

Abstract
Key points
Adenosine is a widespread neuromodulator in the mammalian brain, but whether it affects information processing in sensory system(s) remains largely unknown.
Here we show that adenosine A1 receptors hyperpolarize mitral cells, one class of principal neurons that propagate odour information from the olfactory bulb to higher brain areas, by activation of background K+ channels.
The adenosine‐modulated background K+ channels belong to the family of two‐pore domain K+ channels.
Adenosine reduces spontaneous activity of mitral cells, whereas action potential firing evoked by synaptic input upon stimulation of sensory neurons is not affected, resulting in a higher ratio of evoked firing (signal) over spontaneous firing (noise) and hence an improved signal‐to‐noise ratio.
The study shows for the first time that adenosine influences fine‐tuning of the input–output relationship in sensory systems.
Abstract
Neuromodulation by adenosine is of critical importance in many brain regions, but the role of adenosine in olfactory information processing has not been studied so far. We investigated the effects of adenosine on mitral cells, which are projection neurons of the olfactory bulb. Significant expression of A1 and A2A receptors was found in mitral cells, as demonstrated by in situ hybridization. Application of adenosine in acute olfactory bulb slices hyperpolarized mitral cells in wild‐type but not in adenosine A1 receptor knockout mice. Adenosine‐induced hyperpolarization was mediated by background K+ currents that were reduced by halothane and bupivacaine, which are known to inhibit two‐pore domain K+ (K2P) channels. In mitral cells, electrical stimulation of axons of olfactory sensory neurons evoked synaptic currents, which can be considered as input signals, while spontaneous firing independent of sensory input can be considered as noise. Synaptic currents were not affected by adenosine, while adenosine reduced spontaneous firing, leading to an increase in the signal‐to‐noise ratio of mitral cell firing. Our findings demonstrate that A1 adenosine receptors activate two‐pore domain K+ channels, which increases the signal‐to‐noise ratio of the input–output relationship in mitral cells and thereby modulates information processing in the olfactory bulb.
Keywords: adenosine, neuromodulation, two‐pore domain potassium channels
Key points
Adenosine is a widespread neuromodulator in the mammalian brain, but whether it affects information processing in sensory system(s) remains largely unknown.
Here we show that adenosine A1 receptors hyperpolarize mitral cells, one class of principal neurons that propagate odour information from the olfactory bulb to higher brain areas, by activation of background K+ channels.
The adenosine‐modulated background K+ channels belong to the family of two‐pore domain K+ channels.
Adenosine reduces spontaneous activity of mitral cells, whereas action potential firing evoked by synaptic input upon stimulation of sensory neurons is not affected, resulting in a higher ratio of evoked firing (signal) over spontaneous firing (noise) and hence an improved signal‐to‐noise ratio.
The study shows for the first time that adenosine influences fine‐tuning of the input–output relationship in sensory systems.
Introduction
Adenosine is a ubiquitous neuromodulator involved in many physiological processes such as sleep, memory formation, synaptic plasticity and neuronal excitability, but also in the pathophysiology of the brain (Fredholm et al. 2005; Boison, 2009; Ribeiro & Sebastiao, 2010; Dias et al. 2013). There are four adenosine receptors cloned, A1, A2A, A2B and A3, all of them being G‐protein‐coupled receptors. While A1 and A3 receptors are coupled to Gi proteins, A2A and A2B are linked to Gs proteins. A1 and A2A receptors are widespread in the brain and often exert opposing effects on neuronal performance (Cunha, 2005). In the hippocampus, for example, A1 receptors mediate synaptic attenuation by inhibition of presynaptic calcium channels, while A2A receptors counteract this effect by a protein kinase C‐dependent mechanism (Lopes et al. 2002; Gundlfinger et al. 2007). A1 receptors have also been shown to decrease neuronal excitability by activation of G‐protein‐coupled inwardly rectifying potassium channels (GIRKs; Sickmann & Alzheimer, 2003; Kim & Johnston, 2015). GIRKs are targets of A1 receptors in the retina, where they mediate hyperpolarization and a reduction in action potential firing in ganglion cells elicited by light stimulation of photoreceptor cells (Clark et al. 2009). While the role of adenosine in the retina is well documented (for a review, see Dos Santos‐Rodrigues et al. 2015), it is not known if and how adenosine receptors affect the input–output relation of sensory information in other brain areas dedicated to sensory information processing (Housley et al. 2009; Lohr et al. 2014).
First evidence suggests a neuromodulatory role of adenosine receptors in the olfactory bulb. The A2A receptor, for example, has been demonstrated by radioligand binding, in situ hybridization and antibody staining (Johansson et al. 1997; Rosin et al. 1998; Kaelin‐Lang et al. 1999). Olfactory sensory axons might be the source of adenosine since they release ATP as a cotransmitter (Thyssen et al. 2010). ATP excites mitral cells via P2Y1 receptors (Fischer et al. 2012), is then degraded to adenosine and elicits calcium transients in astrocytes expressing A2A receptors, emphasizing a physiological relevance of adenosine in this brain area (Doengi et al. 2008). In addition, adenosine derived from ATP released from astrocytes modulates slow membrane potential oscillations of olfactory bulb mitral cells (Roux et al. 2015). The mechanism by which adenosine modulates mitral cells and whether adenosine affects odour information processing, however, remain to be shown. We found that activation of A1 receptors hyperpolarizes mitral cells by opening of two‐pore domain potassium (K2P) channels, thereby decreasing spontaneous firing activity. The firing frequency of mitral cells upon synaptic stimulation via olfactory sensory axons, in contrast, remains unaffected by adenosine, resulting in a higher ratio of evoked firing (signal) over spontaneous firing (noise) and hence an improved signal‐to‐noise ratio.
Methods
Ethical approval
Animal rearing, dissection and all experiments were performed according to the European Union's and German animal welfare guidelines and were approved by the local authorities (GZ G21305/591‐00.33; Behörde für Gesundheit und Verbraucherschutz, Hamburg, Germany). They conform to the principles and regulations described by Grundy (2015). Mice of both sexes (age: postnatal days 8–21) were anaesthetized with isoflurane and decapitated.
Animals and preparation procedure
Naval Medical Research Institute (NMRI) outbred mice, OMP‐synapto‐pHluorin‐expressing mice (Bozza et al. 2004) and A1 receptor knockout mice (Johansson et al. 2001) were bred in the institute's animal facility (12 h–12 h light–dark cycle, food and water ad libitum). Olfactory bulbs were dissected and horizontal brain slices were prepared as described before (Fischer et al. 2012). Olfactory bulbs were quickly transferred into a chilled slicing artificial cerebrospinal fluid (ACSF) that contained (in mm): NaCl, 83; NaHCO3, 26.2; NaH2PO4, 1; KCl, 2.5; sucrose, 70; d‐glucose, 20; CaCl2, 0.5; MgSO4, 2.5. Sagittal slices 200 μm thick of the bulbs were cut using a vibrating blade microtome (Leica VT1200S, Bensheim, Germany). Brain slices were stored in ACSF containing (in mm): NaCl, 120; NaHCO3, 26; NaH2PO4, 1; KCl, 2.5; d‐glucose, 2.8; CaCl2, 2; MgCl2, 1. Storage lasted for 30 min at 30°C and 15 min at room temperature before starting experiments. ACSF was continuously gassed with carbogen (95% O2–5% CO2; buffered to pH 7.4 with CO2/bicarbonate).
Drugs
Adenosine, adenosine‐5′‐diphosphate, adenosine‐5′‐triphosphate, barium chloride, halothane and 8‐cyclopentyl‐1,3‐dipropylxanthine (DPCPX) were purchased from Sigma‐Aldrich (Munich, Germany); N 6‐cyclopenthyladenosine (N 6‐CPA), N‐[4‐[[1‐[2‐(6‐methyl‐2‐pyridinyl)ethyl]‐4‐piperidinyl]carbonyl]phenyl]‐methanesulfonamide dihydrochloride (E‐4031), tetrodotoxin citrate (TTX), 10,10‐bis(4‐pyridinylmethyl)‐9(10H)‐anthracenone dihydrochloride (XE 991), 6,12,19,20,25,26‐hexahydro‐5,27:13,18:21,24‐trietheno‐11,7‐metheno‐7H‐dibenzo [b,n]‐[1,5,12,16]tetraazacyclotricosin‐5,13‐diium dibromid (UCL 1684) and 4‐aminopyridine (4‐AP) were from Tocris (Bristol, UK); 2,3‐dioxo‐6‐nitro‐1,2,3,4‐tetrahydrobenzo‐[f]‐quinoxaline‐7‐sulfonamide (NBQX), d‐(−)‐2‐amino‐5‐phosphonopentanic acid (d‐APV) and gabazine hydrobromide were from Abcam (Cambridge, UK); tetramethyl ammonium (TEA) was from Applichem (Darmstadt, Germany); 1‐[(2‐chlorophenyl)diphenylmethyl]‐1H‐pyrazole (TRAM‐34) was from Alomone Labs (Jerusalem, Israel).
Electrophysiological recordings
Mitral cells of the main olfactory bulb were investigated using the patch‐clamp technique (MultiClamp 700B amplifier and pCLAMP 10 software, Molecular Devices, Biberach, Germany). Throughout the experiments, brain slices were superfused with ACSF. In ACSF with 5 mm K+ concentration, equimolar amounts of NaCl were replaced by KCl. Drugs were applied with the perfusion system driven by a peristaltic pump (Reglo, Ismatec, Wertheim, Germany) at a flow rate of 2 ml min−1. Application of drugs was achieved by manually changing the inflow of the pump from drug‐free to drug‐containing ACSF. Application bars reflect the time window during which drug‐containing ACSF was added to the bath, the time being corrected for the time needed for the drug to pass the tubing and enter the bath. Complete bath exchange required approximately 30 s. If not stated otherwise, the whole‐cell configuration was employed using patch pipettes with a resistance of ∼3 MΩ. Recordings were digitized (Digidata 1440A, Molecular Devices, Biberach, Germany) at 10–20 kHz and filtered (Bessel filter, 2 kHz). The standard pipette solution contained (in mm): NaCl, 10; potassium gluconate, 105; K3‐citrate, 20; Hepes, 10; MgCl2, 0.5; Mg‐ATP, 5; Na‐GTP, 0.5; mannitol, 21; phosphocreatine, 5. Pipette solution with reduced K+ concentration contained (in mm): NaCl, 10; K‐gluconate, 110; K3‐citrate, 10; Hepes, 10; EGTA, 0.25; MgCl2, 0.5; Mg‐ATP, 3; Na‐GTP, 0.5. High‐chloride pipette solution contained (in mm): NaCl, 10; potassium gluconate, 52.5; KCl, 55; K3‐citrate, 10; Hepes, 10; EGTA, 0.25; MgCl2, 0.5; Mg‐ATP, 3; Na‐GTP, 0.5. The pipette solutions contained 8 μm Alexa Fluor 594 in most of the experiments. For extracellular recordings of action potentials, patch pipettes were filled with ACSF (resistance ∼3 MΩ) and the cell‐attached configuration was established. For stimulation of axons of olfactory sensory neurons we used ACSF‐filled pipettes with a resistance of ∼2 MΩ. The stimulation pipette was placed in the olfactory nerve layer at a distance of at least 50 μm to the apical dendrite of the recorded mitral cell. Current pulses (100 μA, 50 μs) were applied to evoke synaptic excitation of mitral cells. In control experiments, synaptic currents were entirely blocked by application of 1 μm TTX, confirming that stimulation‐induced currents were synaptically evoked, but not evoked by direct stimulation of the mitral cell.
Measurements of membrane conductance and reversal potential
The membrane conductance was measured in voltage clamp with a holding potential of −70 mV. A hyperpolarizing voltage step of 10 mV and 1 s duration was applied every 10 s and the membrane conductance was calculated from the resulting current shift that was measured at the end of the voltage step. To estimate the reversal potential of the adenosine‐induced membrane potential shift, the membrane potential was recorded in current clamp and hyperpolarized by current steps of 500 ms duration and increasing amplitude (50 pA interval) before and during application of adenosine. The difference between membrane potential values recorded before and during adenosine application at a given current step was calculated, and the values of all experiments averaged and plotted against the membrane potential value before adenosine application. All data were pooled and a linear regression was calculated. The point of intersection of the regression line and the abscissa reflects the reversal potential of the adenosine‐induced membrane potential shift. The current–voltage relationship of the adenosine‐evoked current was obtained by applying voltage ramps (duration: 800 ms) from −20 to −130 mV in the absence and in the presence of adenosine. The difference of the obtained current traces revealed the adenosine‐evoked current. The resulting current–voltage relationship was corrected for a liquid junction potential of −18 mV.
Synapto‐pHluorin recordings
Mice expressing synapto‐pHluorin (syn‐pH) exclusively in olfactory sensory neurons were employed to study the effect of adenosine on vesicle fusion in olfactory sensory nerve terminals as described in Thyssen et al. (2010). Olfactory bulbs in toto (Stavermann et al. 2015) were placed under a confocal microscope (eC1, Nikon, Düsseldorf, Germany) and syn‐pH was excited at 488 nm. Fluorescence was collected using a bandpass filter between 515 and 545 nm. Olfactory sensory axons were stimulated using a stimulation isolator (DS3, Digitimer, Welwyn Garden City, UK). Regions of interest were defined in the glomeruli and changes in the syn‐pH fluorescence were calculated with the baseline fluorescence before stimulation taken as 100%.
A1 receptor in situ hybridization
Probe design
mRNA probes for adenosine receptors were designed based upon cDNA clone MGC:90841 (ADORA1) and MGC:118414 (ADORA2A). For A1 receptor RNA probes, a sequence was amplified by PCR from cDNA isolated from mouse brain (SuperScript III, Thermo Fisher Scientific, Darmstadt, Germany) using the following primer set: forward: 5′‐CGTGTCATGTGAGACCCTTCCTGCTG; reverse: 5′CAGGCTCAATTTCCAAAGCTGGGCTCTG. For A2A receptor RNA probes, the following sequences were used: forward: GAAACGCCCACGCCAGGAAACC; reverse: GCAGACGGTCCTCTCGGGTTAGC. The sequences were tested for correctness and orientation (GATC, Konstanz, Germany) and cloned into pGEM‐T easy vector. Vectors were multiplied, linearized and purified as a basis for labelling and transcription of adenosine receptor RNA probes.
Hybridization procedure for radiolabelled probes
For whole brain in situ hybridization (ISH), radioactive probes based on the sequence stated above were used. Labelling of [35S]UTPs was performed with the Maxiscript Kit (Ambion, Thermo Fisher Scientific) according to the manufacturer's instructions. Brain slices (16 μm) on slides were fixed in 4% paraformaldehyde in PBS for 10 min at 4°C. Acetylation was performed for 10 min in triethanolamine (0.1 m), 0.9% NaCl solution and acetic anhydride (2.5 ml l−1) in PBS, followed by dehydration in ethanol (60/80/90/95/99.8%). Prehybridization buffer (50% deionized formamide, 5× hybridization salts (750 mm NaCl, 25 mm PIPES, 25 mm EDTA, pH 6.8), 5× Denhardt's solution (0.05% Ficoll, 0.05% polyvinylpyrrolidone, 0.1% BSA), 0.02% SDS, 10 mm DTT, 10% dextran sulfate, 250 μg ml−1 herring‐sperm DNS, 250 μg ml−1 yeast tRNA) was incubated on slides at 50°C for 2 h in a humidity chamber. Hybridization was performed with 2 × 106 cpm radioactive probe in prehybridization buffer at 50°C overnight. After rinsing 3 × 5 min in 4× SSC (20× SSC: 1% (w/v) BSA, 1% (w/v) Ficoll‐400, 1% (w/v) polyvinylpyrrolidone), brain slices were treated at 37°C (30 min) with RNAse A and rinsed. For visualization, slides were dehydrated, covered with photo emulsion and exposed to radiographic film for 1 week at 4°C. Visualization of tissue structure was achieved with haematoxylin–eosin staining.
Hybridization procedure for DIG‐labelled probes
DIG‐labelled A1 receptor RNA probes were synthesized by in vitro transcription (DIG‐dUTP RNA Labelling Kit, Roche Diagnostics, Mannheim, Germany) based on the vector above. Probes were purified with lithium chloride precipitation and subsequently tested for integrity and DIG labelling. Fixed brain slices on slides were acetylated in 100 mm triethanolamine and 2.5 ml l−1 acetic anhydride in PBS for 10 min followed by dehydration with ethanol (50/70/95/99.8%) for 3 min each step. Subsequently, slides were covered with prehybridization buffer (62.5% formamide, 25% dextran sulfate, 7.5% NaCl (5000 mm), 2.5%, Denhardt's solution (50%), 1.25% Tris, 0.25% EDTA (500 mm)) at 58°C for 2 h in a humidity chamber. Hybridization was performed in prehybridization buffer to which was added 400 ng ml−1 probe at 58°C in a humidity chamber overnight. Slices were treated with RNAse A (37°C, 30 min) and unspecific binding was blocked in 2× SSC, 0.05% Triton X‐100 and 2% normal goat serum (NGS); they were incubated with horseradish peroxidase‐linked anti‐DIG antibody (37°C, 2 h) and visualized with 50 mg ml−1 5‐bromo‐4‐chloro‐3′‐indolyphosphate p‐toluidine and 70 mg ml−1 nitro‐blue tetrazolium chloride overnight. Images were acquired with a bright field microscope (BX51, Olympus, Hamburg, Germany). Images of in situ hybridization using wild‐type tissue and sense probes as well as A1 knockout tissue and antisense probes served as control. These images were contrast‐enhanced by adjusting histogram levels (white level set to 240, gamma set to 0.67; Photoshop CS6, Adobe, San Jose, CA, USA) to improve visibility of the tissue outline.
Data analysis and statistics
Patch‐clamp recordings were analysed using Mini Analysis (Synaptosoft, Fort Lee, NJ, USA), ClampFit (Molecular Devices, Biberach, Germany) and OriginPro (OriginLab Corp., Northampton, MA, USA). Membrane potential values recorded in current clamp using a low‐chloride pipette solution were corrected for a liquid junction potential of −18 mV. All values are means ± standard error of the mean (SEM) with n representing the number of analysed cells. Statistical analysis was performed using one‐way ANOVA with Fisher's post hoc test or Student's t test. Means were defined as statistically different at error probabilities P < 0.05 (*), P < 0.01 (**) and P < 0.001 (***).
Results
Adenosine A1 receptor gene expression in olfactory bulb mitral cells
We first assessed the expression profile of the A1 adenosine receptor gene (ADORA1) in parasagittal sections of the mouse brain. As described before (Mahan et al. 1991; Reppert et al. 1991), we detected high levels of A1 receptor mRNA in hippocampus and cerebellum. In addition, we found A1 receptor expression in the olfactory bulb (Fig. 1 A). One of the strongest expression signals of A1 in the entire brain was found in the olfactory bulb mitral cell layer. At higher magnification, it became evident that the expression signal in the mitral cell layer correlated to large cell bodies of mitral cells (Fig. 1 B and C). In the external plexiform layer and the basal part of the glomerular layer, cell bodies presumably reflecting tufted and external tufted cells were labelled. The expression of A1 receptor gene, as assessed by in situ hybridization, was very low in the outer part of the glomerular layer and the granule cell layer. A1 adenosine receptor expression was not observed using the sense probe and in A1 receptor knockout mice using the antisense probe, confirming the specificity of the in situ hybridization probes (Fig. 1 D–G).
Figure 1. Adenosine receptor gene expression.
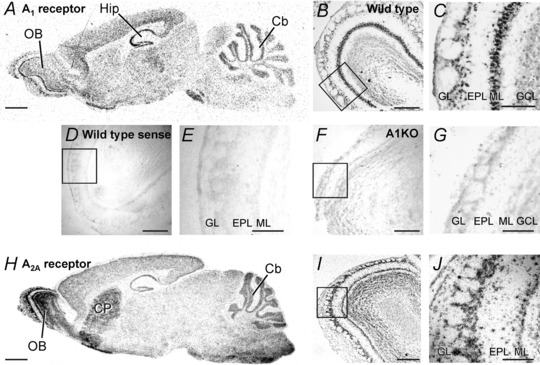
A, in situ hybridization of A1 receptor mRNA in mouse brain. Strong signals were detected in the olfactory bulb (OB), the hippocampus (Hip) and the cerebellum (Cb). B and C, the strongest A1 receptor expression signal in the OB was located in large cells of the mitral cell layer (ML). D–G, no expression signal could be detected in wild‐type mice using sense probes as a control (D and E), and in A1 receptor knockout mice using the antisense probe (F and G). H, in situ hybridization of A2A receptor mRNA. A2A signals were detected in the olfactory bulb, the cerebellum and the caudate putamen (CP). I and J, significant A2A expression was localized in all layers of the OB. EPL, external plexiform layer; GCL, granule cell layer; GL, glomerular layer. Scale bars: 1 mm in A and H; 300 μm in B, D, F and I; 100 μm in C, E, G and J.
Expression of the A2A adenosine receptor gene (ADORA2A) was most prominent in the caudate putamen and the olfactory bulb (Dixon et al. 1996; Svenningsson et al. 1997; Kaelin‐Lang et al. 1999). The expression of A2A in the olfactory bulb was more widespread compared to A1 receptors and was detected in cells of all layers of the olfactory bulb, including mitral cells (Fig. 1 H–J). Hence, high levels of adenosine receptor expression were found in the olfactory bulb and in particular in mitral cells, emphasizing the possible role of adenosine as a neuromodulator in this brain area.
Activation of adenosine A1 receptors hyperpolarizes mitral cells
Physiological data suggest that adenosine might play a role in modulation of neuronal performance in the olfactory bulb (reviewed in Lohr et al. 2014). Therefore, we tested the effect of adenosine on the membrane potential of mitral cells. Mitral cells of wild‐type mice had a mean resting membrane potential of −63.0 ± 0.6 mV (n = 23), which hyperpolarized by 2.6 ± 0.4 mV (n = 35) upon application of 100 μm adenosine for 30 s (Fig. 2 A, left panel). The membrane potential recovered slowly to resting values, presumably due to the slow bath perfusion kinetics. In addition, we cannot exclude the involvement of a sustained component in the adenosine‐stimulated signalling pathway leading to hyperpolarization. In A1 receptor knockout mice, mitral cells did not hyperpolarize due to adenosine application, but instead depolarized by 0.4 ± 0.2 mV (n = 9, P = 0.005), suggesting that A1 receptors mediate the adenosine‐evoked hyperpolarization (Fig. 2 A, right panel). The hyperpolarizing effect of adenosine in wild‐type mice was mimicked by application of the A1 agonist N 6‐CPA (Fig. 2 B). 1 μm of N 6‐CPA evoked a hyperpolarization of 2.8 ± 0.4 mV (n = 13). In the presence of the potent and selective A1 receptor antagonist DPCPX (1 μm), no change in membrane potential was evoked by adenosine (n = 6), confirming that adenosine hyperpolarized mitral cells by activation of A1 receptors (Fig. 2 C). DPCPX alone had no effect on the resting membrane potential and the resting firing rate of mitral cells. The pharmacological profile of the adenosine‐mediated effect on mitral cells, summarized in Fig. 2 D, clearly demonstrates an activation of A1 receptors by adenosine. Previous studies demonstrated release of ATP from olfactory sensory neurons and its degradation to adenosine in glomeruli (Doengi et al. 2008; Thyssen et al. 2010), leading to the hypothesis that the adenosine‐mediated hyperpolarization in mitral cells mainly originates from the apical tuft in the glomerulus. Therefore, we compared the hyperpolarization evoked by adenosine between mitral cells with intact apical dendrite and mitral cells in which the apical tuft had been cut during preparation of brain slices, as visualized by the fluorescent tracer Alexa Fluor 594 added to the pipette solution. We did not find a significant difference in the adenosine‐evoked hyperpolarization between these groups (Fig. 2 E). Adenosine evoked a hyperpolarization of 2.6 ± 0.4 mV (n = 20) in intact mitral cells and 2.6 ± 0.7 mV (n = 15) in mitral cells with cut apical tuft, indicating that the adenosine‐evoked hyperpolarization does not mainly derive from the apical tuft.
Figure 2. Adenosine hyperpolarizes mitral cells by A1 receptor activation.
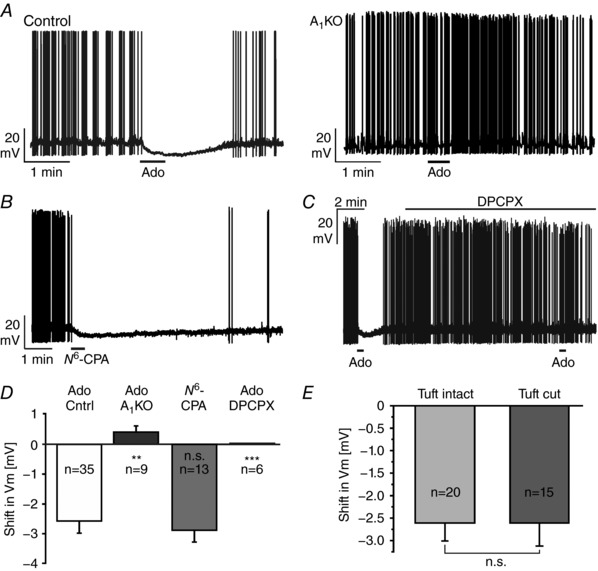
A, 100 μm adenosine (Ado) leads to a hyperpolarization of mitral cells in wild‐type (control) but not in A1 knockout mice (A1KO). B, the A1 receptor agonist N 6‐CPA (1 μm) mimics the effect of adenosine. C, the A1‐selective antagonist DPCPX (1 μm) inhibited the adenosine‐evoked hyperpolarization. D, summary of the pharmacological investigation of the adenosine‐induced membrane potential shift. n.s., not significant. E, no significant difference (n.s.) in the adenosine‐evoked hyperpolarization was found between mitral cells with intact apical dendrite and mitral cells with cut apical dendrite.
A1 receptor activation increases the membrane conductance in mitral cells
The adenosine‐evoked hyperpolarization via A1 receptors could be caused either by the closure of depolarizing non‐specific cation channels or by an opening of hyperpolarizing Cl− or K+ channels. To assess whether channels are opened or closed by application of adenosine, we measured the membrane conductance in voltage‐clamped mitral cells (holding potential: −70 mV) in the presence of the glutamate receptor antagonists NBQX (10 μm) and d‐APV (50 μm), the GABAA receptor antagonist gabazine (10 μm) and TTX (1 μm) to synaptically isolate the recorded mitral cell from its neuronal network. Membrane conductance was assessed by applying voltage steps of −10 mV amplitude. Application of adenosine led to an outward current (Fig. 3 A) and an increase in membrane conductance (Fig. 3 B). Interestingly, the increase in membrane conductance outlasted the outward current and did not reach baseline within 5 min after washout of adenosine (Fig. 3 C). On average, the membrane conductance significantly increased by 21.4 ± 4.4% from 3.24 ± 0.8 to 3.55 ± 0.8 nS (n = 6; P < 0.001) upon application of adenosine, indicating that A1 receptors in mitral cells are linked to the opening of ion channels that mediate hyperpolarization such as Cl− or K+ channels.
Figure 3. Adenosine increases the membrane conductance.
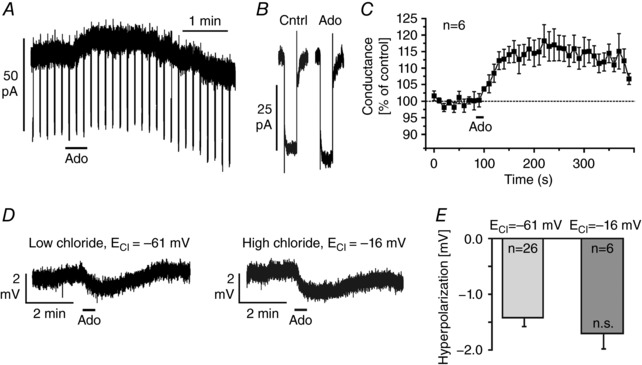
A, membrane conductance was measured by hyperpolarizing voltage steps of −10 mV in presence of TTX (1 μm), NBQX (10 μm), d‐APV (50 μm) and gabazine (10 μm). B and C, application of 100 μm adenosine increased the membrane conductance. D, adenosine‐induced hyperpolarization as measured in whole‐cell current‐clamp configuration using pipette solutions with different chloride concentrations, resulting in calculated chloride reversal potentials (E Cl) of −61 and −16 mV, respectively. E, the adenosine‐evoked hyperpolarization was independent of the chloride reversal potential (n.s., not significant).
We tested the involvement of Cl− channels by using a modified pipette solution in order to shift the reversal potential for Cl−. The calculated Cl− reversal potential using the standard (low Cl−) pipette solution was −61 mV, which is close to the resting membrane potential. We used a high Cl− pipette solution resulting in a calculated Cl− reversal potential of −16 mV. Hence, under the latter conditions, opening of Cl− channels should have led to depolarization. However, application of adenosine elicited a hyperpolarization, regardless of which pipette solution was used (Fig. 3 D). The amplitudes of the hyperpolarizations measured with different pipette solutions were not significantly different, suggesting that Cl− channels make no major contribution to the adenosine‐mediated hyperpolarization (Fig. 3 E).
A1 receptors activate K+ channels
We determined the reversal potential of the adenosine‐mediated membrane potential shift in current‐clamp by stepping to different membrane potentials using the standard pipette solution and defined current steps (50 pA interval) in the absence and in the presence of adenosine (Fig. 4 A). We used a combination of K+ concentrations in the extracellular (5 mm) and pipette solution (140 mm) that resulted in a calculated K+ reversal potential of −86 mV. On average, at membrane potentials more positive than −90 mV, adenosine tended to hyperpolarize the membrane, whereas at membrane potentials more negative than −90 mV, adenosine depolarized the membrane (Fig. 4 B). The reversal potential of the adenosine‐mediated shift in membrane potential as extrapolated from all recordings (n = 5 cells) was −90 mV, close to the calculated K+ reversal potential.
Figure 4. Adenosine activates potassium channels.
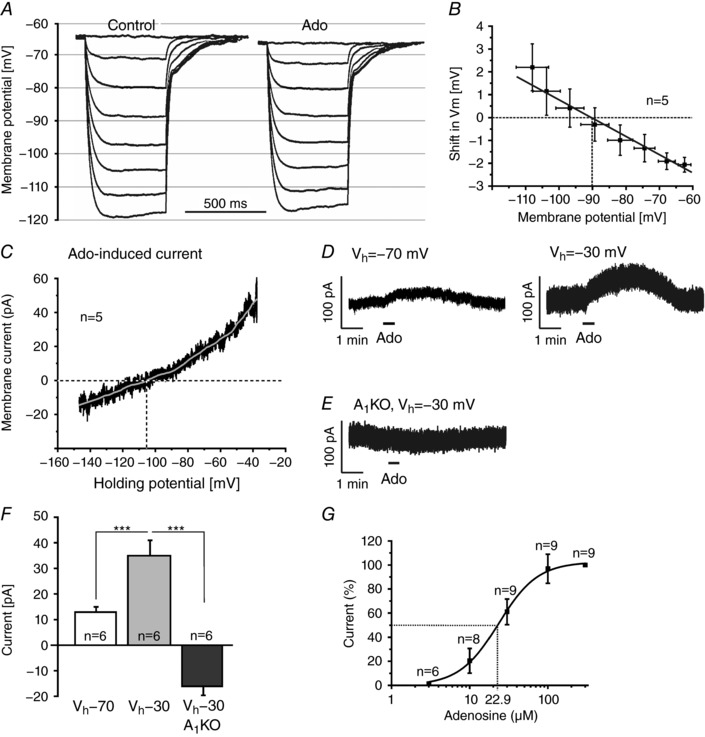
A, shifts in membrane potential evoked by negative current injections (−50 to −350 pA) before (control) and in the presence of adenosine (100 μm). B, extrapolation of adenosine‐evoked membrane potential shifts at different baseline potentials (as measured during the current steps in control) revealed an average reversal potential of −90 mV. C, current–voltage relationship of the adenosine‐evoked current, averaged over recordings from five cells (black trace) and filtered (grey trace; Savitsky‐Golay with second order polynomial and window size of 1501, corresponding to ± 5 mV), revealed a reversal potential of −106 mV. D, outward current evoked by 100 μm adenosine at a holding potential (V h) of −70 mV and −30 mV. E, adenosine failed to evoke outward currents in A1 receptor knockout mice. F, the adenosine‐evoked outward current was significantly larger at V h of −30 mV compared to −70 mV. At −30 mV, adenosine evoked an inward current in A1 knockout mice. G, dose–response relationship of the adenosine‐evoked outward current.
To confirm that adenosine activates a K+ conductance, we measured the ionic current evoked by adenosine in voltage‐clamp experiments at different holding potentials. In these experiments the K+ reversal potential was set to −108 mV to increase the driving force for K+ and, hence, the size of the responses evoked by adenosine. To assess the current–voltage relationship, the membrane potential was first clamped to −20 mV for several minutes to inactivate voltage‐dependent currents, followed by a voltage ramp from −20 to −130 mV within 800 ms. Voltage ramps were performed before and during application of 100 μm adenosine, the difference revealing the adenosine‐evoked current. After correction for the liquid junction potential of −18 mV, this resulted in an adenosine‐evoked outwardly rectifying current that reversed at −106 mV and continued as inward current at more negative membrane potentials (n = 5; Fig. 4 C). To quantify the effect of the membrane potential on the outward current, we measured adenosine‐induced currents at two holding potentials, −70 and −30 mV (Fig. 4 D and G). At a holding potential of −70 mV, adenosine evoked an outward current of 13 ± 2 pA (n = 6; Fig. 4 D, left panel). The outward current significantly increased to 35 ± 6 pA (n = 6, P < 0.001) at a holding potential of −30 mV (Fig. 4 D, right panel). In A1 knockout mice, adenosine failed to induce membrane currents (Fig. 4 E) or resulted in inward currents instead of outward currents, verifying the involvement of A1 receptors. On average, the adenosine‐mediated effect amounted to −16 ± 4 pA (n = 6, P < 0.001 as compared to wild‐type) in A1 knockout mice (Fig. 4 E and F). To achieve a dose–response curve for the adenosine‐evoked response in wild‐type mice, we tested adenosine concentrations ranging from 3 to 300 μm and measured the evoked outward currents (n = 9). The results revealed an EC50 value of 22.9 μm (Fig. 4 G). In summary, these experiments indicate that the adenosine‐mediated current is caused by an A1 receptor‐activated potassium conductance.
Pharmacological characterization of the adenosine‐activated K+ conductance
Activation of GIRK mediated by A1 receptors is a common pathway of adenosinergic neuromodulation (Sickmann & Alzheimer, 2003; Kim & Johnston, 2015). To test the involvement of GIRK in the adenosine‐evoked hyperpolarization of olfactory bulb mitral cells, we applied adenosine in the presence of the GIRK inhibitor Ba2+ in current‐clamp experiments using TTX to suppress neuronal activity. Ba2+ at 1 mm, a concentration sufficient to block GIRK (Sickmann & Alzheimer, 2003; Kim & Johnston, 2015), resulted in depolarization of the membrane of mitral cells and an increase in the frequency of synaptic miniature potentials, which largely recovered following washout of adenosine (Fig. 5 A). However, Ba2+ had no inhibiting effect on the adenosine‐induced hyperpolarization (n = 6). In addition, we used canonical blockers of voltage‐gated K+ channels at concentrations demonstrated to efficiently and selectively block K+ channel subtypes (Gutman et al. 2005; Wei et al. 2005). 4‐AP (10 mm; n = 7) and TEA (10 mm; n = 5) did not significantly reduce adenosine‐evoked hyperpolarizations (Fig. 5 B and C). To reveal the type of K+ channel involved, we additionally tested five inhibitors for different K+ channel subfamilies for their effect on the adenosine‐evoked hyperpolarization (Fig. 5 D and E). Only bupivacaine significantly reduced the hyperpolarization. On average, bupivacaine reduced the adenosine‐evoked hyperpolarization by 41.3 ± 9.0% (n = 7, P = 0.040).
Figure 5. Adenosine activates non‐canonical potassium channels.
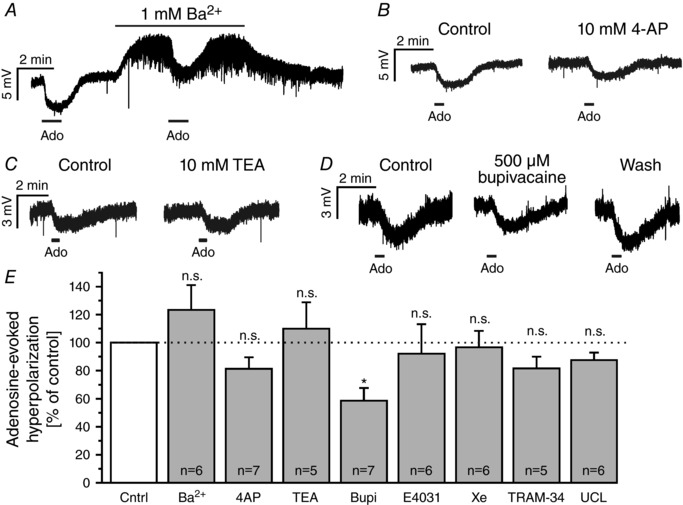
A–D, effect of 1 mm Ba2+ (A), 10 mm 4‐AP (B), 10 mm TEA (C) and 500 μm bupivacaine (D) on the adenosine‐evoked hyperpolarization. E, effects of blockers of inwardly rectifying (1 mm Ba2+), canonical voltage‐gated (10 mm 4‐AP, 10 mm TEA), two‐pore domain (500 μm bupivacaine), erg (10 μm E‐4031), ‘M’‐current (20 μm XE 991) and calcium‐activated (1 μm TRAM‐34, 1 μm UCL 1684) potassium channels. Only bupivacaine significantly reduced the amplitude of adenosine‐evoked hyperpolarization. All recordings were performed in the presence of 1 μm TTX. n.s., not significant.
A1 receptors activate two‐pore domain K+ channels
Bupivacaine has been shown to inhibit some of the K2P channels, a large family of background K+ channels (Enyedi & Czirjak, 2010; Feliciangeli et al. 2015). To confirm the potential involvement of K2P channels we tested halothane, which activates some of the K2P channels, but inhibits other K2P channels such as bupivacaine‐sensitive THIK channels (Patel et al. 2001; Rajan et al. 2001; Bushell et al. 2002). In voltage‐clamp experiments (V h −30 mV), 3 mm halothane evoked a large inward current superimposed by a smaller outward current (Fig. 6 A), demonstrating that a large part of the background current in mitral cells is carried by K2P channels. The halothane‐evoked inward current presumably is the result of inhibition of halothane‐inhibited K2P channels such as THIK‐1, while the small outward current may reflect halothane‐activated K2P channels such as TREK and TASK (Enyedi & Czirjak, 2010; Feliciangeli et al. 2015). In the presence of halothane, the adenosine‐evoked outward current was significantly inhibited by 33.7 ± 10.4% (n = 7, P = 0.043; Fig. 6 B). In addition, bupivacaine reduced the adenosine‐induced outward current by 69.9 ± 8.6% (n = 6, P < 0.001; Fig. 6 C). The effect of bupivacaine was partly reversible after 10 min of washout of bupivacaine. The results indicate that A1 receptors modulate a bupivacaine‐sensitive and halothane‐sensitive background K+ conductance (Fig. 6 D).
Figure 6. Adenosine activates two‐pore domain‐like potassium channels.
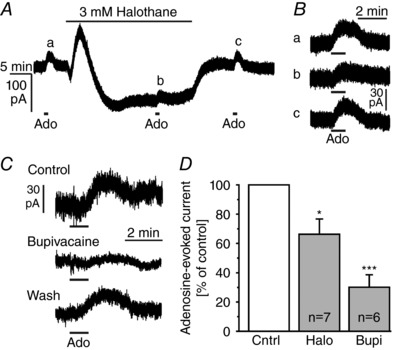
A, effect of the two‐pore domain K+ channel ligand halothane on the membrane current. B, effect of halothane on outward currents evoked by adenosine (100 μm), as measured at the time points a–c indicated in A. C, effect of bupivacaine on the outward current evoked by adenosine (100 μm). D, halothane and bupivacaine significantly reduced the adenosine‐evoked outward currents.
A1 receptors fail to modulate synaptic input from sensory axons
Mitral cells receive synaptic input from olfactory sensory neurons. Synaptic transmission between axons of sensory neurons and mitral cells is subject to extensive neuromodulation by GABA and dopamine (Aroniadou‐Anderjaska et al. 2000; Ennis et al. 2001; Maher & Westbrook, 2008). We were interested in whether adenosine was also capable of modulating synaptic input from olfactory sensory neurons in mitral cells. We measured synaptic currents in mitral cells in response to single pulse stimulation of sensory axons (Fig. 7 A). On average, the amplitude of synaptic currents upon axonal stimulation was −130.7 ± 32.7 pA (n = 6) and was not affected by application of adenosine (Fig. 7 B). As a measure of synaptic vesicle fusion in terminals of olfactory sensory axons, we employed a mouse strain in which olfactory sensory neurons express the vesicle fusion marker syn‐pH (Fig. 7 C). Changes in syn‐pH fluorescence correlate linearly with the transmitter release in receptor axons (Bozza et al. 2004). Fluorescence transients evoked by stimulation of olfactory sensory axons were not reduced in amplitude by application of adenosine (n = 4; Fig. 7 D). The results indicate that the synaptic input from sensory neurons in mitral cells is not influenced by adenosine.
Figure 7. Adenosine has no effect on synaptic input from olfactory sensory neurons.
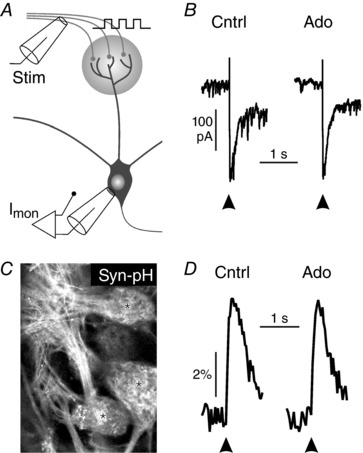
A, schematic drawing of the experimental set‐up. Stim, stimulation; Imon, current monitor. B, mitral cell synaptic currents evoked by single pulse stimulation of sensory axons in the absence and presence of adenosine (100 μm). C, expression of synapto‐pHluorin in olfactory sensory axons. D, adenosine (100 μm) did not alter stimulation‐induced synapto‐pHluorin fluorescence transients.
A1 receptors improve the signal‐to‐noise ratio of mitral cell output signals
Odour information derived from olfactory sensory neurons is processed in the olfactory bulb and propagated by mitral and tufted cells into higher brain centres such as the piriform cortex. The relationship between sensory input signals and mitral cell output signals is one important determinant of odour perception. To assess whether the output signal of mitral cells resulting from sensory stimulation is affected by adenosine, we recorded action currents in the cell‐attached configuration, hence avoiding dialysis of the recorded cell that could compromise excitability. Single stimulation of sensory axons resulted in a biphasic increase in firing rate of the mitral cell (Fig. 8 A). This biphasic increase in firing rate consisted of an early response of approximately 100 ms duration, followed by a late response of approximately 400 ms after a short gap devoid of action potential firing (Fig. 8 A, right panel). The averaged firing rate increased from 5.2 ± 1.6 Hz at baseline (averaged from 19 s preceding the stimulation) to 29.1 ± 6.0 Hz (P < 0.001) during the early response phase (averaged from 0 to 100 ms after stimulation) and to 10.9 ± 3.1 Hz (P < 0.001) in the late response phase (averaged from 100 to 500 ms after stimulation) (n = 10; Fig. 8 B). The latency of the first action potential after stimulation was 8.4 ± 1.3 ms (n = 10) and displayed very little variance within a given experiment (Fig. 8 C), suggesting that the early response resulted from monosynaptic transmission (De Saint Jan et al. 2009) as opposed to disynaptic transmission found upon weak stimulation (Gire et al. 2012). The latency of the first action potential was not altered by adenosine, confirming lack of modulation of the synaptic input from sensory axons (Fig. 8 D).
Figure 8. Synaptic input from olfactory sensory neurons evokes a complex pattern of action potential firing.
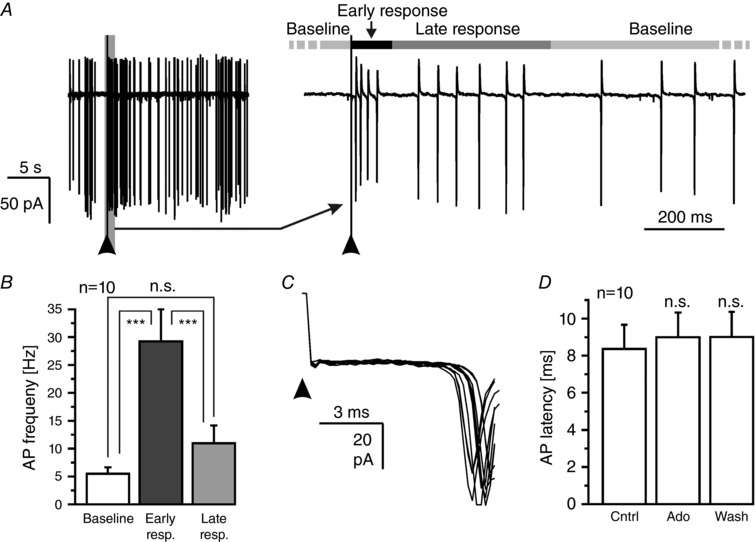
A, mitral cell response (recorded in cell‐attached configuration) upon single pulse stimulation of axons of olfactory sensory neurons (100 μA, 50 μs; indicated by an arrowhead). The response was divided into an early (0–100 ms) and a late response (100–500 ms). B, analysis of the spontaneous firing frequency and the firing frequency during early and late responses. C, 10 consecutive traces (20 s interval) of action current recordings following olfactory sensory axon stimulation measured in the same cell were superimposed to demonstrate little variance in latency. D, the latency between stimulation onset and AP firing was not affected by adenosine (100 μm). n.s., not significant.
We next tested whether adenosine was able to affect the pattern of action potential firing evoked by electrical stimulation of olfactory sensory neurons (Fig. 9 A). Adenosine reduced the baseline firing rate of mitral cells by 37 ± 6% (n = 10, P = 0.002), while it had no significant effect on the firing rate in the presence of DPCPX (n = 6) (Fig. 9 B). Adenosine had no effect on the firing rate during the early response in both the absence and the presence of DPCPX, but decreased the firing rate of the late response in wild‐type mice by 36 ± 5% (n = 10, P = 0.002; Fig. 9 C and D). As adenosine hyperpolarizes the mitral cell membrane by 2–3 mV, we examined whether this hyperpolarization was sufficient to cause the described effect of adenosine on stimulation‐evoked action potential firing. Therefore, we hyperpolarized mitral cells in current‐clamp mode by approximately 3 mV by current injection (−15 to −30 pA) and recorded action potentials evoked by single pulse stimulation of olfactory sensory neurons (Fig. 9 E). In current‐clamp recordings, baseline firing frequency was 5.1 ± 1.3 Hz (n = 5) and increased upon single pulse stimulation to 40.0 ± 6.7 Hz (n = 5) during the early response and to 16.5 ± 2.0 Hz (n = 5) during the late response. The induced hyperpolarization decreased the baseline firing rate by 61 ± 5% (n = 5, P = 0.038) and the late response firing rate by 38 ± 6% (n = 5, P = 0.018), but had no effect on the early response (Fig. 9 F–H). Since spontaneous firing reflects the mitral cell output activity independent of specific odour sensation it can be considered as noise, whereas the early response reflects the signal directly evoked by sensory input and propagated to the cortex upon odour sensation as mimicked by stimulation of sensory axons. Hence, the ratio of early response/spontaneous firing is an estimation of the signal‐to‐noise ratio of the mitral cell output. The signal‐to‐noise ratio of 8.4 ± 1.9 under control conditions increased significantly by 62 ± 23% (n = 10, P = 0.045) upon application of adenosine in the absence but not in the presence of DPCPX (not shown). In addition, the signal‐to‐noise ratio increased by 164 ± 50% (n = 5, P = 0.002) upon current‐induced hyperpolarization (Fig. 9 I). We also calculated the signal‐to‐noise ratio of the late response. Since both baseline firing and firing during the late response were reduced by adenosine, the ratio of late response/baseline was not altered by adenosine (Fig. 9 I). However, it increased upon current‐induced hyperpolarization by 71 ± 27% (n = 5; P = 0.049). The results indicate that A1 receptor activation is able to increase the signal‐to‐noise ratio particularly of the firing pattern directly evoked in mitral cells upon synaptic input from sensory neurons and hence to modulate olfactory information that is propagated from the olfactory bulb to other brain areas such as the piriform cortex.
Figure 9. Adenosine increases the signal‐to‐noise ratio of the input–output relationship of mitral cells.
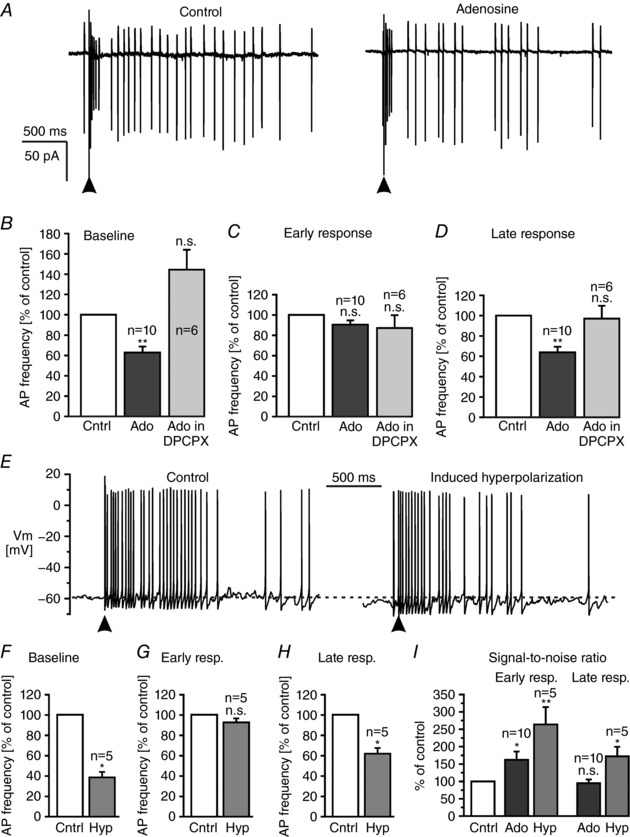
A, cell‐attached recording of action potentials following single pulse stimulation (100 μA, 50 μs; indicated by an arrowhead) of olfactory sensory neurons in the absence (control) and in the presence of 100 μm adenosine. B, effect of adenosine on baseline firing, C and D, early response (C) and late response (D) in control experiments and in the presence of DPCPX. E, effect of hyperpolarization of approximately 3 mV induced by negative current injection on action potential firing evoked by electrical stimulation of olfactory sensory neurons in current clamp recordings. F, hyperpolarization (Hyp) significantly reduced the frequency of baseline firing, while G, firing rate of the early response was not affected. H, hyperpolarization reduced the firing rate of the late response. I, adenosine and current‐induced hyperpolarization increased the signal‐to‐noise ratio of the early response in mitral cells. Adenosine had no effect on the signal‐to‐noise ratio of the late response, while induced hyperpolarization also increased the signal‐to‐noise ratio of the late response. n.s., not significant.
Discussion
The olfactory bulb is a brain region with one of the highest activities of enzymes involved in extracellular ATP degradation and adenosine production (Langer et al. 2008), suggesting an important role of adenosinergic neuromodulation in this sensory pathway. In the present study, we investigated the effect of adenosine on mitral cell membrane properties in the olfactory bulb. Adenosine increased the background K+ conductance by activation of K2P channels, a modulatory pathway not described in neurons before. The increased K+ conductance reduced spontaneous action potential firing (noise), but had no effect on synaptic input in mitral cells derived from olfactory sensory neurons and the subsequent bursting in mitral cells (signal), resulting in an improved signal‐to‐noise ratio.
Adenosine modulates potassium channels
Based on membrane conductance measurements and calculation of the reversal potential of the adenosine‐induced ion conductance, we showed that A1 receptor activation leads to opening of background potassium channels. Our pharmacological data suggest the involvement of K2P channels, which are sensitive to volatile anaesthetics such as chloroform, isoflurane and halothane (Enyedi & Czirjak, 2010; Feliciangeli et al. 2015). Halothane induced a large inward current, suggesting inhibition of K2P channels such as THIK‐1 and THIK‐2, which are highly expressed in the olfactory bulb, including mitral cells (Rajan et al. 2001; Lein et al. 2007), and can form heterodimers (Blin et al. 2014). The halothane‐evoked inward current was superimposed by an outward current, suggesting that in addition to halothane‐inhibited K2P channels, halothane‐activated K2P channels are present in mitral cells, as shown before, for instance, in thalamic neurons (Meuth et al. 2003). Bupivacaine inhibits THIK‐1 (Kang et al. 2014); however, the pharmacology of K2P channels is still poorly understood and the action of bupivacaine alone does not allow for a definite identification of the K2P channel activated by adenosine (Lesage, 2003). For instance, in our experiments barium did not block the adenosine‐evoked hyperpolarization, while in Purkinje neurons a THIK‐1‐like current was reduced by barium (Bushell et al. 2002). Absent barium sensitivity clearly argues against the involvement of GIRK channels, which have been shown to mediate adenosine‐induced hyperpolarization in other types of neurons and are activated by the βγ subunit of G‐proteins coupled to A1 receptors and other metabotropic receptors (Lüscher & Slesinger, 2010). Several other types of potassium channels have been reported in mitral cells including SK channels, Shaker channels, erg channels and A‐type channels (Fadool et al. 2004; Maher & Westbrook, 2005; Hirdes et al. 2009), but blockers of these channels did not affect the adenosine‐mediated hyperpolarization. Modulation of K2P channels by intracellular signalling pathways in neuronal and non‐neuronal cells has been published (Chen et al. 2006, Comoglio et al. 2014; Leist et al. 2017), but modulation of K2P channels by A1 receptors has been described only in retinal Müller glial cells so far (Skatchkov et al. 2006). In addition to A1 receptor expression, we found A2A receptor expression in mitral cells. In A1 knockout mice, adenosine application resulted in slight depolarization instead of hyperpolarization, an effect that may be mediated by A2A receptors, since A2A receptors often exert opposing effects to A1 receptors (Cunha et al. 1994; Rebola et al. 2003).
Adenosine modulates mitral cell activity
The pattern of action potentials propagated by mitral cells to cortical structures upon sensory stimulation is one of the main determinants of odour perception (Gire et al. 2013; Uchida et al. 2014). Synaptic input reaching mitral cells via olfactory sensory axons is not affected by adenosine, in contrast to other neuromodulators such as GABA (via GABAB receptors) and dopamine which reduce neurotransmitter release at olfactory sensory axon terminals (Aroniadou‐Anderjaska et al. 2000; Ennis et al. 2001; Maher & Westbrook, 2008). Firing of mitral cells evoked by synaptic input from olfactory sensory axons comprises two phases, the early response reflecting direct excitation by the olfactory sensory afferents and the delayed response modulated by processing in the bulbar network (Najac et al. 2011). Timing of the early response with respect to the sniffing cycle may determine mitral cell response amplitudes (Smear et al. 2011); however, the latency of the early response to olfactory nerve stimulation is unaltered by adenosine, suggesting that adenosine does not impact temporal odour coding by early response modulation. In contrast, the frequency of spontaneous firing, but also of the firing in the late phase of the response, was reduced. The latter might result from A1 receptor‐sensitive integration of feedback excitation and inhibition (Hayar et al. 2004; Giridhar et al. 2011; Gire et al. 2012; Carey et al. 2015; Liu et al. 2015; Parsa et al. 2015) and the results suggest that the temporal pattern of glomerular network activity is shaped by A1 receptors. Since the temporal pattern of glomerular network activity is decisive for odour perception (Shusterman et al. 2011; Haddad et al. 2013; Tripathy et al. 2013; Nunez‐Parra et al. 2014; Uchida et al. 2014), A1 receptors might thus fine‐tune odour perception. Recently it was reported that the input–output transformation in mitral and tufted cells contributes to the perception of odour concentration invariances (Storace & Cohen, 2017). Tuning of the input–output transformation by neuromodulators such as adenosine might therefore adjust the perception of odour concentration invariances. An increase in signal‐to‐noise ratio of the input–output transformation of mitral cells, comparable to the one described in the present study, has been found by stimulation of noradrenalin receptors in the olfactory bulb, which results in improved odour detection thresholds in behavioural tests (Escanilla et al. 2010; Manella et al. 2017). Adenosine increases the signal‐to‐noise ratio of the evoked firing pattern in mitral cells that are excited by the sensory stimulus; odour‐evoked mitral cell responses, however, consist of a complex ensemble of both excited and inhibited mitral cells (Economo et al. 2016). In contrast to excitatory mitral cell responses, the signal‐to‐noise ratio of inhibitory responses would be attenuated by an adenosine‐mediated reduction of spontaneous firing (noise), suggesting that the action of adenosine on information processing in the olfactory bulb might be even more complex than indicated by the present study. It must be noted that in awake animals, the response pattern of action potential firing upon sensory input, but also the spontaneous action potential firing of mitral cells, can differ from action potential firing in anaesthetized animals or slice preparations. Rinberg et al. (2006) reported an increased spontaneous firing rate of mitral cells in the awake situation compared to anaesthetized mice, while Kollo et al. (2014) found that about one‐third of mitral cells were silent in awake mice, but not in anaesthetized mice. Since K2P channels are affected by anaesthetics such as halothane and isofluorane used in some of the studies using anaesthetized animals, activation and/or inhibition of K2P channels in mitral cells by these anaesthetics may partly account for the differences between awake and anaesthetized mice.
Purinergic signalling has been described as affecting olfactory bulb neurons and glial cells via P2Y receptors and adenosine receptors, but the role of adenosine in odour information processing has not been elucidated before (Rieger et al. 2007; Doengi et al. 2008; Fischer et al. 2012; Roux et al. 2015). It should be noted that in the study by Roux et al. (2015) A1 receptors appeared to increase excitability of mitral cells, whereas in our study activation of A1 receptors reduced excitability, suggesting that adenosinergic modulation of mitral cell activity is intricate and variable.
In conclusion, our study shows that adenosine is able to modulate mitral cell membrane properties, attenuate network activity and tune the input–output relationship of mitral cell signalling. Hence, purinergic signalling might significantly contribute to odour information processing in the olfactory bulb.
Additional information
Competing interest
The authors declare no conflict of interest.
Author contributions
Design of the work: C.L., D.H. and N.R. Acquisition and analysis of data, providing material: C.L., D.H., J.B., K.S., M.B., M.C. N.B., N.R., S.W. and T.F. Writing the manuscript: C.L., D.H., M.C. and N.R. All authors revised and approved the manuscript. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
Financial support by the Deutsche Forschungsgemeinschaft (LO 779/6‐1 and HI 1288/3‐1), the Landesforschungsförderung Hamburg (Read Me) and the Swedish Research Council (Dnr: 2016‐01381) is gratefully acknowledged.
Biography
Natalie Rotermund received her PhD in Neurophysiology from the University of Hamburg. After working in the medical device industry for some years, she decided to return to university and follow her interest in fundamental research. She is currently employed as postdoctoral researcher in the Division of Neurophysiology at the University of Hamburg. Her research focuses on the understanding of purinergic signalling in the central nervous system, with a specialization in electrophysiological and imaging techniques.

Edited by: Yoshihiro Kubo & Takashi Kurahashi
C. Lohr and D. Hirnet contributed equally.
Contributor Information
Christian Lohr, Email: christian.lohr@uni-hamburg.de.
Daniela Hirnet, Email: daniela.hirnet@uni-hamburg.de.
References
- Aroniadou‐Anderjaska V, Zhou FM, Priest CA, Ennis M & Shipley MT (2000). Tonic and synaptically evoked presynaptic inhibition of sensory input to the rat olfactory bulb via GABAB heteroreceptors. J Neurophysiol 84, 1194–1203. [DOI] [PubMed] [Google Scholar]
- Blin S, Chatelain FC, Feliciangeli S, Kang D, Lesage F & Bichet D (2014). Tandem pore domain halothane‐inhibited K+ channel subunits THIK1 and THIK2 assemble and form active channels. J Biol Chem 289, 28202–28212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D (2009). Adenosine‐based modulation of brain activity. Curr Neuropharmacol 7, 158–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozza T, McGann JP, Mombaerts P & Wachowiak M (2004). In vivo imaging of neuronal activity by targeted expression of a genetically encoded probe in the mouse. Neuron 42, 9–21. [DOI] [PubMed] [Google Scholar]
- Bushell T, Clarke C, Mathie A & Robertson B (2002). Pharmacological characterization of a non‐inactivating outward current observed in mouse cerebellar Purkinje neurones. Br J Pharmacol 135, 705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey RM, Sherwood WE, Shipley MT, Borisyuk A & Wachowiak M (2015). Role of intraglomerular circuits in shaping temporally structured responses to naturalistic inhalation‐driven sensory input to the olfactory bulb. J Neurophysiol 113, 3112–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Talley EM, Patel N, Gomis A, McIntire WE, Dong B, Viana F, Garrison JC & Bayliss DA (2006). Inhibition of a background potassium channel by Gq protein α‐subunits. Proc Natl Acad Sci USA 103, 3422–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark BD, Kurth‐Nelson ZL & Newman EA (2009). Adenosine‐evoked hyperpolarization of retinal ganglion cells is mediated by G‐protein‐coupled inwardly rectifying K+ and small conductance Ca2+‐activated K+ channel activation. J Neurosci 29, 11237–11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comoglio Y, Levitz J, Kienzler MA, Lesage F, Isacoff EY & Sandoz G (2014). Phospholipase D2 specifically regulates TREK potassium channels via direct interaction and local production of phosphatidic acid. Proc Natl Acad Sci USA 111, 13547–13552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha RA (2005). Neuroprotection by adenosine in the brain: From A1 receptor activation to A2A receptor blockade. Purinergic Signal 1, 111–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha RA, Johansson B, van der Ploeg I, Sebastião AM, Ribeiro JA & Fredholm BB (1994). Evidence for functionally important adenosine A2a receptors in the rat hippocampus. Brain Res 649, 208–216. [DOI] [PubMed] [Google Scholar]
- De Saint Jan D, Hirnet D, Westbrook GL & Charpak S (2009). External tufted cells drive the output of olfactory bulb glomeruli. J Neurosci 29, 2043–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias RB, Rombo DM, Ribeiro JA, Henley JM & Sebastiao AM (2013). Adenosine: setting the stage for plasticity. Trends Neurosci 36, 248–257. [DOI] [PubMed] [Google Scholar]
- Dixon AK, Gubitz AK, Sirinathsinghji DJ, Richardson PJ & Freeman TC (1996). Tissue distribution of adenosine receptor mRNAs in the rat. Br J Pharmacol 118, 1461–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doengi M, Deitmer JW & Lohr C (2008). New evidence for purinergic signaling in the olfactory bulb: A2A and P2Y1 receptors mediate intracellular calcium release in astrocytes. FASEB J 22, 2368–2378. [DOI] [PubMed] [Google Scholar]
- Dos Santos‐Rodrigues A, Pereira MR, Brito R, de Oliveira NA & Paes‐de‐Carvalho R (2015). Adenosine transporters and receptors: key elements for retinal function and neuroprotection. Vitam Horm 98, 487–523. [DOI] [PubMed] [Google Scholar]
- Economo MN, Hansen KR & Wachowiak M (2016). Control of mitral/tufted cell output by selective inhibition among olfactory bulb glomeruli. Neuron 20, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennis M, Zhou F, Ciombor KJ, Aroniadou‐Anderjaska V, Hayar A & Borrelli E (2001). Dopamine D2 receptor‐mediated presynaptic inhibition of olfactory nerve terminals. J Neurophysiol 86, 2986–2997. [DOI] [PubMed] [Google Scholar]
- Enyedi P & Czirjak G (2010). Molecular background of leak K+ currents: two‐pore domain potassium channels. Physiol Rev 90, 559–605. [DOI] [PubMed] [Google Scholar]
- Escanilla O, Arrellanos A, Karnow A, Ennis M & Linster C (2010). Noradrenergic modulation of behavioral odor detection and discrimination thresholds in the olfactory bulb. Eur J Neurosci 32, 458–468. [DOI] [PubMed] [Google Scholar]
- Fadool DA, Tucker K, Perkins R, Fasciani G, Thompson RN, Parsons AD, Overton JM, Koni PA, Flavell RA & Kaczmarek LK (2004). Kv1.3 channel gene‐targeted deletion produces “Super‐Smeller Mice” with altered glomeruli, interacting scaffolding proteins, and biophysics. Neuron 41, 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feliciangeli S, Chatelain FC, Bichet D, Lesage F (2015). The family of K2P channels: salient structural and functional properties. J Physiol 593, 2587–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer T, Rotermund N, Lohr C & Hirnet D (2012). P2Y1 receptor activation by photolysis of caged ATP enhances neuronal network activity in the developing olfactory bulb. Purinergic Signal 8, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Chen JF, Cunha RA, Svenningsson P & Vaugeois JM (2005). Adenosine and brain function. Int Rev Neurobiol 63, 191–270. [DOI] [PubMed] [Google Scholar]
- Gire DH, Franks KM, Zak JD, Tanaka KF, Whitesell JD, Mulligan AA, Hen R & Schoppa NE (2012). Mitral cells in the olfactory bulb are mainly excited through a multistep signaling path. J Neurosci 32, 2964–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gire DH, Restrepo D, Sejnowski TJ, Greer C, De Carlos JA & Lopez‐Mascaraque L (2013). Temporal processing in the olfactory system: can we see a smell? Neuron 78, 416–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giridhar S, Doiron B & Urban NN (2011). Timescale‐dependent shaping of correlation by olfactory bulb lateral inhibition. Proc Natl Acad Sci USA 108, 5843–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundlfinger A, Bischofberger J, Johenning FW, Torvinen M, Schmitz D & Breustedt J (2007). Adenosine modulates transmission at the hippocampal mossy fibre synapse via direct inhibition of presynaptic calcium channels. J Physiol 582, 263–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stühmer W & Wang X (2005). International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage‐gated potassium channels. Pharmacol Rev 57, 473–508. [DOI] [PubMed] [Google Scholar]
- Haddad R, Lanjuin A, Madisen L, Zeng H, Murthy VN & Uchida N (2013). Olfactory cortical neurons read out a relative time code in the olfactory bulb. Nat Neurosci 16, 949–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayar A, Karnup S, Ennis M & Shipley MT (2004). External tufted cells: a major excitatory element that coordinates glomerular activity. J Neurosci 24, 6676–6685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirdes W, Napp N, Wulfsen I, Schweizer M, Schwarz JR & Bauer CK (2009). Erg K+ currents modulate excitability in mouse mitral/tufted neurons. Pflugers Arch 459, 55–70. [DOI] [PubMed] [Google Scholar]
- Housley GD, Bringmann A & Reichenbach A (2009). Purinergic signaling in special senses. Trends Neurosci 32, 128–141. [DOI] [PubMed] [Google Scholar]
- Johansson B, Georgiev V & Fredholm BB (1997). Distribution and postnatal ontogeny of adenosine A2A receptors in rat brain: comparison with dopamine receptors. Neuroscience 80, 1187–1207. [DOI] [PubMed] [Google Scholar]
- Johansson B, Halldner L, Dunwiddie TV, Masino SA, Poelchen W, Giménez‐Llort L, Escorihuela RM, Fernández‐Teruel A, Wiesenfeld‐Hallin Z, Xu XJ, Hårdemark A, Betsholtz C, Herlenius E & Fredholm BB (2001). Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proc Natl Acad Sci USA 98, 9407–9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin‐Lang A, Lauterburg T & Burgunder JM (1999). Expression of adenosine A2a receptors gene in the olfactory bulb and spinal cord of rat and mouse. Neurosci Lett 261, 189–191. [DOI] [PubMed] [Google Scholar]
- Kang D, Hogan JO & Kim D (2014). THIK‐1 (K2P13.1) is a small‐conductance background K+ channel in rat trigeminal ganglion neurons. Pflugers Arch 466, 1289–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CS & Johnston D (2015). A1 adenosine receptor‐mediated GIRK channels contribute to the resting conductance of CA1 neurons in the dorsal hippocampus. J Neurophysiol 113, 2511–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollo M, Schmaltz A, Abdelhamid M, Fukunaga I & Schaefer AT (2014). ‘Silent’ mitral cells dominate odor responses in the olfactory bulb of awake mice. Nat Neurosci 17, 1313–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer D, Hammer K, Koszalka P, Schrader J, Robson S & Zimmermann H (2008). Distribution of ectonucleotidases in the rodent brain revisited. Cell Tissue Res 334, 199–217. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, Desaki AL, Desta T, Diep E, Dolbeare TA, Donelan MJ, Dong HW, Dougherty JG, Duncan BJ, Ebbert AJ, Eichele G, Estin LK, Faber C, Facer BA, Fields R, Fischer SR, Fliss TP, Frensley C, Gates SN, Glattfelder KJ, Halverson KR, Hart MR, Hohmann JG, Howell MP, Jeung DP, Johnson RA, Karr PT, Kawal R, Kidney JM, Knapik RH, Kuan CL, Lake JH, Laramee AR, Larsen KD, Lau C, Lemon TA, Liang AJ, Liu Y, Luong LT, Michaels J, Morgan JJ, Morgan RJ, Mortrud MT, Mosqueda NF, Ng LL, Ng R, Orta GJ, Overly CC, Pak TH, Parry SE, Pathak SD, Pearson OC, Puchalski RB, Riley ZL, Rockett HR, Rowland SA, Royall JJ, Ruiz MJ, Sarno NR, Schaffnit K, Shapovalova NV, Sivisay T, Slaughterbeck CR, Smith SC, Smith KA, Smith BI, Sodt AJ, Stewart NN, Stumpf KR, Sunkin SM, Sutram M, Tam A, Teemer CD, Thaller C, Thompson CL, Varnam LR, Visel A, Whitlock RM, Wohnoutka PE, Wolkey CK, Wong VY, Wood M, Yaylaoglu MB, Young RC, Youngstrom BL, Yuan XF, Zhang B, Zwingman TA & Jones AR (2007). Genome‐wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. [DOI] [PubMed] [Google Scholar]
- Leist M, Rinné S, Datunashvili M, Aissaoui A, Pape HC, Decher N, Meuth SG & Budde T (2017). Acetylcholine‐dependent upregulation of TASK‐1 channels in thalamic interneurons by a smooth muscle‐like signalling pathway. J Physiol 595, 5875–5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage F (2003). Pharmacology of neuronal background potassium channels. Neuropharmacology 44, 1–7. [DOI] [PubMed] [Google Scholar]
- Liu S, Shao Z, Puche A, Wachowiak M, Rothermel M & Shipley MT (2015). Muscarinic receptors modulate dendrodendritic inhibitory synapses to sculpt glomerular output. J Neurosci 35, 5680–5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr C, Grosche A, Reichenbach A & Hirnet D (2014). Purinergic neuron‐glia interactions in sensory systems. Pflugers Arch 466, 1859–1872. [DOI] [PubMed] [Google Scholar]
- Lopes LV, Cunha RA, Kull B, Fredholm BB & Ribeiro JA (2002). Adenosine A2A receptor facilitation of hippocampal synaptic transmission is dependent on tonic A1 receptor inhibition. Neuroscience 112, 319–329. [DOI] [PubMed] [Google Scholar]
- Lüscher C & Slesinger PA (2010). Emerging roles for G protein‐gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci 11, 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahan LC, McVittie LD, Smyk‐Randall EM, Nakata H, Monsma FJ Jr, Gerfen CR & Sibley DR. (1991). Cloning and expression of an A1 adenosine receptor from rat brain. Mol Pharmacol 40, 1–7. [PubMed] [Google Scholar]
- Maher BJ & Westbrook GL (2005). SK channel regulation of dendritic excitability and dendrodendritic inhibition in the olfactory bulb. J Neurophysiol 94, 3743–3750. [DOI] [PubMed] [Google Scholar]
- Maher BJ & Westbrook GL (2008). Co‐transmission of dopamine and GABA in periglomerular cells. J Neurophysiol 99, 1559–1564. [DOI] [PubMed] [Google Scholar]
- Manella LC, Petersen N & Linster C (2017). Stimulation of the locus coeruleus modulates signal‐to‐noise ratio in the olfactory bulb. J Neurosci 37, 11605–11615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuth SG, Budde T, Kanyshkova T, Broicher T, Munsch T & Pape HC (2003). Contribution of TWIK‐related acid‐sensitive K+ channel 1 (TASK1) and TASK3 channels to the control of activity modes in thalamocortical neurons. J Neurosci 23, 6460–6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najac M, De Saint Jan D, Reguero L, Grandes P & Charpak S (2011). Monosynaptic and polysynaptic feed‐forward inputs to mitral cells from olfactory sensory neurons. J Neurosci 31, 8722–8729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez‐Parra A, Li A & Restrepo D (2014). Coding odor identity and odor value in awake rodents. Prog Brain Res 208, 205–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsa PV, D'Souza RD & Vijayaraghavan S (2015). Signaling between periglomerular cells reveals a bimodal role for GABA in modulating glomerular microcircuitry in the olfactory bulb. Proc Natl Acad Sci USA 112, 9478–9483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AJ, Honoré E, Lesage F, Fink M, Romey G & Lazdunski M (2001). Inhalational anesthetics activate two‐pore‐domain background K+ channels. Nat Neurosci 2, 422–436. [DOI] [PubMed] [Google Scholar]
- Rajan S, Wischmeyer E, Karschin C, Preisig‐Muller R, Grzeschik KH, Daut J, Karschin A & Derst C (2001). THIK‐1 and THIK‐2, a novel subfamily of tandem pore domain K+ channels. J Biol Chem 276, 7302–7311. [DOI] [PubMed] [Google Scholar]
- Rebola N, Sebastião AM, de Mendonca A, Oliveira CR, Ribeiro JA & Cunha RA (2003). Enhanced adenosine A2A receptor facilitation of synaptic transmission in the hippocampus of aged rats. J Neurophysiol 90, 1295–1303. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR, Stehle JH & Rivkees SA (1991). Molecular cloning and characterization of a rat A1‐adenosine receptor that is widely expressed in brain and spinal cord. Mol Endocrinol 5, 1037–1048. [DOI] [PubMed] [Google Scholar]
- Ribeiro JA & Sebastiao AM (2010). Modulation and metamodulation of synapses by adenosine. Acta Physiol (Oxf) 199, 161–169. [DOI] [PubMed] [Google Scholar]
- Rieger A, Deitmer JW & Lohr C (2007). Axon‐glia communication evokes calcium signaling in olfactory ensheathing cells of the developing olfactory bulb. Glia 55, 352–359. [DOI] [PubMed] [Google Scholar]
- Rinberg D, Koulakov A & Gelperin A (2006). Sparse odor coding in awake behaving mice. J Neurosci 26, 8857–8865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosin DL, Robeva A, Woodard RL, Guyenet PG & Linden J (1998). Immunohistochemical localization of adenosine A2A receptors in the rat central nervous system. J Comp Neurol 401, 163–186. [PubMed] [Google Scholar]
- Roux L, Madar A, Lacroix MM, Yi C, Benchenane K & Giaume C (2015). Astroglial connexin 43 hemichannels modulate olfactory bulb slow oscillations. J Neurosci 35, 15339–15352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shusterman R, Smear MC, Koulakov AA & Rinberg D (2011). Precise olfactory responses tile the sniff cycle. Nat Neurosci 14, 1039–1044. [DOI] [PubMed] [Google Scholar]
- Sickmann T & Alzheimer C (2003). Short‐term desensitization of G‐protein‐activated, inwardly rectifying K+ (GIRK) currents in pyramidal neurons of rat neocortex. J Neurophysiol 90, 2494–2503. [DOI] [PubMed] [Google Scholar]
- Skatchkov SN, Eaton MJ, Shuba YM, Kucheryavykh YV, Derst C, Veh RW, Wurm A, Iandiev I, Pannicke T, Bringmann A & Reichenbach A (2006). Tandem‐pore domain potassium channels are functionally expressed in retinal (Müller) glial cells. Glia 53, 266–276. [DOI] [PubMed] [Google Scholar]
- Smear M, Shusterman R, O'Connor R, Bozza T & Rinberg D (2011). Perception of sniff phase in mouse olfaction. Nature 479, 397–400. [DOI] [PubMed] [Google Scholar]
- Stavermann M, Meuth P, Doengi M, Thyssen A, Deitmer JW & Lohr C (2015). Calcium‐induced calcium release and gap junctions mediate large‐scale calcium waves in olfactory ensheathing cells in situ. Cell Calcium 58, 215–225. [DOI] [PubMed] [Google Scholar]
- Storace DA & Cohen LB (2017). Measuring the olfactory bulb input‐output transformation reveals a contribution to the perception of odorant concentration invariance. Nat Commun 8, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P, Le Moine C, Kull B, Sunahara R, Bloch B & Fredholm BB (1997). Cellular expression of adenosine A2A receptor messenger RNA in the rat central nervous system with special reference to dopamine innervated areas. Neuroscience 80, 1171–1185. [DOI] [PubMed] [Google Scholar]
- Thyssen A, Hirnet D, Wolburg H, Schmalzig G, Deitmer JW & Lohr C (2010). Ectopic vesicular neurotransmitter release along sensory axons mediates neurovascular coupling via glial calcium signaling. Proc Natl Acad Sci USA 107, 15258–15263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathy SJ, Padmanabhan K, Gerkin RC & Urban NN (2013). Intermediate intrinsic diversity enhances neural population coding. Proc Natl Acad Sci USA 110, 8248–8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida N, Poo C & Haddad R (2014). Coding and transformations in the olfactory system. Annu Rev Neurosci 37, 363–385. [DOI] [PubMed] [Google Scholar]
- Wei AD, Gutman GA, Aldrich R, Chandy KG, Grissmer S & Wulff H (2005). International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium‐activated potassium channels. Pharmacol Rev 57, 463–472. [DOI] [PubMed] [Google Scholar]