

ABSTRACT
Lantibiotics are a class of lanthionine-containing, ribosomally synthesized, and posttranslationally modified peptides (RiPPs) produced by Gram-positive bacteria. Salivaricin A2 belongs to the type AII lantibiotics, which are generally considered to kill Gram-positive bacteria by binding to the cell wall precursor lipid II via a conserved ring A structure. Salivaricin A2 was first reported to be isolated from a probiotic strain, Streptococcus salivarius K12, but the structural and bioactivity characterizations of the antibiotic have remained limited. In this study, salivaricin A2 was purified and its covalent structure was characterized. N-terminal analogues of salivaricin A2 were generated to study the importance for bioactivity of the length and charge of the N-terminal amino acids. Analogue salivaricin A2(3-22) has no antibacterial activity and does not have an antagonistic effect on the native compound. The truncated analogue also lost its ability to bind to lipid II in a thin-layer chromatography (TLC) assay, suggesting that the N-terminal amino acids are important for binding to lipid II. The creation of N-terminal analogues of salivaricin A2 promoted a better understanding of the bioactivity of this antibiotic and further elucidated the structural importance of the N-terminal leader peptide. The antibacterial activity of salivaricin A2 is due not only to the presence of the positively charged N-terminal amino acid residues, but to the length of the N-terminal linear peptide.
IMPORTANCE The amino acid composition of the N-terminal linear peptide of salivaricin A2 is crucial for function. Our study shows that the length of the amino acid residues in the linear peptide is crucial for salivaricin A2 antimicrobial activity. Very few type AII lantibiotic covalent structures have been confirmed. The characterization of the covalent structure of salivaricin A2 provides additional support for the predicted lanthionine and methyl-lanthionine ring formations present in this structural class of lantibiotics. Removal of the N-terminal Lys1 and Arg2 residues from the peptide causes a dramatic shift in the chemical shift values of amino acid residues 7 through 9, suggesting that the N-terminal amino acids contribute to a distinct structural conformer for the linear peptide region. The demonstration that the bioactivity could be partially restored with the substitution of N-terminal alanine residues supports further studies aimed at determining whether new analogues of salivaricin A2 for novel applications can be synthesized.
KEYWORDS: bioactivity, covalent structure, lanthipeptide, lantibiotic, salivaricin A2
INTRODUCTION
Lantibiotics are a class of lanthionine-containing, ribosomally synthesized, and posttranslationally modified peptides (RiPPs) with antimicrobial activities and are among the most promising candidates of alternative antibiotics for combatting the emergence of multidrug-resistant bacteria (1). Lanthionine (Lan) and/or methyl-lanthionine (MeLan) rings are formed by the thioether linkage between a cysteine residue and a dehydroalanine (Dha) or dehydrobutyrine (Dhb) residue, which is derived from a serine or threonine residue, respectively, by dehydration (2). Generally, lantibiotics can be divided into two groups based on their structures, i.e., the type A lantibiotics, which are elongated and cationic, and the type B lantibiotics, which have relatively rigid and globular structures (3). Type A lantibiotics are further divided into two subclasses based on their biosynthetic machinery. Type AI lantibiotics form the Lan and MeLan residues by the action of two distinct enzymes, dehydratase LanB (4) and cyclase LanC (5), whereas type AII lantibiotics form the two residues by a bifunctional enzyme, LanM (6, 7). Type AI lantibiotics, including nisin and mutacin 1140 (Fig. 1A), possess a broad spectrum of activity against Gram-positive bacteria. The structure of their conserved rings A and B is the lipid II-binding motif, which targets the pyrophosphate, peptidoglycan MurNAc, and the first isoprene of cell wall precursor lipid II (8, 9). The complex formed between nisin or mutacin 1140 and lipid II is extremely tight. It was previously shown that mutacin and nisin could not competitively compete against each other once the lantibiotic/lipid II complex had been formed (10). Type AII lantibiotics are the largest group of lantibiotics and include the lantibiotics lacticin 481 (11), nukacin ISK-1 (12), mutacin II (13), bovicin HJ50 (14), salivaricin A (15), and salivaricin B (16). This class of lantibiotics generally consists of a linear N-terminal region and a globular C-terminal region composed of three intertwined rings.
FIG 1.
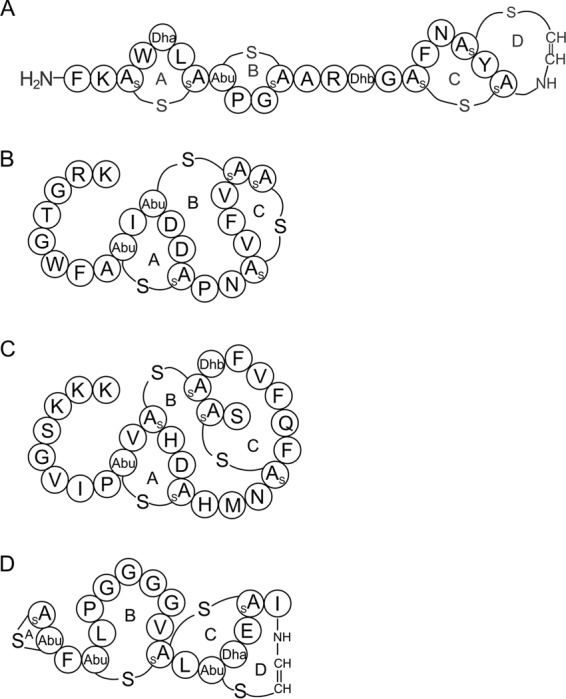
Covalent structures of (A) mutacin 1140, (B) salivaricin A2, (C) nukacin ISK-1, and (D) mersacidin. The lanthionine rings are labeled as rings A, B, C, and D for mutacin 1140 and mersacidin and as rings A, B, and C for salivaricin A2 and nukacin ISK-1. Dehydrated residues from serine and threonine are labeled as Dha and Dhb, respectively. (Methyl-)lanthionine is formed between a cysteine and a dehydrated residue.
The type AII lantibiotic salivaricin A2 (Fig. 1B) was discovered in 2006 and was shown to be active against Micrococcus luteus (17). Later, salivaricin A2 was demonstrated to be encoded at a locus adjacent to that of salivaricin B on a 190-kb megaplasmid in the probiotic strain Streptococcus salivarius K12 (16). However, information about the structure and bioactivity of the lantibiotic is limited. Nukacin ISK-1 (Fig. 1C) is one of the most studied type AII lantibiotics, and its mechanism of action is proposed to be lipid II binding (18). Ring A is believed to be important for lipid II binding, based on a conserved TxS/TxD/EC motif with ring C of the type B lantibiotic mersacidin (Fig. 1D) (19). A previous study showed that mersacidin associates tightly with lipid II micelles and that the addition of isolated lipid II to bacterial culture antagonized the bactericidal activity of the antibiotic (19). The importance of ring A formation, as well as that of a conserved negatively charged aspartic amino acid residue within ring A of nukacin ISK-1, has been shown in a mutagenesis study to be crucial for the antibacterial activity of nukacin ISK-1 (18). Although type AII lantibiotics might share a conserved lipid II-binding motif with type B lantibiotic mersacidin, they do not have any other similarities in their structures. Within the family of type AII lantibiotics, the overall lengths and charges, amino acid composition of rings B and C, and the composition of the N-terminal linear peptide sequence vary significantly (20).
Streptococcus salivarius is a predominant colonizer of oral mucosal surfaces in humans and does not cause disease in healthy individuals (21). Salivaricin A, a type AII lantibiotic consisting of 22 amino acids, is the first characterized lantibiotic produced by Streptococcus salivarius 20P3, and the producing strain is active against most Streptococcus pyogenes strains (15). Salivaricin A2 is a variant of salivaricin A, and it differs from salivaricin A by 2 amino acid substitutions (Ser4Thr and Ile7Phe) (17). Salivaricin A2 was previously isolated from the probiotic strain S. salivarius K12 (BLIS Technologies Ltd., New Zealand) and the m/z value was determined for the peptide by matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) (16). The production and availability of salivaricin A2 from S. salivarius K12 are limited, and the lantibiotic's stability and spectrum of action are still largely unknown. In the same study, the production of salivaricin A2 in the saliva of patients inoculated with the probiotic K12 strain was confirmed by mass spectrometry. However, the study also showed that the presence of salivaricin A2 varied from individual to individual, and thus its effectiveness may vary between patients. It is likely that the production of salivaricin A2 may be the result of a set of complex factors associated with microbial interactions within the oral cavity. Directly using purified salivaricin A2 in the oral cavity may provide better protection against potentially pathogenic strains than using a probiotic-producing strain.
In the present study, salivaricin A2 was purified from Streptococcus salivarius HS0302. We characterized the structure, bioactivity, and stability of the lantibiotic. Solution-phase synthesis was used to generate N-terminal analogues of salivaricin A2 to elucidate the importance of the length and charge of the N-terminal linear peptide for its bioactivity. The study provides a better understanding of salivaricin A2's bioactivity and provides insight into the structural importance of the N-terminal linear peptide.
RESULTS
Isolation and structural characterization of salivaricin A2.
The isolation of salivaricin A2 from S. salivarius HS0302 has enabled studies aimed at understanding its covalent structure and bioactivity. Salivaricin A2 was isolated from a chloroform extraction of culture S. salivarius HS0302 and had a purity greater than 98%, as determined by reversed-phase high-performance liquid chromatography (RP-HPLC) (see Fig. S1 in the supplemental material). The observed mass of salivaricin A2 is in accordance with the predicted mass of 2,368 Da. The truncated analogue salivaricin A2(3-22) was purified using the same method and had the predicted mass of 2,084 Da (Table 1). The previous proposed structure of salivaricin A2 was based on its structural gene sequence, molecular weight, and alignment with other type AII lantibiotics. In this study, we are able to assign the posttranslational modifications (PTMs) present in the peptide. Total correlation spectroscopy (TOCSY) and nuclear Overhauser effect spectroscopy (NOESY) nuclear magnetic resonance (NMR) data on salivaricin A2 and salivaricin A2(3-22) were used to determine the post-translationally modified covalent structure (see Fig. S2, S3, S4, and S5 in the supplemental material). Twenty-two or 20 distinct spin systems for each amino acid were assigned for salivaricin A2 or salivaricin A2(3-22), respectively (see Tables S1 and S2 in the supplemental material). TOCSY and NOESY data sets enabled the sequential assignment of each amino acid spin system. Residues 1 through 9 were identified through an Hαi to HNi + 1 sequential walk (Fig. 2). Residues Abu11 to Asp12, AlaS14 to Val18, and Val20 to AlaS21 were assigned by sequential (i) to (i + 1) NOEs. Residues Abu9 and Abu11 have a distinct chemical shift pattern for a 2-aminobutyric acid moiety of the methyl-lanthionine residue. Residues SAla14, AlaS17, SAla21, and SAla22 have a distinct chemical shift pattern for an alanine moiety of a lanthionine or methyl-lanthionine residue. An HN to Hβ nuclear Overhauser effect (NOE) was detected between residues Abu9 and SAla14, and thus amino acids Abu9 and SAla14 form a methyl-lanthionine residue of ring A. An Hα to Hβ NOE was detected between residues AlaS17 and SAla22, and thus amino acids AlaS17 and SAla22 form a lanthionine residue of ring C. A methyl-lanthionine residue of ring C is believed to be formed between residues Abu11 and SAla21. The ring configuration is supported by Hα to Hβ long-range NOEs between residues Asp12 and AlaS17 or between Asp13 and Phe19. In addition, several other long-range NOEs that support ring B formation were present (Fig. 2). In addition, there were no free thiols detected in the peptide following treatment with CDAP (1-cyano-4-dimethylaminopyridinium tetrafluoroborate) (see Fig. S6 in the supplemental material), which can only happen if residues Abu11 and SAla21 form a methyl-lanthionine residue. The free thiol group from a cysteine residue can be converted to a covalently attached thiocyanate by CDAP, resulting in a 25-Da increase in molecular mass. The mass of the control peptide (SFNSYTC-OH) increased by 25 Da, while salivaricin A2 mass remained unchanged (Table 2), suggesting that all the cysteines within salivaricin A2 are involved in lanthionine or methyl-lanthionine ring formation.
TABLE 1.
Masses of salivaricin A2 analogues determined by ESI-MS
| Salivaricin A2 analogue(s) | Mass (Da) |
|
|---|---|---|
| Expected | Observed | |
| Native | 2,368 | 2,368 |
| A2(3-22) | 2,084 | 2,084 |
| Fmoc-Arg + A2(3-22) | 2,462 | 2,462 |
| Arg + A2(3-22) | 2,240 | 2,240 |
| Fmoc-Lys + A2(3-22) | 2,434 | 2,434 |
| Lys + A2(3-22) | 2,212 | 2,212 |
| Fmoc-Ala-Ala + A2(3-22) | 2,448 | 2,449 |
| Ala + A2(3-22) | 2,155 | 2,156 |
| Ala-Ala + A2(3-22) | 2,226 | 2,226 |
| Lys-Arg + A2(3-22) | 2,368 | 2,368 |
FIG 2.

Summary of the NOEs from the NMR data. Proton interactions are described at the left of each line. The (i + 1) line depicts sequential walk connectivity between adjacent amino acids. The lines (i > 1) depict long-range NOEs between nonadjacent amino acids. The amino acids depicted by the beginning and the end of the line represent the residues that have proton interactions in the NOESY data set. The Abu, SAla, and AlaS abbreviations represent the methyl-lanthionine and lanthionine moieties present in salivaricin A2.
TABLE 2.
Masses of salivaricin A2 and control peptide before and after cyanylation by CDAP
| Substrate | Mass (Da) |
|
|---|---|---|
| Before reaction | After reaction | |
| SFNSYTC | 821 | 846 |
| Salivaricin A2 | 2,368 | 2,368 |
Bioactivity and stability of salivaricin A2.
Compared with the type AI lantibiotic mutacin 1140, which has demonstrated activity against a wide variety of Gram-positive bacteria, salivaricin A2 has a relatively limited spectrum of activity. A small subset of Gram-positive bacteria tested, including M. luteus, Streptococcus pneumoniae, and Corynebacterium spp., were inhibited by the antibiotic (Table 3). Salivaricin A2 was inactive (MIC > 128 μg/ml) against strains of Staphylococcus aureus, Staphylococcus epidermidis, Bacillus subtilis, Bacillus megaterium, and Enterococcus faecalis. The minimum lethal concentration (MLC) of salivaricin A2 was the same as or at most 2-fold higher than its MIC against sensitive strains, indicating that it is a bactericidal antibiotic. The strain of M. luteus was the most sensitive bacterium tested. The strain is often used as an indicator strain for bacteriocin activity. All the Corynebacterium strains were sensitive to salivaricin A2. Salivaricin A2 was active against all six strains of S. pneumoniae tested, but four of the strains were more sensitive to it. Further studies aimed at identifying the differences in the strains may help promote a better understanding of the antibiotic's mechanism of action as well as the differences in sensitivity.
TABLE 3.
MICs and MLCs (μg/ml) for mutacin 1140 and salivaricin A2
| Bacterial strain | Mutacin 1140 |
Salivaricin A2 |
||
|---|---|---|---|---|
| MIC (μg/ml) | MLC (μg/ml) | MIC (μg/ml) | MLC (μg/ml) | |
| M. luteus ATCC 10240 | 0.0625 | 0.0625 | 2 | 2 |
| C. accolens ATCC 49725 | 0.125 | 0.125 | 8 | 8 |
| C. accolens KPL 1818 | 0.125 | 0.125 | 16 | 32 |
| C. accolens KPL 2060 | 0.0625 | 0.0625 | 8 | 16 |
| C. accolens KPL 2061 | 0.25 | 0.25 | 16 | 32 |
| C. pseudodiphtheriticum KPL 1989 | 0.125 | 0.125 | 8 | 8 |
| C. striatum KPL 1959 | 0.5 | 1 | 32 | 32 |
| C. accolens KPL 1855 | 1 | 1 | 16 | 32 |
| S. pneumoniae ATCC 27336 | 0.5 | 0.5 | 32 | 32 |
| S. pneumoniae AI8a | 1 | 1 | 32 | 32 |
| S. pneumoniae AI11a | 1 | 1 | 16 | 16 |
| S. pneumoniae AI14a | 0.125 | 0.125 | 32 | 32 |
| S. pneumoniae AI6 | 1 | 1 | 128 | 128 |
| S. pneumoniae AI7 | 0.25 | 0.25 | 128 | 128 |
| S. mutans JH1140 | 8 | 8 | >128 | >128 |
| S. salivarius HS0302 | 0.5 | 0.5 | 64 | 128 |
| B. subtilis PY79 | 8 | 8 | >128 | >128 |
| B. megaterium ATCC 14581 | 8 | 8 | >128 | >128 |
| S. epidermidis ERIN 30713 | 4 | 8 | >128 | >128 |
| S. aureus ATCC 25923 | 4 | 4 | >128 | >128 |
| E. faecalis ERIN 30002 | 16 | 32 | >128 | >128 |
MICs for the S. pneumoniae strains were tested in the presence of 5% human blood to promote growth.
Compared with mutacin 1140, salivaricin A2 exhibited a nonrapid killing effect against Corynebacterium accolens ATCC 49725 (Fig. 3). During the first 2 h, viable cell counts remained similar for 1× and 2× MIC of salivaricin A2. After 4 h of exposure, at least a 3-fold or 5-fold reduction in viable cells at 1× or 2× MIC, respectively, was observed. At 8 h of exposure, more than a one- or two-log reduction in viable cells was observed following treatment with a 1× or 2× MIC of salivaricin A2, respectively. In comparison to the lantibiotic mutacin 1140, there was at least a log reduction in viable cells following only a 30-min exposure at 1× and 2× MIC. Following a 4-h exposure to mutacin 1140 at 1× and 2× MIC, there was a greater than three-log reduction in viable cell counts. Interestingly, 0.5× MIC of salivaricin A2 inhibited the growth of the bacteria until 18 h, and cells counted at 24 h were more than one log fewer than those in the drugfree control. The 0.5× MIC of mutacin 1140 did not exhibit inhibition effect on growth after 8 h of exposure. Furthermore, the cell density of the subinhibitory concentration of mutacin 1140 cultures was closer to that of the the drugfree control than that of salivaricin A2 at 24 h. Comparing the 0.5× MIC of salivaricin A2 and mutacin 1140, salivaricin A2 appeared to have a more prolonged inhibitory effect at its subinhibitory concentration. The differences in kill kinetics might suggest different mechanisms of action between salivaricin A2 and mutacin 1140.
FIG 3.
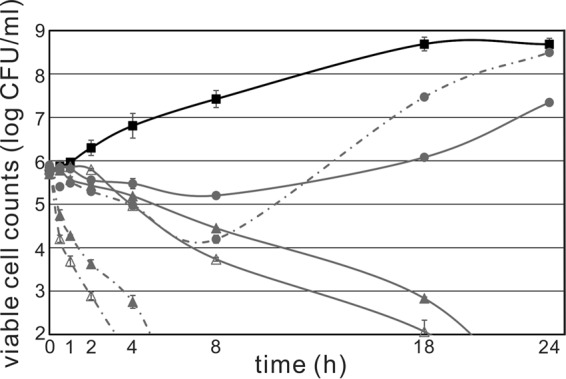
Kill kinetics of salivaricin A2 and mutacin 1140 against C. accolens ATCC 49725. A filled square (■) represents no drug control; a filled circle (●) represents the half-MICs of salivaricin A2 and mutacin 1140; a filled triangle (▲) represents the MICs of salivaricin A2 and mutacin 1140; and an open triangle (△) represents 2× MIC of salivaricin A2 and mutacin 1140. Salivaricin A2 treatments are shown with solid lines, while mutacin 1140 treatments are shown with dashed lines. Some error bars are not visible, since they are smaller than the symbol used.
Salivaricin A2 was stable for at least 1 h at 80°C. The MICs of salivaricin A2 following exposure to temperatures at 50, 60, 70, and 80°C stayed the same as that of the non-heat-treated control (8 μg/ml). In addition to temperature stability testing, the protease stability of salivaricin A2 was tested (see Table S3 in the supplemental material). The protease reaction mixture had no effect on bacterial growth (controls 1 to 4). Pepsin-treated salivaricin A2 had an inhibition effect on bacterial growth similar to that of untreated salivaricin A2, showing that salivaricin A2 was stable following pepsin treatment. Interestingly, trypsin-digested salivaricin A2 did not show any inhibition against bacterial growth. The trypsin digestion mixture was purified by HPLC as described above, and the mass of native and digested salivaricin A2 was determined by electrospray ionization mass spectrometry (ESI-MS) (Table S1 and Fig. S7 and S8 in the supplemental material). The difference in mass between the two compounds was calculated to be 284.20 Da, which is the expected mass of salivaricin A2 minus those of the N-terminal Lys and Arg residues.
N-terminal analogues of salivaricin A2.
The bioactivities of salivaricin A2 N-terminal analogues were determined from an overlay assay in which identical amounts of each analogue and native compound were spotted on the plate containing a lawn of the indicator strain M. luteus ATCC 10240. The bioactivity is expressed in terms of relative activity compared to that of the wild type. A value of 1.0 indicates that the area of the zone of inhibition of the analogue was the same that of as native salivaricin A2. The concentration of each analogue was determined by peak area, using a known amount of native salivaricin A2 for quantification (see Fig. S9 in the supplemental material). Further validation for the purification and quantification came from mass spectrometry data, which showed that the expected and predicted masses for the collected fractions were the same (Table S1 and Fig. S8). Several N-terminal analogues of salivaricin A2 were synthesized and assayed for their bioactivity. The analogues consisted of single Lys, Arg, or Ala substitutions, with and without the 9-fluorenylmethoxy carbonyl (Fmoc) protecting group attached. In addition, Fmoc-Ala1Ala2, Ala1Ala2, Lys1Arg2, and carboxyfluorescein salivaricin A2(3-22) analogs were evaluated (Fig. 4). The salivaricin A2(3-22) and carboxyfluorescein analogues had no activity in the overlay assay. At least a single amino acid has to be added to the N-terminal end to restore some bioactivity. The addition of a single Lys or Arg residue resulted in an area of the zone of inhibition that was approximately half of the wild-type salivaricin A2 inhibition value. Leaving the Fmoc protecting group on the peptide had little effect on the bioactivity of the antibiotic. The addition of a single alanine restored approximately 25% of the inhibition activity, while the addition of the dipeptide Ala1Ala2 residues restored approximately 30% of the inhibition activity. Leaving the Fmoc protecting group on the Ala1Ala2 dipeptide further enhanced the peptide's inhibition activity to approximately 35% of native salivaricin A2 activity. The addition of the Fmoc-Ala1Ala2 group resulted in the loss of all positive charges at the end of the N-terminal leader peptide compared to native compound. The restoration of some of the antibiotic's bioactivity clearly demonstrates the importance of the length of the N-terminal linear peptide for bioactivity. The addition of the Lys1Arg2 residues to the truncated salivaricin A2(3-22) peptide restored the antibiotic back to native salivaricin A2 activity levels (Fig. 4). The positive charge clearly plays a positive role in the activity of the antibiotic. The addition of a single Lys or Arg resulted in approximately a 50% restoration of salivaricin A2 activity compared to only a 25% restoration of activity following the addition of a single Ala residue.
FIG 4.

Bioactivity of N-terminal salivaricin A2 analogues. The activity of each analogue is shown as the ratio of the area of the zone of inhibition of each analogue relative to the area of the zone of inhibition for native salivaricin A2. A value of 1.0 would indicate the same activity as the native compound. M. luteus was used as the indicator strain for antimicrobial activity, and experiments were done in duplicate.
A competition MIC assay was performed to determine if the inactivated analogues can bind to the same target as the native salivaricin A2. MICs of salivaricin A2 against M. luteus remained unchanged when salivaricin A2(3-22) was added at 1×, 2×, and 5× the native salivaricin A2 MIC (i.e., 2 μg/ml). A 5-fold excess of salivaricin A2(3-22) did not shift the MIC value of the native compound, suggesting that the truncated peptide does not bind to the same target in a competitive fashion. The carboxyfluorescein analogue, even though it was shown to not have any inhibitory activity, was also evaluated using the same MIC of the native compound to determine whether it may still bind to the target and have an antagonistic effect. The product did not have any antagonistic activity in the assay. Without any intrinsic inhibitory activity or any antagonistic activity toward the native salivaricin A2, the analogue presumably does not interact with the target. Therefore, fluorescence microscopy studies with the analogue were abandoned.
Salivaricin A2 binds to lipid II with low affinity.
To date, no direct evidence for type AII lantibiotic binding to lipid II has been reported. A thin-layer chromatography (TLC) affinity assay was performed and confirmed the binding of salivaricin A2 to lipid II (Fig. 5 and Fig. S10 in the supplemental material). Peptides like mutacin 1140 and salivaricin A2 do not migrate from where they are spotted on the TLC plate (the origin) and they do not stain with iodine vapors. Lipid II and vancomycin will migrate from the origin and are stained with iodine vapors. Salivaricin A2 could not retain lipid II at the origin when the molar ratio of salivaricin A2 to lipid II was 12:1, but could retain lipid II at the origin when the molar ratio was increased to 24:1 (Fig. 5 and Fig S10). Mutacin 1140, the positive control, can retain lipid II at molar ratios of 3:1 and 12:1; the latter gives a more intense stain at the origin. This represents at least an 8-fold difference in the ability to retain lipid II at the origin. The retention of lipid II at the origin is due to the formation of a complex with salivaricin A2 or mutacin 1140 that prevents its migration on the TLC plate. The difference between salivaricin A2 and mutacin 1140 in their binding affinity to lipid II might partially explain why salivaricin A2 is not as active as mutacin 1140. The truncated analogue salivaricin A2(3-22) lost the ability to retain lipid II even at a molar ratio of 24:1. The loss of lipid II binding may explain why the analogue has no bioactivity and further explain why the analogue does not have any antagonistic activity against the native compound. Vancomycin, used as a negative control in this experiment, is known to bind to the d-Ala-d-Ala moiety of lipid II, but is capable of migrating from the origin (see Fig. S10). As predicted, vancomycin failed to retain lipid II at the origin.
FIG 5.

Origins of the TLC assay. (A) Lane 1: mutacin 1140 and lipid II, 12:1 ratio; lane 2: salivaricin A2 and lipid II, 12:1 ratio; lane 3: salivaricin A2 and lipid II, 24:1 ratio; lane 4: vancomycin and lipid II, 12:1 ratio; lane 5: digested salivaricin A2 and lipid II, 24:1 ratio. (B) Lane 1: lipid II; lane 2: mutacin 1140; lane 3: salivaricin A2; lane 4: vancomycin; lane 5: digested salivaricin A2. Binding of lipid II keeps lipid II at the origin. Panel B serves as negative controls, showing no staining of the peptides or lipid II at the origin. Lipid II and mutacin 1140 were used as a positive control for retaining lipid II at the origin, while lipid II and vancomycin were used as a negative control.
Further analyses of the interactions between salivaricin A2 and lipid II that prevent the migration of lipid II on the TLC plate will be required to understand the molecular interactions between the two compounds. The loss of lipid II binding following the removal of Lys1Arg2, as well as the loss in bioactivity, is supported by an unexpected change in the structure of the linear N-terminal peptide region that is observed in the NMR data. The spectral width of the fingerprint region became narrower in the truncated salivaricin A2(3-22) analogue, and residues Phe7 and Ala8 at the end of the linear peptide region and residue Abu9 at the beginning of ring A had dramatic chemical shift changes. Phe7, Ala8, and Abu9 amide proton frequency shifted by 0.16, 0.80, and 0.24 ppm, respectively (Fig. 6). The dramatic changes in chemical shift values of these residues following the removal of Lys1Arg2 suggest that the linear peptide region is structured and that this structure is lost when the terminal amino acids are removed. Residues Gly3, Thr4, and Gly5 also exhibited dramatic amide proton chemical shifts, but these residues are structurally in the vicinity of the charged amino acids, and the absence of Lys1Arg2 may account for those observed changes. However, residues Phe7, Ala8, and Abu9 would be sufficiently far away in an unstructured peptide such that the removal of Lys1Arg2 should not have a dramatic effect on the amide proton chemical shift values. These changes in the NMR spectra support the loss of a defined structure in the native compound, and the structural change may account for the loss of salivaricin A2's bioactivity and lipid II binding.
FIG 6.

Overlay of TOCSY fingerprint region (NH correlations). The NH correlations of the wild-type sample are shown in black, while the NH correlations for salivaricin A2(3-22) are shown in red. The NH correlation for Trp6 is shown in green, since the residue did not have a significant chemical shift difference between the two samples. Dramatic chemical shifts are observable for all other residues, in particular for Ala8.
DISCUSSION
In this study, we identified a novel strain, named HS0302, of Streptococcus salivarius, which enabled the isolation of salivaricin A2 for bioactivity and structural characterization. NMR analyses confirmed the predicted lanthionine ring configurations within the peptide. The spectrum of activity and the inhibitory activity of salivaricin A2 are narrower and less potent, respectively, compared to the bioactivity of mutacin 1140. Salivaricin A2 was shown to bind and prevent the migration of lipid II on a TLC plate, albeit at a higher molar ratio than that observed for mutacin 1140. A kill kinetics study revealed that salivaricin A2 is bactericidal against C. accolens ATCC 49725, but its kinetics of cell death is slow compared to that of mutacin 1140. Salivaricin A2 was inactivated by trypsin digestion, which yielded the salivaricin A2(3-22) product. Bioactivity could be restored by increasing the length of the N-terminal peptide with alanines, but full bioactivity was only restored when the positively charged amino acids Lys1Arg2 were added to restore the native structure.
S. salivarius colonizes mucosal surfaces within the oral cavity. The S. salivarius K12 strain has been the subject of an immense amount of research demonstrating its use as a probiotic in healthy individuals (22–26). BLIS K12 is now found in a wide array of probiotic supplements distributed worldwide. Salivaricin A2 is one documented bacteriocin produced by this strain that may contribute to its success as a probiotic. The mass and covalent structure of our isolated salivaricin A2 in this study match the mass of salivaricin A2 that has been previously reported (16). Although a covalent structure of salivaricin A2 can be proposed by sequence alignment with other type AII lantibiotics that have been characterized by NMR, including lacticin 481 (27), mutacin II (13), and bovicin HJ50 (20), direct evidence is still needed to confirm its PTMs and lanthionine ring formations. Salivaricin A2 is five amino acids shorter than lacticin 481 and mutacin II and differs significantly in amino acid composition for its C-terminal rings B and C, while bovicin HJ50 contains a rare disulfide bond and is 11 residues longer than salivaricin A2. In this study, NMR studies provided direct observation of the residues involved in forming rings A and C. In addition, several long-range NOEs, in combination with data supporting the absence of free cysteine sulfhydryl groups, support the formation of ring B (Fig. 1). The efficient removal of terminal Lys and Arg residues enabled a direct comparison between the structures of native and truncated compounds. The amide proton spectral width of the linear N-terminal amino acids was narrower with the salivaricin A2(3-22) product compared to the native product, and several amino acids within this region underwent large chemical shift changes. The data suggest that the N-terminal portion of salivaricin A2 is a structured component of the lantibiotic, which is in contrast with a report suggesting that the N-terminal linear peptide is unstructured (20). Our study supports a structural component for the linear N-terminal peptide region that is crucial for the bioactivity of salivaricin A2.
The spectrum of activity of salivaricin A2 appears to be limited among Gram-positive bacteria. Salivaricin A2 is active at micromolar concentration against tested M. luteus, C. accolens, and S. pneumoniae strains, while it did not show any inhibitory activity against other tested Gram-positive bacterial species. Lipid II-binding antibiotics, i.e., mutacin 1140 (28), nisin (29), and mersacidin (30), normally have good activity against Staphylococcus species. However, salivaricin A2 had no activity against tested S. aureus or S. epidermidis strains. Possibly, salivaricin A2's lower affinity to lipid II as determined by TLC assay, in combination with a possible lower efficiency of navigating the cell envelope to reach the lipid II target, is the basis for its reduced activity against Gram-positive bacteria. However, in the kill kinetics assay against a susceptible C. accolens strain, the rate of cell death at inhibitory concentrations is low compared to that of the lipid-II-binding antibiotic mutacin 1140. The differences in the rates of killing as well as the narrower spectrum of activity suggest that salivaricin A2 may have another mechanism of action, or differences in cell envelope may prevent the antibiotic's access to lipid II. Additional studies will be required to further understand the differences in activity.
The structure of mersacidin (Fig. 1D) bound to lipid II has been solved in dodecyl phosphocholine (DPC) micelles by NMR (31). When bound to lipid II, mersacidin alters its backbone structure through its hinge region between Ala12 and Abu13 and exposes the amino group of Lys1 and carboxyl group of Glu17 to the lipid II compound, enabling its interaction. Therefore, it was proposed that the side chain of the Glu residue within ring C is part of the lipid II-binding motif of mersacidin. Recently, the lipid II-binding motif has been solved for the two-component lantibiotic lacticin 3147 (32). The C terminus of LtnA1, which has a high homology to mersacidin, was shown to form a “pyrophosphate cage” with lipid II in a 2:1 ratio. Ring A of type AII lantibiotics has been suggested to be the lipid II-binding motif based on its sequence similarity with ring C of mersacidin. The importance of ring A formation of type AII lantibiotics has been demonstrated for nukacin ISK-1 (18, 33). However, it is not clear from these studies whether the amino acid substitutions were the basis for the loss of activity or whether the point mutations interfered with PTMs like the formation of the lanthionine ring A. Either way, the study did demonstrate the structural importance of ring A for its bioactivity. The importance of N-terminal positively charged residues of nukacin ISK-1 has also been studied. In one site-saturation mutagenesis study (33), each of the first three Lys residues was replaced by Gly or Ser, resulting in mutants with activity similar to that of the wild-type strain, and each of the first two Lys residues can be replaced by Ala or Leu or Asn residues with similar activity. However, one negatively charged Glu or Asp residue in these three positions resulted in an inactive mutant. This study indicates that residues 1 to 3 are individually variable and the loss of one positive charge is well tolerated, but it is not clear if more than one positively charged residue can be replaced. Moreover, the overall bioactivity of the mutant strains is also influenced by the productivity of lantibiotic, and thus the bioactivity of purified nukacin ISK-1 analogues is not clear. In another nukacin ISK-1 study (34), replacement of Lys1Lys2Lys3 with Ala1Ala2Ala3 resulted in a 32-fold decrease in activity, which was the same activity as that of the truncated peptide without the N-terminally charged amino acids. The positive charged amino acids are crucial for nukacin ISK-1 bioactivity, while in our study we also show that the positive charged amino acids improve bioactivity. However, we also show that alanine substitutions can restore bioactivity and that the positive charged amino acids are not absolutely crucial for salivaricin A2 activity. The reason for the differences in observations is not clear. Positively charged residues within the N-terminal linear peptide of bovicin HJ50 were suggested to not be essential for its bioactivity (20). The function of the N-terminal region of type AII lantibiotics still remains unclear and is only exacerbated by the variability in length and amino acid composition of the N-terminal region within type AII lantibiotics. Taken together with our observation that removal of N-terminal Lys-Arg resulted in dramatic amide chemical shifts in several residues and the loss of lipid II-binding activity, our study shows that ring A is not the only requirement for lipid II-binding and bioactivity. The length of the linear peptide may promote the formation of a structured linear peptide. Given the dramatic changes in chemical values for Phe7Ala8 and Abu9, we believe that the length and the positive charges within Lys-Arg may further facilitate the interaction with the negatively charged Asp residues in ring A, but more evidence is needed to confirm this hypothesis.
In summary, the study furthered our understanding of the bioactivity of salivaricin A2 and provided a complete characterization of its covalent structure. The study also provides further evidence that the N-terminal portion of type AII lantibiotics is crucial for their bioactivity. Salivaricin A2 is capable of binding to lipid II, but given the antibiotic's limited spectrum of activity against Gram-positive bacteria and the slow killing of susceptible bacteria compared to that of another lipid II-binding antibiotic, mutacin 1140, there is still much to learn about the compound's inhibitory activity. Future studies on salivaricin A2 will be done to further evaluate whether lipid II is the antibiotic's biological target and to determine the basis for the reduced spectrum of activity against Gram-positive bacteria. Future studies will be aimed at characterizing the mechanism of action or possible actions of salivaricin A2.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Streptococcus salivarius HS0302 was isolated as a contaminant on a Todd-Hewitt yeast extract (THY) agar plate (containing 30 g/liter Todd-Hewitt broth, 3 g/liter yeast extract, and 15 g/liter agar). The strain was identified to be S. salivarius by 16S rRNA alignment (see Fig. S11 in the supplemental material). Bacterial strains used in this study are listed in Table 4. Streptococcus salivarius HS0302, M. luteus ATCC 10240, Bacillus subtilis PY79, Bacillus megaterium ATCC 14581, Staphylococcus epidermidis ERIN 30713, Enterococcus faecalis ERIN 30062, Staphylococcus aureus ATCC 25923, S. pneumoniae ATCC 27336, and S. pneumoniae AI6 and AI7 were cultured in THY (containing 30 g/liter Todd-Hewitt broth and 3 g/liter yeast extract) or on THY agar plates. All Corynebacterium species strains are fatty acid synthase-deficient, and the THY broth or agar was supplemented with 1% of Tween 80 to promote bacterial growth. S. pneumoniae AI8, AI11, and AI14 were cultured in THY broth supplemented with 5% human blood, or on blood agar plates. All bacterial strains were grown at 37°C.
TABLE 4.
Bacterial strains used in this study
| Strain | Description | Reference and/or sourcea |
|---|---|---|
| S. salivarius HS0302 | Producing strain for salivaricin A2 | This study |
| M. luteus ATCC 10240 | Indicator strain for overlay assay | ATCC |
| S. aureus ATCC 25923 | ATCC | |
| S. mutans JH1140 (ATCC 55676) | Producing strain for mutacin 1140 | ATCC |
| S. pneumoniae ATCC 27336 | ATCC | |
| S. pneumoniae AI6 | MSU | |
| S. pneumoniae AI7 | MSU | |
| S. pneumoniae AI8 | MSU | |
| S. pneumoniae AI11 | MSU | |
| S. pneumoniae AI14 | MSU | |
| C. accolens ATCC 49725 | ATCC | |
| C. accolens KPL 1818 | FI, 35 | |
| C. accolens KPL 2060 | FI, 35 | |
| C. accolens KPL 2061 | FI, 35 | |
| C. pseudodiphtheriticum KPL 1989 | FI, 35 | |
| C. striatum KPL 1959 | FI, 35 | |
| C. accolens KPL 1855 | FI, 35 | |
| B. subtilis PY79 | 37 | |
| B. megaterium ATCC 14581 | ATCC | |
| S. epidermidis ERIN 30713 | BCM | |
| E. faecalis ERIN 30002 | BCM |
MSU, Department of Pathobiology and Population Medicine at Mississippi State University; FI, Department of Microbiology at the Forsyth Institute; BCM, Department of Molecular Virology and Microbiology at Baylor College of Medicine.
Purification of salivaricin A2 from S. salivarius HS0302.
Purification of salivaricin A2 was performed as previously described for mutacin 1140 (38). Briefly, S. salivarius strain HS0302 was grown in a modified THY soft agar medium containing 30 g/liter Todd Hewitt broth, 3 g/liter yeast extract, 1 g/liter KH2PO4, 0.1 g/liter K2HPO4, 0.3 g/liter MgSO4, 0.005 g/liter FeSO4, 0.005 g/liter MnSO4, and 0.3% agar. The medium was stab inoculated with S. salivarius HS0302 and incubated at 37°C for 3 days. Following incubation, the soft agar culture was frozen at −80°C overnight. The culture was thawed the next day at 65°C in an oven. The liquified soft agar was centrifuged at 20,000 × g for 30 min at 4°C in 250-ml centrifuge bottles to remove the agar and cellular debris (Beckman J2-21 centrifuge). The supernatant was mixed with an equal volume of chloroform and shaken vigorously. The mixture was again centrifuged at 20,000 × g at 4°C for 30 min. After centrifugation, the interface between the aqueous and chloroform layers was collected and allowed to air dry overnight. The dried sample was suspended in 20 ml per liter extraction of 35% acetonitrile (ACN)/water (vol/vol) with 0.1% trifluoroacetic acid (TFA) and subsequently centrifuged at 20,000 × g for 10 min. The supernatant was collected and passed through a 0.22-μm filter before loading on a Bio-Rad Duoflow chromatography system with either a semiprep C18 column (Agilent Zorbax, octyldecyl silane [ODS], 5 μm, 10 mm × 250 mm; Agilent Technologies, Santa Clara, CA) or an analytical column (C18, 5 μm, 4.6 mm × 250 mm) (38). All solvents for HPLC contained 0.1% TFA. The sample was separated through a water/acetonitrile gradient starting from 90% to 70% water over 30 min and an isocratic flow at 70% water for 7 min, followed by a linear gradient from 70% to 10% water over 25 min. Salivaricin A2 eluted from the column at ∼48% water. Each HPLC fraction was collected and dried. The molecular weight of the collected product was determined by electrospray ionization mass spectrometry (ESI-MS) on a ThermoFisher DecaXP ion trap mass spectrometer in positive mode. Briefly, around 1 μg of dried sample was resuspended in 100 μl of 50% ACN/water (vol/vol) and applied by direct infusion in positive mode.
Determination of the covalent structure of salivaricin A2.
CDAP (1-cyano-4-dimethylaminopyridinium tetrafluoroborate) and TCEP [Tris (2-carboxyethyl)phosphine] were used to determine free thiols (39). The reaction was used to determine whether salivaricin A2 has any free thiols from cysteine residues, as previously described (40). Briefly, CDAP was dissolved in 0.1 N hydrochloric acid. Peptide (SFNSYTC-OH) was used as a positive control. The reaction mixtures were purified by ZipTip (Pierce C18 tips, product 87782; ThermoFisher Scientific) and confirmed by ESI-MS on a ThermoFisher DecaXP ion trap mass spectrometer in positive mode.
NMR analysis of salivaricin A2 was performed as described previously (41). Salivaricin A2 was dissolved in deuterated dimethyl sulfoxide (DMSO-d6) and the NMR data were collected on a Bruker Advance III-HD spectrometer operating at a proton frequency of 850 MHz, using a triple resonance CryoProbe (TCI). The 1H resonances were assigned according to standard methods (42) using correlation spectroscopy (COSY), TOCSY (43), NOESY (44) and 13C-HSQC (heteronuclear single quantum coherence) spectroscopy (45) experiments. NMR experiments were collected at 25°C. The TOCSY experiment was acquired with a 60-ms mixing time using the Bruker DIPSI-2 spinlock sequence. The NOESY experiment was acquired with 400- and 500-ms mixing times. Phase-sensitive indirect detection for NOESY, rotating-frame nuclear Overhauser effect spectroscopy (ROESY), TOCSY, and COSY experiments was achieved using the standard Bruker pulse sequences. Peaks were assigned using NMRView software (46).
Bioactivity and kill kinetics.
Determination of MIC was performed following a modified version of the broth microdilution method described in standard M07-A8 by the Clinical and Laboratory Standards Institute (47). The purified salivaricin A2 was dried and weighed on an analytical balance (Adventurer Pro AV114C; Ohaus Corporation, USA) and suspended in 35% acetonitrile with 0.1% TFA. The MIC is the lowest concentration of compound that inhibits the visible growth of the bacteria after 24 h of incubation at 37°C. The MLC was determined by plating 50 μl of culture medium on THY plates at 2× and 1× of the determined MIC. The concentration in which no CFU formed after 24 h was determined to be the MLC. A type AI lantibiotic, mutacin 1140, was used as a comparison. The MICs and MLCs were determined against M. luteus ATCC 10240, S. aureus ATCC 25923, S. salivarius HS0302, Bacillus subtilis PY79, Bacillus megaterium ATCC 14581, Staphylococcus epidermidis ERIN 30713, Enterococcus faecalis ERIN 30002, 6 strains of S. pneumoniae, and 7 strains of Corynebacterium spp. Experiments were done in triplicate.
The bactericidal activity of salivaricin A2 was further elucidated using a time-kill study against C. accolens ATCC 49725 and compared to that of mutacin 1140. The preparation of inoculum was based on the CFU counted at 48 h. The bacterium was inoculated onto fresh THY broth containing 1% Tween 80 and allowed to grow to an optical density at 600 nm (OD600) of 0.6. The culture was then diluted to an OD600 of 0.132, after which a 15-fold dilution was performed. The dilution resulted in a 106 CFU/ml concentration of cells. A 5-ml aliquot of the prepared inoculum was added to 5 ml of the growth medium, resulting in an initial 5 × 105 CFU/ml cell density. The growth medium was supplemented with salivaricin A2 or mutacin 1140 to yield a final concentration equivalent to its 0.5×, 1×, or 2× MIC. At the initial predicted 5 × 105 CFU/ml cell density, the MICs of salivaricin A2 and mutacin 1140 were determined to be 16 μg/ml and 0.125 μg/ml, respectively. Cultures without antibiotic supplementation were used as a negative control. The 50-ml tubes containing 10 ml of the culture were placed in a shaking incubator at 37°C and 200 rpm (Stuart orbital incubator SI500). For each sample, 100 μl of the culture was removed at time points of 0 h, 0.5 h, 1 h, 2 h, 4 h, 8 h, 18 h, and 24 h. The culture was 10× serially diluted in fresh medium before being spread onto a THY agar plate supplemented with 1% Tween 80. The cultures were allowed to grow for 48 h at 37°C before the CFU were calculated. Plates with around 30 to 300 colonies were counted. CFU/ml for each time point at each drug concentration was calculated and all experiments were done in triplicate.
Lipid II binding assay.
A lipid II binding assay using a thin-layer chromatography (TLC) procedure was performed as previously described (48). Briefly, the mobile solvent used consisted of butanol-acetic acid-water-pyridine in a 15:3:12:10 volume ratio. The reaction mixture was made in 10 μl of mobile solvent consisting of 0.2 mM lipid II and 0.6 mM, 2.4 mM, or 4.8 mM peptide in glass vials. Peptides included native salivaricin A2 and salivaricin A2(3-22). Mutacin 1140 and vancomycin were used as positive and negative controls, respectively. Vancomycin binds to lipid II, but also migrates from the origin on the TLC plate. Additional controls included lipid II and each peptide spotted separately. The reaction mixture was incubated for 2 h at room temperature before spotting approximately 10 μl on the TLC plate. The spotted samples were allowed to air dry before the plate was inserted into a chamber containing the mobile solvent. The mobile solvent was allowed to run until it reached approximately 2 cm from the top of the plate. The plate was removed and air dried before being stained in an iodine chamber for visualization.
Stability assays.
The protease stability of salivaricin A2 was tested as previously described with minor modifications (49). Briefly, for the trypsin treatment, a 2× stock solution of sodium phosphate buffer (0.134 M, pH 7.6) and a 10× trypsin stock solution (5.19 mg of trypsin in 1 ml of 1 mM HCl solution) were used. For the pepsin treatment, a 2× stock solution of 20 mM HCl and a 10× pepsin stock solution (0.8 mg of pepsin in 1 ml double-distilled water [ddH2O]) were used. Each reaction mixture consisted of 200 μl 10× protease stock solution, 1 ml 2× stock solution, 780 μl ddH2O, and 20 μl DMSO. Then, 100 μl of the reaction mixture was aliquoted into each 1.8-ml centrifuge tube (catalog no. 89000-028; VWR). Using a prepared 10-μg/μl stock solution of salivaricin A2 in 35% acetonitrile, 64 μg of salivaricin A2 was added to each tube and incubated for 4 h (pepsin) or 0.5 h (trypsin) at 37°C. After the specified incubation period, a 5 × 105 CFU/ml cell density inoculum of C. accolens ATCC 49725 was made as described above, and 900 μl of the inoculum was added to each of the tubes to give a final volume of 1 ml. The final sample concentration of salivaricin A2 was 4× MIC. All tubes were incubated at 37°C for 4 h, and viable cells were determined by a serial dilution as described above. The following controls were used: 100 μl of reaction mixture containing the protease inhibitor phenylmethylsulfonyl fluoride (PMSF), 100 μl reaction mixture, 100 μl reaction mixture without protease, 100 μl THY broth with 64 μg salivaricin A2, and 100 μl of THY broth. All experiments were done in duplicate.
Temperature stability experiments were performed as previously described (49) with some modifications. First, 12.8 μg of salivaricin A2 was dried in 1.8-ml centrifuge tubes before being suspended in 10 μl of 0.1 M sodium phosphate buffer at pH 7.0. The samples were incubated at room temperature, 50°C, 60°C, 70°C, and 80°C for 1 h. MIC assays were done against C. accolens ATCC 49725 in duplicate, as previously described, to determine whether the antibiotic was stable at the tested temperatures.
Synthesis and characterization of the N-terminal analogues of salivaricin A2.
The first two amino acids at the N terminus of salivaricin A2 were cleaved after 30 min of treatment with trypsin, and salivaricin A2(3-22) was purified using the purification method described above by HPLC on an analytical column (Agilent Zorbax, OFD, C18, 5 μm, 2.1 mm x 250 mm). The sample was separated through a linear water/ACN gradient from 90% to 20% water over 30 min. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) was used as a carboxyl-activating agent for the coupling of primary amines to yield amide bonds as previously reported (9, 36) with some minor modifications. The EDC coupling reaction mixture consisted of 100 μl dimethylformamide (DMF), 100 nmol salivaricin A2(3-22), 500 nmol compound with one free carboxyl group, 500 nmol EDC, and 500 nmol HOAt (1-hydroxy-7-azabenzotriazole). Compounds included Boc-Ala-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Arg-OH, Fmoc-Lys-OH, Boc-Ala-Ala-OH, and Fmoc-Ala-Ala-OH. All compounds were obtained from Sigma, except for Fmoc-Ala-Ala-OH, which was obtained from Peptides & Elephants (Germany). The reaction mixtures were incubated overnight at room temperature, except for Boc-Ala-OH, which was incubated for only 4 h. The Fmoc group was removed by adding piperidine to a final concentration of 20% (vol/vol) and incubating for 30 min. The Boc group was readily removed by adding 35% acetonitrile/water (vol/vol) with 0.1% TFA. The Pbf group was removed by adding excess 10% TFA/water (vol/vol) and incubating at room temperature for 2 h. To restore the native structure of salivaricin A2, Fmoc-Arg(Pbf)-OH was coupled to salivaricin A2(3-22), followed by Fmoc deprotection and ice-cold ether precipitation. The precipitated peptide was dried on a SpeedVac (catalog no. 7810010; Labconco) and was used for the second coupling reaction with Fmoc-Lys(Boc)-OH. Twice the reaction volume of ice-cold ether was added to each of the reaction samples, and the samples were stored on ice for 2 h before centrifuging at 20,000 × g (Eppendorf centrifuge 5424) for 10 min. The pellet from each sample was brought up to a final volume of 1 ml of 35% ACN with 0.1% TFA and run on a C18 RP-HPLC analytical column with a modified gradient as described above. HPLC fractions were collected between 50 to 30% water and were analyzed by ESI-MS as described above. Each fraction with desired mass was rerun on the HPLC column. The amount of each sample was quantified by comparing the peak area of each sample to the peak area of a standard amount of salivaricin A2.
An antagonism assay was used to determine the bioactivity of N-terminal analogues of salivaricin A2 as previously described with minor modifications (38). Briefly, the indicator strain M. luteus was inoculated onto 5 ml of THY, grown to an OD600 of 0.6, and diluted to an OD600 of 0.2. Subsequently, 400 μl of bacterial culture was added to every 10 ml of molten THY top agar (0.75% agar) that was below 55°C, which was poured over the surface of each THY agar plate. After solidification, 10 μg, 5 μg, and 2.5 μg of native salivaricin A2 and salivaricin A2 analogues were spotted onto the plate. Once the spots were dry, the plates were inverted and incubated overnight at 37°C. The area of zone of inhibition of 10 μg of each analogue against M. luteus in overlay assay was measured and compared with 10 μg of native salivaricin A2. All of the antagonism bioassay experiments were done in duplicate.
Supplementary Material
ACKNOWLEDGMENTS
We would like to acknowledge Rama Krishna and the support of the UAB Cancer Center NMR Shared Facility. We thank Katherine Lemon, Department of Microbiology at the Forsyth Institute, for the use of her Corynebacterium strains and we also would like to thank Robert Britton, Department of Molecular Virology and Microbiology at Baylor College of Medicine, for use of his Enterococcus faecalis and Staphylococcus epidermidis strains. We also thank Eefjan Breukink, Department of Chemistry at Utrecht University, for the lipid II.
We have no conflicts of interest to declare.
The NMR experiments were performed at the UAB Cancer Center High-Field NMR Facility.
The NMR experiments were supported by the NIH grants 1P30 CA-13148 and 1S10 RR022994-01A1.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02528-17.
REFERENCES
- 1.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Cotter PD. 2013. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Product Rep 30:108–160. doi: 10.1039/C2NP20085F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chatterjee C, Paul M, Xie L, van der Donk WA. 2005. Biosynthesis and mode of action of lantibiotics. Chem Rev 105:633–684. doi: 10.1021/cr030105v. [DOI] [PubMed] [Google Scholar]
- 3.Jung G. 1991. Lantibiotics—ribosomally synthesized biologically active polypeptides containing sulfide bridges and α,β-didehydroamino acids. Angew Chem Int Ed 30:1051–1068. doi: 10.1002/anie.199110513. [DOI] [Google Scholar]
- 4.Kluskens LD, Kuipers A, Rink R, de Boef E, Fekken S, Driessen AJ, Moll GN. 2005. Post-translational modification of therapeutic peptides by NisB, the dehydratase of the lantibiotic nisin. Biochemistry 44:12827–12834. doi: 10.1021/bi050805p. [DOI] [PubMed] [Google Scholar]
- 5.Rink R, Kluskens LD, Kuipers A, Driessen AJ, Kuipers OP, Moll GN. 2007. NisC, the cyclase of the lantibiotic nisin, can catalyze cyclization of designed nonlantibiotic peptides. Biochemistry 46:13179–13189. doi: 10.1021/bi700106z. [DOI] [PubMed] [Google Scholar]
- 6.Uguen P, Le Pennec JP, Dufour A. 2000. Lantibiotic biosynthesis: interactions between prelacticin 481 and its putative modification enzyme, LctM. J Bacteriol 182:5262–5266. doi: 10.1128/JB.182.18.5262-5266.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thibodeaux CJ, Wagoner J, Yu Y, van der Donk WA. 2016. Leader peptide establishes dehydration order, promotes efficiency, and ensures fidelity during lacticin 481 biosynthesis. J Am Chem Soc 138:6436–6444. doi: 10.1021/jacs.6b00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu STD, Breukink E, Tischenko E, Lutters MA, de Kruijff B, Kaptein R, van Nuland NA. 2004. The nisin–lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nat Struct Mol Biol 11:963–967. doi: 10.1038/nsmb830. [DOI] [PubMed] [Google Scholar]
- 9.Hasper HE, Kramer NE, Smith JL, Hillman JD, Zachariah C, Kuipers OP, Breukink E. 2006. An alternative bactericidal mechanism of action for lantibiotic peptides that target lipid II. Science 313:1636–1637. doi: 10.1126/science.1129818. [DOI] [PubMed] [Google Scholar]
- 10.Smith L, Hasper H, Breukink E, Novak J, Čerkasov J, Hillman JD, Orugunty RS. 2008. Elucidation of the antimicrobial mechanism of mutacin 1140. Biochemistry 47:3308–3314. doi: 10.1021/bi701262z. [DOI] [PubMed] [Google Scholar]
- 11.Piard JC, Kuipers OP, Rollema HS, Desmazeaud MJ, de Vos WM. 1993. Structure, organization, and expression of the lct gene for lacticin 481, a novel lantibiotic produced by Lactococcus lactis. J Biol Chem 268:16361–16368. [PubMed] [Google Scholar]
- 12.Sashihara T, Kimura H, Higuchi T, Adachi A, Matsusaki H, Sonomoto K, Ishizaki A. 2000. A novel lantibiotic, nukacin ISK-1, of Staphylococcus warneri ISK-1: cloning of the structural gene and identification of the structure. Biosci Biotechnol Biochem 64:2420–2428. doi: 10.1271/bbb.64.2420. [DOI] [PubMed] [Google Scholar]
- 13.Krull RE, Chen P, Novak J, Kirk M, Barnes S, Baker J, Caufield PW. 2000. Biochemical structural analysis of the lantibiotic mutacin II. J Biol Chem 275:15845–15850. doi: 10.1074/jbc.275.21.15845. [DOI] [PubMed] [Google Scholar]
- 14.Xiao H, Chen X, Chen M, Tang S, Zhao X, Huan L. 2004. Bovicin HJ50, a novel lantibiotic produced by Streptococcus bovis HJ50. Microbiology 150:103–108. doi: 10.1099/mic.0.26437-0. [DOI] [PubMed] [Google Scholar]
- 15.Ross KF, Ronson CW, Tagg JR. 1993. Isolation and characterization of the lantibiotic salivaricin A and its structural gene salA from Streptococcus salivarius 20P3. Appl Environ Microbiol 59:2014–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hyink O, Wescombe PA, Upton M, Ragland N, Burton JP, Tagg JR. 2007. Salivaricin A2 and the novel lantibiotic salivaricin B are encoded at adjacent loci on a 190-kilobase transmissible megaplasmid in the oral probiotic strain Streptococcus salivarius K12. Appl Environ Microbiol 73:1107–1113. doi: 10.1128/AEM.02265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wescombe PA, Upton M, Dierksen KP, Ragland NL, Sivabalan S, Wirawan RE, Jenkinson HF. 2006. Production of the lantibiotic salivaricin A and its variants by oral streptococci and use of a specific induction assay to detect their presence in human saliva. Appl Environ Microbiol 72:1459–1466. doi: 10.1128/AEM.72.2.1459-1466.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Islam MR, Nishie M, Nagao JI, Zendo T, Keller S, Nakayama J, Sonomoto K. 2012. Ring A of nukacin ISK-1: a lipid II-binding motif for type-A (II) lantibiotic. J Am Chem Soc 134:3687–3690. doi: 10.1021/ja300007h. [DOI] [PubMed] [Google Scholar]
- 19.Brötz H, Bierbaum G, Leopold K, Reynolds PE, Sahl HG. 1998. The lantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipid II. Antimicrob Agents Chemother 42:154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang J, Feng Y, Teng K, Lin Y, Gao Y, Wang J, Zhong J. 2014. Type AII lantibiotic bovicin HJ50 with a rare disulfide bond: structure, structure–activity relationships and mode of action. Biochem J 461:497–508. doi: 10.1042/BJ20131524. [DOI] [PubMed] [Google Scholar]
- 21.Smith DJ, Anderson JM, King WF, Houte J, Taubman MA. 1993. Oral streptococcal colonization of infants. Molec Oral Microbiol 8:1–4. doi: 10.1111/j.1365-2958.1993.tb01197.x. [DOI] [PubMed] [Google Scholar]
- 22.Burton JP, Chilcott CN, Moore CJ, Speiser G, Tagg JR. 2006. A preliminary study of the effect of probiotic Streptococcus salivarius K12 on oral malodour parameters. J Appl Microbiol 100:754–764. doi: 10.1111/j.1365-2672.2006.02837.x. [DOI] [PubMed] [Google Scholar]
- 23.Burton JP, Wescombe PA, Moore CJ, Chilcott CN, Tagg JR. 2006. Safety assessment of the oral cavity probiotic Streptococcus salivarius K12. Appl Environ Microbiol 72:3050–3053. doi: 10.1128/AEM.72.4.3050-3053.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horz HP, Meinelt A, Houben B, Conrads G. 2007. Distribution and persistence of probiotic Streptococcus salivarius K12 in the human oral cavity as determined by real-time quantitative polymerase chain reaction. Oral Microbiol Immun 22:126–130. doi: 10.1111/j.1399-302X.2007.00334.x. [DOI] [PubMed] [Google Scholar]
- 25.Cosseau C, Devine DA, Dullaghan E, Gardy JL, Chikatamarla A, Gellatly S, Yu LL, Pistolic J, Falsafi R, Tagg J, Hancock REW. 2008. The commensal Streptococcus salivarius K12 downregulates the innate immune responses of human epithelial cells and promotes host-microbe homeostasis. Infect Immun 76:4163–4175. doi: 10.1128/IAI.00188-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burton JP, Cowley S, Simon RR, McKinney J, Wescombe PA, Tagg JR. 2011. Evaluation of safety and human tolerance of the oral probiotic Streptococcus salivarius K12: a randomized, placebo-controlled, double-blind study. Food Chem Toxicol 49:2356–2364. doi: 10.1016/j.fct.2011.06.038. [DOI] [PubMed] [Google Scholar]
- 27.van den Hooven HW, Lagerwerf FM, Heerma W, Haverkamp J, Piard JC, Hilbers CW, Rollema HS. 1996. The structure of the lantibiotic lacticin 481 produced by Lactococcus lactis: location of the thioether bridges. FEBS Lett 391:317–322. doi: 10.1016/0014-5793(96)00771-5. [DOI] [PubMed] [Google Scholar]
- 28.Ghobrial OG, Derendorf H, Hillman JD. 2009. Pharmacodynamic activity of the lantibiotic MU1140. Int J Antimicrob Agents 33:70–74. doi: 10.1016/j.ijantimicag.2008.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piper C, Draper LA, Cotter PD, Ross RP, Hill C. 2009. A comparison of the activities of lacticin 3147 and nisin against drug-resistant Staphylococcus aureus and Enterococcus species. J Antimicrob Chemother 64:546–551. doi: 10.1093/jac/dkp221. [DOI] [PubMed] [Google Scholar]
- 30.Kruszewska D, Sahl HG, Bierbaum G, Pag U, Hynes SO, Ljungh Å. 2004. Mersacidin eradicates methicillin-resistant Staphylococcus aureus (MRSA) in a mouse rhinitis model. J Antimicrob Chemother 54:648–653. doi: 10.1093/jac/dkh387. [DOI] [PubMed] [Google Scholar]
- 31.Hsu STD, Breukink E, Bierbaum G, Sahl HG, de Kruijff B, Kaptein R, Bonvin AM. 2003. NMR study of mersacidin and lipid II interaction in dodecylphosphocholine micelles. Conformational changes are a key to antimicrobial activity. J Biol Chem 278:13110–13117. doi: 10.1074/jbc.M211144200. [DOI] [PubMed] [Google Scholar]
- 32.Bakhtiary A, Cochrane SA, Mercier P, McKay RT, Miskolzie M, Sit CS, Vederas JC. 2017. Insights into the mechanism of action of the two-peptide lantibiotic lacticin 3147. J Am Chem Soc 139:17803–17810. doi: 10.1021/jacs.7b04728. [DOI] [PubMed] [Google Scholar]
- 33.Islam MR, Shioya K, Nagao J, Nishie M, Jikuya H, Zendo T, Sonomoto K. 2009. Evaluation of essential and variable residues of nukacin ISK-1 by NNK scanning. Mol Microbiol 72:1438–1447. doi: 10.1111/j.1365-2958.2009.06733.x. [DOI] [PubMed] [Google Scholar]
- 34.Asaduzzaman SM, Nagao JI, Aso Y, Nakayama J, Sonomoto K. 2006. Lysine-oriented charges trigger the membrane binding and activity of nukacin ISK-1. Appl Environ Microbiol 72:6012–6017. doi: 10.1128/AEM.00678-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bomar L, Brugger SD, Yost BH, Davies SS, Lemon KP. 2016. Corynebacterium accolens releases antipneumococcal free fatty acids from human nostril and skin surface triacylglycerols. mBio 7:e01725-15. doi: 10.1128/mBio.01725-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson-Stanford S, Kalli A, Håkansson K, Kastrantas J, Orugunty RS, Smith L. 2009. Oxidation of lanthionines renders the lantibiotic nisin inactive. Appl Environ Microbiol 75:1381–1387. doi: 10.1128/AEM.01864-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stubbendieck RM, Straight PD. 2016. Correction: escape from lethal bacterial competition through coupled activation of antibiotic resistance and a mobilized subpopulation. PLoS Genet 12:e1005807. doi: 10.1371/journal.pgen.1005807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen S, Wilson-Stanford S, Cromwell W, Hillman JD, Guerrero A, Allen CA, Sorg JA, Smith L. 2013. Site-directed mutations in the lanthipeptide mutacin 1140. Appl Environ Microbiol 79:4015–4023. doi: 10.1128/AEM.00704-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pipes GD, Kosky AA, Abel J, Zhang Y, Treuheit MJ, Kleemann GR. 2005. Optimization and applications of CDAP labeling for the assignment of cysteines. Pharm Res 22:1059–1068. doi: 10.1007/s11095-005-5643-3. [DOI] [PubMed] [Google Scholar]
- 40.Escano J, Stauffer B, Brennan J, Bullock M, Smith L. 2014. The leader peptide of mutacin 1140 has distinct structural components compared to related class I lantibiotics. Microbiologyopen 3:961–972. doi: 10.1002/mbo3.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ravichandran A, Gu G, Escano J, Lu SE, Smith L. 2013. The presence of two cyclase thioesterases expands the conformational freedom of the cyclic peptide occidiofungin. J Nat Prod 76:150–156. doi: 10.1021/np3005503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wüthrich K. 1986. NMR with proteins and nucleic acids. Europhys News 17:11–13. doi: 10.1051/epn/19861701011. [DOI] [Google Scholar]
- 43.Braunschweiler L, Ernst RR. 1983. Coherence transfer by isotropic mixing: application to proton correlation spectroscopy. J Magn Reson (1969) 53:521–528. doi: 10.1016/0022-2364(83)90226-3. [DOI] [Google Scholar]
- 44.Kumar A, Ernst RR, Wüthrich K. 1980. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem Biophys Res Commun 95:1–6. doi: 10.1016/0006-291X(80)90695-6. [DOI] [PubMed] [Google Scholar]
- 45.Bodenhausen G, Ruben DJ. 1980. Natural abundance nitrogen-15 NMR by enhanced heteronuclear spectroscopy. Chem Phys Lett 69:185–189. doi: 10.1016/0009-2614(80)80041-8. [DOI] [Google Scholar]
- 46.Johnson BA, Blevins RA. 1994. NMR View: a computer program for the visualization and analysis of NMR data. J Biomol NMR 4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 47.Clinical and Laboratory Standards Institute. 2009. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard—eighth edition. CLSI document M07-A8. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 48.Escano J, Ravichandran A, Salamat B, Smith L. 2017. Carboxyl analogue of mutacin 1140, a scaffold for lead antibacterial discovery. Appl Environ Microbiol 83:e00668-17. doi: 10.1128/AEM.00668-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ellis D, Gosai J, Emrick C, Heintz R, Romans L, Gordon D, Smith L. 2012. Occidiofungin's chemical stability and in vitro potency against Candida species. Antimicrob Agents Chemother 56:765–769. doi: 10.1128/AAC.05231-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.