

Abstract
γ -Aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the central nervous system. Inhibition of GABA aminotransferase (GABA-AT), a pyridoxal 5′-phosphate (PLP)-dependent enzyme that degrades GABA, has been established as a possible strategy for the treatment of substance abuse. The raised GABA levels that occur as a consequence of this inhibition have been found to antagonize the rapid release of dopamine in the ventral striatum (nucleus accumbens) that follows an acute challenge by an addictive substance. In addition, increased GABA levels are also known to elicit an anticonvulsant effect in patients with epilepsy. We previously designed the mechanism-based inactivator (1S,3S)-3-amino-4-difluoromethylenyl-1-cyclopentanoic acid (2), now called CPP-115, that is 186 times more efficient in inactivating GABA-AT than vigabatrin, the only FDA-approved drug that is an inactivator of GABA-AT. CPP-115 was found to have high therapeutic potential for the treatment of cocaine addiction and for a variety of epilepsies, has successfully completed a Phase I safety clinical trial, and was found to be effective in the treatment of infantile spasms (West syndrome). Herein we report the design, using molecular dynamics simulations, synthesis, and biological evaluation of a new mechanism-based inactivator, (S)-3-amino-4-(difluoromethylenyl)cyclopent-1-ene-1-carboxylic acid (5), which was found to be almost 10 times more efficient as an inactivator of GABA-AT than CPP-115. We also present the unexpected crystal structure of 5 bound to GABA-AT, as well as computational analyses used to assist the structure elucidation process. Furthermore, 5 was found to have favorable pharmacokinetic properties and low off-target activities. In vivo studies in freely moving rats showed that 5 was dramatically superior to CPP-115 in suppressing the release of dopamine in the corpus striatum, which occurs subsequent to either an acute cocaine or nicotine challenge. Compound 5 also attenuated increased metabolic demands (neuronal glucose metabolism) in the hippocampus, a brain region that encodes spatial information concerning the environment in which an animal receives a reinforcing or aversive drug. This multidisciplinary computational design to preclinical efficacy approach should be applicable to the design and improvement of mechanism-based inhibitors of other enzymes whose crystal structures and inactivation mechanisms are known.
Graphical Abstract
INTRODUCTION
γ-Aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the central nervous system of mammals.1 After its release from presynaptic inhibitory neurons, it can bind to chloride-selective ion channel receptors (GABAA and GABAC)2 and G-protein coupled receptors (GABAB)3 to hyperpolarize the postsynaptic membrane, thereby downwardly controlling neuronal activity. 4 Low levels of GABA are linked to many neurological disorders, including epilepsy,5 Parkinson’s disease,6 Alzheimer’s disease,7 and Huntington’s disease.8
In 1998 a novel strategy was developed for the treatment of cocaine addiction based on inhibiting the activity of γ-aminobutyric acid aminotransferase (GABA-AT), 9 the pyridoxal 5′-phosphate (PLP)-dependent enzyme that degrades GABA to succinic semialdehyde via an aldimine intermediate (1, Scheme 1). GABA-AT inhibition raises GABA levels, which antagonizes the rapid release of dopamine in the nucleus accumbens (NAcc) that occurs as a neurochemical response to cocaine or other drugs of abuse.9 Since then, the anti-epilepsy drug vigabatrin, the only FDA-approved drug that acts as an inactivator of GABA-AT,10 has been successful in the treatment of addiction in animal models for cocaine,9 nicotine,11 methamphetamine, heroin, and alcohol.12 Vigabatrin also was effective in the treatment of cocaine addiction in humans,13,14 with up to 28% of patients achieving abstinence in a 9-week double-blind trial.15 The therapeutic potential of vigabatrin as a treatment for addiction, however, may be problematic because permanent retinal damage has been reported in 25–40% of patients after long-term administration.16
Scheme 1.

Catalytic mechanism of GABA-AT for the degradation of GABA into succinic semialdehyde
In an effort to minimize or eliminate this side effect, we designed a mechanism-based inactivator,17 (1S,3S)-3-amino-4-difluoromethylenyl-1-cyclopentanoic acid (2, Scheme 2), now called CPP-115, which is 186 times more efficient in inactivating GABA-AT than vigabatrin.18 A mechanism-based inactivator is an unreactive compound that requires target enzyme catalysis to convert it to a species that inactivates the enzyme prior to that species leaving the active site. When tested in a multiple-hit rat model of infantile spasms, CPP-115 suppressed spasms at doses over 100-fold lower than those used for vigabatrin and additionally produced longer spasm suppression.19 This compound also was more effective against addictive challenges; when tested in freely moving rats after administration of 20 mg/kg cocaine, CPP-115 was over 300 times more potent than vigabatrin in reducing the release of dopamine in the NAcc.20 Administration of CPP-115 (1 mg/kg) with cocaine given to cocaine-addicted rats showed a similar effect in eliminating their addictive behavior as with administration of vigabatrin at 300 mg/kg with cocaine. CPP-115 also displayed a much larger margin of safety and a considerably lower retinal toxicity liability than vigabatrin.19 To date, CPP-115 has completed a Phase I safety clinical trial without adverse effects. In addition, a child with infantile spasms refractory to all treatments has been treated successfully with CPP-115.21
Scheme 2.

Potential Mechanism of Inactivation of GABA-AT by CPP-115
Originally, CPP-115 was designed to inactivate GABA-AT via a Michael addition mechanism that would lead to a covalent adduct with the enzyme, similar to that with vigabatrin.18 However, it was discovered from the crystal structure of GABA-AT inactivated by CPP-115 that the enzyme forms a noncovalent, tightly-bound complex with CPP-115 via strong electrostatic interactions between the two carboxylate groups in the resulting metabolite with Arg192 and Arg445 in the active site (Scheme 2).22 Inactivation is initiated by Schiff base formation between CPP-115 and the lysine-bound PLP, followed by γ-proton removal and tautomerization, resulting in a highly reactive Michael acceptor (3, Scheme 2). However, before Lys329 can attack this Michael acceptor, catalytic hydrolysis of the difluoromethylenyl group occurs, leading to the PLP-bound dicarboxylate metabolite (4, Scheme 2), which elicits a conformational change in the enzyme and tightly binds to Arg192 and Arg445 via electrostatic interactions. Molecular dynamic simulations and computer modeling indicated a movement of the difluoromethylenyl group of CPP-115 away from Lys329 upon enzyme-catalyzed tautomerization, leaving it too far away from Lys329 for nucleophilic attack; the enzyme catalyzes its hydrolysis instead.22 On the basis of further classical molecular dynamics simulations and computer modeling with GABA-AT, we have identified a design approach that would bring the electrophilic intermediate closer to Lys329 for the encouragement of nucleophilic addition and covalent bond formation. This approach, the unexpected outcome, and the in vitro and in vivo properties of the designed molecule are reported here.
RESULTS AND DISCUSSION
Design of a highly potent GABA-AT inactivator
To avoid or minimize the movement of the difluoromethylenyl group away from Lys329 after tautomerization, we proposed to minimize the flexibility of the ring by incorporating a double bond into the structure of CPP-115, leading to the design of the mechanism-based inactivator (S)-3-amino-4-(difluoromethylenyl)cyclopent-1-ene-1-carboxylic acid (5, Figure 1). Flexible docking followed by classical molecular dynamic simulations of the tautomerized form of 5 within the active site of GABA-AT showed that the incorporation of the endocyclic double bond left the difluoromethylenyl group closer to the Lys329 nitrogen atom (4.6 ± 0.3 A) than in the case of CPP-115 (7.5 ± 0.6 A) (Figure 2). The visualization provided by these molecular dynamics simulation studies provided validation to synthesize and test 5.
Figure 1.
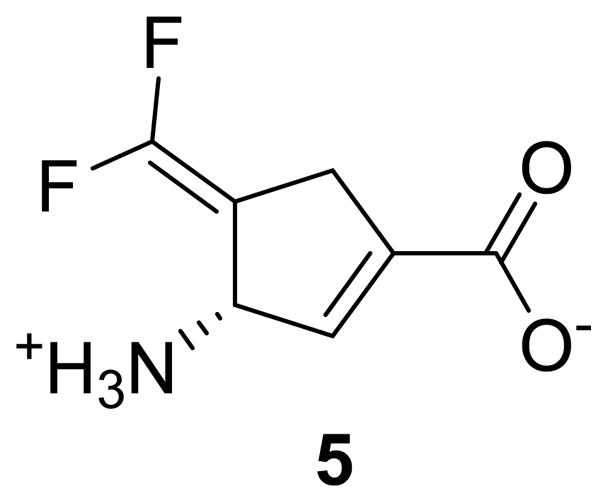
Structure of (S)-3-amino-4-(difluoromethylenyl)cyclopent-1-ene-1-carboxylic acid (5)
Figure 2.

Classical molecular dynamics simulations of CPP-115 and 5 bound to PLP of GABA-AT. Color legend: Yellow or orange-carbon; white-hydrogen; red-oxygen; blue-nitrogen; brown-fluorine; taupe-phosphorus. (A) Distance measurements during the 50 ns of the trajectories between the catalytic Lys329 nitrogen atom and the inhibitors after tautomerization (see Scheme 2). The simulation of CPP-115’s tautomer stabilized after about 4 ns of simulation as a result of a conformational change within the ring, which increased the distance between the CF2 group and Lys329 to about 7.5A. This distance with 5 was only 4.6A (B) Superimposition of the average equilibrated poses of CPP-115 (orange) and 5 (yellow); average distances from the ε-nitrogen of Lys329 to the CF2 carbon atom are shown, calculated using the last 40 ns of the trajectories.
To eliminate differences in ring dynamics as a reason for the shorter CF2-Lys329 distances, Principal Component Analyses (PCA) of the ring carbons’ motions during the last 40 ns of the simulations were performed for both CPP-115 and 5. These multivariate analyses highlight the major multiatom conformational changes from other main molecular motions within a MD trajectory. Even though 5 was determined to be noticeably more rigid, the analyses ultimately suggested there were similar dynamic modes associated with the five-membered rings of both CPP-115 and 5; therefore, dynamics of the five-membered rings cannot be the reason for the different distances from Lys329 (see Supporting Information Figures S1 and S2). Figure 3A shows the projection of trajectories on the common subspace defined by the top two Principal Components (PC1 and PC2) for the two inhibitors in the tautomerized complexes. The larger overlap between CPP-115 and 5 in both complexes is evidence for the similar conformational space shared by both inhibitors in the complexes.
Figure 3.

(A) Projection of trajectories on the common subspace defined by the top two Principal Components (PC1 and PC2) for CPP-115 and 5 in the tautomerized complexes after equilibration. Most of the variance is centered around only one main conformation for each structure. (B) Free energy landscape of inhibitors along the first two PCs and average structures corresponding to energy minima in the tautomerized complex (CPP-115 is in orange and 5 in yellow)
On the other hand, we found that the average structures corresponding to the energy minimum of the free energy landscape of CPP-115 and 5 (along PC1 and PC2) in the tautomerized complex reveal an envelope conformation for CPP-115 and a more planar conformation for 5 (Figure 3B). Visual inspection of the trajectories also confirms the presence of these two average structures. Interestingly, the PCA using the entire trajectory (50 ns) reveals two well separated clusters for CPP-115, which identifies a movement of the equatorial carboxylate group of CPP-115 to an axial carboxylate around 4 ns, where the significant Lys329-CF2 distance difference starts to appear (Figure 2A and Supporting Information Figures S1 and S2). Density Functional Theory (DFT) calculations of the optimized structures (B3LYP/6-311++G(d, p)) corresponding to the isolated conformations with axial and equatorial carboxylate groups in implicit water indicate a 0.075 kcal/mol slight stability in favor of the axial conformation, which is also the more prominent form in the enzyme active site (Supporting Information Figure S3; for detailed procedure, see Supporting Information). This change in relative orientation between the carboxylate and amino pharmacophoric groups in the CPP-115 tautomer causes a realignment of the molecule within the binding site to accommodate the interaction of the carboxylate with Arg192, which pulls the difluoromethylene group away from Lys329. In the case of 5, the endocyclic double bond prevents the ring from adopting an envelope conformation because of geometric constraints, which prevents any major conformational changes between the original and tautomerized versions of the molecule. This major conformational difference between CPP-115 and 5 explains the Lys329-inhibitor distance difference, in which the difluoromethenyl group in 5 is closer to the catalytic Lys329 than that in CPP-115 (Figure 2 and Supporting Information Figure S4).
Even though these computational studies encompass only geometric factors, and not electronic ones like pKa changes that might impact reaction rates, the calculations suggest that 5 would be more likely than CPP-115 to inactivate GABA-AT by a covalent mechanism; consequently, a synthesis for 5 was devised.
Synthesis of 5
The synthesis of 5, shown in Scheme 3, started from CPP-115 (2).18 The carboxylic acid and amino groups were protected (6), and then the α-carbon was selenated, resulting in a 7:3 inseparable mixture of diastereomeric selenides (7). The protecting groups were removed to afford 9; it was found that the purity of 9 was crucial for the final purity of 5. Oxidative elimination of the phenylselenyl group in 9 under mild conditions gave a 5:3 isomeric mixture of 5 and 10. Many attempts to separate 5 from 10 by chromatography were unsuccessful, but it was discovered in the process that 10 was much more reactive than 5. Consequently, a strategy involving selective modification and removal of more reactive 10 using a soft thiol nucleophile (2-mecaptobenzoic acid) was successfully developed.
Scheme 3.

Synthesis of (S)-3-amino-4-(difluoromethylenyl)cyclopent-1-ene-1-carboxylic acid (5) from CPP-115 (2).
In vitro activity of 5
In vitro studies showed that 5 was an exceedingly potent inactivator of GABA-AT. In fact, the inactivation occurred so rapidly that the inhibition constant (KI) and the rate constant of enzyme inactivation (kinact) for the inactivation of GABA-AT by 5 could not be determined accurately using a Kitz and Wilson replot,23 even under nonoptimal conditions that were originally done by Pan et al. for CPP-115.18 A recently developed progress curve analysis method24 was then used to measure the kinetic constants (Supporting Information Figure S5), which allowed us to perform the measurements under optimal conditions. This same method was used to measure the kinetic constants of 4-amino-5-fluoropentanoic acid, vigabatrin, and CPP-115, and we compared the results with those obtained previously using the Kitz and Wilson procedure, as a means to validate this new approach (Supporting Information Figure S6). The results showed that 5 had a higher binding affinity to GABA-AT than CPP-115 (KI values of 5 and CPP-115 were 9.69 ± 1.50 μM and 58.8 ± 6.9 μM, respectively), and 5 also inactivated GABA-AT with a larger rate constant than CPP-115 (kinact values of 5 and CPP-115 were 3.32 ± 0.23 min−1 and 2.05 ± 0.09 min−1, respectively). Overall, the efficiency constant for 5 (kinact/KI = 342 ± 58 mM−1min−1) is 9.8 times larger than that for CPP-115 (kinact/KI = 34.9 ± 4.4 mM−1min−1); therefore, 5 is 9.8 times more efficient as an inactivator of GABA-AT than CPP-115!
Because 5 was designed to act by irreversible (covalent) inhibition, time-dependent reactivation of GABA-AT after treatment with the compound was conducted to test if the mechanism involved both irreversible and reversible components, as was observed for CPP-115.22 After GABA-AT activity was completely abolished by 10–1000 equivalents of 5 with 4 h incubation, the treated enzyme was dialyzed, and aliquots at different time intervals (7, 24, and 48 h) were collected and assayed for return of enzyme activity. At 10, 40, and 100 equivalents of 5, activity returned to 30%, 25%, and 15%, respectively, after 24 h of dialysis, and these values remained the same at 48 h (Figure 4, Left). With 1000 equivalents of 5, no enzyme activity returned, even after 48 h of dialysis. In comparison with CPP-115, the same experiment gave approximately the same results. The return of a small amount of enzyme activity from 5- and CPP-115-inactivated GABA-AT at lower concentrations of inactivators indicates that the inactivation includes both an irreversible and a reversible component, as was the case for CPP-115.22 Additionally, the turnover number (the extrapolated number of molecules needed to inactivate the enzyme fully) for 5 was found to be 1.08 ± 0.08 compared to a value of 1.03 ± 0.01 for CPP-115, and 5 demonstrated nonpseudo-first order kinetics, as was previously observed with CPP-115,21 also suggesting both irreversible and reversible components of inactivation (Figure 4, right). The similarities in percent reactivation and turnover number of the two molecules indicate that the increase in activity of 5 does not arise from more efficient turnover by the enzyme.
Figure 4.

(Left) Time-dependent reactivation of inactivated GABA-AT by varying concentrations of 5 and CPP-115 after 24 h incubation with the enzyme; increasing the equivalents of 5 led to further inactivation of the enzyme. (Right) Determination of the turnover number for 5 and CPP-115. The remaining % enzyme activity obtained from graph A was plotted against the equivalents of 5 used; linear regression was used on the linear portion of the curves to obtain the X-intercept, which was the turnover number.
Crystal structure of 5 bound to GABA-AT
To gain a better understanding of the mechanism of inactivation of GABA-AT by 5, we obtained an X-ray crystal structure of the inactivated enzyme, refined to a resolution of 1.95 A in space group P1211; the final Rwork/Rfree values were 0.169/0.215, respectively (Supporting Information Table S2). Unexpectedly, the refined models showed, as in the case of CPP-115,22 a noncovalent, tight-binding inhibitor complex where the difluoromethylene group has been hydrolyzed to a carboxylate (Figure 5; see Figure S7 in Supporting Information for the simulated annealing omit map (Fo−Fc) around the ligand moiety (5) at 2.5 μ and the refined 2Fo−Fc map around the final adduct at 1.0 μ). On the basis of our computer model (Figure 2B), which showed that the electrophilic difluoromethylenyl group of 5 after tautomerization was closer to Lys329 than that of CPP-115 after tautomerization (4.6 A for 5 and 7.5 A for CPP-115), we thought a covalent reaction with Lys329 would occur, as was seen when vigabatrin inactivated GABA-AT,25 but there was no evidence of this in the crystal structure model. Apparently, 4.6 A is still too far from Lys329 for nucleophilic attack.
Figure 5.

Structure of GABA-AT inactivated by 5. As with CPP-115, the difluoromethylene group of 5 has been hydrolyzed to a carboxylate moiety. Inactivation of the enzyme appears to be caused by tight binding between the enzyme and a stable PLP-5 noncovalent complex, rather than covalent modification. Color legend: PLP-5 carbons-yellow; protein carbons-white; oxygens-red; nitrogens-blue; phosphorus-taupe. The crystal structure has been deposited in the Protein Data Bank, PDB code 6B6G.
Effect of active site water molecules
To rationalize a hydrolysis mechanism, we considered the case of aspartate aminotransferase with substrate and substrate analogues,26 for which the active-site lysine (Lys258 in Asp-AT) has been proposed to be the base that deprotonates a water molecule and catalyzes hydrolysis of the tautomerized aldimine intermediate. For GABA-AT this would correspond to hydrolysis of 1 from GABA (B = Lys329 in Scheme 1) on the way to succinic semialdehyde and, by analogy, hydrolysis of intermediate 3 from CPP-115 (B = Lys329 in Scheme 2) leading to inactivated enzyme (4). This hydrolysis mechanism in GABA-AT is supported by the distribution of water molecules around the ε-nitrogen atom of Lys329 in the tautomerized CPP-115 complex, as observed by classical MD simulations. A comparison of the radial distribution function of water molecules around the Lys329 nitrogen with that for adjacent Lys330 and a surface lysine residue (Lys207) demonstrates that Lys329 has a close, well-ordered hydration sphere (1.8 A) followed by two less ordered hydrogen spheres around 2.9 A and 3.2 A, respectively (Figure 6). By comparison, neither Lys330 nor Lys207 has a close well-ordered hydration sphere, indicating a higher probability that Lys329 can induce a water-mediated nucleophilic attack. A similar result was seen for the radial distribution function of 5 in the tautomerized complex. Therefore, having the electrophilic difluoromethylenyl group closer to Lys329 also puts it closer to the catalytic water molecule, which enhances the hydrolysis of the difluoromethylenyl group and increases the efficiency of inactivation with 5, as shown in the MD simulation result (Figure 2).
Figure 6.

Radial distribution function (rdf) of water around Lys329, Lys330, and Lys207 in the CPP-115-GABA-AT complex. A well-ordered hydration sphere can be observed around 1.8A of the terminal nitrogen atom of Lys329, whereas a more disordered one exists for all three lysines between 2.5 and 4A.
Tautomeric forms of the structure of 5-inactivated GABA-AT
On the basis of the crystal structure in Figure 5 and the in vitro activity (Figure 4), the mechanism of inactivation of GABA-AT by 5 closely resembles that by CPP-115 (Scheme 2). However, the structure of 5-inactivated GABA-AT theoretically could be any one (or more than one) of eight tautomeric forms (Figure 7). Although identification of the structure of the bound tautomeric form does not shed any more light on the inactivation mechanism, it presents an interesting challenge, which we attempted to resolve by various computational methods. The electron density of the crystal structure of 5 bound to GABA-AT (Supporting Information Figure S7B) reveals that the ring has an sp3 carbon atom adjacent to the carboxylate that was initially the difluoromethylenyl group. Of the eight theoretical tautomeric forms of the enzyme-bound inactivator-PMP structure (11a–h, Figure 7), only three of them would accommodate an sp3 atom in the ring at that position (11a–c). On the basis of the crystal structure (Figure 5 and Supporting Information Figure S7), it is not possible to differentiate these three tautomeric forms. Consequently, we performed DFT calculations with a large B3LYP/6-311++G(d,2p) basis set on interaction models of each tautomer with residues within a 5 A radius according to their X-ray crystallographic distances to determine their interaction energies (Supporting Information Figure S8; see details in Supporting Information). Basis set superimposition error-corrected gas phase interaction energies for the models of tautomers 11a–c at each nearby residue are shown in Table 1. The DFT calculations show that the tautomer with the lowest interaction energies is 11c.
Figure 7.

Eight theoretical tautomers of 5-inactivated GABA-AT. 11d through 11h are irrelevant based on crystallographic information (the indicated ring carbon is not planar); energy calculations suggest that either 11a or 11c are the most likely tautomeric forms.
Table 1.
Basis set superimposition error-corrected gas phase interaction energies of tautomer models with individual residues (in kcal/mol), calculated at the B3LYP/6-311++G(d,3p) level (X-ray crystallographic pose), where ++ are diffusion functions on both heavy atoms and hydrogens, d,3p are the polarization functions added to the heavy atoms and hydrogens, 6-311 is the split valance basis set used for the calculations, and B3LYP is the DFT method.
| Residue | 11a | 11b | 11c |
|---|---|---|---|
|
| |||
| Arg192 | −161.88 | −157.34 | −167.24 |
| Arg445 | −141.41 | −133.53 | −141.23 |
| Gln301 | −14.28 | −11.69 | −17.73 |
| Glu270 | 117.26 | 119.29 | 116.88 |
| Ile72 | 1.09 | 0.28 | 1.27 |
| Lys329 | −128.49 | −127.97 | −124.82 |
| Phe189 | −2.14 | 0.12 | −1.67 |
| Phe351 | −6.99 | −8.53 | −7.55 |
| Thr353 | −2.48 | −4.36 | −1.94 |
|
| |||
| Total | −339.32 | −323.73 | −344.03 |
The difference in the total interaction energy between 11a and 11c is 4.71 kcal/mol, but the difference in interaction energy from the interaction with Arg192 alone is 5.36 kcal/mol. To determine if the stabilization energy from the Arg192 interaction, resulting from electron delocalization and hyperconjugation, accounts for the difference in the total interaction energy of tautomer 11a vs 11c, natural bond orbital (NBO) analysis was carried out. NBO analysis, which provides information about the interactions between the filled (bonding or lone pair) Lewis type NBOs (donor) and the empty (antibonding or Rydberg) non-Lewis NBOs (acceptor) (Supporting Information Figure S9; see Supporting Information for details), shows a difference of 4.84 kcal/mol in their stabilization energies in favor of 11c, supporting the importance of the enhanced interaction of 11c with Arg192 in stabilizing the entire tautomer complex. These calculations do not take into account the kinetics for the formation of 11a–c, which is beyond the scope of this work. The exact identity of the final metabolite tautomer, whether 11a or 11c, does not affect the overall mechanistic result, namely, that 5 inactivates GABA-AT in a manner similar to CPP-115, except that tautomerization to a more stable complex can occur.
As a result of inactivation, the catalytic lysine (Lys329) is freed from its covalent linkage to the PLP, and the final adduct forms salt bridges with Arg445 and Arg192 (Figure 5). Gln301 also is within hydrogen bonding distance to the newly formed carboxylate of the inactivated enzyme adduct. Glu270, which forms a salt bridge with Arg445 in the native structure, has been displaced by the newly formed carboxylate of the inactivated enzyme adduct. In all of the tautomer cases, Glu270 has highly repulsive interactions (11a = 117.26 kcal/mol, 11b = 119.29 kcal/mol, 11c = 116.88 kcal/mol) because of its proximity to the new negatively charge carboxylate group of the tautomers. On the basis of this information, we suggest that the two strong salt bridge interactions between the ligand and the arginine residues (Arg192 and Arg445) overcome this repulsive interaction. These calculations also explain how the ligand is capable of replacing the native Arg445-Glu270 salt-bridge interaction with newly formed salt bridge interactions between the ligand and the two arginine residues.
Potential off-target activity and favorable ADMET profile of 5
Unlike vigabatrin,27 CPP-115 was reported not to inactivate or inhibit off-target enzymes, such as aspartate aminotransferase (Asp-AT) and alanine aminotransferase (Ala-AT),22 which could have contributed to its larger margin of safety than vigabatrin. Therefore, the activity of 5 was also tested with these off-target enzymes. The results showed that 5 also does not cause time-dependent inhibition and is only a very weak reversible inhibitor of both Asp-AT and Ala-AT with an IC50 > 4 mM (Supporting Information Figures S10 and S11, respectively). Another important PLP-dependent off-target enzyme is ornithine aminotransferase (OAT).28 CPP-115 was reported to be a moderate inactivator of OAT with a KI value of 0.116 mM and a kinact value of 0.097 min−1.29 Therefore, compound 5 also was tested for activity against OAT; the results showed that 5 is a potent inactivator of OAT with a KI value of 0.0033 mM and a kinact value of 0.025 min−1 (Supporting Information Figure S12). A comparison of the kinact/KI value of 5 (7.6 mM−1min−1) with that of CPP-115 (0.84 mM−1min−1) shows that 5 is 9.0 times more efficient as an inactivator of OAT than CPP-115, consistent with its 9.8 times higher efficiency as an inactivator of GABA-AT. Acute inhibition of human OAT has been shown to be beneficial for the treatment of hepatocellular carcinoma.29 Given that 5 is 44 times more effective at inhibiting GABA-AT than OAT, and considering that the effective dose of 5 should be quite low, we do not expect side effects from this inactivation.
hERG is a potassium ion channel that contributes to the electrical activity of the heart, which coordinates the heart’s beating.30 This channel is sensitive to drug binding, and when its ability to conduct electrical current across the cell membrane is compromised, it can result in potentially fatal cardiac adverse effects; therefore, it is important to avoid hERG inhibition during drug development.30 Like CPP-115,22 5 does not inhibit the activity of the hERG channel, even at 10 μM concentration (Supporting Information Figure S13).
Microsomal cytochromes P450 (CYPs) are major enzymes that are involved in drug metabolism, accounting for ~ 75% of all drug metabolism.31 Inhibition or activation of CYPs can lead to undesirable drug-drug interactions. As with CPP-115,22 5 does not inhibit or induce the seven most common CYPs (1A, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A) that are involved in >95% of the reactions in drug metabolism,31 even at 10 μM concentration (Supporting Information Figure S14). Furthermore, plasma protein binding is only 27%, indicating a high percentage of free drug in plasma.
Compound 5 was also evaluated for its metabolic stability in human liver microsomes (HLM) by incubating 5 with microsomes and monitoring its disappearance over time using LC-MS/MS. Terfenadine was run under the same conditions as a positive control. No metabolism of 5 was observed over the 90 min assay time.
To determine if 5 affects other important pharmacological targets, the Cerep SpectrumScreen (Eurofins/Cerep Panlabs) of 176 pharmacological targets showed no significant activity with any of the targets at 3 μM concentration. It also was negative in the Ames test for mutagenicity (Eurofins/Cerep Panlabs), and, on the basis of the Irwin (primary observation) test (Porsolt) in mice, the no observed adverse effect level (NOAEL) was 6 mg/kg by oral administration.
Effect of 5 on cocaine and nicotine addiction in rats
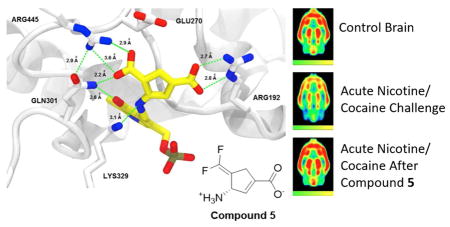
Addiction is a consequence of the release of dopamine in the NAcc when an addictive substance is ingested or an addictive behavior occurs. The effect of 5 on the release of dopamine at the NAcc in freely moving rats was determined using in vivo micropositron emission tomography (microPET) imaging (Figure 8).9 In the central nervous system, especially the corpus striatum, there is a high concentration of dopamine D2 receptors, as seen by the red in each image in Figure 8, which corresponds to [11C]-raclopride binding; [11C]-raclopride competes with dopamine for the same receptor sites located on postsynaptic dopamine terminals. MicroPET was used to measure the dissociation of the [11C]-raclopride tracer from dopamine receptors caused by either cocaine- or nicotine-induced increases in synaptic dopamine levels. When animals received cocaine (n = 8) or nicotine (n = 6), the striatal dopamine levels rapidly increased;32 these elevations effectively displaced [11C]-raclopride from the receptors, as seen in Figure 8 (middle frame; the intensity of the red is much lower). If the same animals (on a different day) received 5 (1.0 mg/kg) prior to cocaine or nicotine, there was no significant change in [11C]-raclopride binding (Figure 8, bottom frame); the degree of redness is equal to that of the controls (Figure 8, top frame), indicating that there was no increase in dopamine levels that would effectively compete with [11C]-raclopride binding. Therefore, 5 blocks both cocaine- and nicotine-induced elevations of dopamine, thereby blocking the addictive reward response. These experiments were carried out at three low doses of 5, 1.0 mg/kg, 0.1 mg/kg, and 0.01 mg/kg. The results in Table 2 indicate that a 10 mg/kg dose of cocaine reduces [11C]-raclopride binding by 36%, which represents an increase in dopamine release. A 0.01 mg/kg dose of 5 administered 30 minutes prior to cocaine reduced [11C]-raclopride binding by 20%, 0.1 mg/kg dose of 5 reduced [11C]-raclopride binding by 12%, and 1.0 mg/kg dose of 5 reduced [11C]-raclopride binding by 5% (comparable to control). CPP-115 was shown earlier to be 300–600 times more potent in inhibiting cocaine-induced dopamine release than vigabatrin; a 0.5 mg/kg dose of CPP-115 was comparable to a 300 mg/kg dose of vigabatrin.20 We have now shown that, in vitro, 5 is 9.8 times more potent than CPP-115 and inhibits dopamine release at a dose of 0.01 mg/kg following a cocaine challenge. Compound 5 is the most potent compound we have tested to date for antiaddictive properties. If this potency translates to humans, it would mean therapeutic doses in the very low milligram range, which would have the benefit of low liability potential from idiosyncratic toxicity.33
Figure 8.

Effects of an acute dose of 5 and cocaine or nicotine on 11C-raclopride uptake in rat brain. (Top) Basal amounts of [11C]-raclopride binding to dopamine receptors in the animal’s brain; red indicates higher concentrations. (Middle) The increase in dopamine levels after cocaine- or nicotine challenge displaces a significant amount of [11C]-raclopride from the receptors. (Bottom) Administration of 5 before cocaine or nicotine administration prevents [11C]-raclopride displacement, suggesting that dopamine levels remained constant.
Table 2.
Effect of 5 on 11C-raclopride binding following a cocaine challenge. 5 suppressed dopamine release caused by cocaine administration in a dose-dependent manner.
| Entry | Condition | ST-CB/CBa | %=b |
|---|---|---|---|
|
| |||
| 1 | Control | 3.75 ± 0.47 | |
| 2 | Test/retest | 3.81 ± 0.33 | 2 |
| 3 | Control/Blocked (cld rac) | 0.46 ± 0.50 | −88 |
| 4 | Cocaine (10 mg/kg) | 2.39 ± 0.21 | −36 |
| 5 | Cocaine/5 (0.01 mg/kg) | 3.01 ± 0.44 | −20 |
| 6 | Cocaine/5 (0.1 mg/kg) | 3.31 ± 0.52 | −12 |
| 7 | Cocaine/5 (1.0 mg/kg) | 3.56 ± 2.11 | −5 |
Striatum (dopamine-rich brain region)-Cerebellum (dopamine devoid brain region)/Cerebellum. This is to control for nonspecific binding of the radiotracer, which in this case is very low because of its high specificity for D2 receptors.
Percent inhibition of cocaine-induced increase in synaptic dopamine
Effect of 5 on rat brain hippocampus glucose metabolism
[18F]-2′-Fluoro-2′-deoxy-D-glucose (18FDG) and microPET were used to examine the regional effects of 5 on cocaine-induced increases in glucose metabolism. 18FDG is an analogue of glucose that is taken up into neurons (or any cells in the human body) just like glucose. However, after 18FDG is phosphorylated, the corresponding 6′-phosphate cannot be further metabolized in the glycolytic pathway and remains in cells. Consequently, human PET studies have used 18FDG for decades to map the brain.34 For example, if an individual in a PET scanner performs a specific task with one hand while 18FDG is injected intravenously, neurons in the brain underlying that hand’s ability to perform that task incorporate this radiolabeled sugar while other surrounding neurons do not. When a human or a rat receives a psychostimulant like cocaine, dopamine floods the synapse, causing postsynaptic neurons to fire frantically. Because this neuronal firing requires energy in the form of glucose, the result is that brain glucose metabolism increases in specific brain regions. The effect of 5 on cocaine-induced increases in glucose metabolism in freely moving rats was determined using statistical parametric mapping, in which all of the images from the cocaine-only animals were added together and then compared to the images obtained from the same animals that received 5 and cocaine. A statistical threshold (p < 0.00001) was set, and a statistical parametric map, an image showing all the pixels that were statistically different between the two conditions, was generated and then overlaid on an MRI of the rat brain (Figure 9). In the cocaine-only animals, an enormous activation in the hippocampus, a bilateral structure, was observed with one large bright red spot on each side (Figure 9, left). However, in the cocaine/5 animals, no activation in the hippocampus was detected (Figure 9, right). This is the largest attenuation by ingestion of a compound that we have ever observed. Vigabatrin and CPP-115 previously went through similar tests; they both blocked cocaine-induced increases in striatal dopamine but did not completely block the hippocampal metabolism as 5 did.
Figure 9.

Statistical parametric map images showing the effects of 5 on increased metabolic demands in the hippocampus. (Left) Cocaine administration causes dopamine release and a strong activation of the hippocampus, based on an increased uptake of [18F]-2′-fluoro-2′-deoxy-D-glucose (18FDG) (red color). (Right) Administration of 5 before the cocaine challenge blocks dopamine release and prevents the activation of the hippocampus (no red color in the hippocampus region)
Cocaine- and nicotine-induced increases in striatal dopamine dose-dependently produce a conditioned place preference, which results in animals ‘learning’ to associate a specific environment with the drug they receive, which leads to dopamine release when they return to that environment, even in the absence of the stimulant. When the striatum is activated by an elevation in dopamine levels (subsequent to a cocaine or nicotine challenge), projections to the hippocampi cause it to activate. The hippocampus plays a pivotal role in spatial memory; therefore, it is important for encoding environmental conditions during drug exposure. Because 5 blocked cocaine-induced increases in striatal dopamine, it is not surprising that it also inhibited increased metabolic demands in the hippocampus.
CONCLUSION
On the basis of molecular dynamics simulations with CPP-115 (2) bound to GABA-AT and molecular docking, we have designed an exceedingly potent mechanism-based GABA-AT inactivator, (S)-3-amino-4-(difluoromethylenyl)cyclopent-1-ene-1-carboxylic acid (5), which was predicted to have the generated reactive group much nearer to Lys329 for potential covalent modification, unlike that of CPP-115. Flexible docking alone did not show a difference in the distance between the difluoromethylenyl group and Lys329 with CPP-115 or 5 bound; the desired difference was observed only after classical molecular dynamics simulations. In vitro results show that 5 is 9.8 times more efficient as an inactivator of GABA-AT than CPP-115, previously the most potent GABA-AT inactivator; CPP-115 displays high therapeutic potential as a treatment for addiction and has been found to be effective in treating infantile spasms.19 The crystal structure, unexpectedly, showed that the irreversible inhibition of GABA-AT by 5 is the result of a noncovalent, tight-binding complex, just like CPP-115. The crystal structure electron density map, along with calculations using density functional theory and natural bond orbital analysis, were used to elucidate an interesting question, namely, which of the eight possible tautomeric forms of the adduct is most likely the structure bound to the enzyme in terms of interaction energies.. Given the inability of X-ray crystallography to differentiate tautomeric forms, these combined computational approaches were able to predict tautomer 11c was most likely the bound structure. Compound 5 had little off-target activity, with the notable exception of inactivating OAT, which has been previously shown to be a viable approach to inhibit the growth of hepatocellular carcinoma.29 In vivo studies in freely moving rats showed that 5 is superior to CPP-115 and vigabatrin in suppressing the release of dopamine in the NAcc following a cocaine- or nicotine challenge. Compound 5 also promoted the highest attenuation we have observed of dopamine-induced increases in metabolic demand within the hippocampus, a brain region previously demonstrated to encode spatial conditions of the environment (conditioned place preference) associated with drug-induced increases in dopamine. The studies described here demonstrate the combined power of mechanistic enzymology and molecular dynamic simulations with molecular docking to be important in the design and improvement of mechanism-based inhibitors of enzymes whose crystal structures and inactivation mechanisms are known.
EXPERIMENTAL SECTION
General Procedures
CPP-115 was synthesized at IRIX Pharmaceuticals for Catalyst Pharmaceuticals, which generously provided it; other chemicals were obtained from Sigma-Aldrich and used as received unless specified. All syntheses were conducted with anhydrous conditions under an atmosphere of argon, using flame-dried glassware and employing standard techniques for handling air-sensitive materials, unless otherwise noted. All solvents were distilled and stored under an argon or nitrogen atmosphere before use. 1H NMR and 13C NMR spectra were taken on a Bruker AVANCE III 500 spectrometer, an Agilent DDR2 400 MHz spectrometer, or an Agilent DD2 500 MHz spectrometer with an Agilent 5 mm HFX probe at 26 °C using DMSO-d6 or D2O as solvents, recorded in δ (ppm) and referenced to DMSO-d6 (2.50 ppm for 1H NMR and 39.52 ppm for 13C NMR) or D2O (4.79 ppm for 1H NMR). High resolution mass spectra (HRMS) were measured with an Agilent 6210 LC-TOF (ESI, APCI, APPI) mass spectrometer.
Methyl (1S,3S)-3-((tert-butoxycarbonyl)amino)-4-(difluoromethylenyl)cyclopentane-1-carboxylate (6)
To dry methanol (27 mL) was added acetyl chloride (2.49 mL, 35 mmol) at 0 °C, and the mixture was stirred for 10 min. To the resulting solution was added CPP-115 hydrochloride salt (2, 1.50 g, 7.0 mmol), and stirring continued for 24 h at room temperature. Triethylamine (6.84 mL, 49 mmol) and di-tert-butyl dicarbonate (1.93 mL, 8.4 mmol) were then added, and the resulting solution was stirred for 20 h at room temperature. The reaction mixture was concentrated and redissolved in ethyl acetate. The organic solution was washed with 2 N HCl, saturated NaHCO3, and brine. The organic layer was dried over Na2SO4, followed by filtration and evaporation to afford 6 (1.99 g, 6.83 mmol, 97%) as a white solid.; 1H NMR (500 MHz, 60 °C, DMSO-d6) δ 6.89 (s, 1H), 4.57 (s, 1H), 3.63 (s, 3H), 2.86 (m, 1H), 2.55–2.51 (m, 1H), 2.23 (ddt, J = 10.0, 7.1, 2.9 Hz, 1H), 1.79 (m, 1H), 1.39 (s, 9H).; 13C NMR (126 MHz, 60 °C, DMSO-d6) δ 173.74, 154.78, 152.80, 150.54, 148.27, 92.12, 91.98, 91.84, 77.86, 51.72, 49.45, 40.31, 36.51, 28.31, 28.16.; 19F NMR (470 MHz, 60 °C, DMSO-d6) δ −89.49 (d, J = 55.9 Hz), −92.89 (d, J = 57.7 Hz).; HRMS (M+Na+) calcd for C13H19F2NNaO4 314.1174, found 314.1179.
Methyl (3S)-3-((tert-butoxycarbonyl)amino)-4-(difluoromethylenyl)-1-(phenylselanyl)cyclopentane-1-carboxylate (7)
To a solution of KHMDS (14.96 mL of 1M solution in THF, 14.96 mmol) and dry THF (10 mL) at −78 was added a solution of 6 (1.98 g, 6.80 mmol) in dry THF (10 mL) slowly via syringe. The reaction mixture was stirred at −78 °C for 90 min. A solution of phenylselenyl chloride (1.43 g, 7.48 mmol) in dry THF (2 mL) was added, and stirring was continued at −78 °C for 75 min. The reaction mixture was then allowed to warm to 0 °C, stirred at 0 °C for 3 h, warmed to room temperature, and stirred at room temperature for 2 h. Saturated aqueous ammonium chloride and ethyl acetate were added, and the organic layer was washed with saturated aqueous ammonium chloride and dried over Na2SO4. Filtration and evaporation gave a crude mixture, which was purified by silica gel column chromatography (hexane/EtOAc) to afford a 7:3 diastereomeric mixture (7, 2.12g, 4.75 mmol, 70%) as a pale brown syrup; 1H NMR (500 MHz, 60 °C, DMSO-d6) δ 7.58–7.37 (m, 5H), 6.97 (s, 1H), 4.83 (s, 0.7H), 4.47 (s, 0.3H), 3.64 (s, 2.2H), 3.57 (s, 0.8H), 2.96–2.88 (m, 1H), 2.68–2.63 (m, 0.7H), 2.43–2.36 (m, 0.7H), 2.23–2.14 (m, 1.3H), 2.00–1.95 (m, 0.3H), 1.39 (s, 9H); 13C NMR (126 MHz, DMSO-d6) δ 172.09, 171.96, 154.41, 153.76, 151.48, 150.64, 149.21, 136.79, 136.76, 129.48, 129.28, 128.91, 128.84, 126.53, 126.20, 91.00, 90.91, 90.85, 90.76, 90.70, 90.61, 77.88, 59.38, 52.11, 51.85, 51.84, 50.01, 48.64, 42.72, 40.67, 35.76, 34.17, 34.15, 27.90, 27.55.; 19F NMR (470 MHz, 60 °C, DMSO-d6) δ−88.29 (d, J = 51.2 Hz), −89.34 (d, J = 52.9 Hz), −91.00 (d, J = 54.8 Hz).; HRMS (M+Na+) calcd for C19H23F2NNaO4Se 470.0654, found 470.0660 (the most abundant Se isotope was selected).
(3S)-3-((tert-Butoxycarbonyl)amino)-4-(difluoromethylenyl)-1-(phenylselanyl)cyclopentane-1-carboxylic acid (8)
To a solution of 7 (1.40 g, 3.13 mmol) in methanol (14 mL) and water (4 mL) at 0 °C was added lithium hydroxide (225 mg, 9.39 mmol). The reaction mixture was allowed to warm to room temperature and was stirred for 20 h. Ethyl acetate was added, and the organic solution washed with 10% citric acid and brine. The organic layer was then dried over Na2SO4, filtered, and concentrated. The crude mixture was subjected to silica gel column chromatography (hexane/ethyl acetate) to afford 8 (1.25 g, 2.89 mmol, 92%) as a white powder. 1H NMR (500 MHz, 60 °C, DMSO-d6) δ 12.61 (s, 1H), 7.64–7.54 (m, 2H), 7.46–7.42 (m, 1H), 7.41–7.35 (m, 2H), 6.94 (s, 1H), 4.81 (s, 0.7H), 4.48 (s, 0.3H), 2.97–2.83 (m, 1H), 2.67–2.56 (m, 0.7H), 2.35 (dd, J = 16.9, 2.7 Hz, 0.7H), 2.20–2.10 (m, 1.3H), 1.91 (dd, J = 12.9, 8.3 Hz, 0.3H), 1.42–1.35 (m, 9H).; 13C NMR (126 MHz, DMSO-d6) δ 173.76, 173.56, 154.71, 154.64, 153.93, 151.65, 150.69, 149.38, 136.87, 136.81, 129.60, 129.39, 129.20, 129.14, 127.06, 126.55, 91.67, 91.52, 91.40, 91.37, 91.26, 91.11, 78.06, 52.38, 49.99, 48.69, 48.42, 42.95, 40.68, 36.08, 34.26, 28.15.; 19F NMR (470 MHz, 60 °C, DMSO-d6) δ−88.52 (d, J = 52.9 Hz), −89.53 (d, J = 53.2 Hz), −91.25 (d, J = 53.2 Hz), −91.45 (d, J = 55.3 Hz).; HRMS (M+Na+) calcd for C18H21F2NNaO4Se 456.0502, found 456.0498 (the most abundant Se isotope was selected).
(3S)-3-Amino-4-(difluoromethylenyl)-1-(phenylselanyl)cyclopentane-1-carboxylic acid (9)
To a solution of 8 (1.31 g, 3.03 mmol) in CH2Cl2 (13 mL) at 0 °C was added trifluoroacetic acid (3.2 mL), and the reaction was stirred for 4 h at the same temperature. The mixture was concentrated and dried in vacuo. The crude residue was subjected to cation exchange column chromatography (Dowex 50W-X8, 5% aqueous pyridine as eluent) to afford 9 (990 mg, 2.98 mmol, 98%) as an off-white solid. 1H NMR (500 MHz, D2O) δ 7.70–7.63 (m, 2H), 7.53–7.46 (m, 1H), 7.45–7.40 (m, 2H), 4.61 (t, J = 6.7 Hz, 0.7H), 4.44 (m, 0.3H), 3.02–2.84 (m, 1.3H), 2.71–2.57 (m, 1H), 2.49 (dd, J = 14.7, 7.8 Hz, 0.7H), 2.29 (dd, J = 14.7, 7.4 Hz, 0.7H), 2.09 (dd, J = 14.4, 5.3 Hz, 0.3H).; 13C NMR (126 MHz, D2O) δ 182.36, 181.63, 158.52, 158.05, 156.23, 155.76, 153.93, 153.46, 140.18, 139.99, 132.61, 132.55, 132.25, 132.20, 129.96, 129.65, 91.48, 91.42, 91.33, 91.26, 91.22, 91.13, 91.07, 60.00, 58.39, 52.13, 52.09, 52.03, 44.28, 43.38, 38.91, 38.30.; 19F NMR (470 MHz, D2O) δ−84.06 (d, J = 42.6 Hz), −84.38 – −84.58 (m), −84.72 (ddd, J = 45.2, 4.5, 2.4 Hz), −85.08 (ddd, J = 44.6, 5.8, 2.7 Hz).; HRMS (M+H+) calcd for C13H14F2NO2Se 334.0158, found 334.0155 (the most abundant Se isotope was selected).
(S)-3-Amino-4-(difluoromethylenyl)cyclopent-1-ene-1-carboxylic acid Hydrochloride (5)
To a solution of 9 (100 mg, 0.30 mmol) and NaHCO3 (55 mg, 0.66 mmol) in water (2 mL) at 0 °C was added sodium periodate (71 mg, 0.33 mmol). The reaction mixture was allowed to warm to room temp and stirred for 6 h. The reaction mixture was directly applied to a cation exchange column (Dowex 50W-X8, 2 N HCl as eluent) to afford a crude mixture. The crude mixture was subjected to C-18 reverse phase column chromatography (water/methanol) to afford a mixture of 5 and 10 (62 mg, 0.29 mmol, 96%) as a white powder (Note: an aliquot of 2 N HCl was added when concentrating the sample to make sure the solution was strongly acidic). To a solution of the mixture of 5 and 10 (51 mg, 0.24 mmol) in methanol (2 mL) and water (0.5 mL) at 0 °C was added thiosalicylic acid (112 mg, 0.72 mmol). The reaction mixture was allowed to warm to room temperature and stirred for 5 h. After the reaction was confirmed as complete by 19F NMR spectrometry, the reaction mixture was concentrated, and the residue was suspended in water. The suspension was filtered through a cotton plug, and the filtrate was subjected to C-18 reverse phase column chromatography (water/methanol) to afford 5 (23 mg, 0.11mmol, 76% from the content of 5 in the previous isomeric mixtures) as a white powder; 1H NMR (500 MHz, D2O) δ 6.29 (s, 1H), 5.16 (s, 1H), 3.37 (m, 2H).; 13C NMR (126 MHz, D2O) δ 174.42, 158.05, 155.76, 153.46, 150.08, 132.06, 89.80, 89.64, 89.59, 89.43, 57.84, 57.79, 34.85.; 19F NMR (376 MHz, D2O) δ−83.86 (ddd, J = 42.8, 6.0, 3.2 Hz), −84.12 (ddd, J = 43.0 4.9, 2.7 Hz).; HRMS (M-H−) calcd for C7H6F2NO2 174.0372, found 174.0374.; HPLC purity (100% by UV absorbance at 210 nm).
Analysis of Sample Purity by HPLC
An Agilent 1260 infinity HPLC system was used, which consists of a variable wavelength detector (G1314A), a thermostatted column compartment (G1316A), an autosampler (G1329B), an evaporative light scattering detector (ELSD, G4261A), a quaternary pump (G1311B), and a C-18 reverse phase column (Agilent Poroshell 120, 2.7 μm, 4.6 mm × 50 mm). The experiments were run with 5 μL (0.5 mg/mL in water) injections, and sample elution was monitored by UV absorbance at 210 nm and by ELSD in a linear gradient experiment (water/acetonitrile with 0.05% trifluoroacetic acid, gradient system: from initial 2% acetonitrile to 100% acetonitrile in 7 min, then 100% acetonitrile for 3 min).
Enzyme and Assays
GABA-AT (1.48 mg/mL) was purified from pig brain by a procedure described previously.35 Succinic semialdehyde dehydrogenase (SSDH) was purified from GABase, a commercially available mixture of SSDH and GABA-AT, using a known procedure. 36 GABA-AT activity was assayed using a published method.37 GABase (Pseudomonas fluorescens) and succinic semialdehyde were purchased from Sigma-Aldrich. The final assay solution consisted of 10 mM GABA, 1.2 mM NADP+, 5 mM α-ketoglutarate, 5 mM β-mercaptoethanol, and excess SSDH in 50 mM potassium pyrophosphate buffer, pH 8.5. The change in UV absorbance at 340 nm at 25 °C caused by the conversion of NADP+ to NADPH was monitored. The enzyme assays for the determination of kinact and KI values were recorded with a Shimadzu UV-1800 UV/Vis spectrophotometer, using a 1 mm width, 10 mm path length, 45 mm height quartz microcuvette. The enzyme assays for the GABA-AT inactivation and dialysis experiment were recorded with a BioTek Synergy H1 microplate reader.
Determination of the kinact and KI Values
The activity of the GABA-AT was measured under the conditions described in the Enzyme and Assays section in the presence of different concentrations of inactivators, ranging from 1 to 200 μM for 5, and from 50 to 1600 μM for CPP-115 (2). The curves of GABA-AT activity caused by inactivation were fitted to Equation 124 using GraphPad Prism 6™ software to afford the kobs values at each inactivator concentration.
| Equation 1 |
where vi is the initial velocity, vs is the steady state velocity, t is time, a0 is the initial absorbance and kobs is the observed rate of inactivation. The kobs values were plotted against concentrations of the compound, and the best fit curve was then fitted into Equation 224 to afford KI and kinact values.
| Equation 2 |
where [I] is the inactivator concentration, S is the substrate (GABA) concentration applied, Km is the Michaelis-Menten constant of the substrate (GABA). The Km value of GABA with GABA-AT used for the calculation was 1.3 mM.38
Inactivation of GABA-AT by 5 and CPP-115, Dialysis of the Inactivated Enzyme, and Determination of Turnover Number
The reversibility experiment was conducted following a previously reported procedure.22 To the GABA-AT buffer (0.443 mg/mL, 10 μL) was added 70 μL of the inactivator buffer solution (50 mM potassium pyrophosphate, pH 8.5, 5 mM α-ketoglutarate, 5 mM β-mercaptoethanol) so that the final concentration of the inactivator was 10, 40, 100, and 1000 μM (5) or 10, 100, and 1000 μM (CPP-115), and the final concentration of GABA-AT was 1 μM. A control sample was also prepared with 10 μL of the GABA-AT buffer and 70 μL potassium pyrophosphate buffer solution. The sample solutions were incubated for 4 h at room temperature in the dark. The remaining enzyme activity was measured by taking 5 μL from the solution. The inactivated and the control GABA-AT solution were transferred to a D-Tube™ Dialyzer Mini (MWCO 12–14 kDa) and dialyzed against the dialysis buffer (500 mL, 50 mM potassium pyrophosphate, pH 8.5, 0.1 mM α-ketoglutarate, 0.1 mM pyridoxal 5′-phosphate) at 4 °C. The enzyme activity was measured at 7, 24, and 48 h. The turnover number was determined using a previously reported assay.22
Inhibition of Aspartate Aminotransferase by 5
Microtiter plate wells were loaded with 90 μL of an assay mixture containing 100 mM potassium phosphate at pH 7.4, 5.55 mM α-ketoglutarate, 1.11 mM NADH, 5.55 mM L-aspartate, 11.1 units of malic dehydrogenase, and various concentrations of 5. After incubation of the mixture at room temperature for a few min, 10 μL of Asp-AT (3.0 units/mL in 100 mM potassium phosphate at pH 7.4) was added. The plate was shaken at room temperature for 1 min, and the absorbance was measured at 340 nm every 6 s for 90 min. All assays were performed in duplicate (Supporting Information Figure S10).
Inhibition of Alanine Aminotransferase by 5
The assay was identical to that with aspartate aminotransferase except L-alanine was used as the substrate, and lactate dehydrogenase was the enzyme in the buffer (Supporting Information Figure S11).
Time- and Concentration- Dependent Inhibition of Ornithine Aminotransferase by 5
These assays were performed using a modification of the procedure by Juncosa, Lee, and Silverman.39 OAT (0.25 μg) was incubated with various concentrations of 5 (0.5 μM, 2 μM, 5 μM, 10 μM, 20 μM) in 100 mM potassium pyrophosphate buffer, pH 8.0, containing 1 mM α-ketoglutarate in a total volume of 20 μL at room temperature. At time intervals, 80 μL of assay solution, preincubated at 37 °C for 10 min, containing PYCR1 (0.5 μg), 12.5 mM α-ketoglutarate, 1 mM NADH, 0.03 mM PLP, and 25 mM L-ornithine in 100 mM potassium pyrophosphate buffer, pH 8.0, were added to the incubation mixture and assayed for OAT activity at 37 °C for 20 min. All assays were performed in duplicate, and the remaining OAT activity at each preincubation time at each inhibitor concentration was averaged. The natural logarithm of the percentage of the remaining OAT activity was plotted against the preincubation time at each inhibitor concentration to obtain the kobs (slope) value for each concentration. The kobs is the rate constant describing the inactivation at each inhibitor concentration. kobs is replotted against the inhibitor concentration using nonlinear regression analysis (GraphPad Prism 6™; GraphPad Software Inc.). KI and kinact were estimated from Equation 3,
| Equation 3 |
where kinact is the maximal rate of inactivation, KI is the inhibitor concentration required for half-maximal inactivation, and [I] is the preincubation concentration of 5 (Supporting Information Figure S12).
Inhibition of the hERG Channel
This experiment was performed by Eurofins Panlabs (Redmond, WA 98052, USA). hERG CHO-K1 cell line was used. The test concentrations were 0.1 μM, 1 μM, and 10 μM. The incubation time was 5 min at room temperature, cumulatively. The detection method used an automated whole-cell patch clamp. The experiments were duplicated, and the % inhibition of the tail current was averaged (Supporting Information Figure S13).
Inhibition of Microsomal Cytochromes P450
These experiments were performed by Eurofins Panlabs (Redmond, WA 98052, USA). CYP1A inhibition (HLM, phenacetin substrate), CYP2B6 inhibition (HLM, bupropion substrate), CYP2C8 inhibition (HLM, paclitaxel substrate), CYP2C9 inhibition (HLM, diclofenac substrate), CYP2C19 inhibition (HLM, omeprazole substrate), CYP2D6 inhibition (HLM, dextromethorphan substrate), and CYP3A inhibition (HLM, midazolam and testosterone substrates) were tested. The test concentration was 10 μM. The incubation time was 10 min at 37 %. The detection method was HPLC-MS/MS. The experiments were duplicated, and the % inhibition of the control values was averaged (Supporting Information Figure S14).
Metabolic Stability in Human Liver Microsomes
This experiment was performed by SAI Life Sciences Ltd. (Hyderabad, India). The test concentration of 5 was 1 μM. The incubation time was 0, 45, and 90 min at 37 %. The detection method was by LC-MS/MS. Terfenadine was run under the same conditions as a positive control.
MicroPET Imaging
Adult male rats (Sprague-Dawley, 200 – 250 g, n = 16) were obtained from Taconic Farms. Animals were maintained on a 12/12 light-dark cycle. Scanning was performed using a Siemens Inveon. All emission scans were corrected for attenuation. Animals received baseline microPET scans using either 11C-raclopride or 18FDG as described previously. 40 Uptake of both radiotracers occurred while animals were awake and freely moving. Immediately prior to microPET scanning, all animals were anesthetized and maintained under isoflurane.
Crystallization of GABA-AT with 5
GABA-AT inactivated with 5 (10 mM) was concentrated to 10 mg/mL using a 30,000 molecular weight cut off (MWCO) Amicon-Ultra centrifugal filter device (Millipore, MA), and the concentrated protein was buffer exchanged into the crystallization buffer (40 mM sodium acetate, pH 5.5). Crystals were grown using previously published conditions.22 Crystals with the best morphology appeared in the well solution containing 0.1 M ammonium acetate, 0.1 M Bis-Tris pH 5.5, and 17% PEG 10000 after heterogeneous seeding with crystals of active GABA-AT and incubation at room temperature for a week. Crystals of GABA-AT inactivated by 5 with the best morphology were transferred into a cryo-protecting solution (Well Solution supplemented with 25% (v/v) glycerol) before being flash cooled in liquid nitrogen.
Data Collection and Processing
Monochromatic data sets were collected at the LS-CAT, Advanced Photon Source (APS) at Argonne National Laboratory (ANL). Diffraction data were collected at a wavelength of 0.98 A at 100 K using a Dectris Eiger 9M. All collected data sets were indexed and integrated using programs Autoproc41 and Aimless42. The best data set was processed at a resolution of 1.95 A. Data collection statistics are summarized in Supporting Information Table S2.
Structure Solution, Model Building and Refinements
The structure of GABA-AT inactivated by 5 was solved by molecular replacement using PHASER in the Phenix software suit.43 The initial search model was based on a previously published structure of GABA-AT (PDB Code: 1OHV). Models were rebuilt using Coot and refined using the program Phenix43 and analyzed using the programs Coot44 and UCSF Chimera.45 Final refinement statistics are presented in Supporting Information Table S2 (PDB code: 6B6G). Structural figures were made using UCSF Chimera.
Construction of the Enzyme-Inhibitor Complex for MD Simulations
A docking protocol was employed to generate the initial GABA-AT complex corresponding to PLP bound CPP-115 and 5 for the classical MD simulation. The protein structure chosen for this study was the X-ray crystal structure of porcine GABA-AT covalently bound to vigabatrin (PDB ID: 1OHW) with a resolution of 2.3 A.25 The missing residues from the beginning and the end of the dimer sequence were systematically added using UCSF chimera interface to Modeller.46 The two covalently bound vigabatrin inhibitor molecules were removed from the two active sites, and a PLP molecule was covalently linked to the catalytic Lys329 residue of one of the monomers in its X-ray crystallographic pose using UCSF chimera (preserving X-ray crystallographic hydrogen bonds). The protonation states of the amino acid residues were determined using the H++ server. 47 The structures of non-tautomerized CPP-115 and 5 corresponding to the initial complex were optimized using Gausian09 software48 at a HF/6-31+(G) level of theory. The inhibitor and protein structures were further refined using AutoDockTools-1.5.6 available with the MGLTools software package.49 Refined inhibitor structures were docked into the GABA-AT active site using Autodock 4.249 software with the Lys329 residue as a flexible sidechain. A grid box centered on the active site was generated using Autogrid 4.2 software with a grid spacing of 0.375 A and dimensions of 45 × 45 × 45 points along the x, y, and z axes. Lamarckian genetic algorithm (GA) was used for the conformational search with a GA population size of 150 and a maximum number of evaluations of 2,500,000. The 100 generated poses were clustered according to their rmsd values (1.5 A cut-off), and the lowest energy conformation of the best cluster was selected for the MD simulation of the initial complex.
Molecular Dynamics Simulations
The inhibitor structures for initial and tautomerized complexes in this study were considered to be in their N-protonated ketoenamine conformation.50 The electrostatic potential energies (ESP) of the geometrically optimized inhibitors (HF/6-31+G) were calculated at the HF/6-31+G level of theory. Then these energies were used to derive the partial atomic charges of the inhibitors using the restricted electrostatic potential square fit (RESP) method employed in the antechamber module available in the Amber12 program.51 This is the method used to derive the partial atomic charges of the original Amber force fields. The inhibitor atoms were treated with general Amber force field (GAFF), and the automatically assigned GAFF atom types were manually adjusted to accurately represent their chemical environment before deriving the parameters. The parameters and topology files were generated using tLEaP and antechamber modules of the Amber12 program. Then these parameters and topology files were converted to GROMACS compatible format using ACPYPE.52 The force field parameters and the topology for covalently bound PLP were derived similarly. The Amber consistent parameters (well validated) for the FeS cluster were taken from the literature.53 The partial atomic charges and the parameters for the inhibitors and the PLP are listed in the Supporting Information (Figures S31–S34). The modified Amber ff99SB-ILDN force field with parameters for the FeS cluster and the covalently bound PLP were used to simulate the protein. Parameters for the GABA-AT dimer were derived using the pdb2gmx module available in the GROMACS 5.1.2 software package. All of the MD simulations were performed using GROMACS 5.1.2 software.54 The extended single point charge model (SPC/E) was used as the water model. The catalytic Lys329 residue was treated as deprotonated in initial and tautomerized complexes, as suggested by literature findings.55 The protein-inhibitor complex was immersed in a dodecahedron box filled with SPC/E water where the boundaries are extending at least 1.7 nm in all directions from the edges of the protein. Then NaCl was added to the system up to a concentration of 0.15 M (physiological NaCl concentration) to neutralize the charge of the system. The solvated system was energy minimized with the steepest descent followed by the conjugate gradient method until it converged with a maximum force no greater than 500 KJmol−1nm−1. The resulting energy minimized periodic system was the starting configuration for the MD simulation. Prior to the production MD simulation, the system was subjected to equilibration in two steps. First, it was equilibrated at the NVT (constant number of particles, volume, and temperature) ensemble for 1 ns. Then the resulting system was equilibrated at the NPT (constant number of particles, pressure, and temperature) ensemble for 5 ns. Temperature and pressure were controlled at 310 K and 1 bar by the V-rescale thermostat (time constant of 0.4 ps) and the Parrinello-Rahman barostat (time constant of 2 ps), respectively. In both of the equilibration steps positions of the heavy atoms were restrained by applying a force constant of 1000 KJmol−1nm−2. After that, the position restraints were gradually reduced from 500 KJmol−1nm−2 to 100 KJmol−1nm−2 over two runs (1 ns each). Finally, a production MD simulation of the equilibrated system was carried out at the NPT ensemble for 50 ns (time step of 2 fs) without positional restraints (duplicates for initial and tautomerized complexes). During the NPT equilibration, the V-rescale thermostat was replaced by the most accurate Nose-Hoover thermostat and used for the rest of the simulation protocol. The long-range electrostatic interactions were treated with the particle mesh Ewald (PME) method while Coulomb and van der Waals interactions were cut off at 1.2 nm. Bond lengths of the atoms were restrained using the Linear Constraint Solver (LINCS) algorithm. After energy minimization the final conformations of the inhibitor structures were isomerized in place and used as the starting points for the next simulations (simulation of the tautomerized complex).
Catalytic Lys329-Inhibitor Distance Measurements
To determine the important distance measurements between the catalytic Lys329 nitrogen atom and the reactive centers of the inhibitors in the tautomerized complexes, the distance and mindist tools available in the GROMACS software package were used.
Principal Component Analysis (PCA)
Principal component analysis (PCA) was performed to study the collective motion of the five-membered ring heavy atoms in CPP-115 and 5. A detailed procedure can be found elsewhere. 56 Briefly, the covariance matrix (M) was constructed from the position coordinates of the heavy atoms of the five-membered ring after all of the conformations were least square fitted to the average structure according to Equation 4,
| Equation 4 |
where < > represents the mean across all time steps, and T superscript represents the trans pose. Then this symmetric matrix was subjected to eigen decomposition to identify the eigenvectors and corresponding eigenvalues. Each eigen vector corresponds to a specific motion of the five-membered ring. To perform this task and the rest of the analysis, the covar and anaeig tools available in the GROMACS software package and in-house python scripts were utilized. The sham tool available in the GROMACS software package was used for free energy landscape generation.
Distribution of Water Around the Catalytic Lys329 Residue
The distribution of water molecules around the catalytic Lys329 nitrogen atom in the tautomerized complex was examined by means of the radial distribution function (rdf). Rdf expresses the distribution of atoms or molecules around a reference point with respect to the bulk. The GROMACS rdf tool was used to determine the rdf.
BSSE Corrected Interaction Energy Calculations
Theoretical calculations were performed to examine the interactions of the three tautomers (11a, 11b, and 11c) in their X-ray crystallographic poses with the enzyme active site that can be built into the electron density of the GABA-AT-Compound 5 X-ray crystal structure. Interaction models were constructed by selecting the amino acid residues within a 5 A radius from the tautomers (11a, 11b, and 11c) and placing them based on their X-ray crystallographic distances. The amino acid residues were simplified to only their side chains, and the PLP moiety of the tautomers was replaced with CH3 groups in their orientations (Supporting Information Figure S8). For consistency Lys329 of all of the models was considered to be in its protonated state. Basis set superposition error (BSSE) corrected gas phase interaction energies of individual residues of each model were calculated using DFT with a large B3LYP/6-311++G(d,2p) basis set (hydrogens were optimized at the same level of theory before the calculation; atomic coordinates and energy values are given in the Supporting Information, Tables S3-S5). The counterpoise technique was employed to estimate the BSSE.
Supplementary Material
Acknowledgments
The authors are grateful to the National Institutes of Health (Grant R01 DA030604 to R.B.S.) for financial support. We would like to thank Catalyst Pharmaceuticals (Coral Cables, FL) and Park Packing Co. (Chicago, IL) for their generosity in providing CPP-115 and fresh pig brains, respectively, for this study. K.T. is grateful to Shionogi & Co., Ltd. for their financial support of his sabbatical leave. This work made use of the IMSERC at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF NNCI-1542205), the State of Illinois, and the International Institute for Nanotechnology (IIN). The authors also acknowledge the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation (NSF) grant number ACI-1548562, for providing the computer resources for the computer modeling studies. X-ray diffraction data collection used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). The authors thank Dr. Joseph Brunzelle at LS-CAT for help on data collection. The authors are grateful to Professor Michael Toney (University of California, Davis) for providing helpful references.
Footnotes
Supporting Information: Additional computational analyses (including principal component analyses) and experimental data, including inhibition plots, off-target activities, NMR spectra, crystallographic data and computational parameters.
References
- 1.Karlsson A, Fonnum F, Malthe-Sorenssen D, Storm-Mathisen J. Biochem Pharmacol. 1974;23(21):3053–3061. doi: 10.1016/0006-2952(74)90281-0. [DOI] [PubMed] [Google Scholar]
- 2.Bormann J. Trends Neurosci. 1988;11(3):112–116. doi: 10.1016/0166-2236(88)90156-7. [DOI] [PubMed] [Google Scholar]
- 3.Bowery NG, Bettler B, Froestl W, Gallagher JP, Marshall F, Raiteri M, Bonner TI, Enna SJ. Pharmacol Rev. 2002;54(2):247–264. doi: 10.1124/pr.54.2.247. [DOI] [PubMed] [Google Scholar]
- 4.Obata K. P Jpn Acad B-Phys Biol Sci. 2013;89(4):139–156. doi: 10.2183/pjab.89.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yogeeswari P, Sriram D, Vaigundaragavendran J. Curr Drug Metab. 2005;6(2):127–139. doi: 10.2174/1389200053586073. [DOI] [PubMed] [Google Scholar]
- 6.Nishino N, Fujiwara H, Noguchi-Kuno SA, Tanaka C. Jpn J Pharmacol. 1988;48(3):331–339. doi: 10.1254/jjp.48.331. [DOI] [PubMed] [Google Scholar]
- 7.Aoyagi T, Wada T, Nagai M, Kojima F, Harada S, Takeuchi T, Takahashi H, Hirokawa K, Tsumita T. Chem Pharm Bull. 1990;38(6):1748–1749. doi: 10.1248/cpb.38.1748. [DOI] [PubMed] [Google Scholar]
- 8.Iversen LL, Bird ED, Mackay AV, Rayner CN. J Psychiatr Res. 1974;11:255–256. doi: 10.1016/0022-3956(74)90099-5. [DOI] [PubMed] [Google Scholar]
- 9.Dewey SL, Morgan AE, Ashby CR, Jr, Horan B, Kushner SA, Logan J, Volkow ND, Fowler JS, Gardner EL, Brodie JD. Synapse. 1998;30(2):119–129. doi: 10.1002/(SICI)1098-2396(199810)30:2<119::AID-SYN1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 10.Waterhouse EJ, Mims KN, Gowda SN. Neuropsychiatr Dis Treat. 2009;5:505–515. doi: 10.2147/ndt.s5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dewey SL, Morgan AE, Ashby CR, Jr, Horan B, Kushner SA, Logan J, Volkow ND, Fowler JS, Gardner EL, Brodie JD. Synapse. 1999;31(1):76–86. doi: 10.1002/(SICI)1098-2396(199901)31:1<76::AID-SYN10>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 12.Gerasimov MR, Ashby CR, Jr, Gardner EL, Mills MJ, Brodie JD, Dewey SL. Synapse. 1999;34(1):11–19. doi: 10.1002/(SICI)1098-2396(199910)34:1<11::AID-SYN2>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 13.Brodie JD, Figueroa E, Laska EM, Dewey SL. Synapse. 2005;55(2):122–125. doi: 10.1002/syn.20097. [DOI] [PubMed] [Google Scholar]
- 14.Brodie JD, Figueroa E, Dewey SL. Synapse. 2003;50(3):261–265. doi: 10.1002/syn.10278. [DOI] [PubMed] [Google Scholar]
- 15.Brodie JD, Case BG, Figueroa E, Dewey SL, Robinson JA, Wanderling JA, Laska EM. Am J Psychiatry. 2009;166(11):1269–1277. doi: 10.1176/appi.ajp.2009.08121811. [DOI] [PubMed] [Google Scholar]
- 16.Wild JM, Chiron C, Ahn H, Baulac M, Bursztyn J, Gandolfo E, Goldberg I, Goni FJ, Mercier F, Nordmann JP, Safran A, Schiefer U, Perucca E. CNS Drugs. 2009;23(11):965–982. doi: 10.2165/11317650-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 17.(a) Silverman RB. Meth Enzymol. 1995;249:240–283. doi: 10.1016/0076-6879(95)49038-8. [DOI] [PubMed] [Google Scholar]; (b) Silverman RB. Mechanism-Based Enzyme Inactivation: Chemistry and Enzymology. I and II. CRC Press; Boca Raton, FL: 1988. [Google Scholar]
- 18.Pan Y, Qiu J, Silverman RB. J Med Chem. 2003;46(25):5292–5293. doi: 10.1021/jm034162s. [DOI] [PubMed] [Google Scholar]
- 19.Silverman RB. J Med Chem. 2012;55(2):567–575. doi: 10.1021/jm201650r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pan Y, Gerasimov MR, Kvist T, Wellendorph P, Madsen KK, Pera E, Lee H, Schousboe A, Chebib M, Brauner-Osborne H, Craft CM, Brodie JD, Schiffer WK, Dewey SL, Miller SR, Silverman RB. J Med Chem. 2012;55(1):357–366. doi: 10.1021/jm201231w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doumlele K, Conway E, Hedlund J, Tolete P, Devinsky O. Epilepsy Behav. 2016;6:67–69. doi: 10.1016/j.ebcr.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee H, Doud EH, Wu R, Sanishvili R, Juncosa JI, Liu D, Kelleher NL, Silverman RB. J Am Chem Soc. 2015;137(7):2628–2640. doi: 10.1021/ja512299n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitz R, Wilson IB. J Biol Chem. 1962;237:3245–3249. [PubMed] [Google Scholar]
- 24.Salminen KA, Leppanen J, Venalainen JI, Pasanen M, Auriola S, Juvonen RO, Raunio H. Drug Metab Dispos. 2011;39(3):412–418. doi: 10.1124/dmd.110.036376. [DOI] [PubMed] [Google Scholar]
- 25.Storici P, De Biase D, Bossa F, Bruno S, Mozzarelli A, Peneff C, Silverman RB, Schirmer T. J Biol Chem. 2004;279(1):363–373. doi: 10.1074/jbc.M305884200. [DOI] [PubMed] [Google Scholar]
- 26.(a) Rishavy MA, Cleland WW. Biochemistry. 2000;39(25):7546–7551. doi: 10.1021/bi000458d. [DOI] [PubMed] [Google Scholar]; (b) Wright SK, Rishavy MA, Cleland WW. Biochemistry. 2003;42(27):8369–8376. doi: 10.1021/bi030092f. [DOI] [PubMed] [Google Scholar]; (c) Toney MD. Biochim Biophys Acta. 2011;1814:1407–1418. doi: 10.1016/j.bbapap.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kirsch JF, Eichele G, Ford GC, Vincent MG, Jansonius JN. J Mol Biol. 1984;174:497–525. doi: 10.1016/0022-2836(84)90333-4. [DOI] [PubMed] [Google Scholar]
- 27.Okumura H, Omote M, Takeshita S. Arzneimittelforschung. 1996;46(5):459–462. [PubMed] [Google Scholar]
- 28.Sarhan S, Knodgen B, Grauffel C, Seiler N. Neurochem Res. 1993;18:539–549. doi: 10.1007/BF00967259. [DOI] [PubMed] [Google Scholar]
- 29.Zigmond E, Ya’acov AB, Lee H, Lichtenstein Y, Shalev Z, Smith Y, Zolotarov L, Ziv E, Kalman R, Le HV, Lu H, Silverman RB, Ilan Y. ACS Med Chem Lett. 2015;6(8):840–844. doi: 10.1021/acsmedchemlett.5b00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanguinetti MC, Tristani-Firouzi M. Nature. 2006;440(7083):463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- 31.Guengerich FP. Chem Res Toxicol. 2008;21(1):70–83. doi: 10.1021/tx700079z. [DOI] [PubMed] [Google Scholar]
- 32.Dewey SL, Chaurasia CS, Chen C-E, Volkow ND, Clarkson FA, Porter SP, Straughter-Moore RM, Alexoff DL, Tedeschi D, Russo NB, Fowler JS, Brodie JD. Synapse. 1997;25(4):393–8. doi: 10.1002/(SICI)1098-2396(199704)25:4<393::AID-SYN11>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 33.Stepan AF, Walker DP, Bauman J, Price DA, Baillie TA, Kalgutkar AS, Aleo MD. Chem Res Toxicol. 2011;24:1345–1410. doi: 10.1021/tx200168d. [DOI] [PubMed] [Google Scholar]
- 34.Gjedde A. Neuromethods. 2014;90:341–364. [Google Scholar]
- 35.Koo YK, Nandi D, Silverman RB. Arch Biochem Biophys. 2000;374(2):248–254. doi: 10.1006/abbi.1999.1623. [DOI] [PubMed] [Google Scholar]
- 36.Silverman RB, Bichler KA, Leon AJ. J Am Chem Soc. 1996;118(6):1241–1252. [Google Scholar]
- 37.Scott EM, Jakoby WB. J Biol Chem. 1959;234(4):932–936. [PubMed] [Google Scholar]
- 38.Yuan H, Silverman RB. Bioorg Med Chem. 2006;14(5):1331–1338. doi: 10.1016/j.bmc.2005.09.067. [DOI] [PubMed] [Google Scholar]
- 39.Juncosa JI, Lee H, Silverman RB. Anal Biochem. 2013;440(2):145–149. doi: 10.1016/j.ab.2013.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patel VD, Lee DE, Alexoff DL, Dewey SL, Schiffer WK. Neuroimage. 2008;41(3):1051–66. doi: 10.1016/j.neuroimage.2008.02.065. [DOI] [PubMed] [Google Scholar]
- 41.Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, Womack T, Bricogne G. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Evans PR, Murshudov GN. Acta Crystallogr D Biol Crystallogr. 2013;69(Pt 7):1204–14. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. Acta Crystallogr D Biol Crystallogr. 2002;58(Pt 11):1948–54. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 44.Emsley P, Cowtan K. Acta Crystallogr D Biol Crystallogr. 2004;60(Sp.Iss.1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 45.Paettersen E, Goddard T, Huang C, Couch G, Greenblatt D, Meng E, Ferrin T. Journal of Comput Chem. 2004;25(13):1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 46.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;13:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 47.Anandakrishnan R, Aguilar B, Onufriev AV. Nucleic Acid Res. 2012;40(W1):W537–541. doi: 10.1093/nar/gks375. ( http://biophysics.cs.vt.edu/H++) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJx. Gaussian 09, Revision A.02. Gaussian, Inc; Wallingford CT: 2016. [Google Scholar]
- 49.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. J Comput Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Banik SD, Chandra A. J Phys Chem B. 2014;118:11077–11089. doi: 10.1021/jp506196m. [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Wang W, Kollman PA, Case DA. J Mol Graph Model. 2006;25(2):247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 52.Sousa Da Silva AW, Vranken WF. BMC Research notes. 2012;5:367. doi: 10.1186/1756-0500-5-367. ( https://doi.org/10.1186/1756-0500-5-367) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alexandra TPC, Ana FST, Maria JR. J Comput Chem. 2013;34:1540–1548. [Google Scholar]
- 54.Pronk S, SzilardPRoland S, Per L, Par B, Rossen A, Michael RS, Jeremy CS, Peter MK, David VD, Berk H, Erik L. Bioinformatics. 2013;29(7):845–854. [Google Scholar]
- 55.Lisa MJ, Jack FK. Biochemistry. 1995;34:3990–3998. [Google Scholar]
- 56.(a) Mueller Stein SA, Loccisano AE, Firestine SM, Evanseck JD. Ann Rep Comput Chem. 2006;2:233–261. [Google Scholar]; (b) Bro R, Smilde AK. Analytical Methods. 2014;6:2812–2831. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.