

Abstract
Background
Glial cells are essential in maintaining synaptic function. In glutamatergic synapses astrocytes remove the products of neural activity (i.e. potassium, glutamate and excess water) from the synaptic cleft and redistribute them across the glial network; these products of neural activity can then be recycled for neuronal use or released into the vascular compartment. This type of highly coupled cell network -or syncytium- maintains the balance of synaptic activity by restoring the basal levels of such molecules in the synaptic cleft. Previous studies have reported alterations of glia related genes in Major Depressive Disorder, including some genes related to syncytial function.
Methods
We used RNA isolated from hippocampal tissues of 13 MDD subjects and 10 healthy controls to broadly examine gene expression using microarrays. Hippocampal RNA samples were isolated by laser capture microdissection from human tissue sections carefully avoiding contamination from neighboring structures. Once RNA quality was validated RNA was labeled and hybridized to microarrays.
Results
Analysis of microarray data identified mRNA transcripts involved in glial syncytial function that were downregulated in MDD subjects compared to controls, including potassium and water channels (KCNJ10, AQP4), gap junction proteins (GJA1) and glutamate transporters (SLC1A2, SLC1A3). These gene expression differences were confirmed by qPCR.
Conclusions
The downregulation of these genes related to the syncytial network activity of glial cells is consistent with the hypothesis that synaptic homeostasis is disrupted thereby disrupting hippocampal synaptic function in MDD patients. Such glial gene expression changes could contribute either to the onset or perpetuation of depressive symptoms and hence, represent targets for novel therapeutics.
Introduction
Over the last century several theories have emerged in an effort to explain the etiology of major depressive disorder (MDD). These theories have led to the development of diverse therapeutic treatment strategies, each with inconsistent efficacy (Murrough and Charney 2012). Moreover, the sum of current evidence suggests that MDD may be the result of multifactorial pathogenic processes that lead to disrupted affect, cognitive performance and neuroendocrine homeostatic processes. These clinical manifestations are underlined by numerous functional alterations and genetic variability, making it unlikely that such heterogeneity could be captured comprehensibly by a single theoretical model of depression. For example, current antidepressant therapies are largely based on the use of monoaminergic-related compounds, but the overall treatment efficacy remains controversial, and may be as low as 25% (Pigott, Leventhal et al. 2010). Other mechanisms explored include the alterations of the HPA axis, which are found in about 50% of patients (Halbreich, Asnis et al. 1985, Akil, Haskett et al. 1993, Checkley 1996, Medina, Seasholtz et al. 2013) and the involvement of growth factors and neuroimmune molecules, but the therapeutic applications of these mechanisms are still under investigation. More recently research efforts point towards the glutamatergic system as a new target for the management of treatment-resistant depression (Machado-Vieira, Manji et al. 2009, Bunney and Bunney 2012, Mathews, Henter et al. 2012). Although most glutamate-related alterations seem to be brain region-dependent and somewhat inconsistent, one finding that appears in a more consistent manner is the downregulation of glial glutamate transporters (Choudary, Molnar et al. 2005, Bernard, Kerman et al. 2011, Medina, Burke et al. 2013). These membrane proteins are located mainly in the astrocytes surrounding the synaptic cleft and constitute a pivotal element on the regulation of glutamatergic neurotransmission (Torp, Danbolt et al. 1994, Danbolt 2001, Amara and Fontana 2002). Furthermore, the reports involving the downregulation of glutamate transporters in MDD indicate that other components of the glial compartment may be compromised as well (Choudary, Molnar et al. 2005, Bernard, Kerman et al. 2011).
Information processing in the brain is a function traditionally attributed to neurons, due to their excitable properties and specialized connectivity. However, an increasing body of evidence suggests that glial cells also contribute to the modulation of neural activity, by receiving and integrating neurochemical signals and by altering the neuronal response to such stimuli (Vernadakis 1996, Parpura, Heneka et al. 2012, Verkhratsky, Rodriguez et al. 2012). To competently modulate neuronal functions, glial cells require properties and mechanisms that permit them to quickly and efficiently communicate with both neurons as well as other glial cells. Kuffler et al (1966) described how glial cells were widely coupled through cell to cell junctions, and such observations led to the idea of a syncytium-like organization of the glial compartment in the brain. By operating as a highly coupled network glia cells allow rapid communication across this network while permitting the restoration of homeostatic status of neural tissue after synaptic activity (Albers and Siegel 1999). The effectiveness of the glial syncytium depends on the coordinated activity of a series of membrane molecules that include gap junction forming proteins, water and ion channels and membrane transporters, as well as the structural protein complexes associated with them. The search for a specific role of glial cells in psychiatric disorders, including MDD is a novel approach to this disease that may reveal new options for patient treatment.
Previous studies from our consortium have revealed alterations of genes related to the glial syncytial function in other brain regions, where they showed up in canonical pathway analysis or by targeted searches of neurotransmitter systems (Choudary, Molnar et al. 2005, Bernard, Kerman et al. 2011). However, in those studies no functional association between these particular set of genes is mentioned. Our approach comes from the collation of separate pieces of evidence that pointed towards a specific astrocytic function that is altered in MDD, namely the syncytial function. We elect to use hippocampal tissue as this structure is a key component of the limbic system and has a central role in the regulation of emotion and cognition and a large body of evidence emphasizes the role of hippocampal dysfunction in psychiatric symptoms. For a review see Drevets, Price (2008).
We hypothesize that the decreased capability of the glial syncytial function is an important component of pathological changes underlying MDD clinical features.
Materials and Methods
Human subjects
Human brain tissues from 13 MDD subjects and 10 controls were obtained from the Brain Donor Program at the University of California, Irvine by agreement with the Pritzker Neuropsychiatric Consortium. The program obtains informed consent and medical information from the subjects’ next of kin, including physical health, medication and recreational substance use, to be added to the medical records and coroner’s investigation reports. The subjects included in the MDD group met diagnostic criteria from the Diagnostics and Statistical Manual of Mental Disorders (DSM-IV). In the control group there was no evidence of psychiatric or neurological disorders nor any in their first degree relatives. The information on serial numbers, general demographics, cause of death, pH, agonal factor scores (AFS) and post mortem intervals in hours (PMI) for the subjects used in the study is summarized in table 1.
Table 1.
The table shows the serial numbers, general demographics, cause of death, pH, agonal factor scores (AFS) and post mortem intervals in hours (PMI) for the subjects used in the study. SMC: sudden medical condition. LTMC: long-term medical condition. Acc/overdose: accidental overdose. C: Caucasian.
| Subject ID | Dx | Gender | Age | Cause of Death | AFS | pH | PMI | Race |
|---|---|---|---|---|---|---|---|---|
| 4354 | MDD | M | 55 | SMC | 0 | 7.1 | 20.8 | C |
| 2944 | MDD | M | 52 | SMC | 0 | 6.8 | 16 | C |
| 2267 | MDD | M | 19 | Suicide | 0 | 7.1 | 18 | C |
| 3204 | MDD | M | 46 | SMC | 0 | 6.5 | 24 | C |
| 3426 | MDD | M | 63 | SMC | 0 | 7.1 | 29 | C |
| 4282 | MDD | M | 51 | SMC | 0 | 6.5 | 29 | C |
| 4590 | MDD | M | 55 | Suicide | 0 | 6.9 | 25 | C |
| 4626 | MDD | M | 64 | Suicide | 0 | 7.0 | 12 | C |
| 3168 | MDD | M | 39 | Suicide | 0 | 6.7 | 28 | C |
| 4188 | MDD | M | 65 | Suicide | 0 | 6.8 | 27 | C |
| 4232 | MDD | M | 54 | Suicide | 0 | 6.7 | 25 | C |
| 3071 | MDD | M | 49 | Acc/overdose | 0 | 7.0 | 31 | C |
| 3169 | MDD | M | 35 | Acc/overdose | 0 | 7.0 | 25 | C |
| 3572 | Control | M | 49 | SMC | 0 | 6.6 | 28 | C |
| 2805 | Control | M | 45 | SMC | 0 | 6.86 | 21 | C |
| 1834 | Control | M | 40 | LTCM | 0 | 6.7 | 12 | C |
| 2619 | Control | M | 48 | SMC | 0 | 6.7 | 20 | C |
| 3520 | Control | M | 74 | SMC | 0 | 7.2 | 19 | C |
| 3993 | Control | M | 73 | SMC | 0 | 6.7 | 27 | C |
| 4235 | Control | M | 42 | SMC | 0 | 7.0 | 20 | C |
| 4327 | Control | M | 56 | SMC | 0 | 6.6 | 9 | C |
| 4387 | Control | M | 53 | SMC | 0 | 6.6 | 14 | C |
| 4638 | Control | M | 78 | SMC | 0 | 6.7 | 22 | C |
Previous post-mortem studies conducted by our group concluded that tissue pH and a patient’s agonal state are the strongest influences on brain gene expression patterns (Tomita, Vawter et al. 2004). To minimize that effect, we used brain samples with a pH over 6.5 and an agonal factor of zero, indicating a quick death. As for other factors such as treatment and alcohol comorbidity, we acknowledge the possibility of this having an effect on the results, but the nature of the sample collection restrains the sample size so we chose to include all subjects available that passed the pH/agonal factor criteria to avoid losing analysis power.
Human brain tissue preparation
Following the method described by Jones et al (1992) the brains were removed during autopsy, cooled down and cut in the coronal plane into 0.75 cm slabs, which were then frozen and stored at −80°C. To ensure the selection of samples adequate for gene expression analysis, brain tissue pH and agonal factors were evaluated following the procedure described by Tomita et al (2004).
Smaller tissue blocks containing the hippocampus were dissected from the frozen brain slabs, sectioned on a cryostat 10 μm, mounted onto Superfrost®/Plus glass slides (Fisher Scientific, USA), and stored at −80 °C. Since the successful analysis of gene expression in the brain across individuals and disease is enhanced when similar brain structures are compared, prior to this study a detailed anatomical alignment of the each subject’s hippocampus was performed using a combination of classic histological techniques and in situ hybridization for the mineralocorticoid receptor (MR) as anatomical descriptors of human hippocampal anatomy to optimize the processes of laser capture microdissection, RNA isolation and microarray analyses. (Krolewski, Medina et al. 2010, Medina, Seasholtz et al. 2013).
Laser Capture Microdissection and Microarray analysis
Based on the anatomical alignment, tissue sections containing coronal sections of the hippocampus approximately midway of its rostrocaudal axis were selected for each subject to perform laser capture microdissection and RNA isolation.
Samples were processed for laser capture microdissection using an Arcturus Autopix LCM 1110 (Life Technologies, Carlsbad, CA), according to the protocol described by Bernard et al (2009). Briefly, adjacent tissue sections were processed for in situ hybridization using mineralocorticoid receptor (MR) cRNA probes. MR hybridization signals were then used as an anatomical guide to align hippocampi across patient samples (Medina, Seasholtz et al. 2013), carefully excluding the anatomical structures that surround the hippocampus, such as the enthorrinal cortex or the choroid plexus of the neighboring ventricles, which would considerably affect the gene expression patterns observed in the microarray experiments (Figure 1). Tissues were captured on Arcturus CapSure HS LCM caps (Life Technologies, Carlsbad, CA). RNA extraction was performed using PicoPure RNA isolation kit and amplified with 1.5 round RiboAmp Plus Protocol from Applied Biosystems (Life Technologies, Carlsbad, CA) followed by CTP and UTP biotin labeling using Enzo Bioarray High Yield RNA transcription labeling kit (Enzo Life Sciences, Farmingdale, NY). Biotin-tagged cRNA samples were then applied to the Illumina Human HT-12 v4.0 array (Illumina Inc., Hayward, CA) and processed following manufacturer instructions. All microarrays were scanned on the Bead Array Reader (Illumina Inc, Hayward, CA). Intensity data was quantile normalized using Genome Studio software (Illumina Inc, Hayward, CA) and diff scores with p values were obtained. Fold changes were calculated by exporting data into Microsoft Excel 2007 software (Microsoft, Redmond, WA, USA).
Figure 1.
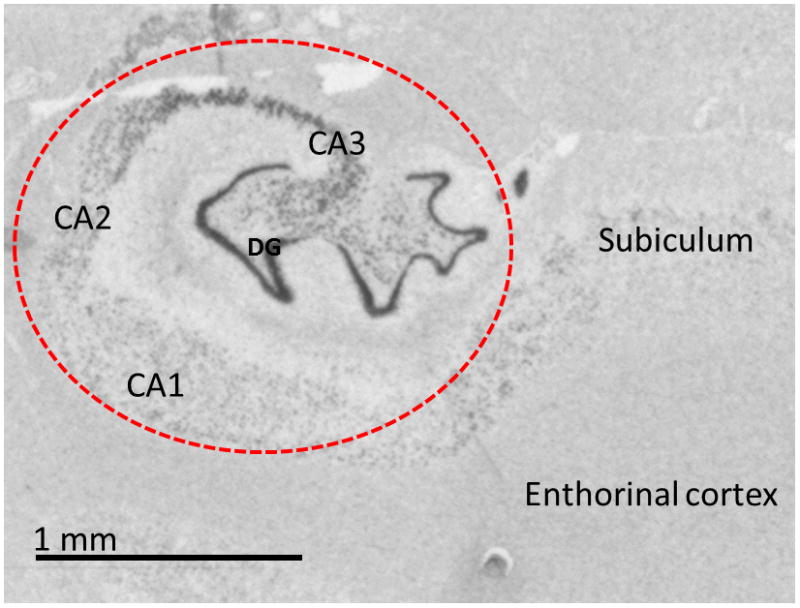
In situ hybridization image for the mineralocorticoid receptor (MR). The pattern of expression of MR mRNA was used as an anatomical template for the laser capture microdissection to ensure the exclusion of neighboring structures that could affect the content of the mRNA extracts. The red dotted line represents the area captured for each subject in this study.
Previous expression profiling studies from our group have shown altered expression of some glial syncytial function-related genes in different brain areas in MDD (Choudary, Molnar et al. 2005, Bernard, Kerman et al. 2011, Medina, Burke et al. 2013). However, since this specific function does not appear in pathway analysis tools, we propose a model of molecular interaction (figure 2) based on literature findings that have shown close functional interactions between these gene products and literature reports of the relationship between individual syncytium related molecules and the presence of depressive symptoms (Nagelhus, Horio et al. 1999, Amiry-Moghaddam, Williamson et al. 2003, Zeng, Sun et al. 2007, Banasr and Duman 2008). The selected genes were considered to be significantly altered in the disease state compared to controls if diff scores were 13 or higher or −13 or lower in the base of negative values, corresponding to P values that were p=0.05 or lower. The diff score is a value provided by the Illumina® software GenomeStudio that provides directionality to the p-value based on the difference between the average signal in the reference group vs. the comparison group. The formula is: DiffScore = 10*sgn(μcond-μref)*log10p; For a p-value of 0.05, DiffScore = ± 13; For a p-value of 0.01, DiffScore = ± 22; For a p-value of 0.001, DiffScore = ± 33. P values used in this study are always two-tailed. The results were then confirmed using qPCR. Fold change (FC) values were also calculated to assess the magnitude of the change in the disease subjects.
Figure 2.

mRNA expression levels of glial syncytium related genes in the human hippocampus. qPCR results show a downregulation of all genes studied in MDD subjects compared to controls. Error bars: SEM.
Real time PCR
We used qPCR to validate the differential gene expression results from selected genes from our microarray study. qPCR primers were designed using MacVector software (MacVector Inc, Cary, NC). Total RNA (1 ug) from each RNA sample was converted into cDNA using iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA). Quantitative PCR reactions included 1 μl of cDNA as a template and all reactions were performed in duplicate using SsoAdvanced SYBR green supermix according to the manufacturer’s instructions (Bio-Rad Laboratories, Hercules, CA). Beta actin mRNA was used as housekeeping mRNA to normalize the expression level of the genes of interest, and no differences were observed between the two groups. Values for each gene were evaluated using the 2−(ΔΔCt) method (Livak and Schmittgen 2001) and expressed as fold change from the control samples.
Results
Syncytial related genes are downregulated in MDD
The data obtained from GenomeStudio showed a total of 1114 genes differentially expressed between the control and MDD groups. For this particular experiment we selectively searched the results for genes related to glial function that had previously shown on canonical pathway analyses and targeted neurotransmitter system searches of previous microarray results in different brain areas of individuals of this cohort (Choudary, Molnar et al. 2005, Bernard, Kerman et al. 2011, Medina, Burke et al. 2013).
Microarray results indicated that a group of key mRNA transcripts involved in glial syncytial function are downregulated in MDD subjects compared to controls. The molecules affected included two glutamate transporters, SLC1A2 and SLC1A3, (p=0.01, FC −1.61 and p=0.002, FC=−1.39 respectively), the water channel AQP4 (p=0.004, FC=−1.61), the gene GJA1 (encoding for connexin 43, p=0.0002, FC=−1.64) and KCNJ10, encoding for the inward rectifying potassium channel Kir4.1 (p=0.04, FC=−1.24).
qPCR assays confirmed the downregulation of SLC1A2 (p=0.002, FC=−1.51); SLC1A3, (p=0.001, FC −1.58); AQP4 (p=0.003, FC=−1.59); GJA1 (p=0.004, FC=−1.59) and showed a trend for KCNJ10 (p=0.08, FC=−1.32). These results are summarized in figure 2 and table 2. The magnitude of the changes observed in the qPCR results was similar to that observed in the microarray experiment. We performed Pearson’s correlation analysis of the fold change values for microarray and qPCR results, which showed a strong correlation between the two values for all genes except for SLC1A3, where even though both experiments showed a downregulation of its expression the fold change observed for qPCR was larger than the one observed in the microarray experiment (see supplemental material).
Table 2.
Summary of microarray and qPCR results for genes associated with the astrocyte syncytial function.
| Gene Name | Microarray Diff Score | Microarray Fold change | QPCR P value | QPCR Fold change |
|---|---|---|---|---|
| AQP4 | −23.93 | −1.61 | 0.003 | −1.59 |
| GJA1 | −36.2 | −1.64 | 0.004 | −1.59 |
| SLC1A2 | −19.54 | −1.61 | 0.002 | −1.51 |
| SLC1A3 | −26.92 | −1.39 | 0.001 | −1.58 |
| KCNJ10 | −13.6 | −1.24 | 0.08 | −1.32 |
Discussion
Traditionally CNS research studies had focused their attention upon neuronal functions to explain the pathophysiology of mood disorders, with emphasis in neurotransmitter systems and stress pathways. However, there is growing evidence in the literature of a substantial role of glia cells in the pathogenesis of MDD (Evans, Choudary et al. 2004, Choudary, Molnar et al. 2005, Bernard, Kerman et al. 2011, Ordway, Szebeni et al. 2012, Medina, Burke et al. 2013). Furthermore, animal studies have shown that pharmacological ablation of glia induces depressive behaviors (Banasr and Duman 2008), and the depressive behaviors induced by chronic unpredictable stress in rats can be mimicked by blocking glial gap junctions (Sun, Liu et al. 2012). Our results support the hypothesis of glial dysfunction in MDD, and in particular suggest that the syncytial mechanisms that help maintain homeostasis of the neural tissue are compromised in MDD patients.
The glial syncytium maintains the homeostatic conditions that are critical for normal neuronal activity in the central nervous system. It is known however that a variety of neurological diseases result from genetic or autoimmune disruption of the glial pathways that are specialized to support neuronal activity, particularly those glial mechanisms that provide for potassium homeostasis and for the transport and release of obligatorily associated osmotic water (Rash 2010). The neurological diseases associated to dysfunction of the glial syncytium, such as neuromyelitis optica and multiple sclerosis are known to present with a depressive phenotype (Akaishi, Nakashima et al. 2015, Pan, Zhao et al. 2015). This idea supports our hypothesis that glial dysfunction may actively contribute to the onset or perpetuation of depressive states.
We hypothesize that the disruption of the functionality of the glial network caused by the downregulation of syncytial function-related genes affects its ability to maintain homeostasis of the brain tissue, and therefore may contribute to the establishment of psychiatric symptoms.
In the brain and especially in astrocytes Connexin 43, the product of GJA1 is the main protein forming gap junction channels. Giaume (1996) suggested that the functionality of gap junctions in astrocytes, including their conductance and permeability are controlled by endogenous compounds released from neural and endothelial cells. Therefore, a certain degree of plasticity can be expected from the gap junction communication system in astrocyte networks and the system is then vulnerable to the changes elicited by pathological states in both the glial cells and other brain cell populations.
Since the syncytial function of astrocytes is based on the presence and functionality of gap junctions, the downregulation of GJA1 mRNA expression observed in MDD subjects may alter the effectiveness of the astrocyte network to buffer the products of synaptic activity. Future studies designed to assess GJA1 protein expression will be vital to perform in order to more substantively examine this hypothesis.
A fundamental homeostatic function performed by astrocytes is called “potassium siphoning”, a term that refers to the sequestration and long-distance disposal of the potassium released during each action potential (Rash 2010). This process allows the restoration of potassium balance in the peri-synaptic regions. The potassium released to the extracellular space by neural activity is passed on to the glia through strongly rectifying heteromeric Kir4.1 channels (encoded by KCNJ10). The potassium is then redistributed through glial network via gap junctions, following the astrocytes’ resting membrane potential gradient, from the strongly negatively charged astrocytes surrounding the synaptic sites towards less negatively charged astrocytes located close to the blood vessels, where it is then released through weakly rectifying homomeric Kir4.1 channels (Butt and Kalsi 2006). Furthermore, Amiry-Moghaddam et al (2003) showed that the efficiency of potassium clearance is interdependent with the water flux mediated by the water channel AQP4, which is colocalized with kir4.1 and appears to have a synchronic function with it.
In this fashion, the glial syncytium acts as a water/potassium sink that keeps the neural homeostasis by maintaining the electrochemical balance of the synapse. Potassium balance is pivotal for several neuronal processes, such as maintaining membrane potential, the activity of voltage gated channels, modulating synaptic transmission and keeping an electrogenic gradient for neurotransmitter transport (Kofuji and Newman 2004). Our results indicate that several genes related to potassium syphoning (KCNJ10 (p=0.08), GJA1 (p-0.004) and AQP4 (p=0.003) are downregulated in MDD subjects compared to controls, and this decrease may alter the efficiency of potassium syphoning and therefore the ability of glial cells to modulate hippocampal neurotransmission.
Finally, the downregulation of glutamate transporters in MDD has been previously reported (Choudary, Molnar et al. 2005, Bernard, Kerman et al. 2011, Medina, Burke et al. 2013). The uptake and redistribution of synaptic glutamate is performed by astrocytes through the membrane glutamate transporters EAAT1 and EAAT2, encoded by SLC1A3 and SLC1A2 respectively (Danbolt 2001, Amara and Fontana 2002). The glutamate recovered from synaptic activity can then be redistributed across the astrocyte network via gap junctions.
There are several limitations to this study. Here we show a difference in the expression levels of glial syncytial function related transcripts between MDD subjects and a control group, and some of these genes are known to be associated with neurological syndromes that present a depressive phenotype. However, it is not possible to establish from these results if the decreased expression of syncytial related transcripts is a direct cause or a consequence of the depressive state. Further studies on different experimental paradigms would be necessary to establish causality, as postmortem studies present limitations in this regard, since the conditions of the disease state cannot be controlled.
When performing gene expression studies several variants should be taken into consideration as they can potentially alter the results of the study. These include gender, age, pH, agonal factors and post mortem interval (PMI). In our study the cohort used was composed of all male subjects, with an agonal factor score of zero and pH over 6.5. There are no significant differences in age or pH between the two groups. When analyzing the PMI for both groups there seemed to be a trend for longer PMIs for the MDD group (p=0.07). To examine this PMI trend, we ran correlation analysis for each gene in all patients as well as in each group. Regardless, we did not find a significant correlation between PMI and the qPCR values for any gene in our study. The results of this correlation analysis can be found in the supplemental material section.
Furthermore, the acquisition of brain tissue of proper quality for gene expression studies affects the sample size and limits analysis of other covariants such as alcoholism comorbidity, suicidality and patient treatment that may affect the results; also, in our particular case we did not have an adequate group of female samples, so further studies will be required to address the important question of gender in MDD, as the disease largely affects women. Nevertheless, considering that a large group of MDD affected patients is still considered unresponsive to current therapeutic options, this study provides an insight in to a different approach in the exploration of the pathophysiology disease.
To summarize, the ability of the glial syncytium to maintain the homeostasis of the synapses depends on the coordinated activity of gap junction proteins, glutamate transporters, and potassium and water channels. Combining the downregulation of genes related to water transport and potassium syphoning as well as gap junction communication, our findings present a picture of moderate glial dysfunction that could contribute to a chronic imbalance of synaptic activity, and particularly affect the functionality of glutamatergic synapses in the hippocampus of MDD subjects. Such gene expression changes in MDD may underline the onset or contribute to the perpetuation of depressive symptoms and present an opportunity for identifying novel therapeutic targets that could potentially broaden the availability of treatment options for MDD patients.
Supplementary Material
Figure 3.

Syncytial function of glia cells surrounding a glutamatergic synapse. Glutamate exocytosis in to the synaptic cleft is accompanied by the release of K+ ions and changes of volume in the extracellular compartment. To limit the neurotransmitter activity and restore the basal conditions of the synapse the released elements must be collected and redistributed through the astrocytic network. Glutamate uptake is a function of the glial transporters SLC1A2 and SLC1A3 whereas the inward rectifying channel KCNJ10 and AQP4 are in charge of the internalization of K+ and water respectively. And all these small molecules can then be redistributed to the neighboring astrocytes through the connexin channels (GJA1). The recovered glutamate can be added to the metabolic amino acid pool of the astrocytes or recycled for neurotransmission and any excess K+ and water can be released to the neighboring blood vessels through a weakly rectifying KCNJ10 subtype and AQP4.
Acknowledgments
The authors want to thank Ms Jennifer Fitzpatrick and Mr Li Hui for their technical support.
Footnotes
Financial disclosures:
The authors are members of the Pritzker Neuropsychiatric Disorders Research Consortium, which is supported by the Pritzker Neuropsychiatric Disorders Research Fund L.L.C. A shared intellectual property agreement exists between this philanthropic fund and the University of Michigan, Stanford University, the Weill Medical College of Cornell University, HudsonAlpha Institute of Biotechnology and the University of California at Irvine, to encourage the development of appropriate findings for research and clinical applications. HA was funded by NIH grant R01 MH14261.
References
- Akaishi T, Nakashima I, Misu T, Fujihara K, Aoki M. Depressive state and chronic fatigue in multiple sclerosis and neuromyelitis optica. J Neuroimmunol. 2015;283:70–73. doi: 10.1016/j.jneuroim.2015.05.007. [DOI] [PubMed] [Google Scholar]
- Akil H, Haskett RF, Young EA, Grunhaus L, Kotun J, Weinberg V, Greden J, Watson SJ. Multiple HPA profiles in endogenous depression: effect of age and sex on cortisol and beta-endorphin. Biol Psychiatry. 1993;33(2):73–85. doi: 10.1016/0006-3223(93)90305-w. [DOI] [PubMed] [Google Scholar]
- Albers RW, Siegel GJ. In: Secondary Transport Systems. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. Siegel ABGJ, Albers RW, Wayne Albers R, Fisher Stephen K, Uhler Michael D, editors. Philadelphia: Lippincott-Raven; 1999. [Google Scholar]
- Amara SG, Fontana AC. Excitatory amino acid transporters: keeping up with glutamate. Neurochem Int. 2002;41(5):313–318. doi: 10.1016/s0197-0186(02)00018-9. [DOI] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Williamson A, Palomba M, Eid T, de Lanerolle NC, Nagelhus EA, Adams ME, Froehner SC, Agre P, Ottersen OP. Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of alpha-syntrophin-null mice. Proc Natl Acad Sci U S A. 2003;100(23):13615–13620. doi: 10.1073/pnas.2336064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banasr M, Duman RS. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol Psychiatry. 2008;64(10):863–870. doi: 10.1016/j.biopsych.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard R, Kerman IA, Meng F, Evans SJ, Amrein I, Jones EG, Bunney WE, Akil H, Watson SJ, Thompson RC. Gene expression profiling of neurochemically defined regions of the human brain by in situ hybridization-guided laser capture microdissection. J Neurosci Methods. 2009;178(1):46–54. doi: 10.1016/j.jneumeth.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard R, Kerman IA, Thompson RC, Jones EG, Bunney WE, Barchas JD, Schatzberg AF, Myers RM, Akil H, Watson SJ. Altered expression of glutamate signaling, growth factor, and glia genes in the locus coeruleus of patients with major depression. Mol Psychiatry. 2011;16(6):634–646. doi: 10.1038/mp.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunney BG, Bunney WE. Rapid-acting antidepressant strategies: mechanisms of action. Int J Neuropsychopharmacol. 2012;15(5):695–713. doi: 10.1017/S1461145711000927. [DOI] [PubMed] [Google Scholar]
- Butt AM, Kalsi A. Inwardly rectifying potassium channels (Kir) in central nervous system glia: a special role for Kir4.1 in glial functions. J Cell Mol Med. 2006;10(1):33–44. doi: 10.1111/j.1582-4934.2006.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checkley S. The neuroendocrinology of depression and chronic stress. Br Med Bull. 1996;52(3):597–617. doi: 10.1093/oxfordjournals.bmb.a011570. [DOI] [PubMed] [Google Scholar]
- Choudary PV, Molnar M, Evans SJ, Tomita H, Li JZ, Vawter MP, Myers RM, Bunney WE, Jr, Akil H, Watson SJ, Jones EG. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc Natl Acad Sci U S A. 2005;102(43):15653–15658. doi: 10.1073/pnas.0507901102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65(1):1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Drevets WC, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Funct. 2008;213(1–2):93–118. doi: 10.1007/s00429-008-0189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans SJ, Choudary PV, Neal CR, Li JZ, Vawter MP, Tomita H, Lopez JF, Thompson RC, Meng F, Stead JD, Walsh DM, Myers RM, Bunney WE, Watson SJ, Jones EG, Akil H. Dysregulation of the fibroblast growth factor system in major depression. Proc Natl Acad Sci U S A. 2004;101(43):15506–15511. doi: 10.1073/pnas.0406788101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaume C, McCarthy KD. Control of gap-junctional communication in astrocytic networks. Trends Neurosci. 1996;19(8):319–325. doi: 10.1016/0166-2236(96)10046-1. [DOI] [PubMed] [Google Scholar]
- Halbreich U, Asnis GM, Shindledecker R, Zumoff B, Nathan RS. Cortisol secretion in endogenous depression. I. Basal plasma levels. Arch Gen Psychiatry. 1985;42(9):904–908. doi: 10.1001/archpsyc.1985.01790320076010. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. 2004;129(4):1045–1056. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krolewski DM, Medina A, Kerman IA, Bernard R, Burke S, Thompson RC, Bunney WE, Jr, Schatzberg AF, Myers RM, Akil H, Jones EG, Watson SJ. Expression patterns of corticotropin-releasing factor, arginine vasopressin, histidine decarboxylase, melanin-concentrating hormone, and orexin genes in the human hypothalamus. J Comp Neurol. 2010;518(22):4591–4611. doi: 10.1002/cne.22480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuffler SW, Nicholls JG, Orkand RK. Physiological properties of glial cells in the central nervous system of amphibia. J Neurophysiol. 1966;29(4):768–787. doi: 10.1152/jn.1966.29.4.768. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Machado-Vieira R, Manji HK, Zarate CA. The role of the tripartite glutamatergic synapse in the pathophysiology and therapeutics of mood disorders. Neuroscientist. 2009;15(5):525–539. doi: 10.1177/1073858409336093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews DC, Henter ID, Zarate CA. Targeting the glutamatergic system to treat major depressive disorder: rationale and progress to date. Drugs. 2012;72(10):1313–1333. doi: 10.2165/11633130-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina A, Burke S, Thompson RC, Bunney W, Jr, Myers RM, Schatzberg A, Akil H, Watson SJ. Glutamate transporters: A key piece in the glutamate puzzle of major depressive disorder. J Psychiatr Res. 2013;47(9):1150–1156. doi: 10.1016/j.jpsychires.2013.04.007. [DOI] [PubMed] [Google Scholar]
- Medina A, Seasholtz AF, Sharma V, Burke S, Bunney W, Jr, Myers RM, Schatzberg A, Akil H, Watson SJ. Glucocorticoid and mineralocorticoid receptor expression in the human hippocampus in major depressive disorder. J Psychiatr Res. 2013;47(3):307–314. doi: 10.1016/j.jpsychires.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrough JW, Charney DS. Is there anything really novel on the antidepressant horizon? Curr Psychiatry Rep. 2012;14(6):643–649. doi: 10.1007/s11920-012-0321-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagelhus EA, Horio Y, Inanobe A, Fujita A, Haug FM, Nielsen S, Kurachi Y, Ottersen OP. Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Muller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia. 1999;26(1):47–54. doi: 10.1002/(sici)1098-1136(199903)26:1<47::aid-glia5>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Ordway GA, Szebeni A, Chandley MJ, Stockmeier CA, Xiang L, Newton SS, Turecki G, Duffourc MM, Zhu MY, Zhu H, Szebeni K. Low gene expression of bone morphogenetic protein 7 in brainstem astrocytes in major depression. Int J Neuropsychopharmacol. 2012;15(7):855–868. doi: 10.1017/S1461145711001350. [DOI] [PubMed] [Google Scholar]
- Pan J, Zhao P, Cai H, Su L, Wood K, Shi FD, Fu Y. Hypoxemia, Sleep Disturbances, and Depression Correlated with Fatigue in Neuromyelitis Optica Spectrum Disorder. CNS Neurosci Ther. 2015;21(7):599–606. doi: 10.1111/cns.12411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpura V, Heneka MT, Montana V, Oliet SH, Schousboe A, Haydon PG, Stout RF, Jr, Spray DC, Reichenbach A, Pannicke T, Pekny M, Pekna M, Zorec R, Verkhratsky A. Glial cells in (patho)physiology. J Neurochem. 2012;121(1):4–27. doi: 10.1111/j.1471-4159.2012.07664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigott HE, Leventhal AM, Alter GS, Boren JJ. Efficacy and effectiveness of antidepressants: current status of research. Psychother Psychosom. 2010;79(5):267–279. doi: 10.1159/000318293. [DOI] [PubMed] [Google Scholar]
- Rash JE. Molecular disruptions of the panglial syncytium block potassium siphoning and axonal saltatory conduction: pertinence to neuromyelitis optica and other demyelinating diseases of the central nervous system. Neuroscience. 2010;168(4):982–1008. doi: 10.1016/j.neuroscience.2009.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JD, Liu Y, Yuan YH, Li J, Chen NH. Gap junction dysfunction in the prefrontal cortex induces depressive-like behaviors in rats. Neuropsychopharmacology. 2012;37(5):1305–1320. doi: 10.1038/npp.2011.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita H, Vawter MP, Walsh DM, Evans SJ, Choudary PV, Li J, Overman KM, Atz ME, Myers RM, Jones EG, Watson SJ, Akil H, Bunney WE., Jr Effect of agonal and postmortem factors on gene expression profile: quality control in microarray analyses of postmortem human brain. Biol Psychiatry. 2004;55(4):346–352. doi: 10.1016/j.biopsych.2003.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torp R, Danbolt NC, Babaie E, Bjoras M, Seeberg E, Storm-Mathisen J, Ottersen OP. Differential expression of two glial glutamate transporters in the rat brain: an in situ hybridization study. Eur J Neurosci. 1994;6(6):936–942. doi: 10.1111/j.1460-9568.1994.tb00587.x. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Rodriguez JJ, Parpura V. Neurotransmitters and integration in neuronal-astroglial networks. Neurochem Res. 2012;37(11):2326–2338. doi: 10.1007/s11064-012-0765-6. [DOI] [PubMed] [Google Scholar]
- Vernadakis A. Glia-neuron intercommunications and synaptic plasticity. Prog Neurobiol. 1996;49(3):185–214. doi: 10.1016/s0301-0082(96)00012-3. [DOI] [PubMed] [Google Scholar]
- Zeng XN, Sun XL, Gao L, Fan Y, Ding JH, Hu G. Aquaporin-4 deficiency down-regulates glutamate uptake and GLT-1 expression in astrocytes. Mol Cell Neurosci. 2007;34(1):34–39. doi: 10.1016/j.mcn.2006.09.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.