

Abstract
Key points
CA3 pyramidal cells display input‐specific differences in the subunit composition of synaptic NMDA receptors (NMDARs).
Although at low density, GluN2B contributes significantly to NMDAR‐mediated EPSCs at mossy fibre synapses.
Long‐term potentiation (LTP) of NMDARs triggers a modification in the subunit composition of synaptic NMDARs by insertion of GluN2B.
GluN2B subunits are essential for the expression of LTP of NMDARs at mossy fibre synapses.
Abstract
Single neurons express NMDA receptors (NMDARs) with distinct subunit composition and biophysical properties that can be segregated in an input‐specific manner. The dynamic control of the heterogeneous distribution of synaptic NMDARs is crucial to control input‐dependent synaptic integration and plasticity. In hippocampal CA3 pyramidal cells from mice of both sexes, we found that mossy fibre (MF) synapses display a markedly lower proportion of GluN2B‐containing NMDARs than associative/commissural synapses. The mechanism involved in such heterogeneous distribution of GluN2B subunits is not known. Here we show that long‐term potentiation (LTP) of NMDARs, which is selectively expressed at MF–CA3 pyramidal cell synapses, triggers a modification in the subunit composition of synaptic NMDARs by insertion of GluN2B. This activity‐dependent recruitment of GluN2B at mature MF–CA3 pyramidal cell synapses contrasts with the removal of GluN2B subunits at other glutamatergic synapses during development and in response to activity. Furthermore, although expressed at low levels, GluN2B is necessary for the expression of LTP of NMDARs at MF–CA3 pyramidal cell synapses. Altogether, we reveal a previously unknown activity‐dependent regulation and function of GluN2B subunits that may contribute to the heterogeneous plasticity induction rules in CA3 pyramidal cells.
Keywords: hippocampus, synaptic plasticity, NMDA receptors, subunit composition
Key points
CA3 pyramidal cells display input‐specific differences in the subunit composition of synaptic NMDA receptors (NMDARs).
Although at low density, GluN2B contributes significantly to NMDAR‐mediated EPSCs at mossy fibre synapses.
Long‐term potentiation (LTP) of NMDARs triggers a modification in the subunit composition of synaptic NMDARs by insertion of GluN2B.
GluN2B subunits are essential for the expression of LTP of NMDARs at mossy fibre synapses.
Introduction
The subunit composition of the tetrameric NMDA receptors (NMDARs) determines important functional properties, such as open probability and decay kinetics (Paoletti, 2011). GluN2A and GluN2B subunits are the most prevalent subunits in the brain. Their presence with GluN1 either in heterodimers (GluN1–GluN2A or GluN1–GluN2B) or heterotrimers (GluN1–GluN2A–GluN2B) accounts in large part for the diversity in NMDAR subtypes found in the brain. The GluN2C and GluN2D subunits are abundantly expressed at early stages of development, or in selected neuronal populations in the adult brain (Monyer et al. 1994; Paoletti et al. 2013). In the adult, particularly in the forebrain, synaptic NMDARs are predominantly either di‐heteromeric GluN1–GluN2A or tri‐heteromeric GluN1–GluN2A–GluN2B (Al‐Hallaq et al. 2007; Gray et al. 2011; Rauner & Köhr, 2011; Tovar et al. 2013).
It is well established that a switch in the subunit composition of synaptic NMDARs occurs during postnatal development from NMDARs containing primarily GluN2B to NMDARs containing both GluN2B and GluN2A (Sheng et al. 1994; van Zundert et al. 2004; Bellone & Nicoll, 2007; Rodenas‐Ruano et al. 2012; K. Williams et al. 1993). In neonatal synapses, but not in mature synapses, the GluN2 subunit composition of NMDARs can be rapidly and bidirectionally modulated in an activity‐dependent manner through a mechanism involving postsynaptic Ca2+ rise and mGluR5 activation (Bellone & Nicoll, 2007; Matta et al. 2011). At mature synapses both GluN2A‐ and GluN2B‐containing NMDARs are present, perhaps with each having distinct functional roles (Yashiro & Philpot, 2008; Paoletti et al. 2013). Although the exact contribution of GluN2B and GluN2A to long‐term potentiation (LTP) and long‐term depression (LTD) has been debated (Paoletti et al. 2013), synaptic GluN2B‐containing NMDARs are thought to play an important part in NMDAR‐dependent plasticity processes, because they exhibit higher fractional Ca2+ current (Sobczyk & Svoboda, 2007) and they are preferentially tethered to the plasticity protein calcium/calmodulin‐dependent protein kinase II (CaMKII) (Barria & Malinow, 2005).
Beyond their well‐established role in synaptic plasticity, NMDARs themselves undergo long lasting depression or potentiation (Rebola et al. 2010; Hunt & Castillo, 2012). Mossy fibre–CA3 pyramidal cell (MF–CA3) synapses express a postsynaptic form of LTP which is selective for NMDARs (Kwon & Castillo, 2008; Rebola et al. 2008). LTP of NMDARs (LTP‐NMDAR) serves as a metaplastic switch making MF–CA3 synapses competent for generating NMDAR‐dependent LTP of AMPA receptor (AMPAR) excitatory postsynaptic currents (EPSCs) (Rebola et al. 2011), and controls heterosynaptic plasticity of associative/commissural (A/C) synapses (Hunt et al. 2013). Immunocytochemical labelling with subunit‐specific antibodies suggests that the subunit composition of synaptic NMDARs differs at A/C fibre–CA3 pyramidal cell (A/C–CA3) synapse and MF–CA3 synapses, with GluN2B being only scarcely expressed in the stratum lucidum (Fritschy et al. 1998; Watanabe et al. 1998). The impact and dependence of LTP‐NMDAR on this heterogeneous distribution of GluN2 subunits in CA3 pyramidal cells (PCs) is not known. We have examined the subunit composition of NMDARs at MF–CA3 synapses, its consequences for LTP‐NMDAR and whether LTP‐NMDAR is accompanied by a change in subunit composition. Using a combination of pharmacological and transgenic approaches, we have found that, although much less abundant than in A/C synapses, GluN2B significantly contributes to NMDAR‐mediated excitatory postsynaptic currents (NMDAR‐EPSCs) at MF–CA3 synapses. The present data indicates that GluN2B is essential for the expression of LTP of NMDARs at MF synapses. In addition, we observe that LTP‐NMDAR leads to an increased content of GluN2B of MF NMDARs. Hence, activity‐dependent insertion of GluN2B in synaptic NMDARs can be observed at mature synapses. This change in subunit composition could be key for the metaplastic switch to conventional LTP of AMPAR‐EPSCs.
Methods
Ethical approval
Animal anaesthesia and euthanasia procedures were carried out in accordance with the Animal Protection Association of ethical standards and the French legislation concerning animal experimentation (authorization no. A33 12 061 and no. A33‐063‐917) and according to the guidelines of the University of Bordeaux/CNRS Animal Care and Use Committee. Wild‐type (C57bl6J) and mutant (Grin2A‐KO, Grin2D‐KO and Grin2bfl/fl) mice of both sexes aged P19–35 were used in this study. Mice were given access to water and food at libitum. All efforts were made to minimize the animals' pain and suffering and to reduce the number of animals used in this study.
The authors understand the ethical principles under which The Journal of Physiology operates and the present work complies with the animal ethics checklist outlined in Grundy (2015).
In vivo postnatal viral injection
Stereotaxic viral injections were performed in P21 Grin2bfl/fl mice (gift from Prof H. Monyer, University of Heidelberg, Germany). The use of this mouse line allowed us to study the consequences of GluN2B removal in CA3 PCs in the adult brain without interfering with its role in development (Akashi et al. 2009). Mice were anaesthetized in a Plexiglas chamber filled with 4% gaseous isoflurane (Temsega, Pessac, France). Mice were then placed on a Kopf stereotaxic apparatus equipped with ear bars and a specialized nose cone for continuous inhalation of 1.5% isoflurane and kept warm (37°C) during all the surgery via a heating pad connected to a DC temperature controller provided with a feedback system (FHC, Bowdoin, ME, USA). Before surgery, the mouse reflexes were tested to ensure that they were in the surgical plane of anaesthesia, eyes were protected from the light by a lubricant and the analgesic buprenorphine (30 μg ml−1) was injected subcutaneously.
The skull was completely exposed by incising with a scalpel through the skin along the medial line and then removing the connective tissue. The head was adjusted so that the height of the skull surface at bregma and lambda was the same (±0.04). Stereotaxic coordinates of CA3 PC layers were calculated from bregma using an atlas (lateral ±2.0 mm; antero‐posterior −2 mm; dorso‐ventral (Z) −1.8–2.0 mm). The injections sites were precisely located with a binocular microscope.
We used a rAAV2.9 (titre 1.3311) virus expressing Cre recombinase, in order to induce the recombination of the double floxed gene, together with green fluorescent protein (GFP) allowing us to visualize the infected neuron. Injections were performed via a 10 μl nano‐fil syringe from World Precision Instrument (Stevenage, UK) using a 34 gauge metal needle and the volume was controlled with an injection pump. A volume of 350 nl per hemisphere at 50 nl min−1 was injected. For thorough diffusion of the injected volume, the needle was left inside the brain for an additional 5 min after the end of the injections. Then the needle was carefully retracted, the skull was cleaned with sterile saline and the skin sutured and cleaned with betadine. After surgery, mice received intraperitoneal injection of saline solution in order to be rehydrated and returned to the home cage; they were used for electrophysiological recording 14–20 days later (Kaspar et al. 2002; Gray et al. 2011).
Slice preparation
Parasagittal hippocampal slices (310–330 μm thick) were obtained from P19–21 mice of both sexes for GluN2A and GluN2D KO mice and ∼35 days‐old for Grin2bfl/fl mice infected with the rAAV2.9‐Cre‐GFP at P19–21. Mice were anaesthetized with intraperitoneal ketamine–xylazine injection (75 and 10 mg per kg, respectively). After checking their reflexes, decapitation was performed. The brain was quickly removed and immersed in ice‐cold cutting solution (the composition is given below). The hemispheres were separated by a sagittal cut in the corpus callosum and each one was glued and mounted on the vibrating blade microtome (Leica VT 1200S, Leica Microsystems, Wetzlar, Germany). Brain slices were made with a razor blade with an angle of ∼17 deg at 0.05 mm s−1.
All the preparation was performed under oxygenation (95% O2 and 5% CO2), in an ice‐cold cutting solution. For P19–21 mice, the cutting solution was composed of (in mm): 220 sucrose, 2 KCl, 1.3 NaH2PO4, 12 MgCl2, 0.2 CaCl2, 10 glucose, 26 NaHCO3. For ∼P35 the following solution was used (in mm): 87 NaCl, 25 NaHCO3, 25 glucose, 75 sucrose, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, and 7 MgCl2. Right after slicing, slices were transferred into a bath at 35°C during 30 min for recovery, and were then placed at room temperature for the rest of the day.
Electrophysiological recordings
Slices were transferred to a recording chamber in which they were continuously superfused with an oxygenated extracellular medium (95% O2 and 5% CO2) containing (mm): 125 NaCl, 2.5 KCl, 2.3 CaCl2, 1.3 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, 20 glucose, pH 7.4. Bicuculline (10 μm) and CGP 55845 (3 μm) were present in all experiments in order to block GABAA and GABAB receptors, respectively. Recordings were performed at room temperature in voltage‐clamp or current‐clamp mode from CA3 PCs using borosilicate pipettes (Harvard apparatus 1.5 mm OD × 0.86 mm ID) pulled with a micropipette puller (P97, Sutter Instruments, Novato, CA, USA), which had resistances between 3 and 4.5 MΩ. The patch electrodes were filled with a solution containing (mm): 140 CsCH3SO3, 2 MgCl2, 4 NaCl, 5 phospho‐creatine, 2 Na2ATP, 0.2 EGTA, 10 Hepes, 0.33 GTP, pH 7.3 adjusted with CsOH.
Evoked EPSCs were recorded in CA3 PCs in the whole‐cell patch clamp mode by stimulating with a glass pipette either positioned in the stratum radiatum to stimulate A/C fibres or in the dentate gyrus to stimulate MFs. At the end of each experiment (2S,1'S,2'S)‐2‐(Carboxycyclopropyl)glycine (LCCG‐I) (10 μm), an mGluR2 agonist, was used to verify the MF origin of the EPSCs.
MF–CA3 EPSCs were evoked by minimal intensity stimulation according to the protocol described previously (Marchal & Mulle, 2004; Sachidhanandam et al. 2009). A glass microelectrode was placed in the hilus of the dentate gyrus to stimulate MFs. While recording from a CA3 PC, the stimulating electrode was moved to a position where a clear‐cut EPSC with fixed latency was evoked. Stimulation intensity was adjusted just above the sharp threshold for activation of a synaptic response. Using such low minimal stimulations, no prominent polysynaptic activation was observed. MF stimulation was assessed by its large dynamic range of short‐term facilitation (40 ms paired pulse ratio > 3 or 1 Hz/0.1 Hz ratio > 3). When pooling together all the cells recorded during this study, the average value obtained for the amplitude of unitary EPSC‐AMPA was of 85 ± 11 pA (n = 45).
Pharmacologically isolated (20 μm 2,3‐dihydroxy‐6‐nitro‐7‐sulfamoyl‐benzo[f]quinoxaline‐2,3‐dione (NBQX)) NMDAR‐mediated EPSCs were recorded at +40 mV and at room temperature. To record NMDAR/AMPAR ratios, AMPAR‐EPSCs were first recorded in the voltage‐clamp mode at −70 mV in the presence of bicuculline (10 μm) and CGP 55845 (3 μm).
NMDAR‐mediated EPSC decay time was calculated using a double exponential fit of recorded EPSCs and tau weighted was calculated as follows:
In experiments with exogenous zinc applications, tricine (10 mm) was used to buffer zinc, and free zinc concentrations (3 nm to 1 μm range) were calculated using the following equation: [Zn]free = [Zn]added/200 (Paoletti et al. 1997; Fayyazuddin et al. 2000; Nozaki et al. 2011).
For experiments in which LTP‐NMDAR at MF–CA3 synapses was evaluated, we have used trains consisting of 6 or 10 bursts of 6 stimuli at 50 Hz repeated every 140 ms (Rebola et al. 2008).
Drugs
All drugs were obtained from Tocris Cookson (Bristol, UK), Sigma‐Aldrich (St Louis, MO, USA) or Ascent Scientific (Bristol, UK). The protein kinase C active fragment (PKM) used in the experiment presented in Fig. 3 G was purchased from Sigma‐Aldrich. PKMζ used in experiments presented in Fig. 4 was purchased from ProQinase (Freiburg, Germany).
Figure 3. LTP of NMDARs recruits GluN2B‐containing NMDARs at MF synapses.
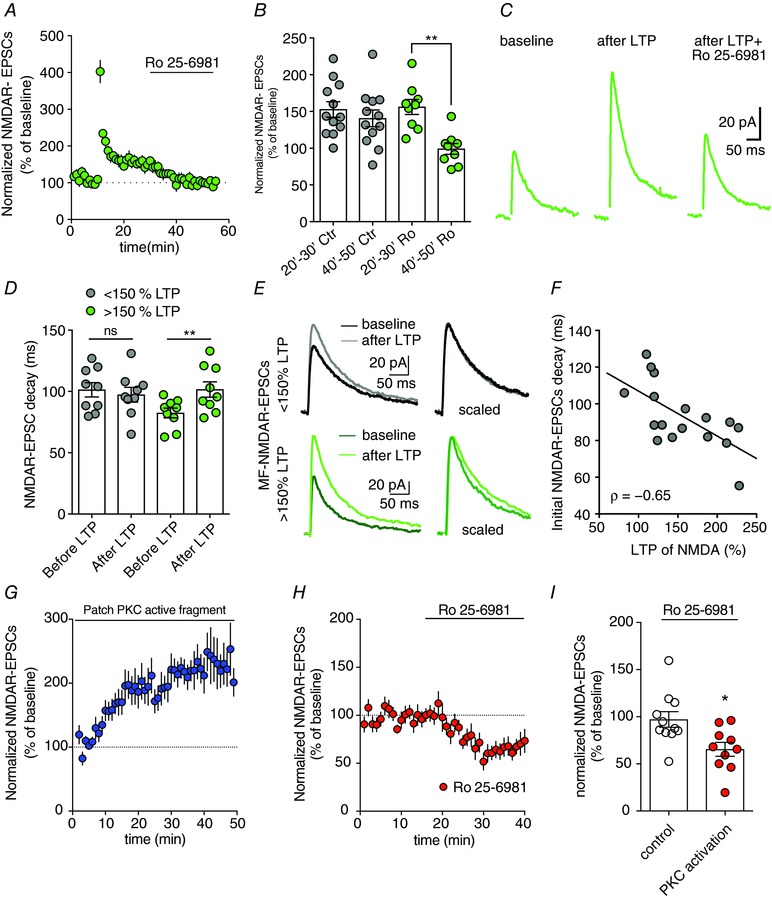
A–C, average time course, summary plot and representative traces reflecting the increased inhibition of NMDAR‐EPSCs by Ro 25–6981 at MF–CA3 synapses after induction of LTP‐NMDAR. Ro‐256981 was applied at 30 min and the average Ro‐256981 effect was quantified between 40 and 50 min. In control cells Ro‐256981 was not applied (** P = 0.039, Wilcoxon's matched‐pairs signed rank test LTP 20–30 Ro25‐6981 and LTP 40–50 Ro25‐6981). D and E, MF–CA3 synapses displaying the larger LTP‐NMDAR displayed NMDAR‐EPSCs with slower decay kinetics after LTP induction (* P = 0.02, Wilcoxon matched‐pairs signed rank test). F, negative correlation between NMDAR‐EPSC decay kinetics and LTP‐NMDAR amplitude (Spearman r = −0.6, P = 0.0074). MF–CA3 synapses showing slower initial NMDAR‐EPSC kinetics display reduced LTP levels possibility reflecting already GluN2B‐containing potentiated MF–CA3 synapses. G‐perfusion of the PKC active fragment via patch pipette induces a progressive potentiation of synaptic NMDARs at MF–CA3 synapses. H and I, average time course and summary plot illustrating the inhibition of NMDAR‐EPSCs at MF–CA3 synapses by Ro25‐6981 after previous PKC activation (see panel G) (* P = 0.01 Mann–Whitney test). [Color figure can be viewed at wileyonlinelibrary.com]
Figure 4. LTP‐NMDAR at MF–CA3 synapses requires the presence of GluN2B subunits.
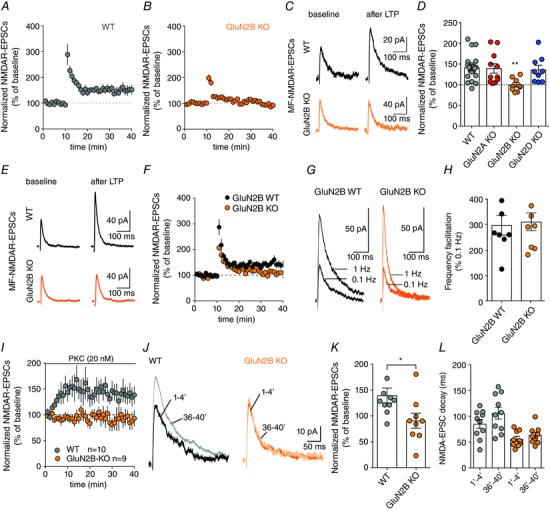
A, time course of LTP‐NMDAR observed at MF–CA3 synapses after application of a brief train of synaptic stimulation in wild‐type mice. B and C, average time course and representative traces illustrating that LTP‐NMDAR at MF–CA3 synapses is impaired when GluN2B subunits are removed. D, summary plot of the amplitude of LTP‐NMDAR in WT and after the selective removal of GluN2A, GluN2B or GluN2D subunits (** P = 0.002, Kruskal–Wallis test followed by Dunn's multiple comparisons test). E and F, sample traces and time course illustrating that LTP‐NMDAR at MF–CA3 synapses cannot be induced in GluN2B KO mice even when the induction protocol was increased to 10 bursts of stimulations (* P < 0.05 KO vs. WT, Mann–Whitney test). G and H, sample traces and summary bar graphs illustrating that short term plasticity such as frequency facilitation is not altered by the removal of GluN2B subunit. In all panels, values are presented as mean ± SEM of n experiments. I, time course of NMDAR‐EPSC peak amplitude observed at MF–CA3 synapses after infusion of PKMζ into CA3 PCs through the intracellular solution in WT and GluN2B KO neurons. J, representative traces illustrating that potentiation effect of PKMζ NMDAR‐EPSCs is absent when GluN2B subunits are removed. K, summary plot of PKMζ effect in NMDAR‐EPSC peak amplitude in WT and GluN2B KO neurons (* P = 0.03, Mann–Whitney test). L, bar graph resuming the effect of PKMζ infusion in NMDAR‐EPSC decay kinetics in WT and GluN2B KO CA3 PCs. [Color figure can be viewed at wileyonlinelibrary.com]
Data acquisition and analysis
Images were acquired via RS Image 1.9.2 using an Olympus BX50WI microscope linked to a Photometrics Cool Snap HQ camera. Recordings were made via Patchmaster 2.71 using an EPC10 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany), filtered at 0.5–1 kHz, digitized at 5 kHz, and stored on a personal computer. Analysis was performed using Neuromatic (www.neuromatic.thinkrandom.com) written within the Igor Pro 6.0 environment (WaveMetrics, Lake Oswego, OR, USA). Values are presented as the mean ± SEM. Either a paired (Wilcoxon matched‐pairs signed rank test) or an unpaired non parameteric test (Mann‐Whitney and Kruskal‐Wallis) was used to assess statistical differences between values using Prism 6 (GraphPad Software, San Diego, CA, USA). P < 0.05 was considered to be statistically significant. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those generally employed in the field. No data point (outliers) was removed from the collected datasets. n represents a single measurement from a single cell. We considered each different cell as a biological replication.
Results
Distinct NMDAR subunit composition at MF–CA3 and A/C–CA3 synapses
To examine the subunit composition of synaptic NMDARs in CA3 PCs, we recorded NMDAR‐EPSCs (+40 mV and 1.3 mm Mg2+) while stimulating either mossy or A/C fibres (MF–CA3 or A/C–CA3, respectively) and tested the effect of different NMDAR subunit‐selective antagonists. High‐affinity zinc inhibition of NMDARs is conferred by the presence of GluN2A subunits (Paoletti & Neyton, 2007). Application of 300 nm zinc significantly reduced NMDAR‐EPSC amplitude (48.1 ± 3.9% of control, n = 16, P < 0.0001, Wilcoxon's signed rank test to theoretical value of 100%) at MF–CA3 synapses, indicating the presence of GluN2A subunits (Fig. 1 A, B and E). Importantly, recording NMDAR‐EPSCs for 30 min, corresponding to the period of drug application, did not induce any significant change of the amplitude of synaptic NMDA currents (90.1 ± 7.4% of control, n = 13, P = 0.204, Wilcoxon's signed rank test to theoretical value of 100%). NMDAR‐EPSCs evoked by stimulating A/C fibres (A/C NMDAR‐EPSCs) were significantly less inhibited by 300 nm zinc (inhibition to 66.3 ± 4.3% of control, n = 10, P = 0.014 as compared to MF–CA3 inhibition; Fig. 1 A, B and E). Zinc binding to GluN2A increases the proton affinity of NMDARs thus reducing peak currents and simultaneously increasing glutamate affinity resulting in the slowing down of NMDAR‐currents. In agreement, NMDAR‐EPSCs recorded in the presence of 300 nm zinc decayed with a slower time course at MF–CA3 (control: 75.7 ± 4.6 ms, n = 15; 300 nm zinc: 96.0 ± 7.3 ms, n = 15; P = 0.008) and A/C synapses (control: 83.8 ± 6.6 ms, n = 10; 300 nm zinc: 119.3 ± 15.6 ms, n = 10; P = 0.020). Ro25‐6981 (1 μm), a selective inhibitor of GluN2B‐containing NMDARs (Fischer et al. 1997) significantly inhibited NMDAR‐EPSCs at A/C synapses (51.3 ± 3.9% of control, n = 15, P < 0.0001, Wilcoxon's signed rank test to theoretical value of 100%; Fig. 1 C–E), with a concurrent decrease in NMDAR‐EPSCs decay kinetics (control: 102.4 ± 8.6 ms, n = 13; Ro25‐6981: 79.1 ± 6.7 ms, n = 13; P = 0.0007). In contrast, Ro25‐6981 had a much more modest effect on NMDAR‐EPSCs recorded at MF–CA3 synapses (amplitude: 83.1 ± 5.4% of control, n = 23, P = 0.0002, Wilcoxon's signed rank test to theoretical value of 100%; decay kinetics: control: 84.5 ± 6.0 ms, n = 22; Ro25‐6981: 67.1 ± 3.6 ms, n = 22; P < 0.0001; Fig. 1 C–E). Similar results were obtained for zinc (300 nm) and Ro25‐6981 (1 μm) when recordings of NMDAR‐EPSCs were performed at −50 mV and with low extracellular Mg2+ (0.3 mm) (data not shown). These results indicate that the subunit composition of synaptic NMDARs differs at A/C and MF synapses of CA3 PCs, with MF–CA3 synapses presenting low levels of GluN2B‐containing NMDARs.
Figure 1. Differential GluN2‐subunit composition of NMDARs in CA3 pyramidal cell synapses.
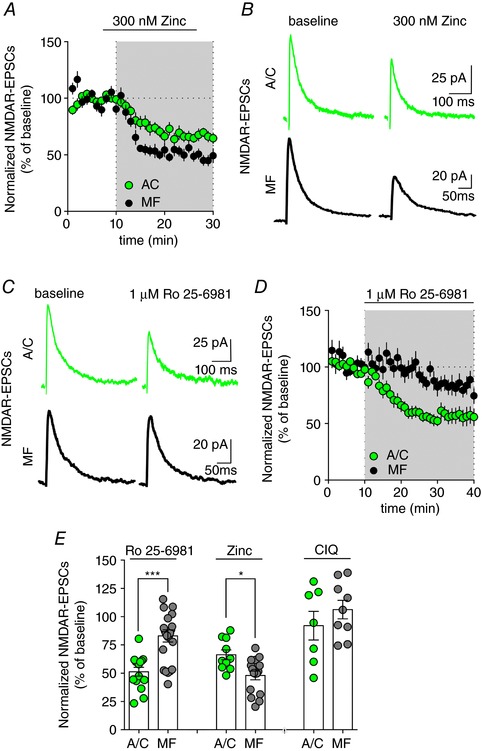
A and B, time course and representative traces illustrating the differential inhibition of NMDAR‐EPSCs by 300 nm zinc at A/C and MF synapses. C and D, the selective GluN2B‐containing NMDAR antagonist Ro 25–6891 (1 μm) induces a significantly higher inhibition of NMDAR‐EPSCs at A/C versus MF synapses. E, bar graph summarizing the impact of the subunit‐selective GluN2B‐ (1 μm Ro 25–6981) and GluN2A‐ (300 nm zinc) containing NMDAR antagonists as well as of the selective GluN2C/D potentiator (CIQ) on A/C and MF NMDAR‐EPSCs. Ro 25–6981, *** P = 0.0002; zinc, * P = 0.014; CIQ, P = 0.44; Mann–Whitney test MF vs. respective A/C group. [Color figure can be viewed at wileyonlinelibrary.com]
Previous studies have suggested that MF–CA3 synapses express GluN2D/C subunits (Thompson et al. 2002; Berg et al. 2013). We have tested this hypothesis using the recently developed allosteric potentiator selective for GluN2C‐ and GluN2D‐containing NMDARs, (3‐chlorophenyl)(6,7‐dimethoxy‐1‐((4‐methoxyphenoxy)methyl)‐3,4‐dihydroisoquinolin‐2(1H)‐yl)methanone (CIQ; 5 μm; Mullasseril et al. 2010). CIQ did not modify NMDAR‐EPSCs recorded at both MF–CA3 synapses and A/C synapses (MF–CA3: 106.3 ± 8.2%, n = 9; A/C–CA3: 92.0 ± 12.6%, n = 7; P = 0.58 A/C, P = 0.57 MF, Wilcoxon's signed rank test to theoretical value of 100%; Fig. 1 E).
The presence of synaptic tri‐heteromeric NMDARs, which contain more than one type of GluN2 subunit, significantly modifies the effectiveness of selective antagonists making it difficult to estimate levels and functional impact of the different GluN2 subunits using only pharmacological approaches (Hansen et al. 2014; Stroebel et al. 2014). To overcome this limitation, we have estimated the participation of the different GluN2 subunits to synaptic NMDARs at MF–CA3 synapses using GluN2‐specific KO mice. In line with the pharmacological data, removal of GluN2A subunits resulted in a marked slowing down of MF–CA3 NMDAR‐EPSCs (GluN2A WT: 70.7 ± 16.5 ms, n = 9; GluN2A KO: 239.0 ± 42.5 ms, n = 10; P = 0.0053; Fig. 2 A, C and D) confirming the presence of GluN2A subunits at MF synapses. Because the remaining synaptic current displays slow decay kinetics, this suggests that in GluN2A KO mice, GluN2A subunits are most likely replaced by di‐heteromeric GluN1–GluN2B NMDARs at MF–CA3 synapses. In agreement, Ro25‐6981 (1 μm) application strongly inhibited MF–CA3 NMDAR‐EPSCs (35.6 ± 8.9% of control, n = 6, P = 0.031, Wilcoxon's signed rank test to theoretical value of 100%) in GluN2A KO mice. Knock‐out of the GluN2A subunit resulted in a significantly smaller NMDAR/AMPAR ratio as compared with WT littermates (GluN2A WT: 0.52 ± 0.06, n = 16; GluN2A KO: 0.30 ± 0.03, n = 23; P = 0.0023) (Fig. 2 A and B). The decrease in NMDAR/AMPAR ratio may result either from a decrease in density/open probability of synaptic NMDARs or from an increase in the AMPAR component. To differentiate between the two possibilities we compared the absolute amplitudes of MF‐EPSCs evoked using a minimal stimulation protocol. In agreement with a selective reduction in the NMDAR component, the amplitude of AMPAR‐EPSCs was similar in GluN2A WT and GluN2A KO mice (GluN2A WT: 126.2 ± 23.0 pA, n = 9; GluN2A KO: 129.9 ± 18.3 pA, n = 12; P = 0.9) while the amplitude of NMDAR‐EPSCs was significantly smaller in GluN2A KO mice (GluN2A WT: 40.4 ± 5.8 pA, n = 9; GluN2A KO: 24.6 ± 3.7 pA, n = 12; P = 0.9).
Figure 2. Selective GluN2‐subunit KO mice reveal the differential participation of GluN2A and GluN2B in MF‐NMDA EPSCs.
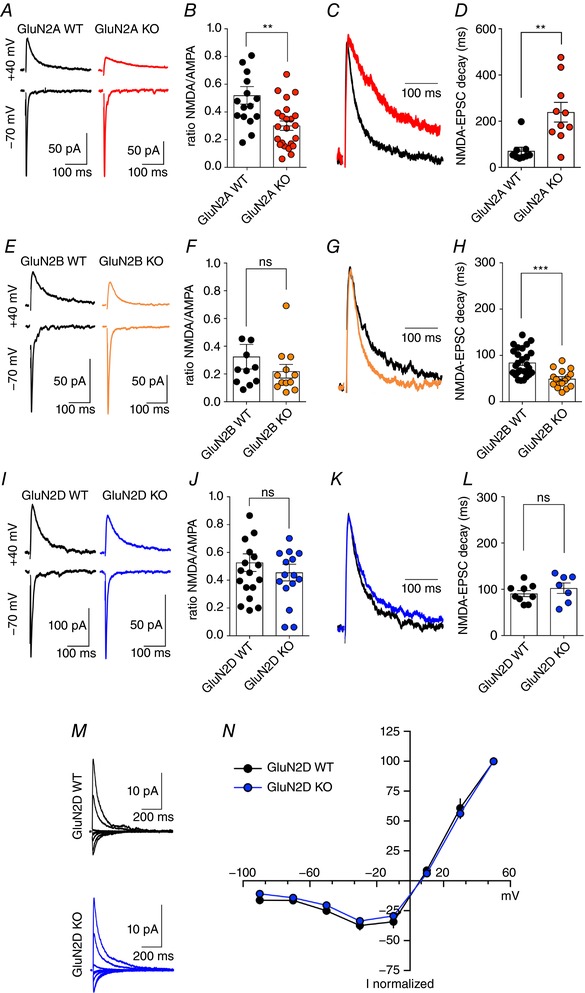
A and B, sample traces and summary plot illustrating the reduced NMDAR/AMPAR ratio in GluN2A KO mice at MF synapses, suggesting decreased levels of synaptic NMDARs (** P = 0.0023, Mann–Whitney test). C, peak scaled MF NMDAR‐EPSCs obtained from WT and GluN2A KO mice. D, summary graph illustrating the significant slowing down of MF NMDAR‐EPSCs when removing GluN2A subunits (** P = 0.0053, Mann–Whitney test). E and F, sample traces and summary plot showing similar NMDAR/AMPAR ratios in WT and GluN2B KO mice at MF synapses (P = 0.19, Mann–Whitney test). G, peak scaled MF NMDAR‐EPSCs obtained from WT and GluN2B KO mice. H, summary graph illustrating that despite similar NMDAR/AMPAR ratios, GluN2B subunits contribute significantly to NMDAR‐EPSC kinetics at MF synapses (*** P < 0.0001, Mann–Whitney test). I–L, genetic removal of GluN2D subunits does not significantly alter NMDAR/AMPAR ratio nor MF NMDAR‐EPSCs kinetics. Decay values represent the weighted decay calculated after using a double exponential fit of recorded NMDAR‐EPSCs (see Methods). M and N, sample traces and summary graph illustrating that the I–V curve recorded in GluN2D KO mice does not differ from that recorded in control littermate WT mice (GluN2D WT n = 6; GluN2D KO n = 8). [Color figure can be viewed at wileyonlinelibrary.com]
In contrast to GluN2A KO mice, homozygous genetic knockout of GluN2B subunits results in mouse death at postnatal day 0 (P0) (Kutsuwada et al. 1996). To overcome this limitation we infected floxed‐GluN2B transgenic mice at P21 with an AAV virus encoding Cre recombinase. This resulted in the removal of the GluN2B subunit in a small number of CA3 PCs. MF–CA3 NMDAR‐EPSCs were insensitive to inhibition by Ro25‐6981 in these cells (GluN2B WT: 87.8 ± 6.2% of control, n = 12; GluN2B KO: 103.3 ± 3.9% of control, n = 10; P = 0.03). Surprisingly, and in contrast with the modest effect of Ro25‐6981 in WT mice, MF–CA3 NMDAR‐EPSCs were markedly faster in GluN2B KO neurons (GluN2B WT: 89.35 ± 8.0 ms, n = 13; GluN2B KO: 47.5 ± 4.4 ms, n = 18; P < 0.0001; GluN2B WT + Cre: 84.5 ± 7.7 ms, n = 16; Fig. 2 E, G and H). These results suggest that, although of relatively low abundance, GluN2B subunits significantly impact on the functional properties of NMDARs at MF–CA3 synapses. Not surprisingly, removal of GluN2B subunits also accelerated NMDAR‐EPSCs recorded at A/C synapses (GluN2B WT: 110.0 ± 12.8 ms, n = 8; GluN2B KO: 72.4 ± 7.1 ms, n = 10; P = 0.03). Removing GluN2B subunits did not result in a significant modification of the NMDAR/AMPAR ratio (GluN2B WT: 0.37 ± 0.09, n = 12; GluN2B KO: 0.22 ± 0.05, n = 12; P = 0.19; Fig. 2 E and F). We completed our genetic approach by testing the consequences of GluN2D ablation on MF–CA3 NMDAR‐EPSCs. We have found that in GluN2D KO mice neither the NMDAR/AMPAR ratio (GluN2D WT: 0.53 ± 0.06, n = 19; GluN2D KO: 0.46 ± 0.06, n = 16; P = 0.62), nor the decay kinetics of MF–CA3 NMDAR‐EPSCs (GluN2D WT: 90.6 ± 6.6 ms, n = 9; GluN2D KO: 102.3 ± 11.3 ms, n = 7; P = 0.29) were altered as compared to slices from WT littermates (Fig. 2 I and L). In addition, the I–V curves for MF–CA3 NMDAR‐EPSCs did not reveal any difference in magnesium sensitivity between WT and GluN2D KO mice (Fig. 2 M and N). Overall, both our genetic and our pharmacological data strongly suggest the absence of GluN2D subunits at MF–CA3 synapses. Surprisingly, however, inhibition of NMDAR‐EPSCs by Ro25‐6981 was significantly increased in GluN2D KO mice (GluN2D WT: 88.0 ± 12.8% of control, n = 8; GluN2D KO: 47.4 ± 4.4% of control, n = 7; P = 0.0019), indicating a higher content of GluN2B at MF–CA3 synapses in these mice. A possible interpretation of these results is that the removal of GluN2D during development results in a delayed maturation of the GluN2 composition of synaptic NMDARs at MF–CA3 synapses (Yamasaki et al. 2014).
Activity‐induced changes in NMDAR subunit composition at MF–CA3 synapses
During development, the subunit composition of synaptic NMDARs switches from a predominance of GluN2B‐containing to GluN2A‐containing receptors (Paoletti et al. 2013). At neonatal synapses, this developmental change in subunit composition of synaptic NMDARs is under rapid, bidirectional control depending on the pattern of synaptic activity (Bellone & Nicoll, 2007). MF–CA3 synapses were recently shown to express LTP selective for NMDAR‐EPSCs (LTP‐NMDAR) (Kwon & Castillo, 2008; Rebola et al. 2008). We thus examined whether LTP‐NMDAR was accompanied by a change in the subunit composition and properties of NMDARs. LTP‐NMDAR, induced with six bursts of five MF stimuli at 50 Hz, lasted for at least 50 min (Rebola et al. 2008) (Fig. 3 A). Twenty minutes after LTP induction, we applied subunit‐selective antagonists to test for a potential switch in subunit composition. Strikingly, Ro25‐6981 (1 μm), which inhibits MF–CA3 NMDAR‐EPSCs by only 10–15% in control conditions (Fig. 1), led to a pronounced inhibition of NMDAR‐EPSCs after LTP induction (63.7 ± 2.6% of control, n = 9, P = 0.0072, Mann–Whitney test between LTP 40–50 Ctr and LTP 40–50 Ro25‐6981) (Fig. 3 A–C) followed by a significant acceleration of MF–CA3 NMDAR‐EPSCs (decay kinetics: control: after LTP 92.2 ± 5.0 ms, n = 8; after LTP + Ro25‐6981: 72.8 ± 3.2 ms, n = 8; P = 0.02). The recruitment of GluN2B subunits is expected to slow down NMDAR‐EPSCs whereas no significant change of NMDAR‐EPSC kinetics was observed after LTP when all data were pooled (control: 91.9 ± 4.1 ms; after LTP: 99.5 ± 4.3 ms, n = 18; P = 0.15). However, a detailed analysis revealed that MF–CA3 synapses presenting robust potentiation (>50%) displayed NMDAR‐EPSCs with a significantly slower decay (Fig. 3 D and E) in agreement with the recruitment of GluN2B subunits. Interestingly, the level of LTP was negatively correlated with the initial decay of MF–CA3 NMDAR‐EPSCs (Fig. 3 F). These results suggest that at MF–CA3 synapses, the initial synaptic content in GluN2B subunits is quite variable and that LTP‐NMDAR preferentially occurs at synapses with lower densities of GluN2B subunits. LTP‐NMDAR not only increases the density of NMDARs, but also induces an enrichment in GluN2B subunits with a consequent slowing down of NMDAR‐EPSC decay kinetics.
At MF–CA3 synapses, PKC activation is necessary for LTP‐NMDAR and its pharmacological activation is sufficient to potentiate NMDAR‐EPSCs (Kwon & Castillo, 2008). We thus tested if PKC activation was sufficient to induce changes in the subunit composition of NMDARs. We included PKM (the active catalytic subunit of PKC) (0.3 μm) in the patch pipette solution and we continuously recorded NMDAR‐EPSCs from CA3 PCs. Infusion of PKM in CA3 PCs induced a clear potentiation of NMDAR‐EPSCs, which stabilized after 30–40 min (Fig. 3 G). The sensitivity of MF–CA3 NMDAR‐EPSCs to Ro25‐6981 was tested after reaching a stable baseline. In these conditions Ro25‐6981 induced a significant inhibition of MF–CA3 NMDAR‐EPSCs (34.6 ± 7.3%, n = 10, P = 0.01, Fig. 3 H and I). These results indicate that LTP‐NMDAR results in the enrichment of GluN2B containing NMDARs at MF–CA3 synapses, most likely through a PKC‐dependent mechanism.
Role of NMDAR subunits in LTP‐NMDAR at MF–CA3 synapses
We then tested the dependence of LTP‐NMDAR at MF–CA3 synapses on the different NMDAR subunits. For this, we examined whether LTP‐NMDAR could still be induced in GluN2A, GluN2B and GluN2D KO mice. While short bursts of MF stimulation (6 bursts of 5 MF stimuli at 50 Hz) induced LTP‐NMDAR in GluN2A and GluN2D KO mice, no LTP‐NMDAR was observed after ablation of GluN2B (GluN2A KO: 139.4 ± 11.3%, n = 11; GluN2B KO: 99.5 ± 5.1%, n = 9; GluN2D KO: 136.3 ± 10.6%, n = 9; Kruskal–Wallis test, Dunn's test for multiple comparison, P = 0.002; Fig. 4 A–D). A more robust stimulation protocol (10 bursts of 5 MF stimuli at 50 Hz) failed to induce LTP‐NMDAR in GluN2B KO, suggesting that the lack of plasticity was not simply due to an initial smaller NMDAR‐EPSC peak amplitude (GluN2B WT: 145.5 ± 12.1%, n = 8; GluN2B KO: 107.4 ± 7.3%, n = 7; * P < 0.05 vs. WT, Mann–Whitney test) (Fig. 4 E and F). This conclusion is further supported by the presence of LTP in the GluN2A KO mice where, despite a pronounced decrease in NMDAR/AMPAR ratio as compared to WT mice (Fig. 2) LTP‐NMDAR is still present (Fig. 4 D). Importantly, presynaptic function evaluated by frequency facilitation (percentage increase in EPSC peak amplitude while varying stimulation frequency between 0.1 and 1 Hz) was not altered by removing GluN2B subunits from CA3 PCs (GluN2B WT: 291.3 ± 38.1%, n = 8; GluN2B KO: 304.9 ± 34.2%, n = 9; Fig. 4 G and H). Overall we can conclude that GluN2B subunits are essential for the induction and/or the expression of LTP‐NMDAR in MF–CA3 synapses.
In an attempt to get more insight into the role of GluN2B subunits in LTP‐NMDAR, we directly tested the effect of PKC activation in cells where GluN2B subunits have been selectively removed. PKC activation has been previously shown to occlude LTP of NMDARs suggesting that this kinase is indeed necessary and sufficient to induce potentiation of NMDARs at MF–CA3 synapses (Kwon & Castillo, 2008). Infusion of PKMζ (20 nm; see Tsokas et al. 2016) into CA3 PCs induced a robust potentiation of NMDAR‐EPSC peak amplitude in wild‐type but not in GluN2B KO neurons (WT: 139.6 ± 14.4%, n = 10; GluN2B KO: 90.8 ± 14.5%, n = 9; P = 0.027, Mann–Whitney test; Fig. 4 I–L). Interestingly, the increase in peak amplitude was followed by a tendency for a slowing down of NMDA‐EPSPC decay that was not statistically significant (control (1–4 min): 85.2 ± 8.6 ms, n = 10; after PKC (36–40 min): 106.0 ± 11.44 ms, n = 10; P = 0.064, Mann–Whitney test; Fig. 4 L). Overall this suggests that GluN2B subunits are essential for LTP‐NMDARs at MF synapses by controlling the potentiation of synaptic NMDARs after PKC activation and not by acting at the induction phase.
Discussion
The heterogeneity of expression levels, properties and subunit composition of synaptic NMDARs likely has a considerable impact on the function and plasticity of neuronal circuits. Here we have combined NMDAR subunit‐specific pharmacological tools and transgenic mice to show that the subunit composition of synaptic NMDARs at distinct inputs to single CA3 PCs differs. Importantly, we observe that LTP‐NMDAR requires synaptic GluN2B and that the subunit composition and properties of synaptic NMDARs at MF–CA3 synapses are subject to activity‐dependent plasticity.
Both the GluN2A and GluN2B subunits of NMDARs are expressed in adult brain neurons (Monyer et al. 1994). NMDARs with distinct subunit compositions can segregate to specific synaptic inputs in a given layer V neuron of the neocortex (Kumar & Huguenard, 2003). Whether this is a general feature of NMDARs to meet the needs of neuronal computation is not thoroughly documented. In CA3 PCs, GluN2B appears more abundant at synapses formed by contralateral inputs from other CA3 PCs than those formed by ipsilateral inputs (Kawakami et al. 2003). Functional NMDARs are less abundant at MF–CA3 synapses as compared to A/C synapses in CA3 (Rebola et al. 2011). Our experiments indicate in addition a different subunit composition of synaptic NMDARs at MF–CA3 and A/C–CA3 synapses, with a markedly higher proportion of GluN2B in the latter. The reduced presence of GluN2B subunits at MF–CA3 synapse in basal conditions is in agreement with scarce immunocytochemical labelling of GluN2B subunits in the stratum lucidum (Fritschy et al. 1998; Watanabe et al. 1998) as well as with modest inhibition of the MF–CA3 NMDAR‐EPSCs peak amplitudes by ifenprodil (Wyeth et al. 2014).
Because Ro25‐6981 binds tri‐heteromeric GluN1/GluN2A/GluN2B NMDARs with high affinity but has a reduced maximum inhibition (Paoletti et al. 2013), the limited inhibition by Ro25‐6981 observed may indicate that most GluN2B subunits are included in tri‐heteromeric NMDARs. Decay kinetics are dominated by GluN2A subunits in tri‐heteromeric GluN1–GluN2A–GluN2B NMDARs (Hansen et al. 2014; Stroebel et al. 2014). At MF–CA3 synapses, the decay kinetics of NMDAR‐EPSCs in WT mice is closer to the decay kinetics obtained for GluN1/GluN2A homodimers (in GluN2B KO cells) suggesting that GluN2B subunits are most likely part of tri‐heteromeric GluN1–GluN2A–GluN2B NMDAR assemblies. Interestingly, the kinetics of NMDAR‐EPSCs and their sensitivity to Ro25‐6981 appear to vary within MF–CA3 synapses, suggesting a heterogeneous content of GluN2B. Nonetheless, the acceleration of NMDAR‐EPSCs observed at MF synapses after Ro25‐6981 application suggests that not all NMDARs are tri‐heteromeric. Overall, our data suggest a predominance of tri‐heteromeric NMDARs at MF–CA3 synapses with a small participation of di‐heteromeric NMDARs.
The genetic removal of GluN2B subunits from CA3 PCs accelerates NMDAR‐EPSCs at both MF‐ and A/C–CA3 synapses. Interestingly, in CA3 PCs from GluN2B KO mice, in which a homogeneous distribution of GluN1–GluN2A di‐heteromeric receptors is expected, the decay of NMDAR‐EPSCs was still significantly slower at A/C synapses. The most straightforward explanation for this observation is that distal NMDAR‐EPSCs originating in A/C synapses are subject to higher dendritic filtering as compared to the more proximal MF inputs. This conclusion is further supported by previous work where an artificial EPSC waveform with kinetics (rise 3 ms and decay 30 ms) similar to the expected for GluN1–GluN2A di‐heteromeric receptors induced a pronounced voltage escape during voltage‐clamp experiments along the distal dendrites of pyramidal neurons (Williams & Mitchell, 2008).
LTP‐NMDAR can be readily induced in GluN2A KO mice (and in GluN2D KO mice), but not in GluN2B KO mice, indicating a crucial role for GluN2B in either the induction or the expression of LTP. The reduced NMDAR‐EPSC amplitude and the faster decay combine to decrease the overall charge of NMDAR currents at MF–CA3 synapses in GluN2B KO CA3 PCs. However, increasing the strength of the LTP induction protocol did not mitigate the loss of LTP suggesting that it is not a simple consequence of the decrease in the charge of synaptic NMDAR events. Interestingly, we observed an increased sensitivity to the GluN2B antagonist and a slower decay of NMDAR‐EPSCs following LTP‐NMDAR, strongly supporting the notion that LTP is accompanied by an insertion of GluN2B‐containing NMDARs. In agreement, bypassing the induction phase of LTP‐NMDAR by activating directly the downstream pathway through infusion of active PKC in CA3 neurons resulted in a potentiation of NMDARs in WT but not in GluN2B KO neurons. Hence, GluN2B subunits play a critical role in the expression of LTP‐NMDAR.
The activity‐dependent insertion of GluN2B‐containing NMDARs at MF–CA3 synapses following LTP is rather unexpected. At Schaffer collateral–CA1 pyramidal cell synapses, in neonates and juvenile mice, LTP‐NMDAR is rather paralleled by an increased content of GluN2A (Bellone & Nicoll, 2007; Peng et al. 2009). In mature midbrain dopamine neurons LTP‐NMDAR does not lead to any apparent change in subunit composition (Harnett et al. 2009). Synaptic incorporation of GluN2B‐containing NMDARs is thought to be constitutive and not to require synaptic activity in contrast to GluN2A‐containing NMDARs, which is activity‐dependent (Storey et al. 2011). An increased number of functional NMDARs containing GluN2B was observed at single synapses in dissociated neuronal cultures following prolonged synaptic inactivity, whereas activity led to the insertion of NMDARs with GluN2A subunits (Lee et al. 2010).
PKC activation plays a fundamental role in LTP‐NMDAR at MF–CA3 synapses (Kwon & Castillo, 2008). Interestingly, activation of PKC with intracellular phorbol 12‐myristate 13‐acetate (PMA) application into hippocampal CA1 PCs induces an increase in peak amplitude and slowing down of decay kinetics of NMDA currents, similar to the effect observed during LTP‐NMDAR at MF–CA3 synapses. In CA1 neurons, the PMA effect on NMDAR‐EPSCs is dependent on CaMKII activation and is totally blocked by a Tat‐GluN2B peptide that disrupts GluN2B–CaMKII interaction, whereas a similar peptide targeting GluN2A subunits only reduces the PMA effect by half (Yan et al. 2011). According to such a model, it is tempting to speculate that the preferential interaction between GluN2B subunits and CaMKII could explain the dependence of LTP‐NMDAR at MF–CA3 synapses on the GluN2B subunit.
The dependence of LTP‐NMDAR on the GluN2B subunit seems at odds with our observations that MF–CA3 synapses with faster initial NMDAR‐EPSCs present higher LTP levels. This apparent discrepancy can be related to the fact that in control conditions some MF–CA3 synapses may be already in a potentiated state. MF–CA3 synapses with faster decay kinetics may represent a more homogeneous synaptic population that are in an unpotentiated state. Interestingly, the heterogeneity of NMDARs across MF–CA3 synapses, according to decay kinetics and Ro25‐6981 sensitivity, may reflect these different states of potentiation.
It is well established that synaptic NMDAR composition influences the induction of Hebbian synaptic plasticity of AMPAR‐EPSCs (Barria & Malinow, 2005). Although the exact contribution of GluN2A and GluN2B to synaptic plasticity has been debated, it appears that when the ratio of GluN2A to GluN2B increases, stronger stimulation is required to induce LTP (Yashiro & Philpot, 2008). We speculate that insertion of GluN2B‐containing NMDARs following LTP‐NMDAR at MF–CA3 synapses may facilitate at least in part the metaplastic switch making these synapses competent for expression of Hebbian plasticity of AMPAR‐EPSCs (Rebola et al. 2011).
In addition, following LTP‐NMDAR, MF–CA3 synapses display slower NMDAR‐EPSCs, hence favouring synaptic integration and spike transmission (Rebola et al. 2011). These changes in biophysical properties of NMDAR‐EPSCs may also play a role in heterosynaptic plasticity (Hunt et al. 2013). It remains to be determined if the converse LTD of NMDARs (Hunt et al. 2013) or possible depotentiation mechanisms lead to a reversal of NMDARs to a lower GluN2B to GluN2A ratio.
In conclusion, NMDARs at mature synapses are heterogeneous across inputs in a given neuron, as well as within a specific input such as at MF–CA3 synapses. Even at the adult stage, NMDARs can undergo plasticity and changes in their subunit composition in an activity‐dependent manner, further increasing the heterogeneity between synapses. This heterogeneity may play an important role both in synaptic integration and neuronal computation as well as in the plasticity repertoire which a given neuron can express, hence extending the storage capacity of the brain (Bartol et al. 2015).
Additional information
Competing interests
None declared.
Author contributions
Experiments were designed and planned by MC, BNS, NR and CM. Electrophysiology experiments and analysis were carried out by MC, BNS and NR. AG performed some control immunohistochemistry experiments. MC, BNS, NR and CM wrote the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was supported by the Centre National de la Recherche Scientifique, the Conseil Régional d'Aquitaine, the European Commission (EIF Fellowship awarded to M.C.), and the Fondation pour la Recherche Médicale (to B.N.S.).
Acknowledgements
We are grateful to Hannah Monyer and to Peter Seeburg for making the transgenic mice available to us. We would like to acknowledge Philippe Jean for help in some sterotaxic injections. We thank Elisabeth Normand, Audrey Lacquemant and Amandine Gautier for taking care of the mice and Andrew Penn for fruitful discussions.
Edited by: Jaideep Bains & Katalin Toth
M. Carta and B. N. Srikumar are equal first authors.
N. Rebola and C. Mulle contributed equally.
References
- Akashi K, Kakizaki T, Kamiya H, Fukaya M, Yamasaki M, Abe M, Natsume R, Watanabe M & Sakimura K (2009). NMDA receptor GluN2B (GluR 2/NR2B) subunit is crucial for channel function, postsynaptic macromolecular organization, and actin cytoskeleton at hippocampal CA3 synapses. J Neurosci 29, 10869–10882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Hallaq RA, Conrads TP, Veenstra TD & Wenthold RJ (2007). NMDA di‐heteromeric receptor populations and associated proteins in rat hippocampus. J Neurosci 27, 8334–8343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A & Malinow R (2005). NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron 48, 289–301. [DOI] [PubMed] [Google Scholar]
- Bartol TM, Bromer C, Kinney J, Chirillo MA, Bourne JN, Harris KM & Sejnowski TJ (2015). Nanoconnectomic upper bound on the variability of synaptic plasticity. Elife 4, e10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone C & Nicoll RA (2007). Rapid bidirectional switching of synaptic NMDA receptors. Neuron 55, 779–785. [DOI] [PubMed] [Google Scholar]
- Berg LK, Larsson M, Morland C & Gundersen V (2013). Pre‐ and postsynaptic localization of NMDA receptor subunits at hippocampal mossy fibre synapses. Neuroscience 230, 139–150. [DOI] [PubMed] [Google Scholar]
- Fayyazuddin A, Villarroel A, Le Goff A, Lerma J & Neyton J (2000). Four residues of the extracellular N‐Terminal domain of the NR2A subunit control high‐affinity Zn2+ binding to NMDA receptors. Neuron 25, 683–694. [DOI] [PubMed] [Google Scholar]
- Fischer G, Mutel V, Trube G, Malherbe P, Kew JN, Mohacsi E, Heitz MP & Kemp JA (1997). Ro 25–6981, a highly potent and selective blocker of N‐methyl‐D‐aspartate receptors containing the NR2B subunit. Characterization in vitro . J Pharmacol Exp Ther 283, 1285–1292. [PubMed] [Google Scholar]
- Fritschy JM, Weinmann O, Wenzel A & Benke D (1998). Synapse‐specific localization of NMDA and GABAA receptor subunits revealed by antigen‐retrieval immunohistochemistry. J Comp Neurol 390, 194–210. [PubMed] [Google Scholar]
- Gray JA, Shi Y, Usui H, During MJ, Sakimura K & Nicoll RA (2011). Distinct modes of AMPA receptor suppression at developing synapses by GluN2A and GluN2B: Single‐cell NMDA receptor subunit deletion in vivo. Neuron 71, 1085–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Yuan H & Traynelis SF (2014). Distinct functional and pharmacological properties of triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron 81, 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harnett MT, Bernier BE, Ahn K‐C & Morikawa H (2009). Burst‐timing‐dependent plasticity of NMDA receptor‐mediated transmission in midbrain dopamine neurons. Neuron 62, 826–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt DL & Castillo PE (2012). Synaptic plasticity of NMDA receptors: mechanisms and functional implications. Curr Opin Neurobiol 22, 496–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt DL, Puente N, Grandes P & Castillo PE (2013). Bidirectional NMDA receptor plasticity controls CA3 output and heterosynaptic metaplasticity. Nat Neurosci 16, 1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspar BK, Vissel B, Bengoechea T, Crone S, Randolph‐Moore L, Muller R, Brandon EP, Schaffer D, Verma IM, Lee K‐F, Heinemann SF & Gage FH (2002). Adeno‐associated virus effectively mediates conditional gene modification in the brain. Proc Natl Acad Sci USA 99, 2320–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami R, Shinohara Y, Kato Y, Sugiyama H, Shigemoto R & Ito I (2003). Asymmetrical allocation of NMDA receptor ε2 subunits in hippocampal circuitry. Science 300, 990–994. [DOI] [PubMed] [Google Scholar]
- Kumar SS & Huguenard JR (2003). Pathway‐specific differences in subunit composition of synaptic NMDA receptors on pyramidal neurons in neocortex. J Neurosci 23, 10074–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsuwada T, Sakimura K, Manabe T, Takayama C, Katakura N, Kushiya E, Natsume R, Watanabe M, Inoue Y, Yagi T, Aizawa S, Arakawa M, Takahashi T, Nakamura Y, Mori H & Mishina M (1996). Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor ε2 subunit mutant mice. Neuron 16, 333–344. [DOI] [PubMed] [Google Scholar]
- Kwon H‐B & Castillo PE (2008). Long‐term potentiation selectively expressed by NMDA receptors at hippocampal mossy fiber synapses. Neuron 57, 108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M‐C, Yasuda R & Ehlers MD (2010). Metaplasticity at single glutamatergic synapses. Neuron 66, 859–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchal C & Mulle C (2004). Postnatal maturation of mossy fibre excitatory transmission in mouse CA3 pyramidal cells: a potential role for kainate receptors. J Physiol 561, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta JA, Ashby MC, Sanz‐Clemente A, Roche KW & Isaac JTR (2011). mGluR5 and NMDA receptors drive the experience‐ and activity‐dependent NMDA receptor NR2B to NR2A subunit switch. Neuron 70, 339–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B & Seeburg PH (1994). Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540. [DOI] [PubMed] [Google Scholar]
- Mullasseril P, Hansen KB, Vance KM, Ogden KK, Yuan H, Kurtkaya NL, Santangelo R, Orr AG, Le P, Vellano KM, Liotta DC & Traynelis SF (2010). A subunit‐selective potentiator of NR2C‐ and NR2D‐containing NMDA receptors. Nat Commun 1, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki C, Vergnano AM, Filliol D, Ouagazzal A‐M, Le Goff A, Carvalho S, Reiss D, Gaveriaux‐Ruff C, Neyton J, Paoletti P & Kieffer BL (2011). Zinc alleviates pain through high‐affinity binding to the NMDA receptor NR2A subunit. Nat Neurosci 14, 1017–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P (2011). Molecular basis of NMDA receptor functional diversity. Eur J Neurosci 33, 1351–1365. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Ascher P & Neyton J (1997). High‐affinity zinc inhibition of NMDA NR1‐NR2A receptors. J Neurosci 17, 5711–5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Bellone C & Zhou Q (2013). NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14, 383–400. [DOI] [PubMed] [Google Scholar]
- Paoletti P & Neyton J (2007). NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol 7, 39–47. [DOI] [PubMed] [Google Scholar]
- Peng Y, Zhao J, Gu Q‐H, Chen R‐Q, Xu Z, Yan J‐Z, Wang S‐H, Liu S‐Y, Chen Z & Lu W (2009). Distinct trafficking and expression mechanisms underlie LTP and LTD of NMDA receptor‐mediated synaptic responses. Hippocampus 20, 646–658. [DOI] [PubMed] [Google Scholar]
- Rauner C & Köhr G (2011). Triheteromeric NR1/NR2A/NR2B receptors constitute the major N‐methyl‐D‐aspartate receptor population in adult hippocampal synapses. J Biol Chem 286, 7558–7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebola N, Carta M, Lanore F, Blanchet C & Mulle C (2011). NMDA receptor‐dependent metaplasticity at hippocampal mossy fiber synapses. Nat Neurosci 14, 691–693. [DOI] [PubMed] [Google Scholar]
- Rebola N, Luján R, Cunha RA & Mulle C (2008). Adenosine A2A receptors are essential for long‐term potentiation of NMDAR‐EPSCs at hippocampal mossy fiber synapses. Neuron 57, 121–134. [DOI] [PubMed] [Google Scholar]
- Rebola N, Srikumar BN & Mulle C (2010). Activity‐dependent synaptic plasticity of NMDA receptors. J Physiol 588, 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenas‐Ruano A, Chávez AE, Cossio MJ, Castillo PE & Zukin RS (2012). REST‐dependent epigenetic remodeling promotes the developmental switch in synaptic NMDA receptors. Nat Neurosci 15, 1382–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachidhanandam S, Blanchet C, Jeantet Y, Cho YH & Mulle C (2009). Kainate receptors act as conditional amplifiers of spike transmission at hippocampal mossy fiber synapses. J Neurosci 29, 5000–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN & Jan LY (1994). Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368, 144–147. [DOI] [PubMed] [Google Scholar]
- Sobczyk A & Svoboda K (2007). Activity‐dependent plasticity of the NMDA‐receptor fractional Ca2+ current. Neuron 53, 17–24. [DOI] [PubMed] [Google Scholar]
- Storey GP, Opitz‐Araya X & Barria A (2011). Molecular determinants controlling NMDA receptor synaptic incorporation. J Neurosci 31, 6311–6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroebel D, Carvalho S, Grand T, Zhu S & Paoletti P (2014). Controlling NMDA receptor subunit composition using ectopic retention signals. J Neurosci 34, 16630–16636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CL, Drewery DL, Atkins HD, Stephenson FA & Chazot PL (2002). Immunohistochemical localization of N‐methyl‐D‐aspartate receptor subunits in the adult murine hippocampal formation: evidence for a unique role of the NR2D subunit. Brain Res Mol Brain Res 102, 55–61. [DOI] [PubMed] [Google Scholar]
- Tovar KR, McGinley MJ & Westbrook GL (2013). Triheteromeric NMDA receptors at hippocampal synapses. J Neurosci 33, 9150–9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokas P, Hsieh C, Yao Y, Lesburguères E, Wallace EJC, Tcherepanov A, Jothianandan D, Hartley BR, Pan L, Rivard B, Farese RV, Sajan MP, Bergold PJ, Hernández AI, Cottrell JE, Shouval HZ, Fenton AA & Sacktor TC (2016). Compensation for PKMζ in long‐term potentiation and spatial long‐term memory in mutant mice. Elife 5, 12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zundert B, Yoshii A & Constantine‐Paton M (2004). Receptor compartmentalization and trafficking at glutamate synapses: a developmental proposal. Trends Neurosci 27, 428–437. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Fukaya M, Sakimura K, Manabe T, Mishina M & Inoue Y (1998). Selective scarcity of NMDA receptor channel subunits in the stratum lucidum (mossy fibre‐recipient layer) of the mouse hippocampal CA3 subfield. Eur J Neurosci 10, 478–487. [DOI] [PubMed] [Google Scholar]
- Williams K, Russell SL, Shen YM & Molinoff PB (1993). Developmental switch in the expression of NMDA receptors occurs in vivo and in vitro. Neuron 10, 267–278. [DOI] [PubMed] [Google Scholar]
- Williams SR & Mitchell SJ (2008). Direct measurement of somatic voltage clamp errors in central neurons. Nat Neurosci 11, 790–798. [DOI] [PubMed] [Google Scholar]
- Wyeth MS, Pelkey KA, Petralia RS, Salter MW, McInnes RR & McBain CJ (2014). Neto auxiliary protein interactions regulate kainate and NMDA receptor subunit localization at mossy fiber‐CA3 pyramidal cell synapses. J Neurosci 34, 622–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki M, Okada R, Takasaki C, Toki S, Fukaya M, Natsume R, Sakimura K, Mishina M, Shirakawa T & Watanabe M (2014). Opposing role of NMDA receptor GluN2B and GluN2D in somatosensory development and maturation. J Neurosci 34, 11534–11548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J‐Z, Xu Z, Ren S‐Q, Hu B, Yao W, Wang S‐H, Liu S‐Y & Lu W (2011). Protein kinase C promotes N‐methyl‐D‐aspartate (NMDA) receptor trafficking by indirectly triggering calcium/calmodulin‐dependent protein kinase II (CaMKII) autophosphorylation. J Biol Chem 286, 25187–25200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K & Philpot BD (2008). Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology 55, 1081–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]