

ABSTRACT
G‐protein subunit α‐11 (Gα11) couples the calcium‐sensing receptor (CaSR) to phospholipase C (PLC)‐mediated intracellular calcium (Ca2+ i) and mitogen‐activated protein kinase (MAPK) signaling, which in the parathyroid glands and kidneys regulates parathyroid hormone release and urinary calcium excretion, respectively. Heterozygous germline loss‐of‐function Gα11 mutations cause familial hypocalciuric hypercalcemia type 2 (FHH2), for which effective therapies are currently not available. Here, we report a novel heterozygous Gα11 germline mutation, Phe220Ser, which was associated with hypercalcemia in a family with FHH2. Homology modeling showed the wild‐type (WT) Phe220 nonpolar residue to form part of a cluster of hydrophobic residues within a highly conserved cleft region of Gα11, which binds to and activates PLC; and predicted that substitution of Phe220 with the mutant Ser220 polar hydrophilic residue would disrupt PLC‐mediated signaling. In vitro studies involving transient transfection of WT and mutant Gα11 proteins into HEK293 cells, which express the CaSR, showed the mutant Ser220 Gα11 protein to impair CaSR‐mediated Ca2+ i and extracellular signal‐regulated kinase 1/2 (ERK) MAPK signaling, consistent with diminished activation of PLC. Furthermore, engineered mutagenesis studies demonstrated that loss of hydrophobicity within the Gα11 cleft region also impaired signaling by PLC. The loss‐of‐function associated with the Ser220 Gα11 mutant was rectified by treatment of cells with cinacalcet, which is a CaSR‐positive allosteric modulator. Furthermore, in vivo administration of cinacalcet to the proband harboring the Phe220Ser Gα11 mutation, normalized serum ionized calcium concentrations. Thus, our studies, which report a novel Gα11 germline mutation (Phe220Ser) in a family with FHH2, reveal the importance of the Gα11 hydrophobic cleft region for CaSR‐mediated activation of PLC, and show that allosteric CaSR modulation can rectify the loss‐of‐function Phe220Ser mutation and ameliorate the hypercalcemia associated with FHH2. © 2017 The Authors. Journal of Bone and Mineral Research Published by Wiley Periodicals Inc.
Keywords: PARATHYROID‐RELATED DISORDERS, DISORDERS OF CALCIUM/PHOSPHATE METABOLISM, THERAPEUTICS, CELL/TISSUE SIGNALING, ENDOCRINE PATHWAYS
Introduction
G‐protein subunit alpha‐11 (Gα11) is a major downstream signaling partner of the cell‐surface calcium‐sensing receptor (CaSR), which is a family C G‐protein coupled receptor (GPCR) that plays a pivotal role in the parathyroid and renal regulation of extracellular calcium (Ca2+ e) concentrations.1 Gα11 belongs to the Gq/11 class of G‐proteins that enhance phospholipase C (PLC) activity, thereby leading to formation of: inositol 1,4,5‐trisphosphate (IP3), which induces rapid increases in intracellular Ca2+ (Ca2+ i) concentrations; and enhances diacylglycerol (DAG) formation, which in turn activates protein kinase C and the mitogen‐activated protein kinase (MAPK) signaling cascade.2, 3 These signal transduction events allow the CaSR to respond to small fluctuations in the prevailing Ca2+ e concentrations ([Ca2+]e) and to induce alterations in parathyroid hormone (PTH) secretion and urinary calcium excretion.4
The identification of germline heterozygous mutations of Gα11, which is encoded by the GNA11 gene on chromosome 19p13.3, that result in familial hypocalciuric hypercalcemia (FHH) has demonstrated the importance of this G‐protein subunit in Ca2+ e homeostasis.5 FHH is an autosomal dominant disorder characterized by lifelong elevations of serum calcium concentrations in association with normal or mildly raised serum PTH concentrations and low urinary calcium excretion (calcium‐to‐creatinine clearance ratio <0.01).4 FHH comprises three genetically distinct conditions, designated as FHH types 1 to 3. FHH1 (OMIM #145980) is caused by loss‐of‐function CaSR mutations.1 FHH2 (OMIM #145981) is caused by loss‐of‐function Gα11 mutations; to date, three FHH2‐associated mutations have been reported, comprising two missense mutations, Thr54Met and Leu135Gln, and an in‐frame isoleucine deletion at codon 200 (Ile200del)5, 6 (Supporting Fig. 1). FHH3 (OMIM #600740) is caused by loss‐of‐function mutations of the adaptor protein‐2 sigma subunit‐1 (AP2S1) gene, encoding AP2σ, which is involved in the clathrin‐mediated endocytosis of GPCRs, such as the CaSR.1
Positive allosteric modulators of the CaSR, known as calcimimetics, represent a targeted therapy for patients with symptomatic FHH. Indeed, cinacalcet, a licensed calcimimetic drug, has been used to ameliorate symptomatic hypercalcemia in patients with FHH1 and FHH3.7, 8, 9 However, such effects of cinacalcet in FHH2 patients have not been reported, although cinacalcet has been shown in vitro, to rectify signaling abnormalities associated with FHH2‐causing Gα11 mutations.10 Here, we report the effectiveness of cinacalcet in ameliorating the signaling defects and hypercalcemia due to a previously unreported FHH2‐associated Gα11 mutation, Phe220Ser.
Patients and Methods
Case report
The proband (individual II.1, Fig. 1 A) was a 33‐year‐old male who presented with headaches, constipation, and pruritus. Biochemical investigations revealed hypercalcemia, hypophosphatemia, raised plasma PTH concentrations, and a low urine calcium‐to‐creatinine ratio (Table 1). A DXA scan showed normal bone mineral density values at the spine and hip (Table 1). A skin biopsy, undertaken to investigate the pruritus, demonstrated folliculitis (Table 1). His father, and three of his four children, also had hypercalcemia, constipation, and/or headaches (Table 1). Two of his affected children had eczema, and the proband's father had sustained an osteoporotic vertebral fracture. The familial hypercalcemia and reduced urinary calcium excretion were consistent with FHH; however, CASR or AP2S1 mutations were not identified. Informed consent was obtained from individuals and where appropriate, parents and guardians of children, using protocols approved by the Research Ethics Committee of the Helsinki University Hospital.
Figure 1.
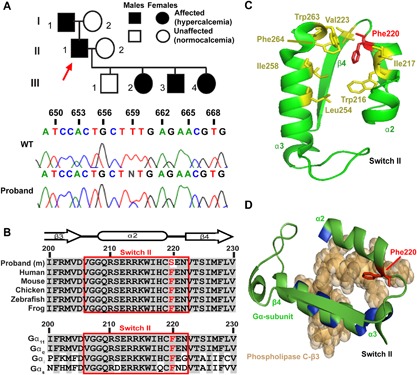
Identification and structural assessment of a Phe220Ser Gα11 mutation in a family with familial hypocalciuric hypercalcemia type 2. (A) The family comprised of 5 affected and 3 unaffected members (top). The proband (individual II.1) is indicated by an arrow. A heterozygous T‐to‐C transition at nucleotide c.659, predicted to result in a missense substitution of the WT Phe to a mutant (m) Ser at codon 220 of the Gα11 protein, was identified in the proband and confirmed to cosegregate with hypercalcemia by Sanger DNA sequence analysis. (B) Multiple protein sequence alignment of residues comprising the β3‐α2 loop, α2‐helix, and α2‐β4 loop that form switch II (red box) of Gα11 orthologs (top) and Gα‐subunit paralogs (bottom). Conserved residues are shown in gray, and WT (Phe, F) and mutant (Ser, S) residues are shown in red. (C) Structural model showing that Phe220 of Gα11 is part of a hydrophobic cluster of amino acids (yellow) within switch II and the α3‐helix, which stabilize the region in a conformation that facilitates effector binding. (D) Close‐up view of Phe220 residue, within the switch II region of the GTPase domain of Gα11, in complex with phospholipase C‐β3 (brown space‐filling model), with directly interacting residues of the α2 and α3 helices colored blue.
Table 1.
Clinical and Biochemical Findings in Available Affected Members of the Family With the Phe220Ser Gα11 Mutation
| Individuals | ||||
|---|---|---|---|---|
| Variable | II.1 | III.2 | III.3 | III.4 |
| Sex | Male | Female | Male | Female |
| Age at diagnosis | 33 years | 7 weeks | At birth | 18 months |
| Associated clinical features | Headaches, pruritus, constipation a | Constipation, scoliosis, headaches b | Premature birth, c eczema, constipation | Constipation, eczema |
| Serum biochemistry d | ||||
| Ionized calcium (mmol/L) | 1.42 | 1.41 | 1.46 | 1.46 |
| Phosphate (mmol/L) | 0.50 | 1.98 | 1.81 | 1.62 |
| Alkaline phosphatase (U/L) | 77 | 275 | 461 | 243 |
| Magnesium (mmol/L) | 0.78 | 1.00 | 0.90 | 0.93 |
| Creatinine (μmol/L) | 76 | 26 | 78 | 26 |
| PTH (ng/L) | 98 | 39 | 100 | 40 |
| 25‐hydroxyvitamin D (nmol/L) | 49 | 50 | 102 | 122 |
| Thyrotropin (TSH) (mU/L) | 2.82 | 1.83 | 5.5 e | 1.53 |
| Urinary calcium excretion | 0.09 f | 0.002 | 0.001 | 0.003 |
A skin biopsy demonstrated folliculitis with keratin within the widened hair follicle and inflammatory cells around the hair follicle. Normal bone mineral density T‐score ≥ –1.0 at lumbar spine and femoral neck.
Individual III.2 (Fig. 1) developed headaches from age 10 years.
Individual III.3 was born prematurely at gestational age 27+3 weeks.
Normal biochemical ranges: ionized calcium, 1.16 to 1.30 mmol/L (adults) and 1.16 to 1.39 mmol/L (age 1 to 12 months); phosphate, 1.50 to 2.60 mmol/L (neonates), 1.50 to 2.50 mmol/L (age 1 to 6 months), 1.20 to 1.80 mmol/L (age 2 to 12 years), and 0.71 to 1.53 mmol/L (adults); alkaline phosphatase activity, 35 to 105 U/L (adults) and 115 to 460 U/L (age 15 days to 1 year); magnesium, 0.71 to 0.94 mmol/L (adults) and 0.7 to 1.0 mmol/L (age 3 months to 17 years); creatinine, 60 to 100 μmol/L (adult males) and 10 to 56 μmol/L (age 8 days to 2 years); PTH, 8 to 73 ng/L; 25 hydroxyvitamin D, >50 nmol/L; thyrotropin; 0.5 to 3.6 mU/L (age >14 weeks) and 0.4 to 7 mU/L (age 11 to 14 weeks); calcium‐to‐creatinine clearance ratio >0.02.
TSH concentration measured in umbilical cord blood (normal range not available for premature infant cord blood TSH).
Calcium‐to creatinine ratio, normal 0.3‐0.7.
Mutational analysis
DNA sequence analyses of GNA11 exons 1 to 7 and adjacent splice sites (NM_002067) was performed using leukocyte DNA and gene‐specific primers (Sigma‐Aldrich, Gillingham, UK), as reported.5, 6 Polymorphic variants were identified from public databases (Supporting Table 1).
Protein sequence alignment and three‐dimensional modeling of Gα11 structure
Protein sequences of Gα11 orthologs were aligned using ClustalOmega (European Bioinformatics Institute [EMBL‐EBI], Cambridgeshire, UK; http://www.ebi.ac.uk/Tools/msa/clustalo/).6 Gα11 three‐dimensional modeling was undertaken using the reported three‐dimensional structures of: Gαq in complex with phospholipase C‐β3 (Protein Data Bank accession no. 3OHM)11; Gαq in complex with the small molecule inhibitor YM‐254890 (Protein Data Bank accession no. 3AH8)12; and Gαq in complex with the regulator of G‐protein signaling 2 (RGS2) (Protein Data Bank accession no. 4EKC and 4QJ3).13, 14 The Gαq protein, which shares 90% identity at the amino acid level with Gα11, was used because crystal structures of Gα11 are not available. Figures were prepared using the PyMOL Molecular Graphics System, Schrodinger.
Cell culture and transfection
Mutations were introduced into the previously described pBI‐CMV2‐GNA11 expression construct by site‐directed mutagenesis using the Quikchange Lightning Kit (Agilent Technologies, Santa Clara, CA, USA) and gene‐specific primers (Sigma‐Aldrich). Wild‐type (WT) or mutant pBI‐CMV2‐GNA11 constructs were transiently transfected into HEK293 cells stably expressing CaSR (HEK‐CaSR) using Lipofectamine 2000 (Life TechnologiesCarlsbad, CA, USA), as described.15, 16 The pBI‐CMV2‐GNA11 bidirectional vector allows for co‐expression of Gα11 and GFP at equivalent levels.5 HEK‐CaSR cells were maintained in DMEM‐Glutamax media (Gibco, Carlsbad, CA, USA) that has a calcium concentration of 1.80mM. The presence of mutations was verified using dideoxynucleotide sequencing with the BigDye Terminator v3.1 Cycle Sequencing Kit (Life Technologies) and an automated detection system (ABI3730 Automated capillary sequencer; Applied Biosystems, Carlsbad, CA, USA).17 Luciferase reporter constructs (pGL4.30‐NFAT, pGL4.33‐SRE) were purchased from Promega. HEK293 cells were used because suitable parathyroid and renal tubular cells are not available, and HEK293 cells have been established as a model for the functional expression of Gα11 proteins.5, 15, 16 HEK‐CaSR cells were cultured in high‐glucose DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum and 1% Geneticin at 37°C, 5% CO2.5 Successful transfection was confirmed by visualizing GFP fluorescence using an Eclipse E400 fluorescence microscope with an epifluorescence filter, and images were captured using a DXM1200C digital camera and NIS Elements software (Nikon, Kingston‐Upon‐Thames, UK).5, 16 The expression of Gα11 and CaSR proteins was confirmed by Western blot analyses using Gα11 (Santa Cruz Biotechnologies), anti‐calnexin (Millipore, Billerica, MA), or anti‐CaSR (Abcam, Cambridge, UK) antibodies. Calnexin, a housekeeping protein, was used as a loading control. The Western blots were visualized using an Immuno‐Star Western C kit (BioRad, Hercules, CA, USA) on a BioRad Chemidoc XRS+ system.6
Fluo‐4 intracellular calcium assay
Ca2+ e‐induced Ca2+ i responses were measured by Fluo‐4 calcium assays adapted from published methods.18 HEK‐CaSR cells were plated in poly‐L‐lysine treated black‐walled 96‐well plates (Corning, Corning, NY, USA), and transiently transfected with 1000 ng/mL pBI‐CMV2‐GNA11. Cells were conditioned in serum‐free media (1.8mM Ca2+ e) overnight, then incubated in Ca2+‐free and Mg2+‐free Hanks Balanced Salt Solution (HBSS) for 1 hour, followed by loading with Fluo‐4 dye, prepared according to manufacturer's instructions (Invitrogen). Cells were loaded for 40 min at 37°C, then either a 20% aqueous solution of 2‐hydoxypropyl‐β‐cyclodextrin (vehicle), or 30nM or 100nM cinacalcet was added. Cells were then incubated for a further 20 min at 37°C.18 Baseline measurements were made and CaCl2 was injected into each well to increase the [Ca2+]e in a stepwise manner from 0.5 to 10mM [Ca2+]e, using an automated system. Changes in Ca2+ i were recorded on a PHERAstar instrument (BMG Labtech, Aylesbury, UK) at 37°C with an excitation filter of 485 nm and an emission filter of 520 nm. The peak mean fluorescence ratio of the transient response after each individual stimulus was measured using MARS data analysis software (BMG Labtech), and expressed as a normalized response. Nonlinear regression of concentration‐response curves was performed with GraphPad Prism (GraphPad Software, Inc., La Jolla, CA, USA) using the normalized response at each [Ca2+]e for each separate experiment for the determination of the EC50 (ie, [Ca2+]e required for 50% of the maximal response). Assays were performed in four to 12 biological replicates for each of the expression constructs. Statistical analysis was performed using the F‐test.5, 6
Luciferase reporter assay
HEK‐CaSR cells were seeded in 48‐well plates and transiently transfected with 100 ng/mL pBI‐CMV2‐GNA11 WT or mutant proteins, 100 ng/mL luciferase construct (either pGL4‐NFAT or pGL4‐SRE) and 10 ng/mL pRL control vector for 48 hours. Cells were incubated in serum‐free media for 12 hours, followed by treatment of cells for 4 hours with 0.1mM to 10mM CaCl2. Cells were lysed and assays performed with Dual‐Glo luciferase (Promega, Madison, WI, USA) on a Veritas Luminometer (Promega), as described.19 Luciferase:renilla ratios were expressed as fold‐changes relative to responses at basal CaCl2 concentrations. For studies with cinacalcet (Cambridge Bioscience, Cambridge, UK), the drug was added to cells, as described.10, 19 All assays were performed using four biological replicates on up to three independent occasions. Statistical analysis was performed by two‐way ANOVA with Tukey's multiple‐comparisons test using GraphPad Prism 6.
Measurement of ERK phosphorylation
HEK‐CaSR cells were seeded in 48‐well plates and transfected with 200 ng/mL WT or mutant Gα11, 48 hours prior to performance of assays. Transfected cells were incubated in serum‐free media 12 hours prior to treatment of cells with 0.1mM to 10mM CaCl2. Cells were then lysed in Surefire lysis buffer, and AlphaScreen Surefire ERK assays (PerkinElmer, Beaconsfield, UK) measuring phosphorylated and total proteins were performed, as described.10, 19 The fluorescence signal in both assays was measured using the PHERAstar FS microplate reader (BMG Labtech).19 The ratio of phosphorylated ERK (pERK) to total ERK measured at each [Ca2+]e were normalized to the mean responses of WT‐expressing cells and expressed as a fold‐change of responses obtained at basal (0.1mM) [Ca2+]e. All assay conditions were performed in four biological replicates on up to four independent occasions. Statistical analysis was performed by two‐way ANOVA with Tukey's multiple‐comparisons test using GraphPad Prism 6.
Web resources
Predicted effect of the mutation was assessed using Polyphen‐2 (http://genetics.bwh.harvard.edu/pph2/)20 and MutationTasting (http://www.mutationtaster.org/).21
Statistics
All studies involved between four and 16 separate transfection experiments. Statistical analyses for Ca2+ i EC50 used the F‐test,19 and two‐way ANOVA with Tukey's multiple‐comparisons test was used for pERK and luciferase reporter assays. Analyses were undertaken using GraphPad Prism, and a value of p < 0.05 was considered significant.
Results
Identification of a novel Phe220Ser Gα11 mutation in a family with FHH2
DNA sequence analysis of GNA11 in the proband identified a heterozygous T‐to‐C transition at nucleotide c.659 (Fig. 1 A), resulting in a missense substitution of the WT Phe to a mutant Ser at residue 220 of the Gα11 protein. Bioinformatic analyses predicted the Phe220Ser variant to be damaging and likely disease‐causing (Polyphen‐2 score = 1, MutationTasting score = 0.99). The absence of this DNA sequence abnormality in >67,200 exomes from the NHLBI‐ESP and Exome Aggregation Consortium cohorts, together with evolutionary conservation of the Phe220 residue in Gα11 orthologs and Gα‐subunit paralogs (Fig. 1 B), also indicated that the Phe220Ser abnormality likely represents a pathogenic mutation rather than a benign polymorphic variant.
The Phe220Ser mutation is located in the switch II region of Gα11 and predicted to impair PLC activation
Homology modeling, using crystal structures of the related Gαq protein,11, 12, 22 revealed the WT Phe220 residue to be located within the α2‐helix of the flexible switch II region of the Gα11 GTPase domain (Fig. 1 B, C; Supporting Fig. 1), and to comprise part of a hydrophobic cluster of amino acids located within a cleft region formed by switch II and the adjacent α3‐helix that interact with PLC upon G‐protein activation (Fig. 1 C, D).11 Substitution of the nonpolar hydrophobic WT Phe220 residue, with the mutant polar hydrophilic Ser220 residue is predicted to disrupt Gα11 function by impairing PLC activation and signaling.
The Gα11 Phe220Ser mutation impairs Ca2+ i responses
The Phe220Ser mutation is predicted to diminish PLC activation, and we therefore assessed its effects on Ca2+ i mobilization, which has previously been shown to be enhanced downstream of PLC by the Gq/11 proteins.23, 24, 25, 26 HEK‐CaSR cells were transiently transfected with pBI‐CMV2‐GNA11 constructs expressing the WT (Phe220) or mutant (Ser220) Gα11 proteins (Supporting Fig. 2). The Ca2+ i responses of the mutant Ser220 Gα11‐expressing HEK‐CaSR cells, revealed that a significantly greater [Ca2+]e was required to achieve half‐maximal (EC50) Ca2+ i responses, when compared with WT‐expressing cells (Fig. 2 A). Thus, the Ser220 mutant‐expressing cells showed a rightward shift in the concentration‐response curve, with a significantly elevated mean EC50 value (p < 0.001) of 2.93mM (95% confidence interval [CI], 2.80 to 3.07mM), compared with 2.40mM (95% CI, 2.27 to 2.53mM) for WT‐expressing cells. The Ca2+ i responses were also assessed by measurement of nuclear factor of activated T‐cells (NFAT), which is a downstream mediator of Ca2+ i signaling.27 NFAT fold‐change responses were determined using a luciferase reporter assay, and these studies showed that HEK‐CaSR cells expressing the Ser220 Gα11 mutant had a significantly reduced fold‐change response (fold‐change = 2.3 ± 0.3) following exposure to 5mM [Ca2+]e, when compared with WT cells (fold‐change = 4.8 ± 0.4, p < 0.001) (Fig. 2 B). These data demonstrated that the Gα11 Phe220Ser mutation impairs CaSR‐mediated Ca2+ i responses, consistent with a loss‐of‐function associated with FHH2.
Figure 2.
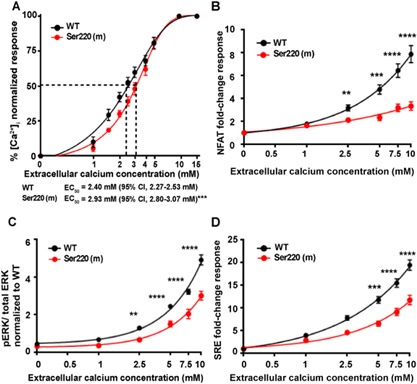
Effect of the mutant Ser220 Gα11 on Ca2+ i and ERK signaling in HEK‐CaSR cells. (A) Ca2+ i responses to changes in [Ca2+]e of HEK‐CaSR cells expressing WT (Phe220) or mutant (Ser220) Gα11 proteins shown as the mean ± SE of 4 to 11 transfections. The Ser220 Gα11 mutant caused a rightward shift in the Ca2+ i concentration‐response curve with significantly increased EC50 value compared to WT cells (***p < 0.0001, F‐test). (B) [Ca2+]e‐induced NFAT reporter responses, which are activated by elevations of Ca2+ i, were assessed in HEK‐CaSR cells expressing WT or mutant Gα11 proteins. Responses at each [Ca2+]e are expressed as a fold‐change of basal [Ca2+]e responses, and shown as mean ± SE of 12 transfections. NFAT luciferase reporter activity increased in all cells in a concentration‐dependent manner, and was significantly reduced in Ser220 cells compared to WT cells. (C) Ca2+ e‐induced pERK responses of cells expressing WT or mutant Gα11 protein, expressed as a ratio to total ERK levels, and shown as mean ± SE of 8 transfections. (D) Ca2+ e‐induced SRE reporter responses of cells expressing WT or mutant Gα11 protein, expressed compared to basal SRE reporter activity, and shown as mean ± SE of 8 transfections. The Ser220 Gα11 mutant was associated with significantly decreased pERK and SRE reporter responses compared to WT. **p < 0.01 and ****p < 0.0001, by a 2‐way ANOVA with Tukey's multiple‐comparisons test.
The Gα11 Phe220Ser mutation impairs ERK phosphorylation
The ERK protein represents a major component of the MAPK signaling cascade, which has previously been shown to be activated by PLC.3, 28 We assessed the effect of the Gα11 Phe220Ser mutation on ERK phosphorylation by measuring changes in pERK in response to increases in Ca2+ e in WT and mutant Ser220 Gα11‐expressing cells using the AlphaScreen assay, and also by assessing serum‐response element (SRE)‐mediated transcriptional activity, which is regulated by ERK signaling.19 The pERK responses of the Ser220 Gα11 mutant were shown to be significantly reduced. Thus, the Ser220 Gα11 mutant led to significant reductions in: pERK‐to‐total ERK ratios (Ser220 response at 5mM Ca2+ e = 3.7 ± 0.4 versus Phe220 = 6.1 ± 0.3, p < 0.0001) (Fig. 2 C); and to SRE reporter fold‐changes (Ser220 fold‐change at 5mM Ca2+ e = 6.5 ± 0.5 versus Phe220 fold‐change = 11.8 ± 0.7, p < 0.0001) (Fig. 2 D). Thus, the Gα11 Phe220Ser mutation impaired CaSR‐mediated ERK phosphorylation.
Phe220 forms part of a hydrophobic cluster of Gα11 residues required for PLC‐mediated signaling
To further determine the mechanistic role of the Phe220 residue and the importance of residue 220 hydrophobicity for Gα11 function and PLC‐mediated signaling (Fig. 1 C), we engineered mutations of Phe220 to Leu and Ala, which are hydrophobic residues, and to Arg and Glu, which are hydrophilic positively‐charged and negatively‐charged residues, respectively, and studied their effect on Ca2+ i and ERK responses following transient expression in HEK‐CaSR cells (Fig. 3). The Leu220 and Ala220 Gα11 mutants did not alter Ca2+ i responses (Fig. 3 A, B; Supporting Fig. 3), or ERK activity, as measured using the pERK AlphaScreen and SRE reporter assays (Fig. 3 C, D). In contrast, the positively‐charged Arg220, and negatively‐charged Glu220 Gα11 mutants, respectively, enhanced and impaired Ca2+ i and ERK signaling responses in HEK‐CaSR cells (Fig. 3, Supporting Fig. 3). Thus, these findings demonstrated that a hydrophobic residue at position 220 is required for signaling via PLC. The enhancement of CaSR‐mediated signaling by the Arg220 Gα11 mutant may be explained by its structural effect on the G11 heterotrimer. Homology modeling, using crystal structures of the related Gq protein in complex with Gβγ,12 revealed that the Arg220 residue faces away from the hydrophobic cluster, into the region at which Gα and Gβγ bind, a hydrophobic region that is necessary for Gα activation29 (Supporting Fig. 4). Disruption of this hydrophobic region has been shown to cause constitutive activation,29 and modeling predicts that the Arg220 residue, which is charged and hydrophilic, disrupts this region, thus enhancing activation by impairing Gαβγ association (Supporting Fig. 4). To investigate the importance of the Gα11 hydrophobic cleft, formed by switch II and the α3‐helix (Fig. 1 C, D), for PLC activation, we engineered mutations of three other hydrophobic residues, Ile217, Val223, and Trp263, within this region, to either an Ala residue, to retain hydrophobicity, or to a Glu residue, to lose hydrophobicity, and studied the effects of these mutants on Ca2+ i and ERK responses following their transient expression in HEK‐CaSR cells (Supporting Fig. 5). This demonstrated that substitution to Ala of only one hydrophobic residue, Ile217, altered PLC‐mediated signaling (Fig. 4 A–C; Supporting Fig. 6). In contrast, substitution of any of the Ile217, Val223, and Trp263 hydrophobic residues to a Glu residue led to a significant reduction in Ca2+ i and ERK responses (Fig. 4 D–F, Supporting Fig. 6). Thus, hydrophobicity of the switch II‐α3 cleft is important for PLC activation.
Figure 3.
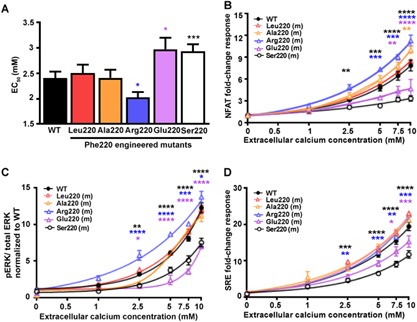
Effect of Gα11 residue 220 hydrophobicity on PLC‐mediated signaling. (A) Histograms showing Ca2+ e‐induced Ca2+ i EC50 values with 95% confidence intervals (CIs) for cells expressing WT Phe220 or residue 220 mutant (m) Gα11 proteins from 6 to 12 transfections. Statistical analyses were performed using the F‐test. The Arg220 Gα11 mutant reduced the EC50 value, whereas the Glu220 and Ser220 Gα11 mutants elevated the EC50 value. (B) Ca2+ e‐induced NFAT reporter responses, which assess Ca2+ i signaling, of cells expressing WT or residue 220 mutant Gα11 proteins. Responses at each [Ca2+]e are expressed as a fold‐change of basal [Ca2+]e responses, and shown as mean ± SE of 4 to 12 transfections. NFAT luciferase reporter activity increased in all cells in a concentration‐dependent manner, however responses were significantly elevated in Arg220 Gα11 mutant, and reduced in Glu220 Gα11 mutant cells, compared to WT cells. (C) Ca2+ e‐induced pERK responses of cells expressing WT or mutant residue 220 Gα11 proteins, expressed as a ratio to total ERK levels. The Arg220 mutant was associated with significantly increased responses, and the Glu220 and Ser220 mutants were associated with decreased responses, when compared to WT. Data are shown as mean ± SE of 4 to 8 independent transfections. (D) Ca2+ e‐induced SRE reporter responses of cells expressing WT or mutant (m) residue 220 Gα11 proteins, expressed compared to basal SRE reporter activity. The engineered Arg220 mutant was associated with significantly increased, and the engineered Glu220 and FHH2‐associated Ser220 mutants resulted in decreased responses, when compared to WT. Data are shown as mean ± SE of 4 to 8 independent transfections. Statistical analyses performed using 2‐way ANOVA with Tukey's multiple‐comparisons test comparing WT with Gα11 mutants Ala220, Arg220, Glu220, and Ser220. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Figure 4.
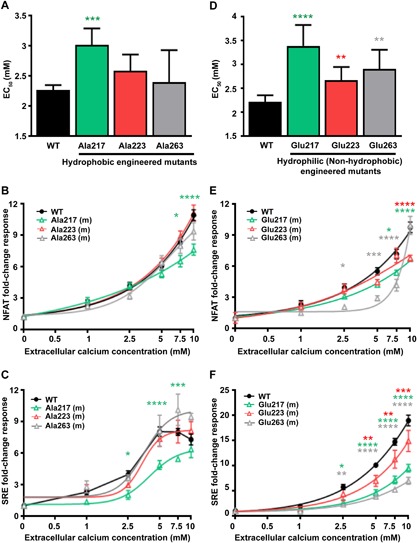
Effect of mutating residues within the Gα11 switch II‐α3 hydrophobic cluster on PLC‐mediated signaling. (A) Histograms showing Ca2+ e‐induced Ca2+ i EC50 values with 95% CI for cells expressing WT and alanine (Ala) Gα11 mutants (m) of residues 217, 223, and 263 Gα11 proteins from 4 to 8 independent transfections. Only the Ala217 Gα11 mutant increased the EC50 value, when compared to WT cells. Statistical analyses were performed using the F‐test. (B, C) Ca2+ e‐induced (B) NFAT and (C) SRE luciferase reporter responses of cells expressing WT or alanine (Ala) Gα11 mutants. Responses at each [Ca2+]e are expressed as a fold‐change of basal [Ca2+]e responses, and shown as mean ± SE of 4 to 8 transfections. NFAT and SRE reporter responses were significantly reduced in Ala217‐expressing cells compared to WT cells. (D) Histograms showing Ca2+ e‐induced Ca2+ i EC50 values with 95% CI for cells expressing WT and glutamic acid (Glu) Gα11 mutants (m) of residues 217, 223, and 263 from 4 to 8 independent transfections. All Glu Gα11 mutants increased the EC50 value, when compared to WT cells. Statistical analyses were performed using the F‐test. (E, F) Ca2+ e‐induced (E) NFAT and (F) SRE luciferase reporter responses of cells expressing WT or glutamic acid (Glu) Gα11 mutants. Responses at each [Ca2+]e are expressed as a fold‐change of basal [Ca2+]e responses, and shown as mean ± SE of 4 to 8 transfections. NFAT and SRE reporter responses were significantly reduced in cells expressing all 3 glutamic acid mutants compared to WT cells. Statistical analyses for B, C, E, and F were performed using 2‐way ANOVA with Tukey's multiple‐comparisons test comparing WT to mutant 217 (green), 223 (red), and 263 (gray) Gα11 proteins. *p < 0.05, ***p < 0.001, ****p < 0.0001.
Cinacalcet rectifies the impaired PLC signaling responses and hypercalcemia caused by the Phe220Ser Gα11 mutation
To determine whether cinacalcet may be an effective therapy for FHH2, we evaluated the effects of different cinacalcet concentrations (0, 30, and 100nM) on the altered PLC signaling responses due to the Phe220Ser Gα11 mutant. Treatment of mutant Ser220 Gα11‐expressing HEK‐CaSR cells with 30nM cinacalcet, failed to normalize the impaired Ca2+ i and SRE reporter responses (Fig. 5 A–C); however, 100nM cinacalcet, reduced the EC50 values, and increased NFAT and SRE reporter activity (Fig. 5 A–C), such that these values were not significantly different from untreated WT cells. These in vitro findings suggested that higher doses of cinacalcet may be required for the treatment of patients harboring the Phe220Ser Gα11 mutation. Cinacalcet was administered to the FHH2 proband as he had symptoms such as headaches, constipation, and pruritus (Table 1), which may have been caused by the hypercalcemia. The proband was initially commenced on 30 mg/day of oral cinacalcet for 3 months, and in keeping with the above cellular studies, which showed a lack of efficacy when using low cinacalcet concentrations, the 30 mg/day dose failed to normalize his elevated serum calcium concentrations (Fig. 5 D). However, increasing the dose of cinacalcet to 60 mg/day, successfully lowered the serum concentrations of: ionized calcium from 1.42 to 1.26 mmol/L (normal range, 1.16 to 1.30 mmol/L) (Fig. 5 D); and PTH from 98 to 58 ng/L (normal range, 8 to 73 ng/L). Although the proband became normocalcemic on cinacalcet, the pruritus, from which he suffered the most, did not resolve, and cinacalcet 60 mg/day was discontinued after 4 months. His hypercalcemia returned upon cessation of cinacalcet therapy (Fig. 5 D).
Figure 5.
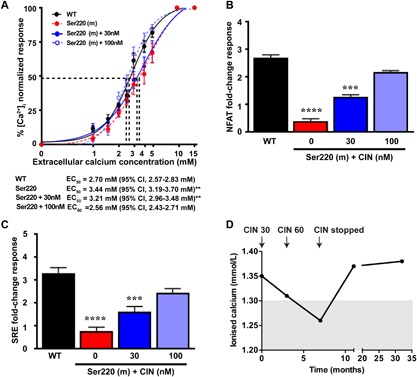
Effect of cinacalcet on the loss‐of‐function and hypercalcemia caused by the Phe220Ser Gα11 mutation. (A) Effect of cinacalcet on Ca2+ i responses to changes in [Ca2+]e of HEK‐CaSR cells transfected with WT or Ser220 Gα11 mutant (m). The Ca2+ i responses to changes in [Ca2+]e are expressed as a percentage of the maximum normalized responses and shown as the mean ± SE of 6 to 12 independent transfections. The Ser220 Gα11 mutant led to a rightward shift in the concentration‐response curve. The addition of 30nM cinacalcet, a concentration previously shown to rectify the loss‐of‐function associated with the reported Leu135Gln Gα11 FHH2 mutant in vitro,10 had no effect on Ser220 responses. However, addition of 100nM cinacalcet rectified the rightward shift of the Ser220 Gα11 mutant, such that it was not different to WT responses. (B, C) Histograms showing (B) NFAT and (C) SRE luciferase reporter activity in response to 5mM Ca2+ e in HEK‐CaSR cells expressing WT or Ser220 mutant constructs, treated with vehicle or cinacalcet (CIN). Data are shown as mean ± SE of 4 independent transfections; 100nM cinacalcet was required to significantly increase NFAT and SRE reporter activity to WT levels. The responses of cells expressing WT or mutant Gα11 proteins were compared using the F‐test for A and 2‐way ANOVA with Tukey's multiple‐comparisons test for B and C. **p < 0.01, ***p < 0.001, ****p < 0.0001. (D) Effect of cinacalcet on the serum ionized calcium concentrations of the FHH2 proband (individual II.1, Fig. 1 A) with the Phe220Ser Gα11 mutation. Arrows indicate initiation of oral cinacalcet at 30 mg/day (CIN 30), at 60 mg/day (CIN 60), and its discontinuation. Cinacalcet treatment decreased serum concentrations of ionized calcium and PTH from 98 ng/L (pretreatment) to 58 ng/L (posttreatment) (normal range, 8 to 73 ng/L). Shaded area indicates normal range.
Discussion
Our studies have identified a previously unreported FHH2‐causing heterozygous Gα11 germline loss‐of‐function mutation that represents the first loss‐of‐function Gα11 mutation to be located within the Gα‐subunit switch regions. The switch regions are comprised of three flexible peptide loops, which are highly conserved among all classes of Gα‐subunits, and undergo substantial conformational changes depending on the guanine nucleotide‐bound state of the Gα subunit.30 In the GTP‐bound state, these peptide loops switch their conformation to one that facilitates coupling with downstream effector proteins.31 The Phe220Ser mutation is located in the switch II region, which is predicted to bind and activate PLC,11 and our in vitro assessment of Ca2+ i and pERK responses in cells expressing the mutant Ser220 Gα11 demonstrated an impairment of signaling that is, at least in part, mediated by PLC.26, 32 Furthermore, our mutagenesis studies showed that Phe220 forms part of a group of hydrophobic residues that are required for coupling to PLC, and that Phe220 is located in a key region within this hydrophobic cleft, such that substitution of the hydrophobic Phe220 residue for a hydrophilic positively (Arg) or negatively (Glu) charged residue resulted, respectively, in a Gα11 gain‐of‐function or loss‐of‐function, thereby indicating that mutations of Phe220 can either retard or enhance the switch II conformational changes that occur upon G‐protein activation.
These studies have involved a comprehensive investigation of the effect of FHH2‐associated Gα11 mutations on PLC signaling at both the cytosolic (ie, Ca2+ i and pERK) and transcriptional (ie, NFAT and SRE reporter) level, and demonstrate that all readouts are affected by FHH2 mutations. The need to measure several signal outputs is increasingly important as recent studies have shown that GPCRs can couple to multiple G‐proteins to enhance their signaling capabilities and respond in diverse tissues.33, 34 Indeed, this is particularly the case in Gα11 mutant cells, where it has been hypothesized that other G‐proteins, in particular Gαq, may compensate for some of the functions of Gα11, giving rise to the milder phenotype observed in FHH2.5, 6 Our studies of the transcriptional events downstream of CaSR activation indicate that Ca2+ i signaling is more severely impaired than ERK signaling, as the Ser220 Gα11 mutant cells have more robust SRE signals than NFAT signals. MAPK signaling is known to occur downstream of several G‐proteins and can be used as a surrogate to assess multiple G‐protein families.32, 35 The differences observed in Ca2+ i signaling compared to ERK signaling in Gα11 Ser220 mutant cells indicates that Gα11 may preferentially signal via Ca2+ i, or that other G‐proteins are able to compensate for a reduction in Gα11‐mediated ERK responses in FHH2 mutant cells.
The Gα11 protein is considered to be ubiquitously expressed,36 thus raising the possibility that germline Gα11 mutations may be associated with non‐calciotropic phenotypes. Indeed, in addition to constipation most of the affected family members had folliculitis or eczematous skin lesions, which may potentially represent a non‐calciotropic phenotype of FHH2. This is plausible as somatic activating Gα11 mutations have been reported to cause cutaneous pigmentation disorders such as dermal melanocytosis,37 and studies of atopic dermatitis, which is characterized by impaired epithelial barrier function and eczematous skin lesions, have revealed that Gq/11‐coupled chemokine receptors facilitate the migration of inflammatory cells within the dermis.38 However, such roles of loss‐of‐function Gα11 mutations in cutaneous disorders remain to be elucidated, and these may be facilitated by studies of additional FHH2 patients and appropriate mouse models.
Our studies have also shown that cinacalcet‐mediated allosteric modulation of the CaSR successfully rectified the loss‐of‐function associated with the Phe220Ser Gα11 mutation in vitro and in vivo. However, a higher concentration of cinacalcet was required to normalize the signaling responses of cells expressing the Gα11 mutant Ser220, when compared to other FHH2‐causing Gα11 mutants.10 These in vitro findings suggest that loss‐of‐function mutations located within the Gα11 switch regions may alter the efficacy of calcimimetic compounds, and it is notable that the hypercalcemic proband with a Phe220Ser Gα11 mutation required an increase in the dose of cinacalcet from 30 to 60 mg daily to normalize the elevated serum calcium concentrations (Fig. 2 C). Moreover, cinacalcet in the patient exerted an inhibitory effect on PTH secretion, as observed by a normalization of serum PTH concentrations. Cinacalcet may be rectifying the impaired signaling by the mutant Phe220Ser Gα11 by increasing signaling through the WT Gα11, which is present endogenously in HEK293 cells and the cells of the heterozygous patient, and/or compensatory signaling by the closely‐related Gαq, which activates the same signaling pathways downstream of CaSR. Importantly, hypocalcemia or adverse effects such as nausea and vomiting, which may affect >25% of patients,39 were not observed in the patient, despite receiving a higher dose of cinacalcet.
In conclusion, we have identified a FHH2‐causing mutation, which affects the Gα11 switch region and disrupts PLC‐mediated signaling, and have also shown that cinacalcet can rectify these signaling disturbances and be used successfully to treat the hypercalcemia caused by this germline loss‐of‐function Gα11 mutation.
Disclosures
FMH and RVT have received grant funding from NPS/Shire Pharmaceuticals and GlaxoSmithKline for unrelated studies involving the use of calcium‐sensing receptor allosteric inhibitors. RVT has also received grants from Novartis Pharma AG and the Marshall Smith Syndrome Foundation for unrelated studies. All other authors confirm no conflict of interest.
Supporting information
Supporting Data S1.
Acknowledgments
This work was supported by the Academy of Finland, Sigrid Jusélius Foundation, and Folkhälsan Research Foundation (to OM); Finska Lakaresallskapet and Helsinki University Hospital Research Funds (to CS‐J); a Wellcome Trust Senior Investigator Award (to RVT); National Institute for Health Research (NIHR) Oxford Biomedical Research Centre Program (to RVT); and NIHR Senior Investigator Award (to RVT).
Author's roles: CMG, FMH, CS‐J, and RVT conceived and coordinated the study; HV, OM, and CS‐J provided the clinical data; CS‐J performed the cinacalcet trial in the proband. CMG and TC performed and analyzed the experiments; CMG, FMH, CS‐J, and RVT wrote the manuscript; and all authors reviewed the results and approved the final version of the manuscript.
References
- 1. Hannan FM, Babinsky VN, Thakker RV. Disorders of the calcium‐sensing receptor and partner proteins: insights into the molecular basis of calcium homeostasis. J Mol Endocrinol. 2016; 57(3):R127–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hofer AM, Brown EM. Extracellular calcium sensing and signalling. Nat Rev Mol Cell Biol. 2003; 4(7):530–8. [DOI] [PubMed] [Google Scholar]
- 3. Wu DQ, Lee CH, Rhee SG, Simon MI. Activation of phospholipase C by the alpha subunits of the Gq and G11 proteins in transfected Cos‐7 cells. J Biol Chem. 1992; 267(3):1811–7. [PubMed] [Google Scholar]
- 4. Hannan FM, Thakker RV. Calcium‐sensing receptor (CaSR) mutations and disorders of calcium, electrolyte and water metabolism. Best Pract Res Clin Endocrinol Metab. 2013; 27(3):359–71. [DOI] [PubMed] [Google Scholar]
- 5. Nesbit MA, Hannan FM, Howles SA, et al. Mutations affecting G‐protein subunit alpha11 in hypercalcemia and hypocalcemia. N Engl J Med. 2013; 368(26):2476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gorvin CM, Cranston T, Hannan FM, et al. A G‐protein subunit‐alpha11 loss‐of‐function mutation, Thr54Met, causes familial hypocalciuric hypercalcemia type 2 (FHH2). J Bone Miner Res. 2016; 31(6):1200–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Howles SA, Hannan FM, Babinsky VN, et al. Cinacalcet for symptomatic hypercalcemia caused by AP2S1 mutations. N Engl J Med. 2016; 374(14):1396–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Festen‐Spanjer B, Haring CM, Koster JB, Mudde AH. Correction of hypercalcaemia by cinacalcet in familial hypocalciuric hypercalcaemia. Clin Endocrinol. 2008; 68(2):324–5. [DOI] [PubMed] [Google Scholar]
- 9. Tenhola S, Hendy GN, Valta H, et al. Cinacalcet treatment in an adolescent with concurrent 22q11.2 deletion syndrome and familial hypocalciuric hypercalcemia type 3 caused by AP2S1 mutation. J Clin Endocrinol Metab. 2015; 100(7):2515–8. [DOI] [PubMed] [Google Scholar]
- 10. Babinsky VN, Hannan FM, Gorvin CM, et al. Allosteric modulation of the calcium‐sensing receptor rectifies signaling abnormalities associated with G‐protein alpha‐11 mutations causing hypercalcemic and hypocalcemic disorders. J Biol Chem. 2016; 291(20):10876–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Waldo GL, Ricks TK, Hicks SN, et al. Kinetic scaffolding mediated by a phospholipase C‐beta and Gq signaling complex. Science. 2010; 330(6006):974–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nishimura A, Kitano K, Takasaki J, et al. Structural basis for the specific inhibition of heterotrimeric Gq protein by a small molecule. Proc Natl Acad Sci U S A. 2010; 107(31):13666–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nance MR, Kreutz B, Tesmer VM, Sterne‐Marr R, Kozasa T, Tesmer JJG. Structural and functional analysis of the regulator of G protein signaling 2‐G alpha(q) complex. Structure. 2013; 21(3):438–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lyon AM, Begley JA, Manett TD, Tesmer JJ. Molecular mechanisms of phospholipase C beta3 autoinhibition. Structure. 2014; 22(12):1844–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nesbit MA, Hannan FM, Howles SA, et al. Mutations in AP2S1 cause familial hypocalciuric hypercalcemia type 3. Nat Genet. 2013; 45(1):93–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hannan FM, Howles SA, Rogers A, et al. Adaptor protein‐2 sigma subunit mutations causing familial hypocalciuric hypercalcaemia type 3 (FHH3) demonstrate genotype‐phenotype correlations, codon bias and dominant‐negative effects. Hum Mol Genet. 2015; 24(18):5079–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Newey PJ, Gorvin CM, Cleland SJ, et al. Mutant prolactin receptor and familial hyperprolactinemia. N Engl J Med. 2013; 369(21):2012–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leach K, Gregory KJ, Kufareva I, et al. Towards a structural understanding of allosteric drugs at the human calcium‐sensing receptor. Cell Res. 2016; 26(5):574–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gorvin CM, Hannan FM, Howles SA, et al. Galpha11 mutation in mice causes hypocalcemia rectifiable by calcilytic therapy. JCI Insight. 2017; 2(3):e91103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen‐2. Curr Protoc Hum Genet. 2013;Chapter 7:Unit7.20. DOI:10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep‐sequencing age. Nat Methods. 2014; 11(4):361–2. [DOI] [PubMed] [Google Scholar]
- 22. Tesmer VM, Kawano T, Shankaranarayanan A, Kozasa T, Tesmer JJ. Snapshot of activated G proteins at the membrane: the Galphaq‐GRK2‐Gbetagamma complex. Science. 2005; 310(5754):1686–90. [DOI] [PubMed] [Google Scholar]
- 23. Bai M, Trivedi S, Lane CR, Yang Y, Quinn SJ, Brown EM. Protein kinase C phosphorylation of threonine at position 888 in Ca2+o‐sensing receptor (CaR) inhibits coupling to Ca2+ store release. J Biol Chem. 1998; 273(33):21267–75. [DOI] [PubMed] [Google Scholar]
- 24. Brown E, Enyedi P, LeBoff M, Rotberg J, Preston J, Chen C. High extracellular Ca2+ and Mg2+ stimulate accumulation of inositol phosphates in bovine parathyroid cells. FEBS Lett. 1987; 218(1):113–8. [DOI] [PubMed] [Google Scholar]
- 25. Shoback DM, Membreno LA, McGhee JG. High calcium and other divalent cations increase inositol trisphosphate in bovine parathyroid cells. Endocrinology. 1988; 123(1):382–9. [DOI] [PubMed] [Google Scholar]
- 26. Chang W, Chen TH, Pratt S, Shoback D. Amino acids in the second and third intracellular loops of the parathyroid Ca2+‐sensing receptor mediate efficient coupling to phospholipase C. J Biol Chem. 2000; 275(26):19955–63. [DOI] [PubMed] [Google Scholar]
- 27. Chakravarti B, Chattopadhyay N, Brown EM. Signaling through the extracellular calcium‐sensing receptor (CaSR). Adv Exp Med Biol. 2012; 740:103–42. [DOI] [PubMed] [Google Scholar]
- 28. Kifor O, Diaz R, Butters R, Brown EM. The Ca2+‐sensing receptor (CaR) activates phospholipases C, A2, and D in bovine parathyroid and CaR‐transfected, human embryonic kidney (HEK293) cells. J Bone Miner Res. 1997; 12(5):715–25. [DOI] [PubMed] [Google Scholar]
- 29. Wall MA, Coleman DE, Lee E, et al. The structure of the G protein heterotrimer Gi alpha 1 beta 1 gamma 2. Cell. 1995; 83(6):1047–58. [DOI] [PubMed] [Google Scholar]
- 30. Oldham WM, Hamm HE. Heterotrimeric G protein activation by G‐protein‐coupled receptors. Nat Rev Mol Cell Biol. 2008; 9(1):60–71. [DOI] [PubMed] [Google Scholar]
- 31. Johnston CA, Siderovski DP. Receptor‐mediated activation of heterotrimeric G‐proteins: current structural insights. Mol Pharmacol. 2007; 72(2):219–30. [DOI] [PubMed] [Google Scholar]
- 32. Kifor O, MacLeod RJ, Diaz R, et al. Regulation of MAP kinase by calcium‐sensing receptor in bovine parathyroid and CaR‐transfected HEK293 cells. Am J Physiol Renal Physiol. 2001; 280(2):F291–302. [DOI] [PubMed] [Google Scholar]
- 33. Masuho I, Ostrovskaya O, Kramer GM, Jones CD, Xie K, Martemyanov KA. Distinct profiles of functional discrimination among G proteins determine the actions of G protein‐coupled receptors. Sci Signal. 2015; 8(405):ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leach K, Sexton PM, Christopoulos A, Conigrave AD. Engendering biased signalling from the calcium‐sensing receptor for the pharmacotherapy of diverse disorders. Br J Pharmacol. 2014; 171(5):1142–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Holstein DM, Berg KA, Leeb‐Lundberg LM, Olson MS, Saunders C. Calcium‐sensing receptor‐mediated ERK1/2 activation requires Galphai2 coupling and dynamin‐independent receptor internalization. J Biol Chem. 2004; 279(11):10060–9. [DOI] [PubMed] [Google Scholar]
- 36. Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol Rev. 2005; 85(4):1159–204. [DOI] [PubMed] [Google Scholar]
- 37. Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010; 363(23):2191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shi G, Partida‐Sanchez S, Misra RS, et al. Identification of an alternative G{alpha}q‐dependent chemokine receptor signal transduction pathway in dendritic cells and granulocytes. J Exp Med. 2007; 204(11):2705–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Block GA, Martin KJ, de Francisco AL, et al. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N Engl J Med. 2004; 350(15):1516–25. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Data S1.