

Abstract
Background & Aims
Consumption of sugar is associated with obesity, type 2 diabetes mellitus, non-alcoholic fatty liver disease, and cardiovascular disease. The conversion of fructose to fat in liver (de novo lipogenesis, DNL) may be a modifiable pathogenetic pathway. We determined the effect of 9 days of isocaloric fructose restriction on DNL, liver fat, visceral fat (VAT), subcutaneous fat, and insulin kinetics in obese Latino and African American children with habitual high sugar consumption (fructose intake more than 50 g/day).
Methods
Children (9–18 years old; n = 41) had all meals provided for 9 days with the same energy and macronutrient composition as their standard diet, but with starch substituted for sugar, yielding a final fructose content of 4% of total kcal. Metabolic assessments were performed before and after fructose restriction. Liver fat, VAT, and subcutaneous fat were determined by magnetic resonance spectroscopy and imaging. The fractional DNL area under the curve value was measured using stable isotope tracers and gas chromatography/mass spectrometry. Insulin kinetics were calculated from oral glucose tolerance tests. Paired analyses compared change from day 0 to day 10 within each child.
Results
Compared with baseline, on day 10, liver fat decreased from a median of 7.2% (inter-quartile range, 2.5%–14.8%) to 3.8% (inter-quartile range, 1.7%–15.5%)(P<.001) and VAT decreased from 123 cm3 (inter-quartile range, 85–145 cm3) to 110 cm3 (inter-quartile range, 84–134 cm3) (P<.001). The DNL area under the curve decreased from 68% (inter-quartile range, 46%–83%) to 26% (inter-quartile range, 16%–37%) (P<0.001). Insulin kinetics improved (P<.001). These changes occurred irrespective of baseline liver fat.
Conclusions
Short-term (9 day) isocaloric fructose restriction decreased liver fat, VAT, and DNL, and improved insulin kinetics in children with obesity. These findings support efforts to reduce sugar consumption. ClinicalTrials.gov no: NCT01200043
Keywords: dietary treatment, NAFLD, pediatric, overweight
INTRODUCTION
High dietary sugar consumption is associated with non-alcoholic fatty liver disease (NAFLD) and excess visceral adipose tissue (VAT),1–3 which are in turn linked to type 2 diabetes mellitus (T2DM), dyslipidemia, and cardiovascular disease (CVD) in adults and children.4–6 NAFLD occurs when hepatic lipid concentration (from peripheral lipolysis or synthesis of new fat by hepatic de novo lipogenesis [DNL]) exceeds the combined rates of hepatic lipid oxidation and export.7, 8 Studies have linked visceral and/or liver fat with metabolic dysfunction, including insulin resistance and T2DM,9–11 and NAFLD is a predictor of type 2 diabetes.12, 13 Recently, a survey in 675 children with biopsy-proven NAFLD showed that 30% had T2DM or prediabetes.14
The link between consumption of sugar, especially fructose, and accumulation of ectopic fat is not well understood, but recent studies suggest that fructose stimulates DNL,2, 15 which may drive the accumulation of liver and/or visceral fat.7, 16 Fructose has been shown to specifically increase carbohydrate response element-binding protein,17 a transcription factor that induces three enzymes of DNL: ATP citrate lyase, acetyl-CoA carboxylase, and fatty acid synthase. We recently demonstrated that in weight-stable healthy men, high fructose intake for a 9-day period was associated with higher DNL and liver fat, compared with a diet with identical energy and macronutrient intake, but in which complex carbohydrate (starch) was substituted for sugar.18 Thus we provided evidence linking fructose-driven DNL with liver fat and demonstrated that short-term reduction in fructose intake was consistently associated with lower levels of liver fat and rates of DNL, even in the absence of weight loss.
In the current study, we hypothesized that short-term fructose restriction in children with obesity and metabolic syndrome who habitually consume high levels of fructose would reduce liver fat and hepatic DNL without change in energy intake or weight. We studied 41 Latino and African-American children with high levels of self-reported sugar intake, feeding them diets that featured isocaloric substitution of starch for most sugar for nine days, resulting in a reduction in total sugar content from 28% to 10%, and fructose from 12% to 4% of total energy intake. In separate publications from this study,19, 20 we reported improvements in glycemia, fasting lipoproteins, blood pressure, and other clinical parameters. Here, we report the effects of isocaloric fructose restriction on liver fat, hepatic DNL, VAT, and subcutaneous adipose tissue (SAT), and their relation to changes in insulin kinetics.
METHODS
Study Design and Population
We recruited non-diabetic African-American and Latino children with obesity and metabolic syndrome who identified as high habitual sugar consumers (>15% sugar, >5% fructose) based upon a food frequency questionnaire and interview by a dietitian.19 As described elsewhere,19 eligibility criteria included ages 8–18 years, BMI z-score ≥1.8, and at least one of the following: systolic blood pressure >95th percentile for age and sex, fasting triglycerides >150 mg/dL, alanine aminotransferase (ALT) >40 U/L, fasting glucose 100–125 mg/dL, fasting insulin >15 μIU/mL, HOMA-IR >4.3,21 or severe acanthosis nigricans. This study protocol was approved by the Institutional Review Boards of the University of California, San Francisco (approval 10-03473) and Touro University-California (approval M-0609) and registered with ClinicalTrials.gov (NCT01200043). Informed written consent/assent were obtained before formal screening was initiated. Comprehensive metabolic assessments were performed before (day 0) and after (day 10) a 9-day dietary intervention.
Metabolic Assessments
Participants and their guardians were instructed to continue their usual home diets and other routines before the study. On days 0 and 10, after fasting at least eight hours, participants underwent metabolic studies at the University of California San Francisco Pediatric Clinical Research Center (Figure 1). Weight and vital signs were measured and urine pregnancy testing was performed in females. Body composition was measured by whole-body dual-energy X-ray absorptiometry (DXA; GE/Lunar Prodigy, Madison WI). A two-hour 75-g oral glucose tolerance test (OGTT) was performed, with glucose, insulin, and C-peptide measurements at t=0, 30, 60, 90, and 120 minutes. Fasting glucose and insulin, and their respective areas under the curve, (AUC) are reported elsewhere.19
Figure 1.
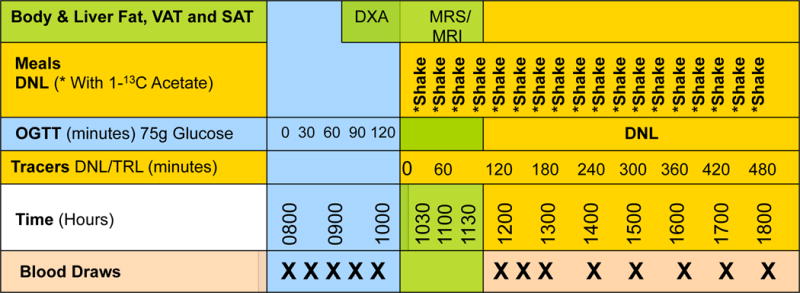
Clinical research design and procedures on day 0 and day 10, depicting the time of oral glucose tolerance testing (OGTT), MR studies, and sodium [1-13C]-acetate administration via liquid meals (shakes) to determine rate of de novo lipogenesis (DNL).
Tracer/Feeding Study
Upon completion of the OGTT, an 8-hour stable isotope tracer/feeding study to measure postprandial DNL was initiated (Figure 1), using liquid meals containing sodium [1-13C]-acetate (Cambridge Isotope Laboratories, Cambridge, MA). After an initial double-sized meal, single-sized meals were fed every half-hour for eight hours. Altogether, the meals provided 67% of estimated daily energy requirement (15% protein, 35% fat, 50% carbohydrate) and 5–7 g of the acetate tracer. On day 0, the fructose content of the liquid meals ranged from 12–18% of energy intake, depending on self-reported usual intake; on day 10, the fructose content was reduced to 4% of energy intake, but overall energy and carbohydrate content matched that of the day 0 test meals. In both cases, the remainder of carbohydrate was provided primarily as glucose polymer. Blood samples were drawn on K2EDTA before the first test meal and every hour thereafter, processed, and frozen at −80°C.
Magnetic Resonance (MR) Imaging and Spectroscopy
During the tracer/feeding study, participants underwent an MR exam on a 3-Tesla scanner (GE Healthcare, Waukesha, WI) to measure liver fat, VAT, and SAT. For the liver fat measures, MR spectroscopy was obtained from a 20cc single voxel (64 acquisitions water-suppressed, 8 acquisitions unsuppressed, with a repetition time of 2500 ms and an echo time of 30 ms), similar to prior reports.22, 23 Spectra were automatically phase-, frequency-, motion-, and T2 relaxation-time corrected (using in-house derived formulas for T2water = −12.4×L/W +31.3ms, and T2lipids = 23.1×L/W + 58.5ms; where L/W is the MR measured lipids/water at TE=30ms).23 Quality was visually confirmed. MR liver fat fractions were calculated from the corrected MR measures of CH2 and CH3 lipids and of water as the total lipids/(total lipids + water).
VAT and SAT volumes were semi-automatically generated based upon either water-suppressed gradient-recalled echo images or on the fat images generated from iterative decomposition and echo asymmetry with least-squares estimation (IDEAL) MR images (10 mm thick) at the disc between lumbar vertebrae 3 and 4. Regions of interest for VAT and SAT were determined by a single reader using a threshold-based contour mapping algorithm written in-house in IDL (Exelis Visual Information Solutions, Inc., Boulder, CO) followed by a manual alteration, as needed.
Outpatient Feeding and Follow-up
Upon completion of the metabolic assessments on day 0, participants were discharged to home with three days of food and detailed instructions. They returned at three-day intervals to pick up food for a total of nine days. On day 10, all day 0 assessments were repeated. As described previously,19 the UCSF-Clinical Research Service Bionutrition Core designed individualized menus for each child and provided all food. Study diets restricted sugar and fructose intake to 10% and 4% of total energy intake, respectively, by substituting an equal number of calories from starch, to match overall proportional carbohydrate consumption in each participant’s self-reported usual diet.19 Total energy content was estimated using Institute of Medicine formulae for weight maintenance in overweight boys and girls24 and adjusted if weight changed more than 2% during outpatient feeding.
DNL and Insulin Kinetics
Samples collected during the tracer/feeding studies underwent ultracentrifugation to isolate triglyceride-rich lipoproteins (TRL; density 1.006), and the palmitate from the TRL-TG fraction was analyzed by gas chromatography/mass spectrometry.25 Fractional DNL (percent of palmitate in circulating triglyceride that was synthesized de novo) was calculated by mass isotopomer distribution analysis.25 Integrated DNL-AUC was calculated during the 8-hour feeding period. Composite insulin sensitivity index (CISI26) and the oral glucose test of insulin sensitivity (OGIS27) were computed using insulin and glucose data from the OGTT. Insulin secretion rates (ISR) were calculated by deconvolution28 and insulin clearance rates determined by dividing the ISR-AUC by the product of insulin volume of distribution (assumed to equal the C-peptide volume of distribution) and the insulin-AUC.29, 30
Statistical Analyses
The primary outcome of the study was change in liver fat, with secondary outcomes of DNL and insulin kinetics. Normal distribution was tested by box-plot, q-norm plot, and Shapiro-Wilk tests. Descriptive statistics were reported as mean ± standard deviation for normally distributed values and as median (1st quartile, 3rd quartile) for non-normally distributed data. Outcome variables on day 0 and day 10 were compared by paired t-test if distributed normally or by Kruskal-Wallis test for non-normally distributed data, including tests for effects of sex or race/ethnicity. Analysis of covariance (ANCOVA) was performed to control for weight change. As reported earlier,19 average weight decreased by 0.9 kg (P=0.01) from day 0 to day 10, of which 0.6 kg was fat-free mass. A post-hoc sensitivity analysis was performed using data from nine participants who did not lose weight during the dietary intervention. To determine the impact of baseline liver fat content on metabolic outcomes, we compared results within and between participants with high liver fat (fat fraction ≥5%) and those with fat fraction <5% by paired and unpaired t-tests or Mann–Whitney U test for outcomes that were not normally distributed. P-values are based on two-tailed tests. Analyses were performed using STATA software version 12.1 (StataCorp, College Station, TX). Investigators remained blinded to key study outcomes including MR data, DNL, DXA, insulin kinetics, and other biochemical outcomes until data collection and analysis were completed. All authors had access to the study data and reviewed and approved the final manuscript.
RESULTS
Participants
As reported previously,19 52 Latino and African-American children were recruited. Two were ineligible, five failed to show on day 0, and two completed day 0 testing but did not return for day 10. This paper reports paired data in 41 children, of which 26 were Latino and 15 African-American; 15 were male and 26 female, median age was 13 years (range 9–18), median BMI z-score was 2.3 (1.9–3.2), and body fat 48.6 (35.3–55.9)%. Daily intake during the nine days of fructose restriction averaged 28±6 kilocalories/kg with an average macronutrient profile of 51±3% carbohydrate, 16±1% protein, and 33±3% fat. Within the carbohydrate fraction, dietary sugar intake decreased from 28±8% to 10±2%, and fructose intake from 12±4% to 4±1%.
MR Measures
At baseline, 25 participants (20 Latino, 5 African-American; p=0.003) had elevated liver fat (fat fraction ≥5%), and 15 (5 Latino, 10 African-American) had low liver fat (fat fraction <5%; Table 1). Paired MR measures were available in 38 participants for liver fat and 40 for VAT and SAT. From day 0 to day 10, liver fat decreased from a median of 7.1 (2.5, 14.8)% to 3.8 (1.7,15.6)% (P<0.001), and VAT decreased from 123 (85,145) cm3 to 110 (84,134) cm3 (P<0.001), while SAT did not change significantly (Fig 2A). Liver fat decreased in all but one of the 38 participants in whom paired data were available (Fig 2B). The decrease in liver fat after adjustment for weight change remained statistically significant (P=0.004). Among the nine participants who did not lose weight (Figs 2E–2H), liver fat decreased from 9.7 (2.5, 20.1)% to 6.3 (2.2, 17.6)% (P=0.02) and VAT from 124 (79, 190) cm3 to 91 (82, 154) cm3 (P=0.06). While males had higher liver fat and VAT on day 0 (p<0.05), loss of fat did not differ significantly by sex, in either absolute or relative terms. Liver fat and VAT were higher in Latinos for both day 0 and day 10 (p<0.003, Kruskal-Wallis; p<0.05, t-test, respectively). However, as a percent of day 0 values, the reductions in liver fat, weight-loss adjusted liver fat, VAT, and SAT were not significantly different between Latinos and African Americans.
Table 1.
Total Group Anthropometric and Metabolic Parameters and Categorized by Low Liver Fat or High Liver Fat
| Total Group; (n = 38 to 41) |
Low Liver Fat (fat fraction <5%); (n = 15) |
High Liver Fat (fat fraction ≥ 5%); (n = 25) |
High vs. Low Liver Fat | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Day 0 | Day 10 | ||||||||||
| Anthropometric Data | Day 0 | Day 10 | P value | Day 0 | Day 10 | P value | Day 0 | Day 10 | P value | P value | P value |
| Age (yrs) | 13.4 ± 2.8 | 13.8 ± 2.0 | 13.0 ± 2.1 | 0.39 | |||||||
| Female/Male | 26/15 | 12/3 | 13/12 | 0.08# | |||||||
| Latino/African American | 26/15 | 5/10 | 20/5 | 0.003# | |||||||
| Height (cm) | 161 ± 8 | 161 ± 7 | 161 ± 9 | 0.88 | |||||||
| BMI z-score | 2.36 ± 0.296 | 2.2 ± 0.2 | 2.4 ± 0.3 | 0.05 | |||||||
| Weight (kg) | 93.1 ± 22.9 | 92.0 ± 23.1 | <0.001 | 86.8 ± 14.8 | 85.8 ± 14.6 | 0.002 | 96.3 ± 26.2 | 95.4 ± 26.4 | <0.001 | 0.15 | 0.2 |
| MR Data | |||||||||||
| Liver Fat (%; n=38) | 7.1 (2.5,14.8) | 3.8 (1.7,15.6) | <0.001 | 2.5 (1.5, 2.8) | 1.4 (0.9, 1.9) | <0.001 | 12.6 (8.7, 25.8) | 11.0 (4.4, 21.4) | <0.001 | <0.001 | <0.001 |
| VAT (cm3; n=40) | 121 ± 40 | 111 ± 38 | <0.001 | 89 ± 24 | 84 ± 24 | 0.03 | 141 ± 35 | 127 ± 36 | 0.002 | <0.001 | <0.001 |
| SAT (cm3; n=40) | 444 (337,553) | 452 (337,555) | 0.10 | 403 (327, 456) | 392 (300, 471) | 0.55 | 474 (355, 589) | 471 (355, 596) | 0.98 | 0.06 | 0.05 |
| DNL Data | |||||||||||
| DNL-AUC (n=40) | 64.8 (46.3,85.3) | 25.9 (16.1,37.7) | <0.001 | 60.3 (36.8, 90.7) | 16.2 (10.3, 25.2) | <0.001 | 66.4 (55.3, 79.8) | 31.3 (25.5, 42.8) | <0.001 | 0.64 | <0.001 |
| Glucose and Insulin Kinetics Data | |||||||||||
| Fasting Glucose (mg/dL; n=41) | 97.5 (92.8,101.5) | 92.6 (88.2,95.6) | <0.001 | 97.2 (92.8, 101.5) | 90.2 (88.2, 97.3) | <0.001 | 98.4 (92, 103.2) | 92.6 (86.9, 95) | 0.03 | 0.69 | 0.89 |
| Fasting Insulin (uU/mL; n=41) | 30.2 (19.7,42.0) | 21.5 (14.0,30.4) | <0.001 | 23.5 ± 11.1 | 16.4 ± 7.5 | 0.002 | 39.0 ± 21.5 | 26.3 ± 10.8 | 0.001* | 0.01* | 0.002 |
| Composite Insulin Sensitivity Index (n=39) | 1.25 (1.05,1.63) | 1.89 (1.61, 2.38) | <0.001 | 1.41 (1.12, 1.70) | 2.04 (1.88, 2.61) | <0.001 | 1.18 (1.00, 1.60) | 1.72 (1.55, 2.31) | <0.001 | 0.02 | 0.049 |
| Oral Glucose Insulin Sensitivity (n=39) | 306 ± 75 | 366 ± 60 | <0.001 | 323 ± 68 | 395 ± 45 | <0.001 | 295 ± 78 | 348 ± 62 | <0.001 | 0.27 | 0.01 |
| Fasting Insulin Secretion Rate (pmol/min; n=39) | 432 ± 209 | 329 ± 148 | <0.001 | 306 ± 95 | 236 ± 82 | 0.000 | 511 ± 223 | 387 ± 152 | 0.001 | 0.001 | 0.001 |
| OGTT Insulin Secretion (pmol/min; n= 39) | 1509 (1081, 1816) | 1260 (884, 1627) | 0.05 | 1121 (898, 1682) | 963 (736, 1185) | 0.04 | 1684 (1303, 2177) | 1495 (1176, 1815) | 0.23 | <0.001 | 0.001 |
| Fasting Insulin Clearance (pools/min; n =39) | 0.42 (0.35,0.53) | 0.49 (0.39,0.58) | 0.06 | 0.42 (0.35, 0.54) | 0.52 (0.39, 0.63) | 0.19 | 0.46 (0.35, 0.53) | 0.49 (0.39, 0.57) | 0.16 | 0.57 | 0.53 |
| OGTT Insulin Clearance (pools/min; n=39) | 0.34 ± 0.11 | 0.40 ± 0.13 | 0.005 | 0.3 ± 0.09 | 0.37 ± 0.14 | 0.02* | 0.39 ± 0.12 | 0.44 ± 0.13 | 0.17* | 0.04 | 0.22 |
Parametric test when normal distribution was achieved; format Mean ± Standard Deviation
Non-parametric test (Kruskal Wallis) applied when normal distribution was not achieved; format Median (Q1, Q3)
Chi-square test applied
Parametric test (paired t-test) applied after data were log transformed to achieve normal distribution
Figure 2.
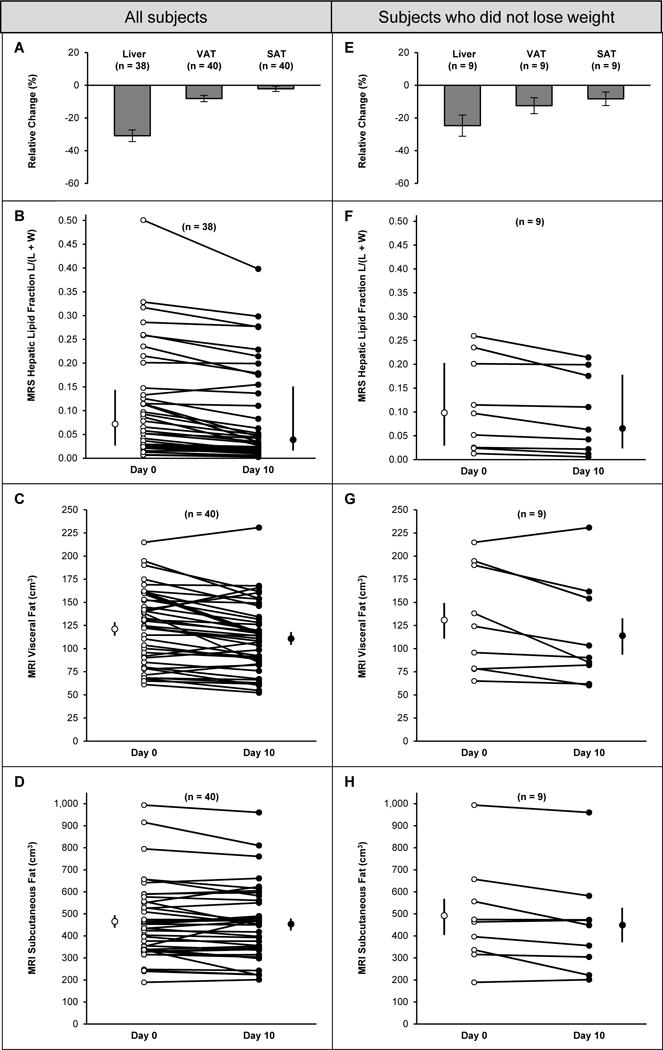
Changes in individual fat compartments in obese children (2A–2D) before and after nine days of isocaloric fructose restriction, and in the subset of 9 children who did not lose weight (2E–2H) during fructose restriction. Figs 2A and 2E depict average changes (mean ± SEM) in liver fat as determined by MR, and visceral (VAT) and subcutaneous (SAT) fat as determined by MR in the entire cohort (2A) and the subgroup of 9 participants who did not lose weight (2E). Figs 2B and 2F depict individual serial measures of liver fat in the entire cohort (2B) and the subgroup of 9 participants who did not lose weight (2F). Figs 2C and 2G depict individual serial measures of VAT. Figs 2D and 2H depict individual serial measures of SAT in the entire cohort (2D) and the subgroup of 9 participants who did not lose weight (2H). Open and closed circles to the left and right of the day 0 and day 10 individual plots depict median and interquartile range (Figs 3B and 3F) or mean ± SEM (Figs 3C, 3D, 3G, 3H). Decreases in liver fat and VAT were statistically significant in the group as a whole (P<0.001 in both cases). In the subgroup who did not lose weight, change in liver fat was statistically significant (P=0.02).
DNL and Insulin Kinetics
Fractional DNL over the 8-hour tracer study decreased significantly after nine days of fructose restriction, with the average values for DNL at each timepoint continuing to diverge for the entire duration of sampling (Fig 3A). DNL-AUC was significantly lower on day 10 (68.4±5.0 vs. 29.7±2.9; P<0.001), decreasing in 37 of 40 participants with paired data (Fig 3B). Results were also statistically significant in the subset of nine participants who did not lose weight (59.9±10.1 vs. 30.1±7.6; P=0.006; Figs 3C, 3D).
Figure 3.
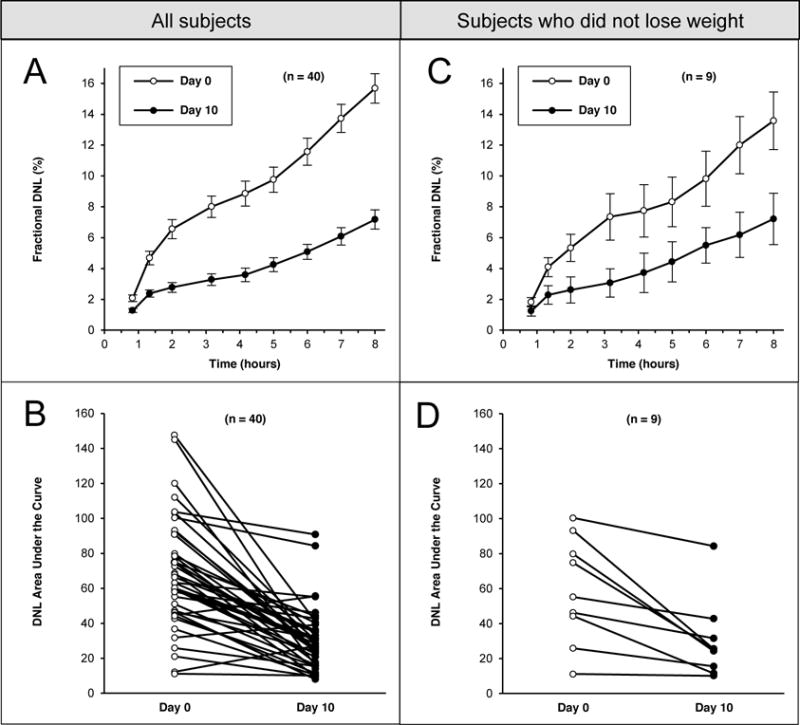
Changes in postprandial fractional de novo lipogenesis (DNL; percent of palmitate in circulating triglyceride that was synthesized de novo) and the integrated DNL-area under the curve (AUC) on days 0 (open circles) and 10 (closed circles) after isocaloric fructose restriction in 40 obese children (3A and 3B), and in the subgroup of 9 children who did not lose weight (3C and 3D) during fructose restriction. On both study days, after an overnight fast, and after the OGTT was complete, participants consumed liquid meals every 20 minutes for six hours, starting at 10:30 AM. Blood samples were obtained hourly during this period. Figs 3A and 3C depict fractional DNL (mean ± SEM) for all subjects (3A) and the subgroup of 9 participants who did not lose weight (3C). Figs 3B and 3D depict individual serial measures of DNL-AUC in the group as a whole (3B) and the subgroup who did not lose weight (3D). Decreases in DNL-AUC were statistically significant in the group as a whole (P<0.001) as well as the subgroup who did not lose weight (P=0.006).
Significant increases were observed in measures of insulin sensitivity (CISI, P<0.001; and OGIS, P<0.001) and OGTT insulin clearance rate (P<0.001) (Table 1). Significant decreases were observed both in fasting ISR (P<0.001), and in ISR during the OGTT (P<0.001). These changes remained significant even after adjustment for weight change (P<0.001).
Comparison of Subjects Based on Baseline Liver Fat Content
At baseline, DNL-AUC did not differ significantly between those with high (≥5%) vs. low liver fat (P=0.64; Table 1). After nine days of fructose restriction, DNL-AUC decreased significantly in both those with high liver fat and those with low liver fat, and the magnitude of decrease did not differ significantly between groups. However, DNL-AUC on day 10 was significantly lower in the low liver fat group, as compared to those with high liver fat. Liver fat and VAT also decreased significantly in both groups. Insulin secretion both during fasting and in response to OGTT decreased significantly in both groups. Insulin clearance rate increased significantly only in the high liver fat group.
DISCUSSION
In the past two decades, the prevalence of NAFLD has more than doubled in adolescents31 and adults32, with current estimates as high as 50% in the U.S.33 Hepatic steatosis, as well as other ectopic fat stores, are implicated in obesity-related metabolic dysfunction that occurs in adolescents34 and adults12 and includes insulin resistance, dyslipidemia, diabetes, and CVD7, 8, 35–38. Multiple cross-sectional studies have linked liver fat and VAT with metabolic complications of obesity, including insulin resistance, T2DM and CVD.6, 9, 10, 36, 38 Fabbrini et al.,10 using sensitive metabolic assessments, found that liver fat was more strongly associated with insulin resistance than was VAT. In this study we demonstrate that as few as nine days of isocaloric fructose restriction significantly reduced liver fat, DNL, and VAT; and improved insulin sensitivity, secretion, and clearance in children with obesity and metabolic syndrome. The improvements in these outcome measures occurred irrespective of baseline liver fat content, sex, or race/ethnicity.
Others have noted that both reduction in glycemic index/load improves liver fat and metabolic function in adolescents with NAFLD.39 Rather, our study demonstrates that isocaloric substitution of starch for sugar, which has the end-effect of increasing glycemic index, improved liver and visceral fat and insulin secretion and sensitivity, and within 10 days. Our data suggest that the effect of fructose on liver fat is specific and mediated through reductions in DNL.
DNL was originally thought to be a minor metabolic pathway in humans.25 However, increased DNL has been demonstrated in adults with NAFLD.35 Using stable isotopes, Donnelly et al.8 showed that in adults with NAFLD, approximately 59% of triglyceride labeled in the liver comes from circulating fatty acids released by peripheral lipolysis, 15% from dietary fat, and 26% from DNL. If fatty acid influx is not matched by hepatic fat oxidation and export, liver fat will accumulate. DNL impacts both sides of this equation, both by generating new lipids and by suppressing hepatic fat oxidation, as the intermediate malonyl-CoA prevents fatty acid transport into mitochondria by inhibiting carnitine palmitoyl transferase-1.40 Fructose consumption has been proposed as a primary contributor to NAFLD4, 41 by increasing DNL. We have recently shown that in healthy adults fed isocaloric diets, DNL and liver fat were higher during high-fructose feeding, when compared with low-fructose feeding.18 Those results, taken together with those of the present study, support the hypothesis that DNL is an important mechanism in the modulation of liver fat.8, 35,42 Moreover, the increases in VAT with high-fructose feeding2 and the decrease in VAT observed with fructose restriction observed in the present study suggest primary links between fructose consumption, DNL, and ectopic fat. The decline of fractional DNL following nine days of fructose restriction argues that the process of DNL is a rational target for dietary intervention.
Although study eligibility was not based on liver fat content, 63% of our participants had high liver fat. Consistent with other reports,43 high liver fat was significantly more prevalent in Latino children compared with African-Americans. However, even with a habitual diet high in fructose, 37% of the children appeared to be protected against NAFLD. On day 0, the subgroup of children with low liver fat had significantly lower fasting insulin levels and higher CISI than those with high liver fat (Table 1). After fructose restriction (day 10), fasting insulin levels remained significantly higher in the group with high liver fat (P=0.002), despite improvements in insulin sensitivity, secretion, and clearance. We noted that both insulin secretion and clearance improved with reduction in liver fat and postprandial DNL after fructose restriction, despite calorically equivalent increases in starch consumption. We have previously shown that persons with hyperinsulinemia have high fasting DNL compared to normoinsulinemic controls.44–46 A key role of hepatic insulin signaling in stimulating DNL has been reported.47, 48 Consistent with these observations, the children with high liver fat, who also had elevated fasting insulin levels, may also have had higher DNL even before feeding. These children likely had ‘round-the-clock’ DNL driven by fructose in the fed state and by hyperinsulinemia in the fasting state, thus providing a potential explanation for why some obese children have elevated liver fat and others do not. Further studies are necessary to test this hypothesis and to characterize the impact of genetic factors on liver fat.
In this paper, we document improvements in liver fat, DNL, insulin kinetics, and to a lesser extent VAT, in obese children when sugar in the diet is replaced with starch; that is, a glucose-for-fructose exchange. Liver and visceral fat are thought to play a prominent role in metabolic dysfunction.6, 7, 12 Previously published results from our study demonstrated reductions in blood pressure and levels of analytes related to prediabetes (e.g. lactate, glucose and insulin).19 In addition we reported improvement in lipoprotein profiles related to atherogenicity (e.g. triglyceride:HDL ratio, LDL size, and Apo-CIII concentration).20 All of these measures were performed in the fasting state, thus the improvements in metabolic function cannot be attributed to acute effect of fructose reduction in the liquid meals during the tracer feeding study on day 10. The improvements in metabolic, lipid, and ectopic fat parameters were accompanied by changes in HOMA-IR and CISI, two measures of peripheral insulin sensitivity. By demonstrating that removal of dietary fructose (the macronutrient most closely associated with hepatic DNL) concomitantly reduces liver fat and improves insulin dynamics irrespective of calories or weight, we are able to suggest a causative mechanism of metabolic dysfunction in these children by linking DNL to both liver fat and insulin resistance. We also demonstrated that despite an increase in the glucose (starch) content of the diet, insulin secretion decreased, thus protecting against beta-cell exhaustion, thought to be important in the pathogenesis of type 2 diabetes;7 and reducing total body insulin burden, thought to contribute to both obesity49 and risk for cardiovascular disease.50 These data also suggest an achievable dietary approach to improve metabolic dysfunction in similarly affected children who are high sugar consumers.
We note the following four limitations of this study. First, the study design did not include a separate external control group. However, including such a control group would have introduced new challenges. For example, studies document that dietary sugar intake by recall is consistently underestimated.51 Had we included an external control group, it is unlikely that we could have accurately matched their true baseline sugar intake, thus raising the possibility of over- or underfeeding sugar to the control group and potentially providing flawed results. Instead, each participant served as his/her own control, which minimized inter-participant variability. Future confirmatory studies should include a control group with specified and monitored fructose intake both prior to and during the experimental diet. Second, despite efforts to maintain baseline weight, overall there was a small but statistically significant weight loss (0.9 kg; 95% CI −1.3, −0.6). While small reductions in weight could improve metabolic health,29 we do not believe that the salutary weight loss in these subjects mitigates our findings related to reductions in liver fat and DNL. As discussed previously,19,20 the weight loss occurred within the first 4 days and then plateaued to reach a new steady state. This result is not consistent with persistent energy deficit at the day 10 visit. As reported previously,19 DXA scanning documented that the weight loss occurred within the fat-free compartment (e.g. water and/or muscle), the loss of either of which would not contribute substantially to improved metabolic health. Moreover, the results remained statistically significant after adjusting for this weight loss using repeated measures ANCOVA. We would be remiss in not acknowledging the possibility that slight changes in macronutrient or fiber content in the fructose-restricted diets19 may have been more satiating than their home baseline diet, which posed a challenge to the study coordinator to persuade participants to increase intake past comfort. Perhaps most importantly, sensitivity analysis documented statistically significant improvements in DNL, liver fat, and VAT in the subgroup of participants who did not lose weight (Figs 2E–2H and 3C and 3D). Third, we did not measure DNL in the fasting state. While such a steady-state measurement would have yielded important information about genetic predisposition toward NAFLD and the existence of ‘round-the-clock DNL’ in susceptible populations, doing so would have required changing from an outpatient to an inpatient protocol, which would have limited recruitment and retention of pediatric participants, which was already quite demanding. Lastly, we acknowledge that our study design does not allow us to speculate on benefits of fructose restriction in normal-weight children or adults, or extrapolate our results to obese individuals whose diets are low in fructose content.
To date, small non-randomized studies in obese children have shown improvements in liver histology and aminotransferase activity after weight loss.52, 53 Rather, in this study we demonstrate that as few as nine days of isocaloric fructose restriction significantly reduced liver fat, DNL, and VAT; and improved insulin sensitivity, secretion, and clearance in children with obesity and metabolic syndrome. The improvements in these outcome measures occurred irrespective of baseline liver fat content or weight change. These results suggest that fructose consumption and hyperinsulinemia are important determinants of DNL and liver fat, at least in high sugar consumers. These short-term data support an intervention focusing on fructose restriction as an approach to both combat NAFLD and improve insulin kinetics. Further studies will be required to determine the efficacy of long-term fructose restriction as a means of preventing or reversing NAFLD and its associated metabolic sequelae. Nonetheless, this study provides evidence that support recent public health efforts to reduce sugar consumption as a means to improve metabolic health.
Acknowledgments
The authors would like to thank all of the participants and parents/caregivers who volunteered for this study. Thanks is also given to all the UCSF Clinical and Translational Sciences Institute (CTSI) Pediatric and Adult CRS Staff (Jean Addis, Sarah Fuerstenau, Erin Matsuda, Grace Mausisa, Abigail Sobejana, Grady Kimes, Erin Miller, Raquel Herrera, Tamara Williamson, John Duda, Caitlin Sheets) who assisted with this study, as well as the Bionutrition staff, Cewin Chao, Jennifer Culp, and Monique Schloetter, who planned and prepared the fructose-restricted diets. A special thank you to Drs. Emily Perito and Patrika Tsai. We also thank Arianna Pham, Davis Tang, Ari Simon, Russell Caccavello, and Artem Dyachenko for laboratory assistance and Dr. Andrew Gilman for assistance with the MRIs. Special acknowledgment is given to Drs. Zea Malawa and Tami Hendriksz, who helped recruit participants. Thanks to Laurie Herraiz, who helped design and implement the dietary protocol. Thanks also to the WATCH clinic coordinators, who helped screen patients and implement this protocol, including Rachel Lipman, Sally Elliott, and Drs. Kelly Jordan and Katrina Koslov.
Funding: This project was supported by the NIH (R01DK089216, P30DK056341), UCSF Clinical and Translational Science Institute (NCATS–UL1-TR00004), and Touro University. The sponsors had no role in study design, the collection, analysis, or interpretation of data, the writing of the report, or in the decision to submit the article for publication.
Grant Support: This project was supported by the NIH (R01DK089216, P30DK056341), UCSF Clinical and Translational Science Institute (NCATS–UL1-TR00004), and Touro University.
Abbreviations
- DNL
de novo lipogenesis
- VAT
visceral adipose tissue
- SAT
subcutaneous adipose tissue
- AUC
area under the curve
- NAFLD
non-alcoholic fatty liver disease
- T2DM
type 2 diabetes mellitus
- CVD
cardiovascular disease
- ATP
adenosine triphosphate
- BMI
body mass index
- ALT
alanine aminotransferase
- HOMA-IR
homeostatic model assessment of insulin resistance
- DXA
dual-energy X-ray absorptiometry
- OGTT
oral glucose tolerance test
- K2EDTA
potassium ethylenediaminetetraacetic acid
- MR
magnetic resonance
- ms
milliseconds
- L/W
lipid/water
- CISI
composite insulin sensitivity index
- OGIS
oral glucose test of insulin sensitivity
- ISR
insulin secretion rate
- ANCOVA
analysis of covariance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: SMN receives funding from Gilead Sciences, Inc. and from Verily Life Sciences for projects outside the submitted work. RHL wrote a book on obesity for the general public in 2012. All other authors report no conflicts of interest.
Author Contributions: JMS, KM, SMN, AG and RHL were responsible for study design and funding, overall study supervision, and writing the draft manuscript. SMN and NJK performed all MR scans and analyses. AEC was responsible for study recruitment and procedures. AEC and SMN performed statistical analysis. VWT was responsible for dietary design, supervision of food preparation, and participant and parent education. MJW organized collection, processing, and storage of clinical specimens and supervised laboratory analyses; GMJ, SP, and MVA analyzed biochemical specimens and performed mass spectrometry analyses. BWP performed modeling analyses on insulin kinetics using data generated by KP. All authors contributed to the interpretation of the results, provided important intellectual input, and approved the final version of this manuscript.
Presented in part at the Endocrine Society, San Diego, CA, USA on March 4, 2015; the International Society of Magnetic Resonance in Medicine, Toronto, Ontario, Canada on May 30, 2015; and the Obesity Society, Los Angeles, CA, USA on Nov. 4, 2015.
References
- 1.Maersk M, Belza A, Stødkilde-Jørgensen H, et al. Sucrose-sweetened beverages increase fat storage in the liver, muscle, and visceral fat depot: a 6-mo randomized intervention study. Am J Clin Nut. 2012;95:283–289. doi: 10.3945/ajcn.111.022533. [DOI] [PubMed] [Google Scholar]
- 2.Stanhope KL, Schwarz JM, Keim NL, et al. Consuming fructose-, not glucose-sweetened beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest. 2009;119:1322–1334. doi: 10.1172/JCI37385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pollock NK, Bundy V, Kanto W, et al. Greater fructose consumption is associated with cardiometabolic risk markers and visceral adiposity in adolescents. J Nutr. 2012;142:251–257. doi: 10.3945/jn.111.150219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim JS, Mietus-Snyder M, Valente A, et al. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol. 2010;7:251–264. doi: 10.1038/nrgastro.2010.41. [DOI] [PubMed] [Google Scholar]
- 5.Stanhope KL, Schwarz JM, Havel PJ. Adverse metabolic effects of dietary fructose: Results from recent epidemiological, clinical, and mechanistic studies. Curr Opin Lipidol. 2013;24:198–206. doi: 10.1097/MOL.0b013e3283613bca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yki-Järvinen H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014;2:901–910. doi: 10.1016/S2213-8587(14)70032-4. [DOI] [PubMed] [Google Scholar]
- 7.Perry RJ, Samuel VT, Petersen KF, et al. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature. 2014;510:84–91. doi: 10.1038/nature13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donnelly KL, Smith CI, Schwarzenberg SJ, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mathieu P, Boulanger MC, Després JP. Ectopic visceral fat: A clinical and molecular perspective on the cardiometabolic risk. Rev Endocr Metab Disord. 2014;15:289–298. doi: 10.1007/s11154-014-9299-3. [DOI] [PubMed] [Google Scholar]
- 10.Fabbrini E, Magkos F, Mohammed BS, et al. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc Natl Acad Sci. 2009;106:15430–15435. doi: 10.1073/pnas.0904944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cali AM, De Oliveira AMKH, Chen S, et al. Glucose dysregulation and hepatic steatosis in obese adolescents: is there a link? Hepatology. 2009;49:1896–1903. doi: 10.1002/hep.22858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sung KC, Kim SH. Interrelationship between fatty liver and insulin resistance in the development of type 2 diabetes. J Clin Endocrinol Metab. 2011;96:1093–1097. doi: 10.1210/jc.2010-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Birkenfeld AL, Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology. 2014;59:713–723. doi: 10.1002/hep.26672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newton KP, Hou J, Crimmins NA, et al. Prevalence of prediabetes and type 2 diabetes in children with nonalcoholic fatty liver disease. JAMA Pediatr. 2016;170(10):e161971. doi: 10.1001/jamapediatrics.2016.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hudgins LC, Parker TS, Levine DM, et al. A dual sugar challenge test for lipogenic sensitivity to dietary fructose. J Clin Endocrinol Metab. 2011;96:861–868. doi: 10.1210/jc.2010-2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacome-Sosa MM, Parks EJ. Fatty acid sources and their fluxes as they contribute to plasma triglyceride concentrations and fatty liver in humans. Curr Opin Lipidol. 2014;25:213–220. doi: 10.1097/MOL.0000000000000080. [DOI] [PubMed] [Google Scholar]
- 17.Kim MS, Krawczyk SA, Doridot L, et al. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J Clin Invest. 2016;126:4372–4386. doi: 10.1172/JCI81993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwarz JM, Noworolski SM, Wen MJ, et al. Effect of a high-fructose weight-maintaining diet on lipogenesis and liver fat. J Clin Endocrinol Metab. 2015;100:2434–2442. doi: 10.1210/jc.2014-3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lustig RH, Mulligan K, Noworolski SM, et al. Isocaloric fructose restriction and metabolic improvement in children with obesity and metabolic syndrome. Obesity. 2016;24:453–460. doi: 10.1002/oby.21371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gugliucci A, Lustig RH, Caccavello R, et al. Short-term isocaloric fructose restriction lowers apoC-III levels and yields less atherogenic lipoprotein profiles in children with obesity and metabolic syndrome. Atherosclerosis. 2016;253:171–177. doi: 10.1016/j.atherosclerosis.2016.06.048. [DOI] [PubMed] [Google Scholar]
- 21.Matthews DR, Hosker JP, Rudenski AS, et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 22.Noworolski SM, Lam M, Merriman RB, et al. Estimation of steatosis with MRS and MRI: validation with histology is confounded by differences in methodology. Radiology. 2012;264:88–96. doi: 10.1148/radiol.12110673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noworolski SM, Tien PC, Merriman R, et al. Respiratory motion-corrected proton magnetic resonance spectroscopy of the liver. Magn Reson Im. 2009;27:570–576. doi: 10.1016/j.mri.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Food and Nutrition Board. Dietary Reference Intakes: Energy, carbohydrate, fiber, fat, fatty acids, cholesterol, protein, and amino acids (macronutrients) Washington, D.C: Institute of Medicine; 2005. [Google Scholar]
- 25.Hellerstein MK, Christiansen M, Kaempfer S, et al. Measurement of de novo hepatic lipogenesis in humans using stable isotopes. J Clin Invest. 1991;87:1841–52. doi: 10.1172/JCI115206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care. 1999;22:1462–1470. doi: 10.2337/diacare.22.9.1462. [DOI] [PubMed] [Google Scholar]
- 27.Mari A, Pacini G, Murphy E, et al. A model-based method for assessing insulin sensitivity from the oral glucose tolerance test. Diab Care. 2001;24:539–548. doi: 10.2337/diacare.24.3.539. [DOI] [PubMed] [Google Scholar]
- 28.Sparacino G, Pillonetto G, Capello M, et al. WINSTODEC: a stochastic deconvolution interactive program for physiological and pharmacokinetic systems. Comput Methods Programs Biomed. 2002;67:67–77. doi: 10.1016/s0169-2607(00)00151-6. [DOI] [PubMed] [Google Scholar]
- 29.Magkos F, Fraterrigo G, Yoshino J, et al. Effects of moderate and subsequent progressive weight loss on metabolic function and adipose tissue biology in humans with obesity. Cell Metab. 2016;23:591–601. doi: 10.1016/j.cmet.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weiss EP, Albert SG, Reeds DN, et al. Calorie restriction and matched weight loss from exercise: independent and additive effects on glucoregulation and the incretin system in overweight women and men. Diab Care. 2015;38:1253–1262. doi: 10.2337/dc14-2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Welsh JA, Karpen S, Vos MB. Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988–1994 to 2007–2010. J Pediatr. 2013;162:496–500. doi: 10.1016/j.jpeds.2012.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams KH, Shackel NA, Gorrell MD, et al. Diabetes and nonalcoholic fatty liver disease: a pathogenic duo. Endocr Rev. 2013;34:84–129. doi: 10.1210/er.2012-1009. [DOI] [PubMed] [Google Scholar]
- 33.Widhalm K, Ghods E. Nonalcoholic fatty liver disease: a challenge for pediatricians. Int J Obes. 2010;34:1451–1467. doi: 10.1038/ijo.2010.185. [DOI] [PubMed] [Google Scholar]
- 34.Fonvig CE, Chabanova E, Andersson EA, et al. 1H-MRS measured ectopic fat in liver and muscle in Danish lean and obese children and adolescents. PLoS One. 2015;10:e0135018. doi: 10.1371/journal.pone.0135018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lambert JE, Ramos–Roman MA, Browning JD, et al. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146:726–735. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burgert TS, Taksali SE, Dziura J, et al. Alanine aminotransferase levels and fatty liver in childhood obesity: associations with insulin resistance, adiponectin, and visceral fat. J Clin Endocrinol Metab. 2006;91:4287–4294. doi: 10.1210/jc.2006-1010. [DOI] [PubMed] [Google Scholar]
- 37.Byrne CD, Targher G. Ectopic fat, insulin resistance, and nonalcoholic fatty liver disease: implications for cardiovascular disease. Arterioscler Thromb Vasc Biol. 2014;34:1155–1161. doi: 10.1161/ATVBAHA.114.303034. [DOI] [PubMed] [Google Scholar]
- 38.Lim S, Meigs JB. Links between ectopic fat and vascular disease in humans. Arterioscler Thromb Vasc Biol. 2014;34:1820–1826. doi: 10.1161/ATVBAHA.114.303035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mager DR, Iñiguez IR, Gilmour S, et al. The effect of a low fructose and low glycemic index/load (FRAGILE) dietary intervention on indices of liverfunction, cardiometabolic risk factors, and body composition in children and adolescents with nonalcoholic fatty liver disease (NAFLD) J Parenter Enteral Nutr. 2015;39:73–84. doi: 10.1177/0148607113501201. [DOI] [PubMed] [Google Scholar]
- 40.McGarry JD. Banting Lecture 2001: Dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes. 2002;51:7–18. doi: 10.2337/diabetes.51.1.7. [DOI] [PubMed] [Google Scholar]
- 41.Ouyang X, Cirillo P, Sautin Y, et al. Fructose consumption as a risk factor for nonalcoholic fatty liver disease. J Hepatol. 2008;48:993–999. doi: 10.1016/j.jhep.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Savage DB, Choi CS, Samuel VT, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest. 2006;116:817–824. doi: 10.1172/JCI27300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kalia HS, Gaglio PJ. The prevalence and pathobiology of nonalcoholic fatty liver disease in patients of different races or ethnicities. Clin Liver Dis. 2016;20:215–224. doi: 10.1016/j.cld.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 44.Schwarz JM, Chiolero R, Revelly JP, et al. Effects of enteral carbohydrates on de novo lipogenesis in critically ill patients. Am J Clin Nutr. 2000;72:940–945. doi: 10.1093/ajcn/72.4.940. [DOI] [PubMed] [Google Scholar]
- 45.Schwarz JM, Mulligan K, Lee J, et al. The effects of recombinant human growth hormone on hepatic lipid and carbohydrate metabolism in HIV-infected patients with fat accumulation. J Clin Endocrinol Metab. 2002;87:942–945. doi: 10.1210/jcem.87.2.8391. [DOI] [PubMed] [Google Scholar]
- 46.Schwarz JM, Linfoot P, Dare D, et al. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am J Clin Nutr. 2003;77:43–50. doi: 10.1093/ajcn/77.1.43. [DOI] [PubMed] [Google Scholar]
- 47.Titchenell PM, Quinn WJ, Lu M, et al. Direct hepatocyte insulin signaling is required for lipogenesis but Is dispensable for the suppression of glucose production. Cell Metab. 2016;23:1154–1166. doi: 10.1016/j.cmet.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. 2008;118:829–838. doi: 10.1172/JCI34275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Templeman NM, Skovsø S, Page MM, et al. A causal role for hyperinsulinemia in obesity. J Endocrinol. 2017;232:R173–R183. doi: 10.1530/JOE-16-0449. [DOI] [PubMed] [Google Scholar]
- 50.Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol. 2016;12:144–153. doi: 10.1038/nrendo.2015.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rangan A, Allman-Farinelli M, Donohoe E, et al. Misreporting of energy intake in the 2007 Australian Children’s Survey: differences in the reporting of food types between plausible, under- and over-reporters of energy intake. J Hum Nutr Diet. 2014;27:450–458. doi: 10.1111/jhn.12182. [DOI] [PubMed] [Google Scholar]
- 52.Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Gastroenterology. 2012;142:1592–1609. doi: 10.1053/j.gastro.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 53.Suzuki A, Abdelmalek MF, Schwimmer JB, et al. Association between puberty and features of nonalcoholic fatty liver disease. Nonalcoholic Steatohepatitis Clinical Research Network. Clin Gastroenterol Hepatol. 2012;10:786–794. doi: 10.1016/j.cgh.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]