

Abstract
Objectives
There are currently few data on the long‐term risk of cancer and death in individuals taking raltegravir (RAL). The aim of this analysis was to evaluate whether there is evidence for an association.
Methods
The EuroSIDA cohort was divided into three groups: those starting RAL‐based combination antiretroviral therapy (cART) on or after 21 December 2007 (RAL); a historical cohort (HIST) of individuals adding a new antiretroviral (ARV) drug (not RAL) to their cART between 1 January 2005 and 20 December 2007, and a concurrent cohort (CONC) of individuals adding a new ARV drug (not RAL) to their cART on or after 21 December 2007. Baseline characteristics were compared using logistic regression. The incidences of newly diagnosed malignancies and death were compared using Poisson regression.
Results
The RAL cohort included 1470 individuals [with 4058 person‐years of follow‐up (PYFU)] compared with 3787 (4472 PYFU) and 4467 (10 691 PYFU) in the HIST and CONC cohorts, respectively. The prevalence of non‐AIDS‐related malignancies prior to baseline tended to be higher in the RAL cohort vs. the HIST cohort [adjusted odds ratio (aOR) 1.31; 95% confidence interval (CI) 0.95–1.80] and vs. the CONC cohort (aOR 1.89; 95% CI 1.37–2.61). In intention‐to‐treat (ITT) analysis (events: RAL, 50; HIST, 45; CONC, 127), the incidence of all new malignancies was 1.11 (95% CI 0.84–1.46) per 100 PYFU in the RAL cohort vs. 1.20 (95% CI 0.90–1.61) and 0.83 (95% CI 0.70–0.99) in the HIST and CONC cohorts, respectively. After adjustment, there was no evidence for a difference in the risk of malignancies [adjusted rate ratio (RR) 0.73; 95% CI 0.47–1.14 for RAL vs. HIST; RR 0.95; 95% CI 0.65–1.39 for RAL vs. CONC] or mortality (adjusted RR 0.87; 95% CI 0.53–1.43 for RAL vs. HIST; RR 1.14; 95% CI 0.76–1.72 for RAL vs. CONC).
Conclusions
We found no evidence for an oncogenic risk or poorer survival associated with using RAL compared with control groups.
Keywords: Raltegravir, survival, risk of cancer, observational treatment comparison, propensity scores
Introduction
After the publication of the results of randomized clinical trials performed against efavirenz‐containing combination antiretroviral therapy (cART), raltegravir (RAL), the first available antiretroviral agent belonging to the class of HIV integrase strand transfer inhibitors (INSTIs), has been approved for both ART‐experienced patients and first‐line therapy in ART‐naïve patients 1, 2, 3, 4, 5, 6. It was widely introduced across Europe starting from 2010 7. Although a satisfactory tolerability profile in patients, including those with underlying comorbidities, and proportionally limited drug–drug interactions have generally been shown, a slight excess of cancers was observed in interim analyses of clinical trials including RAL‐based regimens which was not, however, confirmed in subsequent analyses 8, 9, 10, 11, 12, 13. INSTIs bind at the active site of HIV integrase and block the strand transfer step of integration. Recent in vitro studies showed that suboptimal doses of RAL could lead to the generation of aberrant proviruses during the strand transfer reaction, with significant rearrangements of the host genome, including duplications, inversions, deletions and, occasionally, acquisition of sequences from other chromosomes 14, 15. Based on what is known about the activation of oncogenes in human tumours, it is possible that rearrangements in the host DNA (so far observed only in in vitro models) as a result of potential aberrant HIV DNA integrations could increase the chance that HIV integrations could lead to the development of cancer.
In 2009, a large meta‐analysis was conducted combining data from several randomized RAL clinical trials 16 and found no difference in rates of cancer comparing RAL users and people receiving other treatments. These early results showed that, over the first 2 months of these trials, cancer rates were similar in people who received RAL and in controls. After 2 months in these studies, cancers became more common in RAL users and the number of new cancers then stabilized over time (affecting about 1% of the patients enrolled over the following 20 months). Although there was no evidence for statistical differences in rates of cancer between people receiving RAL and those receiving standard of care, concerns about the earlier findings led to further analyses being conducted in observational studies to monitor long‐term safety profiles in RAL users.
The aim of this analysis was to compare the incidences of malignancies and other comorbidities as well as survival rates in cohorts of individuals initiating RAL‐based and non‐RAL‐based cART regimens in a large European cohort of HIV‐infected patients.
Methods
EuroSIDA is a large, on‐going prospective cohort study of 18 931 individuals living with HIV. The study collects data from 111 hospitals in 34 different countries across Europe, as well as Israel and Argentina 17, 18. Recruitment started in 1994, and data are collected 6‐monthly on standardized case report forms (CRFs). Variables collected include demographic information, CD4 counts, viral load (VL) measurements and start and stop dates for all antiretroviral drugs used. All patients gave informed consent to be included in EuroSIDA at enrolment. Non‐AIDS‐related events are collected in EuroSIDA following the standardized HIV Cohorts Data Exchange Protocol (HICDEP) code for data collection (http://www.hicdep.org/wiki/Hicdep_1.90). Clinicians at EuroSIDA participating sites were asked 6‐monthly to report any event not previously reported to EuroSIDA, including all events since the last follow‐up. There is a specific “List of Definitions” for the relevant events collected in EuroSIDA which follows the accepted World Health Organization International Classification of Diseases (WHO ICD). More detailed information about the study can be found at www.cphiv.dk.
Patients in EuroSIDA were included in the RAL cohort if (1) they started RAL for the first time on or after 21 December 2007 (the RAL authorization date in the European Union); (2) they had at least 1 month's prospective follow‐up in this cohort, and (3) they had a CD4 count and a VL measured within 6 months prior to the start date of RAL. Two control cohorts were chosen a priori and were defined as follows: a ‘historical’ (HIST) and a ‘concurrent’ (CONC) cohort. Patients were included in the HIST comparison cohort if (1) they started a new antiretroviral drug as part of a cART regimen on or after 1 January 2005 and before 21 December 2007 (patients must have had no previous exposure to the new drug, including as part of a different co‐formulation, to be included); (2) they had at least 1 month's prospective follow‐up in this cohort, and (3) they had a CD4 count and a VL measured within 6 months prior to the start date of the new drug. Patients were included in the CONC cohort if (1) they started a new antiretroviral drug other than RAL as part of a cART regimen on or after 21 December 2007, and had no previous exposure to the new drug, including as part of a different co‐formulation; (2) they had at least 1 month's prospective follow‐up in this cohort, and (3) they had a CD4 count and a VL measured within 6 months prior to the start date of the new drug. Baseline for the analysis in the RAL cohort was defined as the date on which the patient first received RAL. In the HIST and CONC cohorts, baseline was the date on which the patient first received the new antiretroviral drug (not RAL). Patients were allowed to contribute data to more than one cohort, but with no overlap in the follow‐up time (Fig. 1). Those who contributed data to the HIST cohort were also eligible for inclusion in the RAL cohort upon initiation of RAL or in the CONC cohort upon initiation of a new antiretroviral drug after 21 December 2007. Patients who contributed data to the CONC cohort were also allowed to ‘switch’ into the RAL cohort upon initiation of RAL. However, in order to be able to evaluate the risk associated with currently being exposed to RAL, patients in the RAL cohort could not switch to the CONC cohort upon initiation of a new antiretroviral drug.
Figure 1.
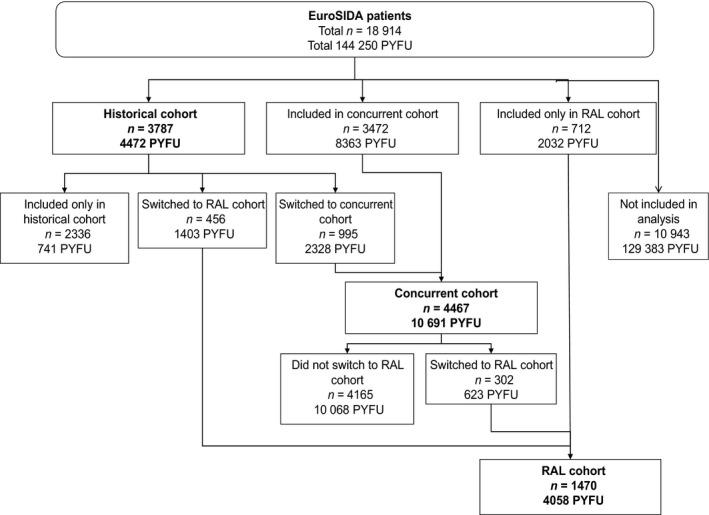
Patients included in the analysis in the raltegravir (RAL), historical and concurrent cohorts. In order to obtain the total number of patients in each cohort, it is sufficient to add the figures in the corresponding boxes, as follows. (1) The RAL cohort: 456 (historical patients who switched to RAL over follow‐up) + 302 (concurrent patients who switched to RAL) + 712 (patients originally included in the RAL cohort) = 1470. (2) The historical cohort: 3787 (patients originally allocated to this group). The concurrent cohort: 3472 (patients originally allocated to this group) + 995 (historical patients who switched to the concurrent cohort over follow‐up) = 4467. PYFU, person‐years of follow‐up.
Analyses were performed on the data set including cumulated data collected from patient journals until December 2014. The data were collected, keyed and quality assured at the Coordinating Centre between January 2014 and December 2014.
Statistical analysis
Four main outcomes were assessed: newly diagnosed malignancies (AIDS‐ and non‐AIDS‐related), clinically important hepatic events, lipodystrophy and mortality. By protocol, a formal comparison of the incidence rates between cohorts was performed only for outcomes with > 30 events in all three comparator cohorts; this target number of events was achieved only for the outcomes of malignancies and overall mortality.
Patient's follow‐up time was defined as follows: only prospectively collected person‐years of follow‐up (PYFU) were included after the date of enrolment in EuroSIDA. Follow‐up time in the RAL cohort was censored at the time of the earliest of any of the following events: discontinuation of RAL, death or last clinic visit. In the HIST cohort, follow‐up time was censored at the earliest of 21 December 2007, discontinuation of any new drugs started on the baseline date, death or last clinic visit. In the CONC cohort, follow‐up time was censored at the earliest of starting RAL (if started), discontinuation of any new drugs started on the baseline date, death or last clinic visit.
For the analysis of the incidence of clinical events, the follow‐up time included was additionally censored at the first occurrence of the specific event of interest if it occurred before the end of the follow‐up period, but not at the first occurrence of any of the other main outcomes. As a consequence of the nature of how the data are collected in our cohort, it is difficult to distinguish between subsequent recurrences and continuations of malignancies, and therefore only first occurrences of each type were analysed in all incidence analyses. A further analysis including all malignancies reported over the follow‐up time, regardless of whether or not they were first occurrences, yielded similar results (not shown).
Baseline characteristics in the three cohorts were compared using univariable and multivariable logistic regression analysis. We also described the incidence of discontinuation of one or more drugs (main reason as reported by the treating physician, for the RAL cohort alone).
The incidence of all outcomes was calculated as the number of events over the follow‐up period divided by PYFU, assuming a Poisson distribution. Univariable and multivariable Poisson regression models were used to estimate rate ratios (RRs) of malignancies and of death between the cohorts. Because of the large number of potential measured confounders, propensity scores (PSs) were used for the adjustment in the multivariable analysis. All factors showing a univariable association with α = 0.1 were included in the PS vector. Quintiles of the PS were fitted in the Poisson regression model as a continuous variable. Results were consistent when we used the alternative approaches of matching, stratifying (data not shown). For all Poisson regression analyses, an intention‐to‐treat (ITT) approach, ignoring treatment switches as well as recurrences of clinical events of the same type, was used. All statistical analyses were performed using sas version 9.4 (SAS Institute, Cary, NC).
Results
Study population
A total of 1470 patients were included in the RAL cohort, 3787 patients were included in the HIST comparison cohort and 4467 patients were included in the CONC cohort (Fig. 1). Table 1a shows a comparison of the demographic characteristics of the patients in the three cohorts at baseline. The majority of patients in all three cohorts were male (approximately 75%) and of white ethnicity (> 84%). The mode of HIV transmission was similar across the three cohorts. Approximately 40% of patients were men who have sex with men (MSM), almost 20% reported heterosexual contact as the mode of transmission, and approximately 30% were persons who inject drugs (PWID). There was some difference in the geographical distribution of the patients between the cohorts; RAL users were mostly split between North Europe (21%), Central West Europe (36%) and South Europe (31%), whereas approximately three‐quarters of patients in the HIST and CONC cohorts were evenly split between North, Central West and South Europe, with the remaining patients distributed between Central East and East Europe, and just 1% in Argentina. The median age was 49 years in the RAL cohort, and 44 years in the HIST and CONC cohorts. Baseline CD4 count was similar across the three cohorts (medians between 392 and 468 cells/μL) but CD4 count nadir was higher in the HIST and CONC cohorts (medians 140 and 175 cells/μL, respectively, compared with 118 cells/μL in the RAL cohort). There was a shorter time from CD4 nadir to enrolment in both of the comparison cohorts compared with the RAL cohort. On average, RAL patients had been enrolled in EuroSIDA for longer than those in the comparison cohorts (median 11 months compared with 7 months in the HIST cohort and 4 months in the CONC cohort). Median baseline VL was 1.7 log10 HIV RNA copies/mL in the RAL cohort, 1.9 log10 copies/mL in the HIST cohort, and 1.8 log10 copies/mL in the CONC cohort. Baseline VL was suppressed to <= 500 copies/mL in 74% of the RAL cohort (60% were suppressed to ≤ 50 copies/mL), 60% of the HIST cohort (53% were suppressed to ≤ 50 copies/mL) and 58% of the CONC cohort (56% were suppressed to ≤ 50 copies/mL), and it was > 10 000 copies/mL in 14% of the RAL cohort, 27% of the HIST cohort and 33% of the CONC cohort. Baseline peak VLs were also similar between the cohorts (median 5.0 log10 copies/mL). Median baseline date for the RAL cohort was February 2010, that for the HIST cohort was September 2006 and that for the CONC cohort was March 2009, and the median follow‐up time was 35 months for the RAL cohort, 13 months for the HIST cohort and 25 months for the CONC cohort. RAL patients had a longer time since HIV‐1‐positive diagnosis (median 17 years compared with 12 years in the HIST cohort and 10 years in the CONC cohort). Only a minority of the study population was ART‐naïve at the date of inclusion in this analysis (4% of the RAL cohort, 11% of the HIST cohort and 30% of the CONC cohort). The prevalences of hepatitis coinfection and comorbidities at baseline are shown in Table 1b.
Table 1.
Baselinea patient characteristics. (a) Baseline comorbidities and prior clinical events according to inclusion in the raltegravir (RAL), historical and concurrent cohorts. (b) Baseline information on hepatitis coinfection status, comorbidities and prior clinical events according to inclusion in the RAL, historical and concurrent cohorts
| RAL | Historical | Concurrent | |
|---|---|---|---|
| (a) | |||
| Total number | 1470 | 3787 | 4467 |
| Gender [n (%)] | |||
| Female | 361 (24.6) | 949 (25.1) | 1229 (27.5) |
| Mode of HIV transmission [n (%)] | |||
| MSM | 640 (43.5) | 1623 (42.9) | 1782 (39.9) |
| Heterosexual contacts | 255 (17.3) | 764 (20.2) | 860 (19.3) |
| PWID | 451 (30.7) | 1129 (29.8) | 1539 (34.5) |
| Other/unknown | 124 (8.4) | 271 (7.2) | 286 (6.4) |
| Ethnicity [n (%)] | |||
| White | 1232 (83.8) | 3274 (86.5) | 3942 (88.2) |
| Asian | 20 (1.4) | 62 (1.6) | 56 (1.3) |
| Black | 90 (6.1) | 239 (6.3) | 282 (6.3) |
| Other/unknown | 128 (8.7) | 212 (5.6) | 187 (4.2) |
| Country of origin [n (%)] | |||
| Same country as clinic | 1142 (77.7) | 2902 (76.6) | 3539 (79.2) |
| Other European country | 82 (5.6) | 212 (5.6) | 225 (5.0) |
| Africa | 94 (6.4) | 243 (6.4) | 284 (6.4) |
| America | 32 (2.2) | 133 (3.5) | 168 (3.8) |
| Asia | 17 (1.2) | 48 (1.3) | 53 (1.2) |
| Other/unknown | 103 (7.0) | 249 (6.6) | 198 (4.4) |
| Geographical region [n (%)] | |||
| South | 458 (31.2) | 1131 (29.9) | 1053 (23.6) |
| Central West | 523 (35.6) | 892 (23.6) | 889 (19.9) |
| North | 315 (21.4) | 1071 (28.3) | 891 (19.9) |
| Central East | 138 (9.4) | 394 (10.4) | 777 (17.4) |
| East | 21 (1.4) | 190 (5.0) | 696 (15.6) |
| Argentina | 15 (1.0) | 109 (2.9) | 161 (3.6) |
| Viral load [n (%)] | |||
| < 500 copies/mL | 1082 (73.6) | 2278 (60.2) | 2584 (57.8) |
| 500–10 000 copies/mL | 179 (12.2) | 486 (12.8) | 411 (9.2) |
| > 10 000 copies/mL | 209 (14.2) | 1023 (27.0) | 1472 (33.0) |
| Age (years) [median (IQR)] | 49 (44, 56) | 44 (39, 50) | 44 (35, 51) |
| CD4 count | |||
| Baseline (cells/μL) [median (IQR)] | 468 (300, 673) | 392 (237, 614) | 404 (255, 630) |
| Nadir (cells/μL) [median (IQR)] | 118 (41, 208) | 140 (50, 225) | 175 (75, 266) |
| Time since nadir (years) [median (IQR)] | 10 (4, 13) | 6 (2, 9) | 4 (0, 10) |
| Viral load (log10 copies/mL) [median (IQR)] | |||
| Baseline | 1.7 (1.6, 2.9) | 1.9 (1.7, 4.2) | 1.8 (1.6, 4.5) |
| Peak | 5.0 (4.3, 5.6) | 4.9 (4.2, 5.5) | 5.0 (4.3, 5.5) |
| Index date (baseline) [median (IQR)] | 2010 (2009, 2012) | 2006 (2005, 2006) | 2009 (2008, 2011) |
| Length of follow‐up (months) [median (IQR)] | 32 (15, 52) | 13 (6, 22) | 25 (11, 45) |
| Time HIV‐1 positive (years) [median (IQR)]b | 17 (13, 21) | 12 (8, 16) | 10 (4, 16) |
| History of ART [n (%)] | |||
| ART‐naïve | 58 (3.9) | 424 (11.2) | 1339 (30.0) |
| (b) | |||
| Total number | 1470 | 3787 | 4467 |
| HBV coinfection status [n (%)]c | |||
| Positive | 92 (6.3) | 287 (7.6) | 279 (6.2) |
| Negative | 1278 (86.9) | 3160 (83.4) | 3672 (82.2) |
| Unknown | 100 (6.8) | 340 (9.0) | 516 (11.6) |
| HCV coinfection status [n (%)]d | |||
| Positive | 376 (25.6) | 954 (25.2) | 1109 (24.8) |
| Negative | 962 (65.4) | 2213 (58.4) | 2622 (58.7) |
| Unknown | 132 (9.0) | 620 (16.4) | 736 (16.5) |
| Prior clinical eventse | |||
| No. of previous AIDS diagnoses [n (%)] | |||
| 0 | 921 (62.7) | 2577 (68.0) | 3350 (75.0) |
| 1 | 311 (21.2) | 760 (20.1) | 764 (17.1) |
| 2 | 144 (9.8) | 270 (7.1) | 240 (5.4) |
| ≥ 3 | 94 (6.4) | 180 (4.8) | 113 (2.5) |
| AIDS‐defining conditions, excluding malignancies [n (%)] | 495 (33.7) | 1086 (28.7) | 1009 (22.6) |
| AIDS dementia complex [n (%)] | 30 (2.0) | 52 (1.4) | 36 (0.8) |
| Candidiasis [n (%)] | 164 (11.2) | 332 (8.8) | 302 (6.8) |
| CMV infection [n (%)] | 32 (2.2) | 62 (1.6) | 42 (0.9) |
| HIV wasting syndrome [n (%)] | 43 (2.9) | 95 (2.5) | 100 (2.2) |
| Mycobacterium tuberculosis infection [n (%)] | 84 (5.7) | 245 (6.5) | 221 (4.9) |
| PCP [n (%)] | 149 (10.1) | 342 (9.0) | 266 (6.0) |
| Toxoplasmosis [n (%)] | 45 (3.1) | 93 (2.5) | 82 (1.8) |
| AIDS‐related malignancy [n (%)] | 123 (8.4) | 238 (6.3) | 191 (4.3) |
| Non‐AIDS‐related malignancy [n (%)] | 99 (6.7) | 108 (2.9) | 101 (2.3) |
| Cardiovascular event [n (%)] | 92 (6.3) | 109 (2.9) | 108 (2.4) |
| Pancreatitis/end‐stage renal disease [n (%)] | 26 (1.8) | 31 (0.8) | 28 (0.6) |
| Hepatic encephalopathy [n (%)] | 41 (2.8) | 74 (2.0) | 65 (1.5) |
| Loss or accumulation of fat [n (%)] | 820 (55.8) | 1802 (47.6) | 1350 (30.2) |
| Ever discontinued a drug because of liver toxicity [n (%)] | 99 (6.7) | 162 (4.3) | 160 (3.6) |
| Ever discontinued a drug because of lipodystrophy/atrophy [n (%)] | 360 (24.5) | 816 (21.5) | 687 (15.4) |
| Time since first AIDS diagnosis (years) [median (IQR)]f | 12 (8, 14) | 9 (5, 10) | 8 (3, 13) |
| Time since last AIDS diagnosis (years) [median (IQR)]f | 11 (7, 14) | 8 (4, 10) | 8 (3, 12) |
CMV, cytomegalovirus; HBV, hepatitis B virus; HCV, hepatitis C virus; IDU, injecting drug use; IQR, interquartile range; PCP, Pneumocystis carinii pneumonia; TB, tuberculosis.
Baseline date in the RAL cohort is defined as the date on which the patient first received RAL, and in the historical and concurrent cohorts, it was the date on which the patient first received the new antiretroviral drug (not RAL).
Missing data for 26 (3.1%) patients in the RAL cohort, 53 (2.0%) patients in the historical cohort and 157 (5.3%) patients in the concurrent cohort.
Hepatitis B virus surface antigen (HBsAg) test results.
Hepatitis C virus antibody (HCVAb) test results.
Prior clinical events refer to any events that occurred before baseline. AIDS‐defining diseases are listed for those that occurred in more than 30 patients.
In those with a previous AIDS (malignancy or nonmalignancy) diagnosis.
Predictors of RAL initiation
Table 2a shows the unadjusted and adjusted odds ratios (aORs) for initiation of RAL vs. a historical agent. In the unadjusted analysis, there were strong associations between the probability of starting RAL and a large number of the factors examined (P < 0.005). The multivariable model showed that, after adjustment for all significant factors, older patients were more likely to receive RAL [aOR per 10‐year increase: 1.42; 95% confidence interval (CI) 1.31–1.54], as were patients with a longer time since CD4 count nadir (aOR per 1 year longer: 1.11; 95% CI 1.04–1.19), patients with a lower baseline VL (aOR per 1 log10 copies/mL higher: 0.80; 95% CI 0.74–0.85), patients with a higher peak VL at baseline (aOR per 1 log10 copies/mL higher: 1.26; 95% CI 1.17–1.36) and patients who had been on ART for a longer period (aOR per 1 year longer: 1.18; 95% CI 1.15–1.24). Patients visiting a clinic in the North of Europe were less likely to receive RAL compared with patients followed up in other regions. Furthermore, higher numbers of previous nucleoside reverse transcriptase inhibitors (NRTIs) and protease inhibitors (PIs) used were linked to increased odds of starting RAL. For example, patients who had previously received three or four PIs were more than twice as likely to start RAL than those who had previously taken only one or two PIs (aOR 1.79; 95% CI 1.50–2.14). Finally, patients who had previously received treatment for an opportunistic infection were less likely to receive RAL (aOR 0.71; 95% CI 0.61–0.84), than patients who had previously experienced loss or accumulation of fat (aOR 0.45; 95% CI 0.39–0.52) (Table 2a). Results for the comparison between RAL recipients and the CONC cohort were similar (Table 2b).
Table 2.
Unadjusted and adjusted odds ratios (ORs) for initiation of raltegravir (RAL) compared with (a) the historical cohort and (b) the concurrent cohort
| Factor | Unadjusted | Adjusted | ||
|---|---|---|---|---|
| OR (95% CI) | P‐value | OR (95% CI) | P‐value | |
| (a) | ||||
| Gender | ||||
| Male | 1.00 | |||
| Female | 0.97 (0.86, 1.10) | 0.672 | ||
| Mode of HIV transmission | ||||
| MSM | 1.00 | 0.329 | ||
| IDU | 0.85 (0.73, 0.98) | |||
| Heterosexual | 1.01 (0.89, 1.15) | |||
| Other | 1.16 (0.95, 1.42) | |||
| Ethnicity | ||||
| White | 1.00 | |||
| Other/unknown | 1.23 (1.07, 1.43) | 0.005 | 1.15 (0.94, 1.40) | 0.168 |
| Geographical region | ||||
| North | 1.00 | < 0.001 | 1.00 | < 0.001 |
| Central West | 1.99 (1.72, 2.31) | 1.68 (1.39, 2.02) | ||
| South/Argentina | 1.30 (1.12, 1.50) | 1.73 (1.42, 2.11) | ||
| Central East/East | 0.93 (0.76, 1.13) | 2.92 (2.21, 3.87) | ||
| HBV coinfection | ||||
| Negative | 1.00 | 0.417 | ||
| Positive | 0.79 (0.64, 0.99) | |||
| Unknown | 0.73 (0.58, 0.90) | |||
| HCV coinfection | ||||
| Negative | 1.00 | < 0.001 | 0.498 | |
| Positive | 0.91 (0.80, 1.03) | 0.78 (0.66, 0.91) | ||
| Unknown | 0.49 (0.40, 0.59) | 0.62 (0.49, 0.78) | ||
| Age (per 10 years older) | 1.74 (1.64, 1.84) | < 0.001 | 1.40 (1.29, 1.52) | < 0.001 |
| CD4 count (per 100 cells/μL higher) | ||||
| Baseline | 1.08 (1.06, 1.10) | < 0.001 | ||
| Nadir | 0.91 (0.87, 0.96) | < 0.001 | 1.11 (1.04, 1.19) | 0.002 |
| Time since CD4 count nadir (per 1 year longer) | 1.17 (1.16, 1.19) | < 0.001 | 1.05 (1.03, 1.07) | < 0.001 |
| Viral load (per log10 copies/mL higher) | ||||
| Baseline | 0.75 (0.71, 0.79) | < 0.001 | 0.80 (0.74, 0.85) | < 0.001 |
| Peak | 1.05 (1.00, 1.11) | 0.069 | 1.26 (1.17, 1.36) | < 0.001 |
| Length of enrolment in EuroSIDA (per 1 year longer) | 1.17 (1.15, 1.19) | < 0.001 | 1.05 (1.03, 1.07) | < 0.001 |
| Prior clinical events (vs. no event) | ||||
| No. of previous AIDS diagnoses | ||||
| 0 | 1.00 | 1.00 | ||
| 1 | 1.14 (1.00, 1.31) | 0.052 | 0.99 (0.83, 1.18) | 0.914 |
| ≥ 2 | 1.48 (1.27, 1.72) | < 0.001 | 1.27 (1.02, 1.58) | 0.033 |
| AIDS‐related malignancy | 1.36 (1.12, 1.65) | 0.002 | 0.87 (0.67, 1.12) | 0.279 |
| Non‐AIDS‐related malignancy | 2.46 (1.90, 3.18) | < 0.001 | 1.31 (0.95, 1.80) | 0.099 |
| Cardiovascular event | 2.25 (1.74, 2.92) | < 0.001 | 1.19 (0.87, 1.64) | 0.280 |
| Loss or accumulation of fat | 1.39 (1.25, 1.55) | < 0.001 | 0.45 (0.39, 0.52) | < 0.001 |
| Discontinued drug because of toxicity | 1.62 (1.28, 2.04) | < 0.001 | 1.15 (0.86, 1.54) | 0.340 |
| Discontinued drug because of lipodistrophy/atrophy | 1.18 (1.04, 1.34) | 0.010 | 0.90 (0.77, 1.05) | 0.165 |
| Prior medication (vs. no medication) | ||||
| OI treatmenta | 1.40 (1.25, 1.57) | < 0.001 | 0.71 (0.61, 0.84) | < 0.001 |
| Lipid‐lowering agents | 1.56 (1.30, 1.88) | < 0.001 | 0.82 (0.67, 1.02) | 0.070 |
| Prior ART | ||||
| Time since started ART (per 1 year longer) | 1.24 (1.21, 1.26) | < 0.001 | 1.18 (1.15, 1.21) | < 0.001 |
| No. of previous treatment failures (per one higher) | 1.47 (1.40, 1.54) | < 0.001 | 0.72 (0.66, 0.80) | < 0.001 |
| No. of previous NRTIs | ||||
| 3–4 | 1.00 | 1.00 | ||
| 0–2 | 0.66 (0.55, 0.79) | < 0.001 | 1.79 (1.42, 2.27) | < 0.001 |
| ≥ 5 | 3.19 (2.77, 3.67) | < 0.001 | 1.89 (1.59, 2.26) | < 0.001 |
| No. of previous NNRTIs | ||||
| 1 | 1.00 | 1.00 | ||
| 0 | 0.32 (0.28, 0.37) | < 0.001 | 0.51 (0.34, 0.77) | 0.001 |
| ≥ 2 | 1.39 (1.20, 1.60) | < 0.001 | 0.95 (0.80, 1.13) | 0.587 |
| No. of previous PIs | ||||
| 1–2 | 1.00 | 1.00 | ||
| 0 | 0.55 (0.46, 0.67) | < 0.001 | 1.42 (1.02, 1.98) | 0.038 |
| 3–4 | 2.66 (2.30, 3.07) | < 0.001 | 1.79 (1.50, 2.14) | < 0.001 |
| ≥ 5 | 4.45 (3.72, 5.32) | < 0.001 | 3.20 (2.47, 4.15) | < 0.001 |
| Ever previously exposed to ART | 3.38 (3.00, 3.81) | < 0.001 | 1.10 (0.71, 1.70) | 0.681 |
| (b) | ||||
| Gender | ||||
| Male | 1.00 | |||
| Female | 0.86 (0.75, 0.98) | 0.021 | ||
| Mode of HIV transmission | ||||
| MSM | 1.00 | 0.254 | ||
| IDU | 0.83 (0.70, 0.97) | |||
| Heterosexual | 0.82 (0.72, 0.93) | |||
| Other | 1.21 (0.97, 1.50) | |||
| Ethnicity | ||||
| White | 1.00 | 1.00 | ||
| Other/unknown | 1.45 (1.24, 1.69) | < 0.001 | 1.15 (0.91, 1.46) | 0.240 |
| Geographical region | ||||
| North | 1.00 | < 0.001 | 1.00 | < 0.001 |
| Central West | 1.66 (1.42, 1.95) | 1.55 (1.29, 1.85) | ||
| South/Argentina | 1.10 (0.94, 1.29) | 1.31 (1.09, 1.59) | ||
| Central East/East | 0.31 (0.25, 0.37) | 0.76 (0.60, 0.96) | ||
| HBV coinfection | ||||
| Negative | 1.00 | < 0.001 | 0.002 | |
| Positive | 0.95 (0.76, 1.19) | 0.63 (0.48, 0.82) | ||
| Unknown | 0.56 (0.45, 0.69) | 0.94 (0.72, 1.23) | ||
| HCV coinfection | ||||
| Negative | 1.00 | < 0.001 | ||
| Positive | 0.92 (0.81, 1.05) | |||
| Unknown | 0.49 (0.40, 0.59) | |||
| Age (per 10 years older) | 1.65 (1.57, 1.74) | < 0.001 | 1.22 (1.13, 1.31) | < 0.001 |
| CD4 count (per 100 cells/μL higher) | ||||
| Baseline | 1.05 (1.03, 1.06) | < 0.001 | ||
| Nadir | 0.76 (0.72, 0.80) | < 0.001 | 0.99 (0.93, 1.05) | 0.715 |
| Time since CD4 count nadir (per 1 year longer) | 1.12 (1.11, 1.14) | < 0.001 | 1.01 (0.99, 1.02) | 0.419 |
| Viral load (per log10 copies/mL higher) | ||||
| Baseline | 0.74 (0.71, 0.77) | < 0.001 | 1.00 (0.93, 1.07) | 0.975 |
| Peak | 1.03 (0.97, 1.08) | 0.349 | 0.96 (0.90, 1.03) | 0.270 |
| Length of enrolment in EuroSIDA (per 1 year longer) | 1.13 (1.12, 1.14) | < 0.001 | 1.00 (0.98, 1.02) | 0.830 |
| Prior clinical events (vs. no event) | ||||
| No. of previous AIDS diagnoses | ||||
| 0 | 1.00 | 1.00 | ||
| 1 | 1.48 (1.28, 1.71) | < 0.001 | 0.97 (0.81, 1.15) | 0.708 |
| ≥ 2 | 2.45 (2.07, 2.90) | < 0.001 | 1.27 (1.01, 1.60) | 0.037 |
| AIDS‐related malignancy | 2.04 (1.64, 2.54) | < 0.001 | 0.92 (0.70, 1.21) | 0.559 |
| Non‐AIDS‐related malignancy | 3.12 (2.39, 4.08) | < 0.001 | 1.89 (1.37, 2.61) | < 0.001 |
| Cardiovascular event | 2.69 (2.06, 3.53) | < 0.001 | 1.35 (0.99, 1.83) | 0.055 |
| Loss or accumulation of fat | 2.91 (2.60, 3.27) | < 0.001 | 0.95 (0.81, 1.11) | 0.516 |
| Discontinued drug because of toxicity | 1.94 (1.53, 2.46) | < 0.001 | 1.09 (0.83, 1.42) | 0.550 |
| Discontinued drug because of lipodistrophy/atrophy | 1.78 (1.56, 2.04) | < 0.001 | 1.04 (0.88, 1.23) | 0.642 |
| Prior medication (vs. no medication) | ||||
| OI treatmenta | 2.51 (2.23, 2.82) | < 0.001 | 0.88 (0.75, 1.03) | 0.109 |
| Lipid‐lowering agents | 1.88 (1.54, 2.28) | < 0.001 | 0.75 (0.60, 0.94) | 0.012 |
| Prior ART | ||||
| Time since started ART (per 1 year longer) | 1.16 (1.15, 1.17) | < 0.001 | 0.99 (0.96, 1.01) | 0.281 |
| No. of previous treatment failures (per one higher) | 2.33 (2.20, 2.47) | < 0.001 | 1.29 (1.17, 1.42) | < 0.001 |
| No. of previous NRTIs | ||||
| 3–4 | 1.00 | 1.00 | ||
| 0–2 | 0.31 (0.26, 0.37) | < 0.001 | 0.89 (0.70, 1.12) | 0.313 |
| ≥ 5 | 3.35 (2.89, 3.88) | < 0.001 | 1.70 (1.43, 2.03) | < 0.001 |
| No. of previous NNRTIs | ||||
| 1 | 1.00 | 1.00 | ||
| 0 | 0.26 (0.23, 0.30) | < 0.001 | 0.35 (0.25, 0.49) | < 0.001 |
| ≥ 2 | 2.39 (2.02, 2.83) | < 0.001 | 1.38 (1.14, 1.68) | < 0.001 |
| No. of previous PIs | ||||
| 1–2 | 1.00 | 1.00 | ||
| 0 | 0.22 (0.18, 0.26) | < 0.001 | 0.34 (0.24, 0.47) | < 0.001 |
| 3–4 | 2.53 (2.19, 2.94) | < 0.001 | 1.52 (1.28, 1.80) | < 0.001 |
| ≥ 5 | 7.04 (5.60, 8.86) | < 0.001 | 2.82 (2.12, 3.74) | < 0.001 |
| Previously taken NRTIs, NNRTIs or PIs | 5.55 (4.91, 6.27) | < 0.001 | 0.54 (0.37, 0.78) | 0.001 |
ART, antiretroviral therapy; CI, confidence interval; IDU, injecting drug use; MSM, men who have sex with men; NRTI, nucleoside reverse transcriptase inhibitor; NNRTI, nonnucleoside reverse transcriptase inhibitor; PI, protease inhibitor.
Multivariable analysis was adjusted for all variables where multivariable ORs are shown. P‐values were obtained using logistic regression.
Prior opportunistic infection (OI) treatment includes treatment for Pneumocystis carinii pneumonia/toxoplasmosis and (brain) Mycobacterium tuberculosis, fungal, hepatitis B virus (HBV), hepatitis C virus (HCV), cytomegalovirus and herpes simplex virus infections, and immunomodulating therapy.
Drug discontinuation in the RAL cohort
A total of 351 patients (24%) discontinued RAL, of whom 39 (11%) discontinued within 3 months. Of the 351 discontinuations, 140 (40%) involved the discontinuation of RAL alone, while in the remaining 211 instances at least one additional drug was discontinued together with RAL (60%). The other drugs most frequently leading to the 351 discontinuations of the RAL regimen were ritonavir (18%), emtricitabine/tenofovir (16%), lamivudine (8%) maraviroc and tenofovir (7%), and atazanavir and efavirenz (6%). Of the 39 discontinuations that occurred within 3 months, 16 (41%) were of RAL alone.
Focussing on these 39 early discontinuations (within 3 months), they were mainly attributable to gastrointestinal toxicity (n = 4) and patient choice (n = 10), whereas the majority of the 312 longer term discontinuations (after the first 3 months) seemed to be attributable to physician decision (including structured treatment interruptions) (n = 71), patient wish (n = 63) or clinicians reporting treatment failure (n = 28), while only 10 were attributable to gastrointestinal toxicity.
Clinical outcomes
A total of 222 malignancies occurred over the follow‐up time, after excluding 13 recurrent events (five in the RAL cohort, two in the HIST cohort and six in the CONC cohort). This included 50 malignancies over the follow‐up time in the RAL cohort, 45 in the HIST cohort and 127 in the CONC cohort. AIDS‐defining malignancies, including Kaposi's sarcoma and non‐Hodgkin's lymphoma, were reported in 14% of diagnoses in the RAL cohort (one case of Kaposi's sarcoma and six cases of non‐Hodgkin's lymphoma), 29% of diagnoses in the HIST cohort (five cases of Kaposi's sarcoma and eight cases of non‐Hodgkin's lymphoma) and 16% of diagnoses in the CONC cohort (10 cases of Kaposi's sarcoma and 10 cases of non‐Hodgkin's lymphoma). The breakdown of all specific cancer locations, including the remaining non‐AIDS‐related events, stratified by cohort, is shown in Supporting Information Table S1.
The incidence of newly diagnosed malignancies (222 events over 23 501 PYFU) was 0.94 (95% CI 0.82–1.08) per 100 PYFU; the incidence of death (197 events/23 936 PYFU) was 0.82 (95% CI 0.71–0.95), that of lipodystrophy (189/23 510 PYFU) was 0.80 (95% CI 0.69–93) and that of hepatic events (82/23 713 PYFU) was 0.35 (95% CI 0.28–0.43). After stratification by cohort, the rates of cancer were 1.1 (95% CI 0.8–1.5), 1.2 (95% CI 0.9–1.6) and 0.8 (95% CI 0.7–1.0) per 100 PYFU in the RAL, HIST and CONC cohorts, respectively. The corresponding mortality rates by cohort remained low at 1.0 (95% CI 0.7–1.3), 0.9 (95% CI 0.6–1.3) and 0.8 (95% CI 0.6–0.9) per 100 PYFU (Table 3).
Table 3.
Overall incidence of outcomes: unadjusted analysis
| RAL | Historical | Concurrent | |||||||
|---|---|---|---|---|---|---|---|---|---|
| No. of events | PYFU | Incidence per 100 PYFU (95% CI) | No. of events | PYFU | Incidence per 100 PYFU (95% CI) | No. of events | PYFU | Incidence per 100 PYFU (95% CI) | |
| Malignancies | 50 | 4505.0 | 1.11 (0.84, 1.46) | 45 | 3748.5 | 1.20 (0.90, 1.61) | 127 | 15 247 | 0.83 (0.70, 0.99) |
| Clinically important hepatic events | 3 | 4583.0 | 0.07 (0.02, 0.20) | 41 | 3750.8 | 1.09 (0.80, 1.48) | 38 | 15 379 | 0.25 (0.18, 0.34) |
| Lipodystrophy | 14 | 4559.9 | 0.31 (0.18, 0.52) | 77 | 3712.0 | 2.07 (1.66, 2.59) | 98 | 15 238 | 0.64 (0.53, 0.78) |
| Mortality | 47 | 4656.2 | 1.01 (0.76, 1.34) | 34 | 3780.2 | 0.90 (0.64, 1.26) | 116 | 15 500 | 0.75 (0.62, 0.90) |
CI, confidence interval; PYFU, person‐years of follow‐up; RAL, raltegravir.
For reasons described in the Methods, adjusted RRs from fitting a Poisson regression are shown only for the endpoints of malignancies and death. The unadjusted analysis for cancer showed a 40% increase in the risk of malignancies for people who started RAL compared with people in the CONC cohort (Table 4a). However, after controlling for a number of potential confounders identified in Table 2, using standard regression adjustment, the estimated RR was closer to the null and not significant (comparing RAL with CONC: unadjusted RR 1.33; 95% CI 0.96–1.85; P = 0.08; adjusted RR 0.98; 95% CI 0.67–1.41; P = 0.90; Table 4a). The adjusted estimate obtained from fitting the quintiles of the PSs as a continuous covariate in the model was consistent with this estimate (RR 0.95; 95% CI 0.65–1.39; P = 0.79). Figure S1 a and b in the Supporting Information show the overlap in the PS distributions in the three cohorts which allowed a meaningful observational comparison using this strategy to control for confounding.
Table 4.
Unadjusted and adjusted relative risks (RRs) of (a) malignancies and (b) death from fitting a Poisson regression analysis using standard adjustment and propensity scores
| Unadjusted | Standard regression adjustment | Propensity scorea quintile regression adjustment | ||||
|---|---|---|---|---|---|---|
| RR (95% CI) | P‐value | RR (95% CI) | P‐value | RR (95% CI) | P‐value | |
| (a) | ||||||
| Historical control comparison | ||||||
| Historical control | 1.00 | 1.00 | 1.00 | |||
| Raltegravir | 0.92 (0.62, 1.38) | 0.703 | 0.73 (0.47, 1.14) | 0.169 | 0.81 (0.53, 1.26) | 0.350 |
| Concurrent control comparison | ||||||
| Concurrent control | 1.00 | 1.00 | 1.00 | |||
| Raltegravir | 1.33 (0.96, 1.85) | 0.086 | 0.95 (0.65, 1.39) | 0.787 | 0.98 (0.67, 1.41) | 0.897 |
| (b) | ||||||
| Historical control comparison | ||||||
| Historical control | 1.00 | 1.00 | 1.00 | |||
| Raltegravir | 1.12 (0.72, 1.74) | 0.608 | 0.87 (0.53, 1.43) | 0.593 | 0.90 (0.56, 1.45) | 0.660 |
| Concurrent control comparison | ||||||
| Concurrent control | 1.00 | 1.00 | 1.00 | |||
| Raltegravir | 1.35 (0.96, 1.89) | 0.084 | 1.14 (0.76, 1.72) | 0.523 | 1.22 (0.83, 1.80) | 0.320 |
Factors included in the vector to construct propensity scores in the two comparisons were: gender, ethnicity, country of origin, geographical region, hepatitis B virus coinfection, hepatitis C virus coinfection, age, CD4 count, time since CD4 count nadir, baseline viral load, length of enrolment in EuroSIDA, comorbidities, loss or accumulation of fat, opportunistic infection treatment use, time since starting antiretroviral therapy, number of previous treatment failures, number of previous nucleoside reverse transcriptase inhibitors, number of previous nonnucleoside reverse transcriptase inhibitors, number of previous protease inhibitors and number of previous drug classes used.
In order to test the hypothesis that the difference in the risk of malignancies between people receiving RAL and control cohorts might vary according to previous history of ART, we stratified the PS analysis to separate people who had previously experienced virological failure to a maximum of two drugs and those who had experienced failure to more than two drugs, and we found no evidence for such an interaction (P = 0.77). Similarly, we found no evidence for an interaction with baseline VL (when using a binary variable with a cut‐off of 400 copies/mL; P = 0.92).
Again, there was no difference in the incidence of death between the RAL and HIST cohorts. In the unadjusted analysis, there was a trend for a greater risk of mortality in the CONC cohort compared with the RAL cohort (RR 1.35; 95% CI 0.96–1.89; P = 0.08). However, this difference was attenuated and not significant after controlling for potential confounders (RR 1.14; 95% CI 0.76–1.72; P = 0.52; Table 4b). After controlling for the confounding factors included in the PS vector, the adjusted RRs were also closer to the value of 1.0 for the comparisons with both the HIST (RR 0.90; 95% CI 0.56–1.45; P = 0.66) and CONC (RR 1.22; 95% CI 0.83–1.80; P = 0.32) cohorts. In the analysis of the unadjusted RRs stratified by PS quintiles, again the results were similar (data not shown). After restricting the analysis to people who developed Hodgkin's or non‐Hodgkin's lymphomas (n = 37 events), the unadjusted RRs were 0.68 (95% CI 0.28–1.64; P = 0.39) when comparing the RAL cohort with the HIST cohort and 2.03 (95% CI 0.89–4.64; P = 0.093) when comparing the RAL cohort with the CONC cohort. The latter was attenuated to a RR of 1.73 (95% CI 0.66–4.57; P = 0.26) after controlling for PSs (quintile adjustment).
Discussion
Soon after the advent of cART, observational studies detected a major reduction in the incidence of Kaposi's sarcoma and non‐Hodgkin's lymphoma following cART initiation among treatment‐naïve HIV‐infected persons 16, 17, 19. The benefit of cART in reducing cancer risk could be explained by suppression of HIV replication, immune function improvement or reduction of inflammation. Some studies also showed a decreased incidence of malignancies not driven by infection with increased cART exposure 20, 21, 22, 23, 24. Therefore, global improvement of immune surveillance against cancer cells was also postulated as a likely mediator of the benefit of cART in reducing cancer risk 25. However, experimental data suggest that specific drugs, such as RAL, may have potential carcinogenic effects 14, 15, 26, 27.
Our analysis, conducted in a large observational cohort of HIV‐infected people receiving RAL, showed no evidence that using RAL was associated with an increased risk of cancer or death compared with other concomitant treatment strategies or a historical control group of individuals on ART. This is largely consistent with a recent meta‐analysis of two large randomized clinical trials of people using RAL (the STARTMRK and BENCHMRK trials) which reported a low risk of adverse events in RAL recipients and no difference in the RR of cancer comparing RAL with efavirenz (RR 0.75; 95% CI 0.30, 1.91) 28, 29. Our analysis extends these results to a longer follow‐up than that observed in these trials.
In a previous meta‐analysis of clinical trials comparing RAL with other agents, 46 participants developed 53 cases of cancer over the follow‐up period 28. Commonly detected cancers included Kaposi's sarcoma, anal or rectal cancer, cancer of the immune system and lymphomas. These were also cancers frequently reported in our analysis, but there was no particular diagnosis that appeared more frequently than expected on the basis of findings in similar HIV‐infected populations. If anything, there was a trend favouring RAL for the incidence of Kaposi's sarcoma (2% in RAL recipients) compared with the HIST (11%) and CONC (8%) cohorts, which should perhaps be investigated in larger studies. Similarly, the incidence of serious hepatic events was found in our analysis to be substantially lower in people using RAL (Table 3). Unfortunately, the small number of events did not allow us to further investigate in a multivariable analysis whether this held true after controlling for baseline imbalances in other factors.
In the trials, cancers were more common in ART‐experienced patients, possibly because of the patients’ weaker immunity, indicated by a generally lower CD4 count at enrolment 28, 29. Cancer typically tends to take years to develop but it is possible that tumours grow faster in people with advanced HIV disease, despite immune restoration, as a consequence of ongoing inflammation and increased coagulation associated with the use of ART 30, 31.
EuroSIDA represents an ideal setting in which to evaluate long‐term outcomes in a heterogeneous population with moderate to extensive pretreatment history and offered the possibility to identify suitable ‘control’ groups of patients who initiated RAL‐sparing treatment regimens. Indeed, well‐known limitations of randomized clinical trials are the selection of patients included (e.g. female patients and populations with lifestyle factors associated with nonadherence and mortality are under‐represented in trials) and lack of data on the long‐term risk of clinical events. Although our follow‐up was a little shorter than 3 years, this is longer than the follow‐up period of any trial previously conducted in people receiving RAL‐based regimens. Thus, with the caveat that other types of bias may be present (mainly because of a lack of randomization), analyses of observational data can provide insights into the risk of these long‐term outcomes. A recent French study (the Racing cohort) in 482 RAL‐treated patients exposed to RAL for >12 months and with available data, in a real‐life setting like ours, reported an elevated rate and variety of comorbidities and a variable adherence rate, but confirmed excellent efficacy and tolerability results, which were comparable to those observed in randomized registration trials 28, 29. In particular, of the 134 reported and treatment‐related adverse events (AEs), the symptoms most frequently described (> 5%) were myalgia (7%) and nausea (5%). A total of 34 serious AEs were reported, of which five were possibly or probably related to RAL. In general, a favourable tolerability profile has been demonstrated for RAL since the earlier registration studies, with headache and gastrointestinal complaints representing the most common reported AEs 28, 32, 33, 34, 35. In contrast, mild neuropsychiatric disorders have been infrequently recorded (usually reported on a subjective basis), and cases of rhabdomyolisis and hypersensitivity reactions have been extremely rare. Finally, there is little evidence that the serum lipid profile is modified by RAL, especially when compared with regimens containing older PIs and also efavirenz. On this basis, it has been suggested by Lee and Carr that RAL should be a preferred option for patients with a pre‐existing risk of cardiovascular diseases, altered serum lipid levels, metabolic syndrome, or changes in body fat composition (i.e. the lipodystrophy syndrome) 36. Manfredi R et al., in an analysis of a large hospital database in Italy, showed that grade 2–4 AEs attributable to RAL were neither directly observed nor reported in the self‐completed record of possible untoward events filled in every month by patients at the time of repeat prescription, and double‐checked by hospital pharmacists and physicians. In contrast, the discontinuation of some companion drugs at the time of RAL introduction and during the 12‐month follow‐up [especially NRTIs, some selected PIs and nonnucleoside reverse transcriptase inhibitors (NNRTIs), and enfuvirtide] had a favourable effect on the tolerability of the RAL‐based regimens 37. In addition, no clinical or laboratory evidence of autoimmune disorders associated with exposure to RAL‐containing cART regimens was found, as also reported in a recent 12‐month observational study 38.
In our analysis, discontinuation was examined in the RAL cohort alone, and we found a risk of discontinuation similar to those reported in the trials. The main reason for stopping RAL appeared to be physician's choice. Although the exact reason reported by the physician under ‘physician's choice’ is not specified in the EuroSIDA data collection report forms, it is conceivable that discontinuations because of toxicity induced by RAL would be filed under the reason ‘toxicity’, not ‘physician's choice’. There was also a nonnegligible frequency of people with missing information regarding the reason for stopping, which reflects the way the date are collected, as reasons are often not known or reported in clinical patient notes. We cannot rule out that some of these might be due to toxicity.
We also found that, at the time of treatment initiation, individuals in the RAL cohort tended to be different from those in the HIST and CONC control groups: RAL recipients were typically older with a longer time since CD4 count nadir, a lower baseline VL and a longer duration of ART. There were also differences by geographical region, previous drug history (RAL recipients had been treated more extensively in the past) and past comorbidities (patients in the RAL cohort were less well). However, once these differences had been taken into account using standard and more sophisticated statistical modelling, we found no difference in the risk of the two main clinical outcomes analysed (risk of malignancy and death) compared with those starting other historical or concurrent cART regimens. Results were similar in people with little and those with more extensive pretreatment drug history, although the power to study such an interaction was low. Further studies with larger numbers of malignancies need to be conducted in order to determine whether the risk of cancer might vary according to the extent of previous virological failure or the extent of exposure to RAL (in vitro risk was highest in those with suboptimal exposure)..
Before drawing firm conclusions, it should be noted that ours are observational cohort study data and hence need to be interpreted practically, realistically and with full knowledge of their limitations and inherent potential biases. While there are extensive data quality programmes in place within EuroSIDA, it remains an observation of routine clinical practice across Europe. As a consequence, although it was reassuring to see that our analyses using a range of techniques aimed at reducing bias caused by confounding, led to similar results, we are not able to exclude the possibility of confounding by indication or other bias introduced by unmeasured confounders (such as traditional lifestyle risk factors for cancer such as smoking) that can only be truly accounted for in a randomized clinical trial. In the specific case of patients who are about to receive chemotherapy, patients are typically switched to RAL to minimize pharmacokinetic interactions. Some of the residual excess risk of death seen in the RAL vs. CONC analysis might be explained by this important unmeasured source of confounding.
Although the list of clinical diagnoses collected in the database follows the accepted classification of the WHO ICD, a central pathological review was not conducted. Specifically, as data are collected in infectious diseases and internal medicine departments, there is a risk of under‐reporting of non‐AIDS‐defining cancers, when there is no linkage with cancer registries. There is also a risk of misclassification, in particular for cancers such as anal cancer, which was particularly frequent in all three cohorts. Furthermore, for the primary endpoint of malignancies, because of the relatively small number of events observed, a certain degree of uncertainty around our estimates remains, calling for additional confirmatory analyses in larger data sets. In our analysis, loss to follow‐up and death from causes other than cancer may act as competing risks which have not be accounted for. The small overall number of cancers did not allow a fully adjusted comparison after restriction to specific diagnoses. However, because direct HIV‐1‐related integrase inhibitor‐induced genome alterations are expected to affect only HIV‐1‐permissive cells, especially CD4 T cells, macrophages and glia cells 14, 15, we restricted the analysis to 37 events of Hodgkin's or non‐Hodgkin's lymphomas, and the unadjusted RRs were similar. Overall, there was no evidence that results varied by level of HIV replication. Neither host DNA or INSTI resistance data were available to evaluate the prevalence of RAL‐induced mutations potentially associated with an increased risk of cancer.
In conclusion, our findings show that use of RAL does not seem to be associated with an increased risk of cancer or reduced survival when compared with the cancer and survival rates seen in people treated with alternative regimens in routine clinical care. With all the caveats of a comparison conducted in observational settings, our data, confirm that RAL is a safe and valid therapeutic option, especially for patients with a history of multiple failures, presenting with a broad spectrum of HIV resistance mutations, or multiple intolerances or contraindications to other antiretroviral drugs and combinations 39.
Supporting information
Table S1. Breakdown of cancer locations by cohort.
Figure S1. (a) Overlap in the distribution of propensity scores comparing Raltegravir and Historical cohorts. (b) Overlap in the distribution of propensity scores comparing Raltegravir and Concurrent cohorts.
Acknowledgment
Funding: EuroSIDA was supported by the European Union's Seventh Framework Programme for research, technological development and demonstration under EuroCoord grant agreement no. 260694. Current support includes unrestricted grants from Bristol‐Myers Squibb, Gilead, GlaxoSmithKline LLC, Janssen R&D, Merck and Co. Inc. and Pfizer Inc. The participation of centres from Switzerland was supported by The Swiss National Science Foundation (Grant 108787). The study is also supported by a grant (grant number DNRF126) from the Danish National Research Foundation.
Conflict of Interest: Amanda Mocroft ‐ she received travel support, honoraria, consultancy and lecture fees from Pfizer, ViiV, Gilead, BI, BMS, Merck and Wragge LLC.
the EuroSIDA Study Group
Members of the multi‐centre EuroSIDA Study Group are as follows (national coordinators in parentheses). Argentina: (M. Losso) and M. Kundro, Hospital J.M. Ramos Mejia, Buenos Aires. Austria: (B. Schmied), Pulmologisches Zentrum der Stadt Wien, Vienna; R. Zangerle, Medical University Innsbruck, Innsbruck. Belarus: (I. Karpov) and A. Vassilenko, Belarus State Medical University, Minsk; V. M. Mitsura, Gomel State Medical University, Gomel; D. Paduto, Regional AIDS Centre, Svetlogorsk. Belgium: (N. Clumeck), S. De Wit and M. Delforge, Saint‐Pierre Hospital, Brussels; E. Florence, Institute of Tropical Medicine, Antwerp; L. Vandekerckhove, University Ziekenhuis Gent, Gent. Bosnia‐Herzegovina: (V. Hadziosmanovic), Klinicki Centar Univerziteta Sarajevo, Sarajevo. Croatia: (J. Begovac), University Hospital of Infectious Diseases, Zagreb. Czech Republic: (L. Machala) and D. Jilich, Faculty Hospital Bulovka, Prague; D. Sedlacek, Charles University Hospital, Plzen. Denmark: G. Kronborg and T. Benfield, Hvidovre Hospital, Copenhagen; J. Gerstoft and T. Katzenstein, Rigshospitalet, Copenhagen; N. F. Møller and C. Pedersen, Odense University Hospital, Odense; L. Ostergaard, Skejby Hospital, Aarhus; L. Wiese, Roskilde Hospital, Roskilde; L. N. Nielsen, Hillerod Hospital, Hillerod. Estonia: (K. Zilmer), West‐Tallinn Central Hospital, Tallinn; J. Smidt, Nakkusosakond Siseklinik, Kohtla‐Järve. Finland: (M. Ristola) and I. Aho, Helsinki University Central Hospital, Helsinki. France: (J.‐P. Viard), Hôtel‐Dieu, Paris; P.‐M. Girard, Hospital Saint‐Antoine, Paris; C. Pradier and E. Fontas, Hôpital de l'Archet, Nice; C. Duvivier, Hôpital Necker‐Enfants Malades, Paris. Germany: (J. Rockstroh), Universitäts Klinik Bonn, Bonn; R. Schmidt, Medizinische Hochschule Hannover, Hannover; O. Degen, University Medical Center Hamburg‐Eppendorf, Infectious Diseases Unit, Hamburg; H. J. Stellbrink, IPM Study Center, Hamburg; C. Stefan, JW Goethe University Hospital, Frankfurt; J. Bogner, Medizinische Poliklinik, Munich; G. Fätkenheuer, Universität Köln, Cologne. Georgia: (N. Chkhartishvili), Infectious Diseases, AIDS & Clinical Immunology Research Center, Tbilisi. Greece: P. Gargalianos, G. Xylomenos and P. Lourida, Athens General Hospital, Athens; H. Sambatakou, Ippokration General Hospital, Athens. Hungary: (J. Szlávik), Szent Lásló Hospital, Budapest. Iceland: (M. Gottfredsson), Landspitali University Hospital, Reykjavik. Ireland: (F. Mulcahy), St. James's Hospital, Dublin. Israel: (I. Yust), D. Turner and M. Burke, Ichilov Hospital, Tel Aviv; E. Shahar and G. Hassoun, Rambam Medical Center, Haifa; H. Elinav and M. Haouzi, Hadassah University Hospital, Jerusalem; D. Elbirt and Z. M. Sthoeger, AIDS Center (Neve Or), Jerusalem. Italy: (A. D'Arminio Monforte), Istituto Di Clinica Malattie Infettive e Tropicale, Milan; R. Esposito, I. Mazeu and C. Mussini, Università Modena, Modena; F. Mazzotta and A. Gabbuti, Ospedale S. Maria Annunziata, Firenze; V. Vullo and M. Lichtner, University di Roma la Sapienza, Rome; M. Zaccarelli, A. Antinori, R. Acinapura and M. Plazzi, Istituto Nazionale Malattie Infettive Lazzaro Spallanzani, Rome; A. Lazzarin, A. Castagna and N. Gianotti, Ospedale San Raffaele, Milan; M. Galli and A. Ridolfo, Osp. L. Sacco, Milan. Latvia: (B. Rozentale), Infectology Centre of Latvia, Riga. Lithuania: (V. Uzdaviniene), Vilnius University Hospital Santariskiu Klinikos, Vilnius; R. Matulionyte, Center of Infectious Diseases, Vilnius University Hospital Santariskiu Klinikos, Vilnius. Luxembourg: (T. Staub) and R. Hemmer, Centre Hospitalier, Luxembourg. The Netherlands: (P. Reiss), Academisch Medisch Centrum bij de Universiteit van Amsterdam, Amsterdam. Norway: (V. Ormaasen), A. Maeland and J. Bruun, Ullevål Hospital, Oslo. Poland: (B. Knysz), J. Gasiorowski and M. Inglot, Medical University, Wroclaw; A. Horban and E. Bakowska, Centrum Diagnostyki i Terapii AIDS, Warsaw; R. Flisiak and A. Grzeszczuk, Medical University, Bialystok; M. Parczewski, K. Maciejewska and B. Aksak‐Was, Medical Univesity, Szczecin; M. Beniowski and E. Mularska, Osrodek Diagnostyki i Terapii AIDS, Chorzow; T. Smiatacz and M. Gensing, Medical University, Gdansk; E. Jablonowska, E. Malolepsza and K. Wojcik, Wojewodzki Szpital Specjalistyczny, Lodz; I. Mozer‐Lisewska, Poznan University of Medical Sciences, Poznan. Portugal: (L. Caldeira), Hospital Santa Maria, Lisbon; K. Mansinho, Hospital de Egas Moniz, Lisbon; F. Maltez, Hospital Curry Cabral, Lisbon. Romania: (R. Radoi) and C. Oprea, Spitalul de Boli Infectioase si Tropicale: Dr. Victor Babes, Bucarest. Russia: (A. Panteleev) and O. Panteleev, St Petersburg AIDS Centre, St Petersburg; A. Yakovlev, Medical Academy Botkin Hospital, St Petersburg; T. Trofimora, Novgorod Centre for AIDS, Novgorod; I. Khromova, Centre for HIV/AIDS and Infectious Diseases, Kaliningrad; E. Kuzovatova, Nizhny Novgorod Scientific and Research Institute of Epidemiology and Microbiology named after Academician I. N. Blokhina, Nizhny Novogrod; E. Borodulina and E. Vdoushkina, Samara State Medical University, Samara. Serbia: (D. Jevtovic), The Institute for Infectious and Tropical Diseases, Belgrade. Slovenia: (J. Tomazic), University Clinical Centre Ljubljana, Ljubljana. Spain: (J. M. Gatell) and J. M. Miró, Hospital Clinic Universitari de Barcelona, Barcelona; S. Moreno and J. M. Rodriguez, Hospital Ramon y Cajal, Madrid; B. Clotet, A. Jou, R. Paredes, C. Tural, J. Puig and I. Bravo, Hospital Germans Trias i Pujol, Badalona; P. Domingo, M. Gutierrez, G. Mateo and M. A. Sambeat, Hospital Sant Pau, Barcelona; J. M. Laporte, Hospital Universitario de Alava, Vitoria‐Gasteiz.
Sweden: (K. Falconer), A. Thalme and A. Sonnerborg, Karolinska University Hospital, Stockholm; A. Blaxhult, Venhälsan‐Sodersjukhuset, Stockholm; L. Flamholc, Malmö University Hospital, Malmö. Switzerland: (A. Scherrer) and R. Weber, University Hospital Zurich, Zurich; M. Cavassini, University Hospital Lausanne, Lausanne; A. Calmy, University Hospital Geneva, Geneva; H. Furrer, University Hospital Bern, Bern; M. Battegay, University Hospital Basel, Basel; P. Schmid, Cantonal Hospital St. Gallen, St. Gallen. Ukraine: A. Kuznetsova, Kharkov State Medical University, Kharkov; G. Kyselyova, Crimean Republican AIDS Centre, Simferopol; M. Sluzhynska, Lviv Regional HIV/AIDS Prevention and Control CTR, Lviv. UK: (B. Gazzard), St. Stephen's Clinic, Chelsea and Westminster Hospital, London; A. M. Johnson, E. Simons and S. Edwards, Mortimer Market Centre, London; A. Phillips, M. A. Johnson and A. Mocroft, Royal Free and University College Medical School (Royal Free Campus), London; C. Orkin, Royal London Hospital, London; J. Weber and G. Scullard, Imperial College School of Medicine at St. Mary's, London; A. Clarke, Royal Sussex County Hospital, Brighton; C. Leen, Western General Hospital, Edinburgh.
The following centres have previously contributed data to EuroSIDA: Infectious Diseases Hospital, Sofia, Bulgaria; Hôpital de la Croix Rousse, Lyon, France; Hôpital de la Pitié‐Salpétière, Paris, France; Unité INSERM, Bordeaux, France; Hôpital Edouard Herriot, Lyon, France; Bernhard Nocht Institut für Tropenmedizin, Hamburg, Germany; 1st I.K.A Hospital of Athens, Athens, Greece; Ospedale Riuniti, Divisione Malattie Infettive, Bergamo, Italy; Ospedale di Bolzano, Divisione Malattie Infettive, Bolzano, Italy; Ospedale Cotugno, III Divisione Malattie Infettive, Napoli, Italy; Dérer Hospital, Bratislava, Slovakia; Hospital Carlos III, Departamento de Enfermedades Infecciosas, Madrid, Spain; Kiev Centre for AIDS, Kiev, Ukraine; Luhansk State Medical University, Luhansk, Ukraine; Odessa Region AIDS Center, Odessa, Ukraine.
EuroSIDA Steering Committee
Steering Committee: J. Gatell, B. Gazzard, A. Horban, I. Karpov, M. Losso, A. d'Arminio Monforte, C. Pedersen, M. Ristola, A. Phillips, P. Reiss, J. Lundgren and J. Rockstroh.
Chair: J. Rockstroh.
Study Co‐leads: A. Mocroft and O. Kirk.
EuroSIDA representatives to EuroCoord: O. Kirk, A. Mocroft, P. Reiss, A. Cozzi‐Lepri, R. Thiebaut, J. Rockstroh, D. Burger, R. Paredes and L. Peters.
EuroSIDA staff
Coordinating Centre staff: O. Kirk, L. Peters, C. Matthews, A. H. Fischer, A. Bojesen, D. Raben, D. Kristensen, K. Grønborg Laut, J. F. Larsen and D. Podlekareva.
Statistical staff: A. Mocroft, A. Phillips, A. Cozzi‐Lepri, L. Shepherd, A. Schultze and S. Amele.
Coauthorships
Please find the SOP for identifying coauthors in the EuroSIDA Study at http://www.chip.dk/Portals/0/files/Eurosida/EuroSIDA/SOP%20coauthorship.pdf?timestamp=1464082845266.
Participation criteria
Please find the criteria for participating in the EuroSIDA study at http://www.chip.dk/Portals/0/files/Eurosida/EuroSIDA/Criteria%20for%20participation%20in%20EuroSIDA%20and%20for%20site%20inactivation.pdf?timestamp=1464080873590.
Contributor Information
A Cozzi‐Lepri, Email: a.cozzi-lepri@ucl.ac.uk.
the EuroSIDA Study Group:
M. Kundro, B. Schmied, I. Karpov, A. Vassilenko, V. M. Mitsura, D. Paduto, N. Clumeck, S. De Wit, M. Delforge, E. Florence, L. Vandekerckhove, V. Hadziosmanovic, J. Begovac, D. Sedlacek, G. Kronborg, T. Benfield, J. Gerstoft, T. Katzenstein, N. F. Møller, C. Pedersen, L. Ostergaard, L. Wiese, L. N. Nielsen, I. Aho, J.‐P. Viard, P.‐M. Girard, E. Fontas, C. Duvivier, J. Rockstroh, R. Schmidt, O. Degen, H. J. Stellbrink, C. Stefan, J. Bogner, N. Chkhartishvili, P. Gargalianos, G. Xylomenos, P. Lourida, J. Szlávik, F. Mulcahy, D. Turner, M. Burke, E. Shahar, G. Hassoun, H. Elinav, M. Haouzi, D. Elbirt, Z. M. Sthoeger, A. D'Arminio Monforte, R. Esposito, I. Mazeu, C. Mussini, F. Mazzotta, A. Gabbuti, V. Vullo, M. Lichtner, M. Zaccarelli, A. Antinori, R. Acinapura, M. Plazzi, A. Lazzarin, A. Castagna, N. Gianotti, M. Galli, A. Ridolfo, V. Uzdaviniene, R. Matulionyte, T. Staub, R. Hemmer, P. Reiss, V. Ormaasen, A. Maeland, J. Bruun, B. Knysz, J. Gasiorowski, M. Inglot, A. Horban, E. Bakowska, A. Grzeszczuk, M. Parczewski, K. Maciejewska, B. Aksak‐Was, M. Beniowski, E. Mularska, T. Smiatacz, M. Gensing, E. Jablonowska, E. Malolepsza, K. Wojcik, I. Mozer‐Lisewska, L. Caldeira, K. Mansinho, F. Maltez, C. Oprea, A. Panteleev, O. Panteleev, A. Yakovlev, T. Trofimora, E. Kuzovatova, E. Borodulina, E. Vdoushkina, D. Jevtovic, J. Tomazic, J. M. Gatell, J. M. Miró, S. Moreno, J. M. Rodriguez, B. Clotet, A. Jou, R. Paredes, C. Tural, J. Puig, I. Bravo, P. Domingo, M. Gutierrez, G. Mateo, M. A. Sambeat, J. M. Laporte, K. Falconer, A. Thalme, A. Sonnerborg, A. Blaxhult, L. Flamholc, A. Scherrer, R. Weber, M. Cavassini, A. Calmy, H. Furrer, M. Battegay, A. Kuznetsova, G. Kyselyova, M. Sluzhynska, B. Gazzard, A. M. Johnson, E. Simons, S. Edwards, A. Phillips, M. A. Johnson, C. Orkin, J. Weber, G. Scullard, A. Clarke, C. Leen, R. Thiebaut, D. Burger, L. Peters, C. Matthews, A. H. Fischer, A. Bojesen, D. Raben, D. Kristensen, K. Grønborg Laut, J. F. Larsen, D. Podlekareva, L. Shepherd, A. Schultze, and S. Amele
References
- 1. Burger DM. Raltegravir: a review of its pharmacokinetics, pharmacology and clinical studies. Expert Opin Drug Metab Toxicol 2010; 6: 1151–1160. [DOI] [PubMed] [Google Scholar]
- 2. Blanco JL, Martinez‐Picado J. HIV integrase inhibitors in ART‐experienced patients. Curr Opin HIV AIDS 2012; 7: 415–421. [DOI] [PubMed] [Google Scholar]
- 3. Adams JL, Greener BN, Kashuba AD. Pharmacology of HIV integrase inhibitors. Curr Opin HIV AIDS 2012; 7: 390–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guidelines for the use of antiretroviral agents in HIV‐1 infected adults and adolescents. Available at http://www.aidsinfo.nih.gov/guidelines (accessed 27 May 2013).
- 5. Wittkop L, Breith D, Da Silva D et al Virological and immunological response in HIV‐1‐infected patients with multiple treatment failures receiving raltegravir and optimized background therapy, ANRS CO3 Aquitaine Cohort. J Antimicrob Chemother 2009; 63: 1251–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gallien S, Braun J, Delaugerre C et al Efficacy and safety of raltegravir in treatment‐experienced HIV‐1‐infected patients switching from enfuvirtide‐ based regimens: 48 week results of the randomized EASIER ANRS 138 trial. J Antimicrob Chemother 2011; 66: 2099–2106. [DOI] [PubMed] [Google Scholar]
- 7. Gu WG. Newly approved integrase inhibitors for clinical treatment of AIDS. Biomed Pharmacother 2014; 68: 917–921. [DOI] [PubMed] [Google Scholar]
- 8. Bonnet F, Chêne G. Evolving epidemiology of malignancies in HIV. Curr Opin Oncol 2008; 20: 534–540. [DOI] [PubMed] [Google Scholar]
- 9. Bower M, Fisher M, Hill T et al CD4 counts and the risk of systemic non‐Hodgkin's lymphoma in individuals with HIV in the UK. Haematologica 2009; 94: 875–880. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baker JV, Peng G, Rapkin J et al CD4+ count and risk of non‐AIDS diseases following initial treatment for HIV infection. AIDS 2008; 22: 841–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Engels EA. Non‐AIDS‐defining malignancies in HIV‐infected persons: etiologic puzzles, epidemiologic perils, prevention opportunities. AIDS 2009; 23: 875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cooper D, Steigbigel R, Lennox J et al Review of cancer incidence in raltegravir trials In: Program and abstracts of the 16th Conference on Retroviruses and Opportunistic Infections, Montreal, Canada, February 8–11 2009. [Abstract 859]. [Google Scholar]
- 13. Teppler H, Brown DD, Leavitt RY et al Long‐term safety from the raltegravir clinical development program. Curr HIV Res 2011; 9: 40–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Varadarajan J, McWilliams MJ, Mott BT, Thomas CJ, Smith SJ, Hughes SH. Drug resistant integrase mutants cause aberrant HIV integrations. Retrovirology 2016; 13: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Varadarajan J, McWilliams MJ, Hughes SH. Treatment with suboptimal doses of raltegravir leads to aberrant HIV‐1 integrations. Proc Natl Acad Sci USA 2013; 110: 14747–14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jacobson LP, Yamashita TE, Detels R et al Impact of potent antiretroviral therapy on the incidence of Kaposi's sarcoma and non‐Hodgkin's lymphomas among HIV‐1‐infected individuals. Multicenter AIDS Cohort Study. J Acquir Immune Defic Syndr 1999; 21 (Suppl 1): S34–S41. [PubMed] [Google Scholar]
- 17. International Collaboration on HIV and Cancer . Highly active antiretroviral therapy and incidence of cancer in human immunodeficiencyvirus‐infected adults. J Natl Cancer Inst 2000; 92: 1823–1830. [DOI] [PubMed] [Google Scholar]
- 18. Clifford GM, Polesel J, Rickenbach M et al Cancer risk in the Swiss HIV Cohort Study: associations with immunodeficiency, smoking, and highly active antiretroviral therapy. J Natl Cancer Inst 2005; 97: 425–432. [DOI] [PubMed] [Google Scholar]
- 19. Polesel J, Clifford GM, Rickenbach M et al Non‐Hodgkin lymphoma incidence in the Swiss HIV Cohort Study before and after highly active antiretroviral therapy. AIDS 2008; 22: 301–306. [DOI] [PubMed] [Google Scholar]
- 20. Burgi A, Brodine S, Wegner S et al Incidence and risk factors for the occurrence of non‐AIDS‐defining cancers among human immunodeficiency virus‐infected individuals. Cancer 2005; 104: 1505–1511. [DOI] [PubMed] [Google Scholar]
- 21. Biggar RJ, Chaturvedi AK, Goedert JJ et al AIDS‐related cancer and severity of immunosuppression in persons with AIDS. J Natl Cancer Inst 2007; 99: 962–972. [DOI] [PubMed] [Google Scholar]
- 22. Hessol NA, Pipkin S, Schwarcz S et al The impact of highly active antiretroviral therapy on non‐AIDS‐defining cancers among adults with AIDS. Am J Epidemiol 2007; 165: 1143–1153. [DOI] [PubMed] [Google Scholar]
- 23. Monforte Ad, Abrams D, Pradier C et al HIV‐induced immunodeficiency and mortality from AIDS‐defining and non‐AIDS‐defining malignancies. AIDS 2008; 21: 2143–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Petoumenos K1, van Leuwen MT, Vajdic CM, Woolley I, Chuah J, Templeton DJ, Law MG Australian HIV Observational Database. Cancer, immunodeficiency and antiretroviral treatment: results from the Australian HIV Observational Database (AHOD). HIV Med 2013. Feb; 14(2): 77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Borges AH, Dubrow R, Silverberg MJ. Factors contributing to risk for cancer among HIV‐infected individuals, and evidence that earlier combination antiretroviral therapy will alter this risk. Curr Opin HIV AIDS 2014; 9: 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Olivero OA, Anderson LM, Diwan BA et al Transplacental effects of 30‐ azido‐20,30‐dideoxythymidine (AZT): tumorigenicity in mice and genotoxicity in mice and monkeys. J Natl Cancer Inst 1997; 89: 1602–1608. [DOI] [PubMed] [Google Scholar]
- 27. National Toxicology Program . Toxicology and carcinogenesis studies of mixtures of 30‐azido‐30‐deoxythymidine (AZT), lamivudine (3TC), nevirapine (NVP), and nelfinavir mesylate (NFV) (Cas Nos. 30516‐87‐1, 134678‐17‐4, 129618‐40‐2, 159989‐65‐8) in B6C3F1 mice (transplacental exposure studies). Natl Toxicol Program Tech Rep Ser 2013; 569: 1–212. [PubMed] [Google Scholar]
- 28. Canada Source's for HIV and hepatitis C information. Treatment Update 173, May/June 2009. Available at http://www.catie.ca/en/treatmentupdate/treatmentupdate-173/cancer/concern-about-cancer-risk-raltegravir (accessed 30 June 2009).
- 29. Mocroft A, Vella S, Benfield TL et al Changing patterns of mortality across Europe in patients infected with HIV‐1. Lancet 1998; 352: 1725–1730. [DOI] [PubMed] [Google Scholar]
- 30. Steigbigel RT, Cooper DA, Teppler H et al Long‐term efficacy and safety of raltegravir combined with optimized background therapy in treat‐ ment‐experienced patients with drug‐resistant HIV infection: week 96 results of the BENCHMRK 1 and 2 Phase III trials. Clin Infect Dis 2010; 50: 605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Borges ÁH, Silverberg MJ, Wentworth D et al Predicting risk of cancer during HIV infection: the role of inflammatory and coagulation biomarkers. AIDS 2013; 27: 1433–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hamlyn E, Fidler S, Stöhr W et al Interleukin‐6 and D‐dimer levels at seroconversion as predictors of HIV‐1 disease progression. AIDS 2014; 28: 869–874. [DOI] [PubMed] [Google Scholar]
- 33. Pugliese P, Poizot‐Martin I, Billaud E et al Observational study of HIV‐1‐infected patients treated with an ARV combination therapy including ralte‐ gravir: the RACING cohort results at month 12. J Int AIDS Soc 2012; 15 (Suppl. 4): 18398. [Google Scholar]
- 34. Lennox JL. The use of HIV‐1 integrase inhibitors in antiretroviral naïve patients. Curr Opin HIV AIDS 2012; 7: 409–414. [DOI] [PubMed] [Google Scholar]
- 35. Rokas KE, Bookstaver PB, Shamroe CL et al Role of raltegravir in HIV‐1 management. Ann Pharmacother 2012; 46: 578–589. [DOI] [PubMed] [Google Scholar]
- 36. Lee FJ, Carr A. Tolerability of HIV integrase in‐ hibitors. Curr Opin HIV AIDS 2012; 7: 422–428. [DOI] [PubMed] [Google Scholar]
- 37. Manfredi R, Calza L, Marinacci G et al Raltegravir use prospectively assessed in a major HIV outpatient clinic in Italy: sample population, virological‐immunological activity, and tolerability profile. Infez Med 2014; 22: 288–295. [PubMed] [Google Scholar]
- 38. Baroncelli S, Mezzaroma I, Fantauzzi A et al No evidence of autoimmune disorders in antiretroviral‐experienced HIV‐1‐infected subjects after long‐ term treatment with raltegravir. Antivir Ther 2013; 18: 321–327. [DOI] [PubMed] [Google Scholar]
- 39. Nozza S, Galli L, Visco F et al Raltegravir, maraviroc, etravirine; an effective protease inhibitor and nucleoside reverse transcriptase inhibitor‐spa‐ ring regimen for salvage therapy in HIV‐infected pa‐ tients with triple‐class experience. AIDS 2010; 24: 924–928. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Breakdown of cancer locations by cohort.
Figure S1. (a) Overlap in the distribution of propensity scores comparing Raltegravir and Historical cohorts. (b) Overlap in the distribution of propensity scores comparing Raltegravir and Concurrent cohorts.