chemosynthetic endosymbioses: adaptations to oxic–anoxic interfaces. Trends Microbiol. 2005;13:439–448. doi: 10.1016/j.tim.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Su J, Jin LL, Jiang Q, Sun W, Zhang FL, Li ZY. Phylogenetically diverse ureC genes and their expression suggest the urea utilization by bacterial symbionts in marine sponge Xestospongia testudinaria. PLoS ONE. 2013;85:e64848. doi: 10.1371/journal.pone.0064848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JD, Glover EA. Lucinidae (Bivalvia) - the most diverse group of chemosymbiotic molluscs. Zool J Linn Soc. 2006;148:421–438. [Google Scholar]
- Taylor MW, Radax R, Steger D, Wagner M. Sponge-associated microorganisms: Evolution, ecology, and biotechnological potential. Microbiol Mol Biol Rev. 2007;71:295–347. doi: 10.1128/MMBR.00040-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MW, Tsai P, Simister RL, Deines P, Botte E, Ericson G, et al. “Sponge-specific” bacteria are widespread (but rare) in diverse marine environments. ISME J. 2012 doi: 10.1038/ismej.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T, Moitinho-Silva L, Lurgi M, Bjork JR, Easson C, Astudillo-Garcia C, et al. Diversity, structure and convergent evolution of the global sponge microbiome. Nat Commun. 2016;7:11870. doi: 10.1038/ncomms11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian R-M, Sun J, Cai L, Zhang WP, Zhou G-W, Qiu J-W, Qian P-Y. The deep-sea glass sponge Lophophysema eversa harbours potential symbionts responsible for the nutrient conversions of carbon, nitrogen and sulfur: Potential symbionts in a glass sponge. Environ Microbiol. 2016;18:2481–2494. doi: 10.1111/1462-2920.13161. [DOI] [PubMed] [Google Scholar]
- Tian RM, Wang Y, Bougouffa S, Gao ZM, Cai L, Bajic V, Qian PY. Genomic analysis reveals versatile heterotrophic capacity of a potentially symbiotic sulfur-oxidizing bacterium in sponge. Environ Microbiol. 2014;16:3548–3561. doi: 10.1111/1462-2920.12586. [DOI] [PubMed] [Google Scholar]
- Tian R-M, Zhang W, Cai L, Wong Y-H, Ding W, Qian P-Y. Genome reduction and microbe-host interactions drive adaptation of a sulfur-oxidizing bacterium associated with a cold seep sponge. mSystems. 2017;2:e00184–16. doi: 10.1128/mSystems.00184-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ultsch A, Moerchen F. ESOM-Maps: tools for clustering, visualization, and classification with Emergent SOM. Dept of Mathematics and Computer Science, University of Marburg; Germany, Marburg: 2005. [Google Scholar]
- Van Soest RWM, Boury-Esnault N, Hooper JNA, Rützler K, de Voogd NJ, Alvarez de Glasby B, et al. World Porifera database 2017 [Google Scholar]
- Venceslau SS, Stockdreher Y, Dahl C, Pereira IAC. The “bacterial heterodisulfide” DsrC is a key protein in dissimilatory sulfur metabolism. Biochim Biophys Acta BBA - Bioenerg. 2014;1837:1148–1164. doi: 10.1016/j.bbabio.2014.03.007. [DOI] [PubMed] [Google Scholar]
- Vogler KG, LePage GA, Umbreit WW. Studies on the metabolism of autotrophic bacteria. respir. Thiobacillus thiooxidans sulfur. 1942;26:89–102. doi: 10.1085/jgp.26.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakimoto T, Egami Y, Nakashima Y, Wakimoto Y, Mori T, Awakawa T, et al. Calyculin biogenesis from a pyrophosphate protoxin produced by a sponge symbiont. Nat Chem Biol. 2014;10:648–655. doi: 10.1038/nchembio.1573. [DOI] [PubMed] [Google Scholar]
- Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, et al. antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:W237–W243. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster NS, Thomas T. The sponge holobiome. mBio. 2016;7(2):e00135–16. doi: 10.1128/mBio.00135-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisz JB, Lindquist N, Martens CS. Do associated microbial abundances impact marine demosponge pumping rates and tissue densities? Oecologia. 2008;155:367–376. doi: 10.1007/s00442-007-0910-0. [DOI] [PubMed] [Google Scholar]
- White JR, Patel J, Ottesen A, Arce G, Blackwelder P, Lopez JV. Pyrosequencing of bacterial symbionts within Axinella corrugata sponges: diversity and seasonal variability. Plos One. 2012;7:e38204. doi: 10.1371/journal.pone.0038204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MC, Mori T, Ruckert C, Uria AR, Helf MJ, Takada K, et al. An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature. 2014;506:58–62. doi: 10.1038/nature12959. [DOI] [PubMed] [Google Scholar]
- Wright JJ, Lee S, Zaikova E, Walsh DA, Hallam SJ. DNA extraction from 0.22 μM Sterivex filters and cesium chloride density gradient centrifugation. J Vis Exp. 2009 doi: 10.3791/1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahel G, Sharp JH, Marie D, Hase C, Genin A. In situ feeding and element removal in the symbiont-bearing sponge Theonella swinhoei: Bulk DOC is the major source for carbon. Limnol Ocean. 2003;48:141–149. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.