

Abstract
Calcium (Ca2+) release from intracellular stores plays a key role in the regulation of skeletal muscle contraction. The type 1 ryanodine receptors (RyR1) is the major Ca2+ release channel on the sarcoplasmic reticulum (SR) of myocytes in skeletal muscle and is required for excitation-contraction (E–C) coupling. This article explores the role of RyR1 in the skeletal muscle physiology and pathophysiology.
Keywords: Calcium, Excitation-contraction coupling, Muscular dystrophy, RyR1, Skeletal muscle
Introduction
Ryanodine receptors (RyRs) are intracellular calcium (Ca2+) release channels located on the endo/sarcoplasmic reticulum (ER/SR) (Flucher et al. 1993), a heterogeneous intracellular compartment consisting of a network of tubules (Chen et al. 2013; Brochet et al. 2005) representing the major Ca2+ reservoir within the cell. There are three subtypes of RyRs in mammalian tissues: RyR1 and RyR2 are required for skeletal muscle and cardiac excitation-contraction coupling (E–C coupling), respectively (Marks et al. 1989; Otsu et al. 1990), and are also expressed in non-muscle tissues (Awad et al. 1997); RyR3, originally identified in the brain (Nakashima et al. 1997), is also widely expressed (Zhang et al. 2011).
RyR1 facilitates the rapid and coordinated release of Ca2+ from SR stores to activate skeletal muscle contraction. EC coupling is the process that converts electrical signals and rising Ca2+ levels into mechanical output (muscle contraction). RyRs are highly regulated for precise control and Ca2+ plays the key signaling role in activating the channel and amplifying the signal (Endo et al. 1970). In this process, depolarization of the plasma membrane activates L-type voltage-gated calcium channels (Cav), which signal RyRs located on the SR to gate open and release Ca2+ to activate muscle contraction (Rios and Brum 1987; Gordon et al. 2000; Tobacman 1996; des Georges et al. 2016). RyR is a 2.2 mega Dalton homotetramer, composed of four ~5000 residue protomers (Marks et al. 1989; Santulli and Marks 2015), making it the largest kfown ion channel (des Georges et al. 2016; Santulli and Marks 2015; Zalk et al. 2015). The narrow transmembrane core and larger cytoplasmic shell result in a mushroom shaped structure (des Georges et al. 2016; Zalk et al. 2015; Hwang et al. 2012). The large shell interacts with other receptors and forms much of the regulatory mechanism for the channel, allowing a range of stimuli to exert precise control over opening (Marks et al. 1989; des Georges et al. 2016; Santulli and Marks 2015; Zalk et al. 2015; Brillantes et al. 1994; Marx et al. 1998, 2000; Lehnart et al. 2005; Huang et al. 2006; Bellinger et al. 2009; Kushnir et al. 2010; Shan et al. 2010; Andersson et al. 2011; Lanner et al. 2010). The core of RyR houses the approximately 90 Å long pore responsible for passage of Ca2+ from the ER/SR to the cytoplasm (des Georges et al. 2016; Yan et al. 2015). This cation channel is actually poorly selective for Ca2+ (~7-fold selective for Ca2+ vs K+) and displays an exceptionally large single channel conductance (Santulli and Marks 2015).
We recently solved the high-resolution structure of RyR1 using cryogenic electron microscopy (cryo-EM) (des Georges et al. 2016; Zalk et al. 2015), confirming that it adopts a fourfold symmetric mushroom-like superstructure, with the large ‘cap’ (about 80% of the mass) located in the cytosol and the ‘stalk’ embedded in the ER/SR membrane, with six transmembrane helices (S1–S6) per protomer surrounding the central pore (des Georges et al. 2016). Each protomer is built around an extended scaffold of alpha-solenoid repeats which include an aminoterminal, a bridging, and a core solenoid (des Georges et al. 2016; Zalk et al. 2015). At the extreme outer corners of the tetramer there are three SPRY domains and two pairs of RyR repeats, RY12 and RY34, the latter containing a regulatory protein kinase A (PKA) phosphorylation site (Marx et al. 2000). The RyR1 pore domain most closely resembles that of the voltage-gated sodium channel (NavAB) and presents a single cytosolic constriction in the ion conduction pathway, at the S6 bundle crossing (Zalk et al. 2015). Glycine residues in the pore-lining helices may operate as “hinges” to facilitate the orientation of the cytoplasmatic extension of S6 in order to modulate the aperture of the channel. In particular, Gly4934 is conserved in all RyR isoforms and in the IP3R.
RyR macromolecular complex
The ER/SR of most cell types contains two types of intracellular Ca2+ release channels: the ryanodine receptors (RyRs) and the inositol 1,4,5-trisphosphate receptors (IP3Rs) (Santulli and Marks 2015; Go et al. 1995; Yuan et al. 2016; Santulli 2017). There is ~40% homology between the RyR and lP3R in the putative transmembrane regions (Marks et al. 1989, 1990; Santulli 2017), a sequence similarity sufficient to indicate that these two channels evolved from a common ancestral cation release channel in unicellular species. The structural homology between RyR1 and IP3R1 is depicted in Fig. 1.
Fig. 1.
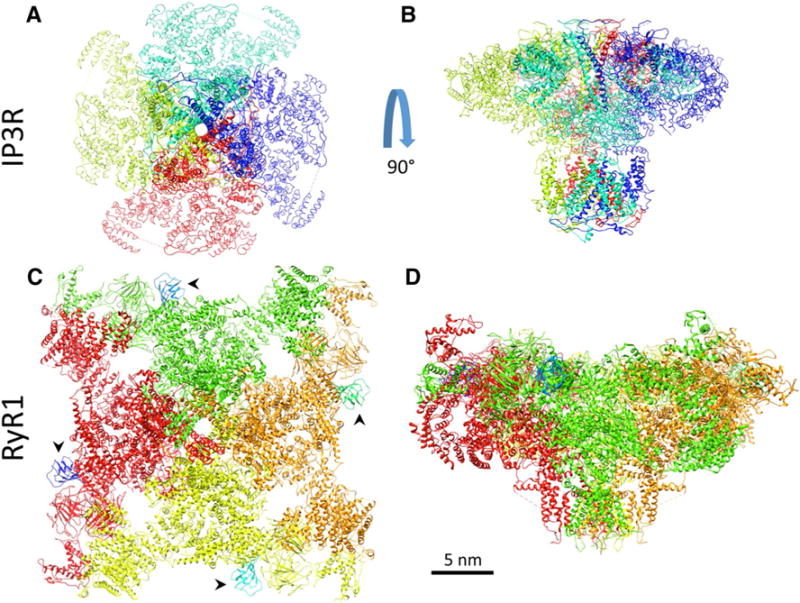
Structural homology between the intracellular Ca2+ release channels IP3R1 (top) and RyR1 (bottom). In a, c channels are viewed from the ER/SR lumen; in c, arrowheads indicate Calstabin
RyR was named based on its purification using the high affinity plant alkaloid ryanodine (Rogers et al. 1948), an agent known to profoundly alter intracellular Ca2+ handling (Fairhurst and Hasselbach 1970). Indeed, when bound to RyR at low concentrations ryanodine locks the channel in a half open state, thereby resulting in depletion of Ca2+ from the SR and subsequent interruption of E–C coupling. This explains the historical use of extracts from the Ryania plant family by natives of South and Central America as poison for blow darts: the release of SR Ca2+ via the locked open RyRs causes tetany, and at high concentrations ryanodine blocks the channel (Rogers et al. 1948). RyR is normally closed at low cytosolic [Ca2+] (~ 100–200 nM); at submicromolar cytosolic [Ca2+] Ca2+ binds to high-affinity binding sites on RyR increasing the open probability (Po) of the channel (Bezprozvanny et al. 1993). Channel activity is maximal at cytosolic [Ca2+] ~10 μM while elevating cytosolic [Ca2+] beyond this point leads to a reduction in Po (Bezprozvanny et al. 1993; Copello et al. 1997; Laver et al. 1995).
The large and complex structure of RyR contains function-modifying phosphorylation sites and protein-binding domains, providing an attractive target for disease intervention (des Georges et al. 2016; Santulli and Marks 2015; Zalk et al. 2015; Brillantes et al. 1994; Marx et al. 1998, 2000, 2001; Lehnart et al. 2005; Kushnir et al. 2010; Marks et al. 2002). RyRs are macromolecular signaling complexes, in which multiple proteins bind to a domain of the channel modulating its function (Marks et al. 1989, 2002). The Ca2+ stabilizing proteins calstabin1 (Calcium channel stabilizing binding protein, previously known as FKBP12) and calstabin2 (FKBP12.6) are peptidyl-propyl-cis–trans isomerases that associate via amphiphilic β-sheet structures with RyR1 and RyR2, respectively, such that one calstabin protein is bound to each RyR monomer (des Georges et al. 2016; Zalk et al. 2015; Jayaraman et al. 1992; Timerman et al. 1993; Xin et al. 1995; Yuan et al. 2014), in order to modulate the channel gating through protein-protein interactions (Brillantes et al. 1994) and prevent pathological intracellular Ca2+ leak that cause diseases (Huang et al. 2006). Calstabin1 and calstabin2 differ at only 18 positions out of 108 residues. We identified the calstabin-binding loop as part of the aminoterminal subdomain of the bridging solenoid (Zalk et al. 2015). Calstabin binding may rigidify the interface between such a subdomain with SPRY1–2, thereby stabilizing the connection with the cytosolic regulatory domains and eventually altering the relative orientation of these domains (Zalk et al. 2015). Highly conserved leucine-isoleucine zipper motifs in RyR2 form binding sites for adaptor proteins that mediate binding of other proteins (Marx et al. 2001; Marks et al. 2002), including kinases (e.g. PKA) (Shan et al. 2010) CaMKIIdelta (Kushnir et al. 2010) and phosphatases (e.g. PP1 and PP2A). Specifically, the adaptor protein mAKAP mediates the binding of PKA and phosphodiesterase PDE43, whereas PP1 and PP2A are targeted to RyR2 via spinophilin and PR130, respectively (Marx et al. 2000; Lehnart et al. 2005). All of the above mentioned proteins regulate the phosphorylation-dephosphorylation of RyR2 in Ser2808 (Shan et al. 2010) in response to stress (Andersson et al. 2011; Shan et al. 2010; Liu et al. 2012; Tester et al. 2007). Other channels are also regulated by stress signals including the voltage-gated Ca2+ channels (Maki et al. 1996). RyRs are also regulated by oxidation and nitrosylation (Shan et al. 2010; Andersson et al. 2011; Santulli 2017; Fauconnier et al. 2010). Other modulatory proteins complex directly and indirectly with RyR, including sorcin (Farrell et al. 2004), calmodulin (Meissner and Henderson 1987), homer (Feng et al. 2002), histidine-rich Ca2+ binding protein (Lee et al. 2001), triadin (Rossi et al. 2014), junctin (Zhang et al. 1997), and calsequestrin (Ohkura et al. 1998).
Intracellular Ca2+ leak
Ca2+ finely regulates innumerable events as muscle contraction, secretion, and gene transcription (Santulli and Marks 2015; Santulli 2017; Ringer 1883; Zetterstrom and Arnhold 1958; Jayaraman and Marks 2000). Cytosolic Ca2+ signals are produced by rapidly increasing the concentration of free Ca2+ ions (Blaustein 1993) by opening channels permeable to Ca2+ either in the surface cell membrane or in the membranes of intracellular organelles containing high Ca2+ concentrations. Amplification of external stimuli by triggering the release of intracellular Ca2+ stores represents a common signaling mechanism in the cell. The key role of RyRs in the rapid and voluminous release of Ca2+ from the SR during E–C coupling is well known. Importantly, RyRs are also crucially involved in maintaining Ca2+ homeostasis in the cell under resting conditions. Stress-induced remodeling of RyRs results in leaky channels and the inappropriate release of Ca2+ from the intracellular stores into the cytosol, contributing to the pathophysiology of diverse disorders including heart failure, cardiac arrhythmias, muscular dystrophy, diabetes, and cognitive dysfunction (Brillantes et al. 1994; Marx et al. 1998, 2000, 2001; Lehnart et al. 2005; Huang et al. 2006; Bellinger et al. 2008, 2009; Kushnir et al. 2010; Shan et al. 2010; Andersson et al. 2011, 2012; Marks et al. 2002; Liu et al. 2012; Tester et al. 2007; Fauconnier et al. 2010; Ward et al. 2003; Umanskaya et al. 2014; Matecki et al. 2016; Santulli et al. 2015a, b, 2017; Xie et al. 2013, 2015).
Skeletal muscle
E–C coupling is similar in skeletal and cardiac muscle but there are important differences (Santulli 2017). Briefly, whereas in the heart a depolarizing Na+ current activates Ca2+ influx via the L-type Ca2+ channel (LCC, Cav1.2), which in turn activates the RyR2 isoform via Ca2+-induced Ca2+ release (Fabiato and Fabiato 1975), the depolarization of skeletal myocytes involves a protein-protein interaction (Rios and Brum 1987) across the junctional cleft between the dihydropyridine receptor (Cav1.1) on specialized invaginations of the sarcolemma (transverse tubules) and RyR1 on the SR membrane (terminal cisternae), leading to Ca2+ release (Nelson et al. 2013). Both morphologic and electrophysiological data are consistent with the concept that four Cav1.1s interact with a single RyR1 tetramer (one Cav1.1 binding to each RyR1 subunit). However, Franzini-Armstrong and Kish determined that a cluster of four Cav1.1 overlie only every other RyR1 tetramer (Franzini-Armstrong and Kish 1995). Reconciling those findings, we have demonstrated coupled gating of RyR1 (Marx et al. 1998), which provides a mechanism by which RyR1 channels that are not associated with Cav1.1 can be regulated. RyRs were initially observed in skeletal muscle, visualized in electron micrographs as large electron-dense masses located along the face of the SR terminal cisternae, which is closely apposed to transverse tubule membranes to form a structure named triad junction (Santulli 2017; Block et al. 1988). Therefore, the RyRs were initially termed triad junctional foot proteins (Wagenknecht et al. 1989; Brandt et al. 1990). Noda and colleagues provided the in vivo evidence for a functional role of RyR1 in E–C coupling, engineering a mouse lacking exon 2 of RyR1 and demonstrating that such a mouse exhibits severe skeletal muscle abnormalities and dies perinatally due to respiratory failure (Takeshima et al. 1994). Subsequent ultrastructural studies of hind limb and diaphragm muscles demonstrated the absence of RyR1-Cav1.1 complexes (Takekura et al. 1995), which are essential for a proper E–C coupling in the skeletal muscle (Nakai et al. 1996).
RyR1 dysfunction has been described in both inherited and acquired muscle disorders (Bellinger et al. 2008; Andersson et al. 2012). Central core disease (CCD) and malignant hyperthermia (MH) represent the best examples of RyR1 channelopathies in the skeletal muscle.
Central core disease (CCD)
CCD is a congenital myopathy first described in 1956 (Magee and Shy 1956), characterized by the presence of tissue cores with reduced oxidative activity in type I myofibers, which results in progressive muscle weakness (Sewry et al. 2002). Common symptoms include hypotonia, delayed motor milestones, and skeletal abnormalities including congenital hip dislocation and scoliosis. Over 60 different RyR1 mutations have been linked to CCD, which presents during infancy as delayed motor development and hypotonia. CCD occurs in 1:100,000 live births, and comprises 16% of total congenital myopathies (Jungbluth 2007).
We now know that RyR1 mutations cause the disorder which should be reclassified as RyR1 myopathies. There are no established therapeutics for RyR1 myopathies (Witherspoon and Meilleur 2016). The phenotypic presentation is quite variable ranging from near normal to neonatal death.
The histopathological appearance of CCD is most closely linked to dominant RyR1 mutations (often missense) clustered (Fig. 2) in disease causing “hot spots” in RyR1 (Quane et al. 1993; Zhang et al. 1993; Lynch et al. 1999; Monnier et al. 2000; Scacheri et al. 2000), whereas RyR1 mutations (often truncating) causing recessive RyR1-related myopathies, including multi-minicore disease, centronuclear myopathies, and congenital fiber-type disproportion, are evenly distributed throughout the entire RYR1 coding sequence (Amburgey et al. 2013; Klein et al. 2012).
Fig. 2.
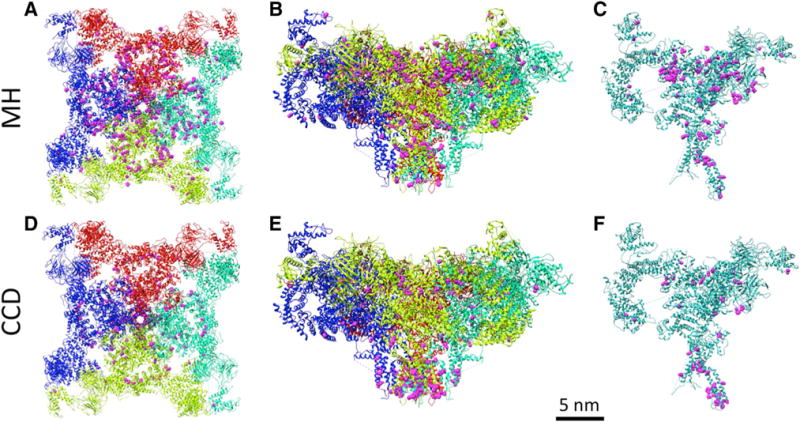
RyR1 with localization of the reported mutations for CCD (a–c) and MH (d–f). a and d are the full tetramer viewed top down from the cytosol, while b and e are rotated 90° to show the narrow transmembrane core and the larger cytoplasmic shell (an additional 45° rotation along the vertical axis was also performed). In c, f one protomer is depicted (following a 60° rotation), demonstrating the high proportion of interprotomer mutation sites (in pink). Interestingly, CCD mutations typically occur in the pore forming C-terminal domain, while MH mutations occur in central and N-terminal clusters
Malignant hyperthermia (MH)
MH is a pharmacogenetic disorder, inherited in an autosomal dominant fashion and causes inhaled anesthetic-induced deaths in otherwise healthy individuals (Censier et al. 1998). MH episodes are typically rapid and severe, reaching core body temperatures of 43 °C, leading to organ failure and death if not rapidly treated. Susceptibility can be determined in vitro by measuring the contractile response to caffeine or halothane in biopsied muscle fibers. Over 100 RyR1 mutations have been associated with MH, involving inappropriate activation of RyR1, which causes uncontrolled release of SR Ca2+ and muscle contractions. MH occurs at a rate of 1:50,000–100,000 adults and 1:15,000 children undergoing anesthesia; some studies have suggested a much more frequent rate of 1:5000 adults with MH susceptible mutations occurring at 1:3000 (Rosenberg et al. 2007; Monnier et al. 2002). The exact prevalence of MH susceptibility is difficult to determine since the syndrome only becomes apparent after exposure to triggering agents including volatile anesthetic agents such as halothane, isoflurane, sevoflurane, desflurane, enflurane and the neuromuscular blocking agent succinylcholine (Larach 2007). A related syndrome referred to as porcine stress syndrome is found in certain lines of domestic swine where stressed pigs undergo stress-induced hyperthermia (Nelson and Bee 1979). Alterations in 3H-ryanodine binding properties in porcine MH samples provided evidence linking RyR1 dysfunction to the disease (Mickelson et al. 1988), which was later confirmed by biophysical experiments (Fill et al. 1990).
Although dantrolene is an established therapeutic that quickly resolves MH episodes, mortality from this event remains at approximately 7% and a validated mechanism of action for dantrolene has yet to be reported (Paul-Pletzer et al. 2002; Zhao et al. 2001). This remains a concern for otherwise healthy individuals harboring these mutations (Fill et al. 1990). Mutations causing MH are autosomal dominant and typically seen (Fig. 2) in the central and N-terminal clusters. Another MH mutation hotspot is at the inter-protomer contacts between the N-terminal domains A and B, which are disrupted in channel opening (Kimlicka et al. 2013).
Notably, there is no clear division between MH and RyR1 myopathies and some RyR1 mutations have been linked to a combined MH and RyR1 myopathy phenotype (Zhou et al. 2007). Importantly, the mutated codons giving rise to MH and RyR1 myopathies tend to cluster in three specific regions of the RyR1 gene (Fig. 2) corresponding to the following domains in the amino acid sequence: regions 1 (C35–R614) and 2 (D2129–R2458) reside in the myoplasmic foot domain of the protein, whereas region 3 (I3916–G4942) is located in the transmembrane/luminal region of the highly conserved carboxy-terminal domain, important for allowing Ca2+ flux through the channel (Zalk et al. 2015). Mutations in RyR1 are also associated with other rare RyR1 related congenital myopathies including centronuclear myopathy, multi-minicore disease, Samaritan myopathy, heat/exercise induced exertional rhabdomyolysis, congenital fiber-type disproportion, late-onset axial myopathy, and atypical periodic paralyses (Bharucha-Goebel et al. 2013; Zvaritch et al. 2009; Ferreiro et al. 2002; Capacchione et al. 2010; Zhou et al. 2010; Inui et al. 1987; Takeshima et al. 1989; Loseth et al. 2013).
Intracellular Ca2+ leak and muscular dystrophy
We recently demonstrated that intracellular Ca2+ leak via RyR1 represents an essential feature of different forms of muscular dystrophy (MD), including Duchenne muscular dystrophy (Bellinger et al. 2009) and limb-girdle (or Erb’s) MD (Andersson et al. 2012). Specifically, RyR1 from a Duchenne muscular dystrophy murine model (mdx mouse) was excessively cysteine nitrosylated and the RyR1 complex was depleted of calstabin1, leading to increased spontaneous RyR1 openings and reduced specific muscle force (Bellinger et al. 2009). Similar findings were obtained when evaluating RyR1 in β-sarcoglycan-deficient mice, an established model of limb-girdle muscular dystrophy (Andersson et al. 2012). Thus, we demonstrated common mechanisms of stress-induced remodeling of RyR1, including post-translational modifications of the channel and dissociation of the stabilizing subunit calstabin1, in two major disorders that weaken the muscular system hampering locomotion and that remain without effective pharmacological treatment. We demonstrated in both cases that stabilizing the RyR1-calstabin1 association using a novel small molecule Rycal called S107 improved muscle function (Bellinger et al. 2009; Andersson et al. 2012), thereby providing an innovative therapeutic target and potential options for the treatment of muscular dystrophy.
In conditions of strenuous muscular stress or in a disease such as heart failure, both of which are characterized by chronic activation of the sympathetic nervous system and increased production of reactive oxygen and nitrogen species (Santulli 2014; Dalla Libera et al. 2005; Santulli and Iaccarino 2016), skeletal muscle function is impaired, possibly due to remodeling of RyR1 and impaired E–C coupling. We have shown in both an animal model as well as in exercising humans that chronic βAR stimulation and depletion of calstabin1 from RyR1 plays a role in contractile failure and muscle fatigue, defined as a decline in ability of a muscle to generate force during sustained exercise (Bellinger et al. 2008). Consistent with these observations, we have demonstrated that the remodeling of RyR1 plays a role in sarcopenia or age-dependent loss of muscle function (Andersson et al. 2011) and we were able to reduce RyR1 dysfunction and improve skeletal muscle function in aged mice (2 years old) by genetically enhancing mitochondrial antioxidant activity (Umanskaya et al. 2014).
Since skeletal muscle dysfunction, as observed in HF or muscular disorders, remains without effective treatment, drugs that restore RyR Ca2+ release function represent promising candidates. In this sense, Rycal treatment could be ideal in conditions that impair both cardiac and skeletal muscle function. Indeed, as well as muscular RyR1 undergoes post-translational modifications in HF (Ward et al. 2003), remodeling of the cardiac RyR2 has been also reported in murine models of Duchenne muscular dystrophy, triggering ventricular arrhythmias (Fauconnier et al. 2010).
RyR1 mutations: clinical significance and structural effects
Over 300 mutations have been mapped to RyRs that are implicated in human diseases and 200 more that do not result in modified channel function. The disease causing mutations are most often found in hotspots, including the N-terminal (~1–600), the central (~2000–2600) and the C-terminal (~4000–5000) regions. High-resolution cryo-EM reconstructions have recently become available making it possible to see how these hotspots are localized, some in the channel pore and others in the inter-protomer and interdomain interfaces (Tung et al. 2010). The phosphorylation domain is another hotspot for disease causing mutations (Yuchi et al. 2012).
Proper post-translational modifications and interaction with other proteins are also critical for RyR function. Several human disorders are linked to improper phosphorylation or oxidation of RyRs including ventilator-induced diaphragmatic dysfunction (VIDD) and Duchenne muscular dystrophy (DMD). VIDD involves diaphragm muscle weakness after extended mechanical ventilation and has been linked to oxidation of RyR (Matecki et al. 2016). RyR1 cysteine-nitrosylation has been shown to have a role in DMD (Bellinger et al. 2009). An age-dependent increase in cysteine-nitrosylation occurs with dystrophic changes in the muscle, depleting the RyR1 macromolecular complex of calstabin1 resulting in Ca2+ leak. This finding links muscle inflammation and Ca2+ leak in the pathogenesis of DMD (Tidball and Villalta 2009). Indeed, in inflamed tissues there is an increased expression of inducible nitric oxide synthase (iNOS), which binds to RyR1 leading to Ca2+ leak and eventually to the activation of Ca2+-dependent proteases (calpains) that promote muscle damage and wasting.
These alterations affect the function of RyRs, but the direct impact on the tetrameric assembly has yet to be shown in structural studies. Due to the critical requirement of the channel for proper muscle function, mutations that severely destabilize or significantly alter the channel structure most likely lead to non-viable embryos. These mutations most often lead to changes in the open probability of the channel, leading to Ca2+ leak. This hypersensitive activation can come from mutations on either the luminal or the cytosolic side of the receptor (Tong et al. 1997; Jiang et al. 2004). One potential explanation is that defects at the interface between the central and N-terminal regions would weaken the interactions stabilizing the receptor in the closed state, leading to increased susceptibility to stimuli (Tateishi et al. 2009; Suetomi et al. 2011). Albeit many disease-associated RyR1 mutations do increase the open probability of the channel, this is far from certain for all RyR1 mutations, in particular with regards to recessive RyR1-related myopathies associated with reduction of the RyR1 protein. Therefore, compounds enhancing the closed probability of the channel would have limited application in conditions where the RyR1 mutations result in reduced rather than enhanced Ca2+ conductance, or where the precise functional consequences of the specific RyR1 mutations are not known.
Adjacent RyRs are known to signal cooperatively as paracrystalline arrays in checkerboard patterns, allowing for simultaneous opening of multiple channels (coupled gating) in response to a stimulus (Marx et al. 1998; Cabra et al. 2016). This provides a mechanism by which RyR channels can effect the rapid and coordinated SR Ca2+ release (via mechanically triggering neighboring channels) that is required for EC coupling. Thus, RyRs act as both signal amplifiers and integrators by triggering neighboring channels both physically and chemically with Ca2+ (Endo et al. 1970; Fabiato 1983).
Acknowledgments
The studies described in this review were supported by NIH grants (R01AR060037 and R01HL061503) and the Fondation Leducq to A.R.M. G.S. is supported by the NIH (K99DK107895). ARM is a consultant and member of the board of ARMGO Pharma that is targeting RyR channels for therapeutic purposes.
References
- Amburgey K, et al. Genotype-phenotype correlations in recessive RYR1-related myopathies. Orphanet J Rare Dis. 2013;8:117. doi: 10.1186/1750-1172-8-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DC, et al. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011;14(2):196–207. doi: 10.1016/j.cmet.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DC, et al. Leaky ryanodine receptors in beta-sarcoglycan deficient mice: a potential common defect in muscular dystrophy. Skelet Muscle. 2012;2(1):9. doi: 10.1186/2044-5040-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad SS, et al. Differential expression of ryanodine receptor RyR2 mRNA in the non-pregnant and pregnant human myometrium. Biochem J. 1997;322(Pt 3):777–783. doi: 10.1042/bj3220777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger AM, Mongillo M, Marks AR. Stressed out: the skeletal muscle ryanodine receptor as a target of stress. J Clin Invest. 2008a;118(2):445–453. doi: 10.1172/JCI34006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger AM, et al. Remodeling of ryanodine receptor complex causes “leaky” channels: a molecular mechanism for decreased exercise capacity. Proc Natl Acad Sci USA. 2008b;105(6):2198–2202. doi: 10.1073/pnas.0711074105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger AM, et al. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med. 2009;15(3):325–330. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny IB, et al. Activation of the calcium release channel (ryanodine receptor) by heparin and other polyanions is calcium dependent. Mol Biol Cell. 1993;4(3):347–352. doi: 10.1091/mbc.4.3.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharucha-Goebel DX, et al. Severe congenital RYR1-associated myopathy: the expanding clinicopathologic and genetic spectrum. Neurology. 2013;80(17):1584–1589. doi: 10.1212/WNL.0b013e3182900380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein MP. Physiological effects of endogenous ouabain: control of intracellular Ca2+ stores and cell responsiveness. Am J Physiol. 1993;264(6 Pt 1):C1367–C1387. doi: 10.1152/ajpcell.1993.264.6.C1367. [DOI] [PubMed] [Google Scholar]
- Block BA, et al. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J Cell Biol. 1988;107(6 Pt 2):2587–2600. doi: 10.1083/jcb.107.6.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt NR, et al. Molecular interactions of the junctional foot protein and dihydropyridine receptor in skeletal muscle triads. J Membr Biol. 1990;113(3):237–251. doi: 10.1007/BF01870075. [DOI] [PubMed] [Google Scholar]
- Brillantes AB, et al. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77(4):513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- Brochet DX, et al. Ca2+ blinks: rapid nanoscopic store calcium signaling. Proc Natl Acad Sci USA. 2005;102(8):3099–3104. doi: 10.1073/pnas.0500059102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabra V, Murayama T, Samso M. Ultrastructural analysis of self-associated RyR2s. Biophys J. 2016;110(12):2651–2662. doi: 10.1016/j.bpj.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capacchione JF, et al. Exertional rhabdomyolysis and malignant hyperthermia in a patient with ryanodine receptor type 1 gene, L-type calcium channel alpha-1 subunit gene, and calsequestrin-1 gene polymorphisms. Anesthesiology. 2010;112(1):239–244. doi: 10.1097/ALN.0b013e3181c29504. [DOI] [PubMed] [Google Scholar]
- Censier K, et al. Intracellular calcium homeostasis in human primary muscle cells from malignant hyperthermia-susceptible and normal individuals. Effect Of overexpression of recombinant wild-type and Arg163Cys mutated ryanodine receptors. J Clin Invest. 1998;101(6):1233–1242. doi: 10.1172/JCI993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Novick P, Ferro-Novick S. ER structure and function. Curr Opin Cell Biol. 2013;25(4):428–433. doi: 10.1016/j.ceb.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copello JA, et al. Heterogeneity of Ca2+ gating of skeletal muscle and cardiac ryanodine receptors. Biophys J. 1997;73(1):141–156. doi: 10.1016/S0006-3495(97)78055-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalla Libera L, et al. Skeletal muscle myofibrillar protein oxidation in heart failure and the protective effect of Carvedilol. J Mol Cell Cardiol. 2005;38(5):803–807. doi: 10.1016/j.yjmcc.2005.02.023. [DOI] [PubMed] [Google Scholar]
- des Georges A, et al. Structural basis for gating and activation of RyR1. Cell. 2016;167(1):145–157 e17. doi: 10.1016/j.cell.2016.08.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo M, Tanaka M, Ogawa Y. Calcium induced release of calcium from the sarcoplasmic reticulum of skinned skeletal muscle fibres. Nature. 1970;228(5266):34–36. doi: 10.1038/228034a0. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol. 1983;245(1):C1–C14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J Physiol. 1975;249(3):469–495. doi: 10.1113/jphysiol.1975.sp011026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairhurst AS, Hasselbach W. Calcium efflux from a heavy sarcotubular fraction. Effects of ryanodine, caffeine and magnesium. Eur J Biochem. 1970;13(3):504–509. doi: 10.1111/j.1432-1033.1970.tb00953.x. [DOI] [PubMed] [Google Scholar]
- Farrell EF, et al. Regulation of cardiac excitation-contraction coupling by sorcin, a novel modulator of ryanodine receptors. Biol Res. 2004;37(4):609–612. doi: 10.4067/s0716-97602004000400015. [DOI] [PubMed] [Google Scholar]
- Fauconnier J, et al. Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 2010;107(4):1559–1564. doi: 10.1073/pnas.0908540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, et al. Homer regulates gain of ryanodine receptor type 1 channel complex. J Biol Chem. 2002;277(47):44722–44730. doi: 10.1074/jbc.M207675200. [DOI] [PubMed] [Google Scholar]
- Ferreiro A, et al. A recessive form of central core disease, transiently presenting as multi-minicore disease, is associated with a homozygous mutation in the ryanodine receptor type 1 gene. Ann Neurol. 2002;51(6):750–759. doi: 10.1002/ana.10231. [DOI] [PubMed] [Google Scholar]
- Fill M, et al. Abnormal ryanodine receptor channels in malignant hyperthermia. Biophys J. 1990a;57(3):471–475. doi: 10.1016/S0006-3495(90)82563-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fill M, et al. Abnormal ryanodine receptor channels in malignant hyperthermia. Biophys J. 1990b;57(3):471–475. doi: 10.1016/S0006-3495(90)82563-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flucher BE, et al. Triad formation: organization and function of the sarcoplasmic reticulum calcium release channel and triadin in normal and dysgenic muscle in vitro. J Cell Biol. 1993;123(5):1161–1174. doi: 10.1083/jcb.123.5.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzini-Armstrong C, Kish JW. Alternate disposition of tetrads in peripheral couplings of skeletal muscle. J Muscle Res Cell Motil. 1995;16(3):319–324. doi: 10.1007/BF00121140. [DOI] [PubMed] [Google Scholar]
- Go LO, et al. Differential regulation of two types of intracellular calcium release channels during end-stage heart failure. J Clin Invest. 1995;95(2):888–894. doi: 10.1172/JCI117739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80(2):853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- Huang F, et al. Analysis of calstabin2 (FKBP12.6)-ryanodine receptor interactions: rescue of heart failure by calstabin2 in mice. Proc Natl Acad Sci USA. 2006;103(9):3456–3461. doi: 10.1073/pnas.0511282103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JH, et al. Mapping domains and mutations on the skeletal muscle ryanodine receptor channel. Trends Mol Med. 2012;18(11):644–657. doi: 10.1016/j.molmed.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Inui M, Saito A, Fleischer S. Purification of the ryanodine receptor and identity with feet structures of junctional terminal cisternae of sarcoplasmic reticulum from fast skeletal muscle. J Biol Chem. 1987;262(4):1740–1747. [PubMed] [Google Scholar]
- Jayaraman T, et al. FK506 binding protein associated with the calcium release channel (ryanodine receptor) J Biol Chem. 1992;267(14):9474–9477. [PubMed] [Google Scholar]
- Jayaraman T, Marks AR. Calcineurin is downstream of the inositol 1,4,5-trisphosphate receptor in the apoptotic and cell growth pathways. J Biol Chem. 2000;275(9):6417–6420. doi: 10.1074/jbc.275.9.6417. [DOI] [PubMed] [Google Scholar]
- Jiang D, et al. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR) Proc Natl Acad Sci USA. 2004;101(35):13062–13067. doi: 10.1073/pnas.0402388101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungbluth H. Central core disease. Orphanet J Rare Dis. 2007;2:25. doi: 10.1186/1750-1172-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimlicka L, et al. Disease mutations in the ryanodine receptor N-terminal region couple to a mobile intersubunit interface. Nat Commun. 2013;4:1506. doi: 10.1038/ncomms2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein A, et al. Clinical and genetic findings in a large cohort of patients with ryanodine receptor 1 gene-associated myopathies. Hum Mutat. 2012;33(6):981–988. doi: 10.1002/humu.22056. [DOI] [PubMed] [Google Scholar]
- Kushnir A, et al. Role of CaMKIIdelta phosphorylation of the cardiac ryanodine receptor in the force frequency relationship and heart failure. Proc Natl Acad Sci USA. 2010;107(22):10274–10279. doi: 10.1073/pnas.1005843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanner JT, et al. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol. 2010;2(11):a003996. doi: 10.1101/cshperspect.a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larach MG, et al. Malignant hyperthermia deaths related to inadequate temperature monitoring, 2007–2012: a report from The North American Malignant Hyperthermia Registry of the Malignant Hyperthermia Association of the United States. Anesth Analg. 2014 doi: 10.1213/ANE.0000000000000421. [DOI] [PubMed] [Google Scholar]
- Laver DR, et al. Cytoplasmic Ca2+ inhibits the ryanodine receptor from cardiac muscle. J Membr Biol. 1995;147(1):7–22. doi: 10.1007/BF00235394. [DOI] [PubMed] [Google Scholar]
- Lee HG, et al. Interaction of HRC (histidine-rich Ca(2+)-binding protein) and triadin in the lumen of sarcoplasmic reticulum. J Biol Chem. 2001;276(43):39533–39538. doi: 10.1074/jbc.M010664200. [DOI] [PubMed] [Google Scholar]
- Lehnart SE, et al. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123(1):25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, et al. Role of leaky neuronal ryanodine receptors in stress-induced cognitive dysfunction. Cell. 2012;150(5):1055–1067. doi: 10.1016/j.cell.2012.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loseth S, et al. A novel late-onset axial myopathy associated with mutations in the skeletal muscle ryanodine receptor (RYR1) gene. J Neurol. 2013;260(6):1504–1510. doi: 10.1007/s00415-012-6817-7. [DOI] [PubMed] [Google Scholar]
- Lynch PJ, et al. A mutation in the transmembrane/luminal domain of the ryanodine receptor is associated with abnormal Ca2+ release channel function and severe central core disease. Proc Natl Acad Sci USA. 1999;96(7):4164–4169. doi: 10.1073/pnas.96.7.4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee KR, Shy GM. A new congenital non-progressive myopathy. Brain. 1956;79(4):610–621. doi: 10.1093/brain/79.4.610. [DOI] [PubMed] [Google Scholar]
- Maki T, et al. Regulation of calcium channel expression in neonatal myocytes by catecholamines. J Clin Invest. 1996;97(3):656–663. doi: 10.1172/JCI118462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks AR, et al. Molecular cloning and characterization of the ryanodine receptor/junctional channel complex cDNA from skeletal muscle sarcoplasmic reticulum. Proc Natl Acad Sci USA. 1989;86(22):8683–8687. doi: 10.1073/pnas.86.22.8683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks AR, et al. Smooth muscle and brain inositol 1,4,5-trisphosphate receptors are structurally and functionally similar. J Biol Chem. 1990;265(34):20719–20722. [PubMed] [Google Scholar]
- Marks AR, et al. Involvement of the cardiac ryanodine receptor/calcium release channel in catecholaminergic polymorphic ventricular tachycardia. J Cell Physiol. 2002;190(1):1–6. doi: 10.1002/jcp.10031. [DOI] [PubMed] [Google Scholar]
- Marx SO, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101(4):365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Marx SO, et al. Phosphorylation-dependent regulation of ryanodine receptors: a novel role for leucine/isoleucine zippers. J Cell Biol. 2001;153(4):699–708. doi: 10.1083/jcb.153.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO, Ondrias K, Marks AR. Coupled gating between individual skeletal muscle Ca2+ release channels (ryanodine receptors) Science. 1998;281(5378):818–821. doi: 10.1126/science.281.5378.818. [DOI] [PubMed] [Google Scholar]
- Matecki S, et al. Leaky ryanodine receptors contribute to diaphragmatic weakness during mechanical ventilation. Proc Natl Acad Sci. 2016;113(32):9069–9074. doi: 10.1073/pnas.1609707113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner G, Henderson JS. Rapid calcium release from cardiac sarcoplasmic reticulum vesicles is dependent on Ca2+ and is modulated by Mg2+, adenine nucleotide, and calmodulin. J Biol Chem. 1987;262(7):3065–3073. [PubMed] [Google Scholar]
- Mickelson JR, et al. Abnormal sarcoplasmic reticulum ryanodine receptor in malignant hyperthermia. J Biol Chem. 1988;263(19):9310–9315. [PubMed] [Google Scholar]
- Monnier N, et al. An autosomal dominant congenital myopathy with cores and rods is associated with a neomutation in the RYR1 gene encoding the skeletal muscle ryanodine receptor. Hum Mol Genet. 2000;9(18):2599–2608. doi: 10.1093/hmg/9.18.2599. [DOI] [PubMed] [Google Scholar]
- Monnier N, et al. Presence of two different genetic traits in malignant hyperthermia families: implication for genetic analysis, diagnosis, and incidence of malignant hyperthermia susceptibility. Anesthesiology. 2002;97(5):1067–1074. doi: 10.1097/00000542-200211000-00007. [DOI] [PubMed] [Google Scholar]
- Nakai J, et al. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 1996;380(6569):72–75. doi: 10.1038/380072a0. [DOI] [PubMed] [Google Scholar]
- Nakashima Y, et al. Molecular cloning and characterization of a human brain ryanodine receptor. FEBS Lett. 1997;417(1):157–162. doi: 10.1016/s0014-5793(97)01275-1. [DOI] [PubMed] [Google Scholar]
- Nelson BR, et al. Skeletal muscle-specific T-tubule protein STAC3 mediates voltage-induced Ca2+ release and contractility. Proc Natl Acad Sci USA. 2013;110(29):11881–11886. doi: 10.1073/pnas.1310571110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson TE, Bee DE. Temperature perturbation studies of sarcoplasmic reticulum from malignant hyperthermia pig muscle. J Clin Invest. 1979;64(4):895–901. doi: 10.1172/JCI109555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkura M, et al. Dual regulation of the skeletal muscle ryanodine receptor by triadin and calsequestrin. Biochemistry. 1998;37(37):12987–12993. doi: 10.1021/bi972803d. [DOI] [PubMed] [Google Scholar]
- Otsu K, et al. Molecular cloning of cDNA encoding the Ca2+ release channel (ryanodine receptor) of rabbit cardiac muscle sarcoplasmic reticulum. J Biol Chem. 1990;265(23):13472–13483. [PubMed] [Google Scholar]
- Paul-Pletzer K, et al. Identification of a dantrolene-binding sequence on the skeletal muscle ryanodine receptor. J Biol Chem. 2002;277(38):34918–34923. doi: 10.1074/jbc.M205487200. [DOI] [PubMed] [Google Scholar]
- Quane KA, et al. Mutations in the ryanodine receptor gene in central core disease and malignant hyperthermia. Nat Genet. 1993;5(1):51–55. doi: 10.1038/ng0993-51. [DOI] [PubMed] [Google Scholar]
- Ringer S. A further Contribution regarding the influence of the different constituents of the blood on the contraction of the heart. J Physiol. 1883;4(1):29–42.3. doi: 10.1113/jphysiol.1883.sp000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios E, Brum G. Involvement of dihydropyridine receptors in excitation-contraction coupling in skeletal muscle. Nature. 1987;325(6106):717–720. doi: 10.1038/325717a0. [DOI] [PubMed] [Google Scholar]
- Rogers EF, Koniuszy FR, et al. Plant insecticides; ryanodine, a new alkaloid from Ryania speciosa Vahl. J Am Chem Soc. 1948;70(9):3086–3088. doi: 10.1021/ja01189a074. [DOI] [PubMed] [Google Scholar]
- Rosenberg H, et al. Malignant hyperthermia. Orphanet J Rare Dis. 2007;2:21. doi: 10.1186/1750-1172-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D, et al. Distinct regions of triadin are required for targeting and retention at the junctional domain of the sarcoplasmic reticulum. Biochem J. 2014;458(2):407–417. doi: 10.1042/BJ20130719. [DOI] [PubMed] [Google Scholar]
- Santulli G. Adrenal signaling in heart failure: something more than a distant ship’s smoke on the horizon. Hypertension. 2014;63(2):215–216. doi: 10.1161/HYPERTENSIONAHA.113.02382. [DOI] [PubMed] [Google Scholar]
- Santulli G, et al. Calcium release channel RyR2 regulates insulin release and glucose homeostasis. J Clin Invest. 2015a;125(5):1968–1978. doi: 10.1172/JCI79273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli G, et al. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci USA. 2015b;112(36):11389–11394. doi: 10.1073/pnas.1513047112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli G, et al. Intracellular calcium release channels: an update. J Physiol. 2017 doi: 10.1113/JP272781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli G, Iaccarino G. Adrenergic signaling in heart failure and cardiovascular aging. Maturitas. 2016;93:65–72. doi: 10.1016/j.maturitas.2016.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli G, Marks AR. Essential roles of intracellular calcium release channels in muscle, brain, metabolism, and aging. Curr Mol Pharmacol. 2015;8(2):206–222. doi: 10.2174/1874467208666150507105105. [DOI] [PubMed] [Google Scholar]
- Scacheri PC, et al. A novel ryanodine receptor gene mutation causing both cores and rods in congenital myopathy. Neurology. 2000;55(11):1689–1696. doi: 10.1212/wnl.55.11.1689. [DOI] [PubMed] [Google Scholar]
- Sewry CA, et al. The spectrum of pathology in central core disease. Neuromuscul Disord. 2002;12(10):930–938. doi: 10.1016/s0960-8966(02)00135-9. [DOI] [PubMed] [Google Scholar]
- Shan J, et al. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J Clin Invest. 2010;120(12):4375–4387. doi: 10.1172/JCI37649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan J, et al. Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J Clin Invest. 2010;120(12):4388–4398. doi: 10.1172/JCI32726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suetomi T, et al. Mutation-linked defective interdomain interactions within ryanodine receptor cause aberrant Ca(2)(+)release leading to catecholaminergic polymorphic ventricular tachycardia. Circulation. 2011;124(6):682–694. doi: 10.1161/CIRCULATIONAHA.111.023259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takekura H, et al. Abnormal junctions between surface membrane and sarcoplasmic reticulum in skeletal muscle with a mutation targeted to the ryanodine receptor. Proc Natl Acad Sci USA. 1995;92(8):3381–3385. doi: 10.1073/pnas.92.8.3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshima H, et al. Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor. Nature. 1989;339(6224):439–445. doi: 10.1038/339439a0. [DOI] [PubMed] [Google Scholar]
- Takeshima H, et al. Excitation-contraction uncoupling and muscular degeneration in mice lacking functional skeletal muscle ryanodine-receptor gene. Nature. 1994;369(6481):556–559. doi: 10.1038/369556a0. [DOI] [PubMed] [Google Scholar]
- Tateishi H, et al. Defective domain-domain interactions within the ryanodine receptor as a critical cause of diastolic Ca2+ leak in failing hearts. Cardiovasc Res. 2009;81(3):536–545. doi: 10.1093/cvr/cvn303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tester DJ, et al. A mechanism for sudden infant death syndrome (SIDS): stress-induced leak via ryanodine receptors. Heart Rhythm. 2007;4(6):733–739. doi: 10.1016/j.hrthm.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball JG, Villalta SA. NO may prompt calcium leakage in dystrophic muscle. Nat Med. 2009;15(3):243–244. doi: 10.1038/nm0309-243. [DOI] [PubMed] [Google Scholar]
- Timerman AP, et al. The calcium release channel of sarcoplasmic reticulum is modulated by FK-506-binding protein. Dissociation and reconstitution of FKBP-12 to the calcium release channel of skeletal muscle sarcoplasmic reticulum. J Biol Chem. 1993;268(31):22992–22999. [PubMed] [Google Scholar]
- Tobacman LS. Thin filament-mediated regulation of cardiac contraction. Annu Rev Physiol. 1996;58:447–481. doi: 10.1146/annurev.ph.58.030196.002311. [DOI] [PubMed] [Google Scholar]
- Tong J, et al. Caffeine and halothane sensitivity of intracellular Ca2+ release is altered by 15 calcium release channel (ryanodine receptor) mutations associated with malignant hyperthermia and/or central core disease. J Biol Chem. 1997;272(42):26332–26339. doi: 10.1074/jbc.272.42.26332. [DOI] [PubMed] [Google Scholar]
- Tung CC, et al. The amino-terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature. 2010;468(7323):585–588. doi: 10.1038/nature09471. [DOI] [PubMed] [Google Scholar]
- Umanskaya A, et al. Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging. Proc Natl Acad Sci USA. 2014;111(42):15250–15255. doi: 10.1073/pnas.1412754111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenknecht T, et al. Three-dimensional architecture of the calcium channel/foot structure of sarcoplasmic reticulum. Nature. 1989;338(6211):167–170. doi: 10.1038/338167a0. [DOI] [PubMed] [Google Scholar]
- Ward CW, et al. Defects in ryanodine receptor calcium release in skeletal muscle from post-myocardial infarct rats. FASEB J. 2003;17(11):1517–1519. doi: 10.1096/fj.02-1083fje. [DOI] [PubMed] [Google Scholar]
- Witherspoon JW, Meilleur KG. Review of RyR1 pathway and associated pathomechanisms. Acta Neuropathol Commun. 2016;4(1):121. doi: 10.1186/s40478-016-0392-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, et al. Imaging atrial arrhythmic intracellular calcium in intact heart. J Mol Cell Cardiol. 2013;64:120–123. doi: 10.1016/j.yjmcc.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, et al. Mitochondrial oxidative stress promotes atrial fibrillation. Sci Rep. 2015;5:11427. doi: 10.1038/srep11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin HB, et al. Affinity purification of the ryanodine receptor/calcium release channel from fast twitch skeletal muscle based on its tight association with FKBP12. Biochem Biophys Res Commun. 1995;214(1):263–270. doi: 10.1006/bbrc.1995.2283. [DOI] [PubMed] [Google Scholar]
- Yan Z, et al. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature. 2015;517(7532):50–55. doi: 10.1038/nature14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Q, et al. Functional role of calstabin2 in age-related cardiac alterations. Sci Rep. 2014;4:7425. doi: 10.1038/srep07425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Q, et al. Maintenance of normal blood pressure is dependent on IP3R1-mediated regulation of eNOS. Proc Natl Acad Sci USA. 2016;113(30):8532–8537. doi: 10.1073/pnas.1608859113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuchi Z, Lau K, Van Petegem F. Disease mutations in the ryanodine receptor central region: crystal structures of a phosphorylation hot spot domain. Structure. 2012;20(7):1201–1211. doi: 10.1016/j.str.2012.04.015. [DOI] [PubMed] [Google Scholar]
- Zalk R, et al. Structure of a mammalian ryanodine receptor. Nature. 2015;517(7532):44–49. doi: 10.1038/nature13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterstrom R, Arnhold RG. Impaired calcium-phosphate homeostasis in newborn infants of diabetic mothers. Acta Paediatr. 1958;47(2):107–112. doi: 10.1111/j.1651-2227.1958.tb07865.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, et al. A mutation in the human ryanodine receptor gene associated with central core disease. Nat Genet. 1993;5(1):46–50. doi: 10.1038/ng0993-46. [DOI] [PubMed] [Google Scholar]
- Zhang L, et al. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem. 1997;272(37):23389–23397. doi: 10.1074/jbc.272.37.23389. [DOI] [PubMed] [Google Scholar]
- Zhang L, et al. Functional SNP in the microRNA-367 binding site in the 3′UTR of the calcium channel ryanodine receptor gene 3 (RYR3) affects breast cancer risk and calcification. Proc Natl Acad Sci USA. 2011;108(33):13653–13658. doi: 10.1073/pnas.1103360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F, et al. Dantrolene inhibition of ryanodine receptor Ca2+ release channels. Molecular mechanism and isoform selectivity. J Biol Chem. 2001;276(17):13810–13816. doi: 10.1074/jbc.M006104200. [DOI] [PubMed] [Google Scholar]
- Zhou H, et al. Molecular mechanisms and phenotypic variation in RYR1-related congenital myopathies. Brain. 2007;130(Pt 8):2024–2036. doi: 10.1093/brain/awm096. [DOI] [PubMed] [Google Scholar]
- Zhou H, et al. Multi-minicore disease and atypical periodic paralysis associated with novel mutations in the skeletal muscle ryanodine receptor (RYR1) gene. Neuromusc Disord NMD. 2010;20(3):166–173. doi: 10.1016/j.nmd.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zvaritch E, et al. Ca2+ dysregulation in Ryr1(I4895T/wt) mice causes congenital myopathy with progressive formation of minicores, cores, and nemaline rods. Proc Natl Acad Sci USA. 2009;106(51):21813–21818. doi: 10.1073/pnas.0912126106. [DOI] [PMC free article] [PubMed] [Google Scholar]