

SUMMARY
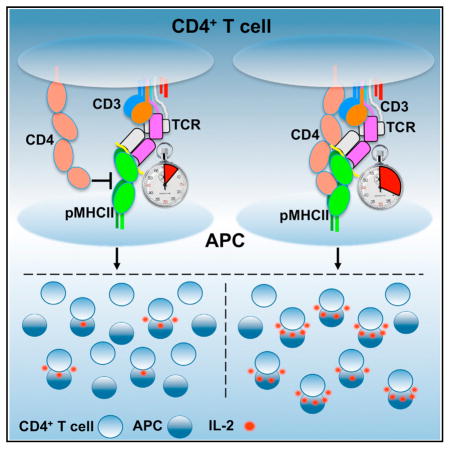
CD4+ T cells convert the time that T cell receptors (TCRs) interact with peptides embedded within class II major histocompatibility complex molecules (pMHCII) into signals that direct cell-fate decisions. In principle, TCRs relay information to intracellular signaling motifs of the associated CD3 subunits, while CD4 recruits the kinase Lck to those motifs upon coincident detection of pMHCII. But the mechanics by which this occurs remain enigmatic. In one model, the TCR and CD4 bind pMHCII independently, while in another, CD4 interacts with a composite surface formed by the TCR-CD3 complex bound to pMHCII. Here, we report that the duration of TCR-pMHCII interactions impact CD4 binding to MHCII. In turn, CD4 increases TCR confinement to pMHCII via reciprocal interactions involving membrane distal and proximal CD4 ectodomains. The data suggest that a precisely assembled macrocomplex functions to reliably convert TCR-pMHCII confinement into reproducible signals that orchestrate adaptive immunity.
In Brief
Glassman, Parrish et al. use functional and biophysical assays to demonstrate that CD4 stabilizes TCR-pMHCII interactions via membrane distal and proximal domains. The data indicate that CD4 docks along a composite surface created by the TCR-CD3-pMHCII axis to confer a uniform macrocomplex architecture upon a diverse TCR repertoire.
INTRODUCTION
CD4+ T cells are remarkable for their sensitivity, specificity, and the range of effector types to which a naive cell can differentiate after detecting a threat (i.e., helper [Th], T follicular helper [Tfh], regulatory [Treg], and memory [Tm]) (Zhu et al., 2010). The quantity and quality of signals generated by the T cell receptor (TCR) are key determinants for CD4+ T cell development, activation, differentiation, and effector cell responses (Allison et al., 2016; Corse et al., 2010; Fazilleau et al., 2009; Gottschalk et al., 2010; Hwang et al., 2015; Savage et al., 1999; Stepanek et al., 2014; Tubo et al., 2013; van Panhuys et al., 2014; Vanguri et al., 2013). But the genesis of these signals remains unclear because the relationship between the TCR and CD4 remains mechanistically undefined.
Each clonotypic TCR provides a CD4+ T cell with specificity for a limited number of peptides presented within class II major histocompatibility complex (pMHCII) molecules on antigen-presenting cells (APCs). The time a TCR spends confined to a pMHCII informs CD4+ T cell responsiveness. For interactions with slow on-rates, such that newly dissociated TCRs and pMHCII diffuse away from each other before rebinding, this equates to their t1/2; however, for TCRs with on-rates that allow rebinding, responsiveness best relates to the aggregate t1/2 (ta) that considers rebinding as part of a total confinement time (Govern et al., 2010; Tubo et al., 2013; Vanguri et al., 2013).
TCR-pMHCII interactions relay information to the immunoreceptor tyrosine-based activation motifs (ITAMs) of the associated CD3γε, CD3δε, and CD3ζζ signaling modules (Gil et al., 2002; Lee et al., 2015); however, transmitting information across the membrane to the ten ITAMs within a TCR-CD3 complex (one per CD3γ, δ, and ε subunit, and three per ζ) is insufficient to generate chemical signals because the complex itself lacks intrinsic kinase activity. Rather, the Src kinase p56Lck (Lck), which non-covalently associates with CD4, primarily phosphorylates the ITAMs (Malissen and Bongrand, 2015).
CD4 is critical for TCR-CD3 signaling to single agonist pMHCII, increases functional responses by 10- to 1,000+-fold and determines how a T cell perceives the potency of a pMHCII (Glaichenhaus et al., 1991; Irvine et al., 2002; Killeen and Littman, 1993; Parrish et al., 2016; Stepanek et al., 2014; Vidal et al., 1999). When a CD4 molecule associated with Lck binds the same pMHCII as a TCR, it is thought to recruit Lck to phosphorylate the ITAMs (Malissen and Bongrand, 2015). In this scenario, CD4 is a constant, binding to a monomorphic region of MHCII regardless of the nature of the peptide embedded therein, and thus regardless of whether or not the TCR is bound to the pMHCII.
But three pieces of evidence raise questions about how, upon TCR-pMHCII engagement, CD4 positions Lck and the ITAMs in a sufficient local concentration for a sufficient duration for phosphorylation to occur; particularly for the weak interactions that drive positive selection and peripheral homeostasis (Glassman et al., 2016; Kao and Allen, 2005; Stepanek et al., 2014; Wang et al., 2001b; Zuñiga-Pflücker et al., 1989 ). First, crystallography data suggest that the TCR-CD3 complex, pMHCII, and CD4 adopt a V-like arch that could place the CD3 ITAMs, and, in particular, the six ITAMs of ζζ, ~100 Å from a CD4-associated Lck (Wang et al., 2001a; Yin et al., 2012). Second, interactions between the CD4 D1 domain and MHCII at the apex of this arch are too weak to measure in solution, and 2D affinity estimates suggest that CD4-MHCII interactions are ~2–3 orders of magnitude weaker than TCR-pMHCII interactions (Hong et al., 2015; Jönsson et al., 2016). Finally, C-terminally truncated CD4 molecules that lack the cysteine clasp, and cannot directly interact with Lck, nevertheless increase TCR-CD3 signaling (Killeen and Littman, 1993; Parrish et al., 2016; Vidal et al., 1999). A unifying understanding of how the TCR-CD3 complex, pMHCII, and CD4 fit and work together thus remains elusive.
Here, we tested predictions made by a V-like macrocomplex versus a more compact architecture in which CD4 docks along a composite surface formed by the TCR-CD3-pMHCII axis (Figure S1A) (Glassman et al., 2016; Kuhns and Badgandi, 2012; Wang et al., 2001a; Yin et al., 2012). In the latter model, TCR-CD3 dwell time on pMHCII would determine the duration with which the CD4 docking site is formed and thus influence CD4 dwell time within a mature macrocomplex. In turn, CD4 docking would extend the TCR-CD3 dwell time on pMHCII. Neither would occur in the V-like arch since TCR-pMHCII and CD4-MHCII interactions are independent of one another. Finally, in the V-like arch CD4 binds MHCII only via the D1 domain, while CD4 would use its length to contact the TCR-CD3-pMHCII axis in a compact macrocomplex. To test these predictions, we varied the duration of TCR-pMHCII interactions with defined altered peptide ligands (APLs), and the duration of CD4-MHCII interactions with a CD4 D1 domain mutant, to study how these changes influenced the interplay between the TCR-CD3 complex and CD4 upon binding pMHCII. We also made mutants in the CD4 D3 domain. The data suggest that reciprocal interactions between the TCR-CD3 complex and CD4 ectodomains around a nucleating pMHCII impact the assembly, stability, and function of the TCR-CD3-pMHCII-CD4 macrocomplex.
RESULTS
CD4 Enhances Functional Responses to Low-Affinity pMHCII
This study was designed to evaluate the interplay between the TCR-CD3 complex and CD4 by testing predictions made by the V-like arch and compact macrocomplex models (Figure S1A). We primarily used the archetypal cytochrome-c-reactive 5c.c7 TCR since there is a wealth of kinetic and functional data for 5c.c7 interactions with the moth cytochrome-c-derived agonist peptide (MCC 88-103) presented within the mouse MHCII I-Ek (dissociation constant [KD] reported at 22.9–43.5 μM), the weak agonist T102S peptide (KD = 206 μM; ~4-fold lower potency), and the immeasurably weak antagonist T102G peptide (Corse et al., 2010; Gottschalk et al., 2010; Huppa et al., 2010).
Prior to testing the models, we functionally characterized CD4. In vitro 5c.c7+ CD4+ T cell responses mirror their in vivo responses to MCC and T102S (Corse et al., 2010), so we stimulated naive transgenic (Tg) 5c.c7+ CD4+ T cells with MCC, T102S, or T102G in the presence of anti-CD8 (control) or anti-CD4 monoclonal antibodies (mAb) in vitro. I-Ek+ M12 cells were used as APCs for consistency with subsequent experiments (Figure S1B). Anti-CD4 reduced responses to MCC and eliminated responses to T102S, while no responses were observed to T102G (Figure 1A). We also evaluated CD69 expression and apoptosis of 5c.c7+ CD4+ CD8− Tg thymocytes in vitro as they are more sensitive to low-affinity ligands. Anti-CD4 reduced responses to T102S as well as T102G (Figures 1B, S1C, and S1D). Moving to more reductionist systems, we evaluated interleukin-2 (IL-2) production by 5c.c7+ CD4+ or CD8α+ 58α−β− T cell hybridomas. Anti-CD4 eliminated responses to MCC and T102S for CD4+ cells, while CD8+ (no CD4) cells did not respond (Figure 1C). These data show that CD4 increases signaling by the 5c.c7 TCR in primary cells and T cell hybridomas responding to agonist and weak pMHCII.
Figure 1. CD4 Enhances TCR-CD3 Signaling.

(A) IL-2 production by CD4+ T cells from 5c.c7 TCR Tg mice cultured with I-Ek+ M12 cells, peptide, and control (α-CD8) or α-CD4 mAb.
(B) Percentage of CD69+ 5c.c7 TCR Tg CD4+CD8− (SP) thymocytes after 14-hr culture with I-Ek+ M12 cells, peptide, and control (α-CD8) or α-CD4 mAb.
(C–E) IL-2 production by (C) 5c.c7+ CD8α+ (no CD4) or CD4+ 58α−β− cultured with I-Ek+ M12 cells, peptide, and control or α-CD4 mAb; (D) 5c.c7+ CD4-Lck+ or CD4Δbind-Lck+ 58α−β− cells cultured with I-Ek+ M12 cells and peptide; (E) 5c.c7+ 58α−β− cells expressing CD4T or CD4TΔbind.
(F) 5c.c7+ CD4T+ 58α−β− cells cultured with Ova:I-Ab plus WT I-Ek or E164A+T167A mutant I-Ek M12 cells.
Data are mean ± SEM of triplicate wells. IL-2 was measured by ELISA at 16 hr. Results shown are representative of two or more independent experiments. See also Figure S1. perceived potency of a pMHCII even when CD4 cannot directly interact with Lck.
Since the frequency of CD4:Lck association influences TCR signaling (Stepanek et al., 2014), we also asked how a CD4-Lck fusion influences the perceived potency of MCC, T102S, and T102G. Previously, we reported that 5c.c7+ CD4-Lck+ 58α−β− cells make IL-2 in response to M12 cells expressing tethered pMHCII with an unexpected hierarchy of T102G > T102S > MCC and also respond to null peptides in a manner that depended on high levels of pMHCII (Parrish et al., 2016). Here, analysis of IL-2 production in response to a peptide titration showed the proper hierarchy of potency (MCC > T102S > T102G), but T102G elicited a greater response than T102S and MCC at the highest peptide concentration (Figure 1D). A small amount of IL-2 was also produced without exogenous peptides, consistent with intrinsic TCR scanning of MHCII (Parrish et al., 2016). Expression of a CD4-Lck mutated in the D1 domain (CD4TΔbind-Lck), where crystal structures show CD4-MHCII interactions and which impairs an ordered spatial relationship between CD4 and the TCR-CD3 subunits upon TCR engagement (Glassman et al., 2016; Wang et al., 2001a), did not facilitate IL-2 production to any of the peptides, so overexpression of Lck did not account for our results.
Since CD4:Lck interactions are not essential for CD4 function, we also interrogated how CD4-MHCII interactions impact TCR signaling in the absence of direct CD4:Lck interactions (Killeen and Littman, 1993; Parrish et al., 2016). We used C-terminally truncated CD4 (CD4T), which lacks the cysteine clasp that mediates CD4:Lck interactions but nevertheless enhances IL-2 production by 5c.c7+CD4T+ 58α−β− T cell hybridomas to ~60%–80% of levels observed with cells expressing full-length CD4, depending on the pMHCII density, and >10-fold relative to cells lacking CD4 (Parrish et al., 2015, 2016). With CD4T, intra-cellular Lck is available to phosphorylate ITAMs but cannot directly tether CD4 to a TCR-CD3-pMHC unit via interactions with the CD3ε proline rich sequence (PRS), the CD3ε basic rich sequence (BRS), or with phosphorylated ITAMs via its SH2 domain (Li et al., 2017; Mingueneau et al., 2008; Xu and Littman, 1993). CD4T was required for IL-2 production in response to MCC and T102S, when compared with CD4TΔbind (Figure 1E). CD4-MHCII interactions via the D1 domain thus impact the
Finally, we asked whether CD4T binds to the same pMHCII as the TCR to elicit IL-2 production, as indicated by previous studies of MHCI or MHCII mutants that impair CD8 or CD4 function, respectively (Connolly et al., 1990; Krogsgaard et al., 2005). M12 cells were transduced with I-Ek or an I-Ek.E164A+T167A mutant that impairs CD4T function, along with tethered OVA:I-Ab that has an intact CD4 binding site but cannot present the MCC peptide (Figure S1E) (König et al., 1992; Krogsgaard et al., 2005). Consistent with prior results, the mutants impaired IL-2 production by 5c.c7+CD4T+ 58α−β− T cell hybridomas in response to MCC and T102S, providing additional evidence that CD4T binds the same pMHCII as the TCR (Figure 1F).
CD4 Increases TCR-Mediated Cell Coupling
Functional T cell responses require coupling to APCs, so we used flow cytometry to evaluate CD4’s role in meditating coupling of 5c.c7+CD4T+ or CD4TΔbind 58α−β− cells to M12 cell expressing tethered pMHCII. 5c.c7+ CD4T+ 58α−β− cells coupled to T102S:I-Ek+ and T102G:I-Ek+ M12 cells at a higher relative frequency than CD4TΔbind cells, and treatment with PP2 to inhibit kinase activity yielded similar results (Figures 2A, 2B, S2A, and S2B) (Bain et al., 2007).
Figure 2. CD4 Enhances Cell Couple Formation and TCR Accumulation.

(A and B) Relative cell coupling between 5c.c7+ CD4T+ or CD4TΔbind+ 58α−β− cells cultured with the indicated tethered pMHCII+ M12 cells in the presence of (A) DMSO as a vehicle control or (B) PP2 to inhibit kinase activity.
(C–F) CD3T+ CD4T+ or CD4TΔbind+ M12 cells expressing the (C) 5c.c7 TCR, (D) 2B4 TCR, (E) B3K506 TCR, or (F) B3K508 TCR were cultured with 58α−β− cells expressing the indicated tethered pMHCII.
Cell coupling is shown relative to the couple frequency observed in the CD4T MCC condition for (A)–(D) or the CD4T 3K condition for (E) and (F). Each data point represents an individual experiment (3–4 experiments per condition); bars represent mean. Data were analyzed using a paired Student’s t test (*p < 0.05, **p < 0.01, ***p < 0.001). See also Figure S2.
(G) Representative TIRFM image showing CD3δTG accumulation on a MCC:I-Ek-coated surface (pseudocolored green) overlayed on a bright-field image. The dashed line represents the region of interest (ROI) exported for analysis. Scale bars represent 5 μm and look up tables indicate mEGFP intensity units.
(H and I) TCR-CD3 complex accumulation. Each dot represents GFP intensity of a single cell for (H) CD3δTG or (I) 2B4βG on immobile pMHCII surfaces with the indicated peptides. n = total number of analyzed cells per condition. Data were tested for normality using a D’Agostino and Pearson omnibus normality test followed by a Mann-Whitney test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Results shown are representative of two experiments.
To further investigate whether the TCR and CD4 mediate coupling on their own we tested (1) whether TCR-pMHCII interactions mediate cell coupling in the absence of signaling or other TCR engagement-associated events (e.g., Nck or Lck interactions with the CD3ε PRS or basic rich sequences) (Gil et al., 2002; Li et al., 2017; Mingueneau et al., 2008; Xu and Littman, 1993); and (2) whether CD4T contributes to cell coupling. The 5c.c7 TCR was expressed with truncated CD3 subunits (CD3T) that lack their intracellular domains, and either CD4T or CD4TΔbind on M12 cells, while our panel of tethered pMHCII were expressed on 58α−β− cells (Figure S2C). Again, the frequency of couple formation followed the expected hierarchy (MCC > T102S > T102G > Hb). MCC-driven coupling was not increased by CD4T relative to CD4TΔbind, and neither CD4T nor CD4TΔbind cells coupled to Hb APCs even with high expression of pMHCII (Figures 2C, S2C, and S2D). 5c.c7 interactions with T102S:I-Ek were sufficient to mediate coupling (CD4TΔbind cells) and CD4T increased these interactions (Huppa et al., 2010). Finally, the affinity of 5c.c7 for T102G:I-Ek was too weak to mediate cell coupling by CD4TΔbind cells, yet when combined with CD4-MHCII interactions cell coupling was achieved. These data show that CD4 can enhance cell coupling mediated by low-affinity TCR-pMHCII interactions at high pMHCII density.
To test whether CD4T impacts coupling with different TCR-pMHCII pairings, we used the 2B4, B3K506, and B3K508 TCRs in the same experimental setup. 2B4 is unique as it lacks five of the eight features typical of cytochrome-reactive TCRs, like 5c.c7, has higher affinity for MCC and T102S (KD ≈5.5–8.7 and 33.8–90 μM, respectively) and drives distinct effector differentiation (Fazilleau et al., 2009; Krogsgaard et al., 2003; Newell et al., 2011; Wu et al., 2002). CD4T increased 2B4-mediated coupling to T102S, and more so to T102G, but not to MCC (Figure 2D), indicating again that CD4 contributes to coupling when TCR-pMHCII affinity falls below some threshold (>33–90 μM). The B3K506 TCR binds the 3K, P5R, and P-1A peptide in I-Ab with affinities above this mark (KD = 7, 11, and 26 μM, respectively), while the B3K508 TCR binds the same pMHCII with a broader range of affinities (29, 93, and >550 μM, respectively), allowing us to further test this idea (Govern et al., 2010). CD4T did not increase cell coupling driven by B3K506 for any peptides but increased B3K508-mediated coupling to P5R and P-1A (Figures 2E, 2F, and S2E); further demonstrating that CD4 can contribute to cell coupling for lower-affinity TCR-pMHCII interactions.
These results could reflect an additive contribution to adhesion if CD4 and the TCR bind pMHCII independently in a V-like arch, although a CD4-MHCII KD >2.5 mM makes this remote. Alternatively, they could reflect a cooperative effect similar to that described for some cytokine receptors and expected in a compact macrocomplex (Spangler et al., 2015).
CD4 Increases TCR Adhesion to pMHCII In Situ
To further analyze CD4’s impact on TCR-pMHCII interactions, we asked whether CD4T increased TCR-CD3TG accumulation on glass coverslips coated with pMHCII monomers relative to CD4TΔbind (Glassman et al., 2016). Bright-field microscopy of 5c.c7+CD3T+CD4T+ M12 cells allowed an unbiased survey of all cells in the field, while TCR-CD3TG intensity was measured by live total internal reflection fluorescence microscopy (TIRFM) for a region of interest (ROI) across the widest point of contact (Figure 2G). CD4TΔbind was used instead of anti-CD4 since prior studies of 5c.c7-MCC:I-Ek interactions in situ reported a slight decline in TCR dwell time with anti-CD4, and one reported a reduction in the total number of TCR-pMHC interactions per cell that was attributed to interference from the antibody (Huppa et al., 2010; O’Donoghue et al., 2013).
Both 5c.c7+CD3T+CD4T+ and CD4TΔbind+ cells had TCR-CD3TG intensity that was proportional to the affinity of TCR-pMHCII interactions (MCC > T102S > T102G > Hb) (Figure 2H). But CD4T dramatically enhanced the intensity on the T102S and T102G surfaces compared to CD4TΔbind. These data indicate that CD4-MHCII interactions enhance weak TCR-pMHCII interactions. Similar results were observed with 2B4+CD3T+CD4T+ and CD4TΔbind+ M12 cells (Figure 2I).
CD4 Mobility on pMHCII Surfaces Is Proportional to TCR-pMHCII Affinity and Density
We next used fluorescence recovery after photobleaching (FRAP) to analyze TCR-CD3 and CD4 dwell time on pMHCII in situ in order to test predictions of the relevant models (Figures S3A and S3B). For example, TCR-pMHCII affinity should not influence independent CD4-MHCII interactions in a V-like arch but should in a compact macrocomplex where TCR-CD3 and CD4 interact around a nucleating pMHCII. TCR+CD3T+CD4T+ or CD4TΔbind+ M12 cells were used to test these predictions in the absence of downstream events associated with TCR engagement.
To establish the system, we calculated the mobile fraction and t1/2 of recovery of 5c.c7-CD3TG molecules as they exchanged in and out of a bleached ROI on cells bound to surfaces coated with pMHCII of increasing affinity (Figures 3A–3C; Movies S1 and S2) (Klammt et al., 2015). This movement would be a measure of on and off rates as well as surface mobility. Surface plasmon resonance (SPR) cannot detect 5c.c7 interactions with T102G:I-Ek, but FRAP showed an increase in TCR-CD3TG t1/2 on the T102G surfaces compared with Hb surfaces that increased further on the T102S and MCC surfaces. The near lack of mobility observed on MCC surfaces showed that rebinding impacts TCR-CD3TG confinement in the bleached area as the t1/2 for a 5c.c7 TCR bound to MCC:I-Ek at 37°C is 2.28 s (Huppa et al., 2010). These data establish that TCR-CD3TG dwell time measured by FRAP follows the expected hierarchy.
Figure 3. TCR-CD3 and CD4 Mobility Decrease as TCR-pMHCII Affinity Increases.

(A–C) FRAP analysis of CD3δTG for 5c.c7+ CD3T+ CD4T+ M12 cells. Images were collected for 2-min postbleach at 3-s intervals. (A) Recovery trace represents mean ± SEM for the indicated number (n) of cells. (B) Mobile fraction (%) and (C) half-life (t1/2) for recovery were determined by curve fitting. Error bars indicate 95% confidence interval.
(D–F) FRAP of CD4TmCh. (D) Recovery trace, (E) mobile fraction (%) and (F) half-life (t1/2) as presented in (A)–(C).
# denotes values with poor fitting with an exponential function. Results shown are representative of at least two experiments. See also Figure S3.
Next, we asked whether TCR-pMHCII affinity does or does not impact CD4T mobility in situ, as predicted by the compact or V-like macrocomplex models, respectively (Figures 3D–3F). While the mobile fraction was minimally impacted by changes in TCR-pMHCII affinity, the CD4TmCh t1/2 increased proportionally to the affinity of TCR-pMHCII interactions on 5c.c7+ CD3T+CD4T+ M12 cells. Similar results were obtained with the 2B4+CD3T+CD4T+ M12 cells, although CD4T mobility was not slowed by MCC to a greater extent than T102S (Figures S3C–S3E).
To determine whether the observed differences in CD4T t1/2 were due to direct interactions versus TCR-CD3 crowding, we compared CD4TmCh and CD4TΔbind.mCh mobility on 5c.c7 cells adhered to MCC:I-Ek where CD4T did not increase TCR-CD3TG intensity (Figure 2H). CD4TΔbind.mCh recovered more rapidly than CD4TmCh, and CD4TmCh intensity increased more than CD4TΔbind.mCh (Figures 4A, 4B, and S4A–S4C). On Hb:I-Ek, where TCR-CD3TG and CD4TmCh versus CD4TΔbind.mCh intensity were equivalent, we observed a small difference in mobile fraction but not t1/2 (Figures 4C, 4D, and S4D–S4F). We also pairwise-matched 5c.c7 cells with TCR-CD3TG intensities that varied by less than 5% on T102S and MCC to compare CD4T dwell times under conditions of equivalent TCR-CD3 crowding and intact CD4-MHCII interactions (Figures 4E and 4F). Here, the CD4TmCh t1/2 was longer on the MCC surface than the T102S surface, while the mobile fraction was equivalent, indicating that TCR-pMHCII dwell time impacts CD4T-pMHCII dwell time (Figures 4G, 4H, and S4G). CD4T mobility is therefore sensitive to TCR-pMHCII interactions.
Figure 4. CD4 Dwell Time Depends upon TCR Engagement.

(A–D) 5c.c7+ CD4TmCh+ or CD4TΔbind. mCh+ M12 cells were adhered to (A and B) MCC:I-Ek or (C and D) Hb:I-Ek-coated surfaces for mCh FRAP analysis as in Figures 3E and 3F.
(E) mEGFP intensity of cells from Figure 3D on T102S or MCC surfaces. Values are post-mCherry bleaching to account for differences in intensity due to CD3δTG::CD4TmCh FRET (Glassman et al., 2016).
(F) mEGFP intensity of pairwise mEGFP intensity-matched cells (<5% variance) from (E).
(G and H) Mobile fraction (G) and mCh t1/2 (H) of pairwise mEGFP-matched cells from (F).
(I–L) mCh t1/2 for TCR+ CD4TmCh+ or CD4TΔbind. mCh+ M12 cells expressing the (I and J) 5c.c7 or (K and L) 2B4 TCR adhered to surfaces coated with 50% or 12.5% MCC:I-Ek diluted in Hb:I-Ek.
Data in (E) and (F) are presented as mean and analyzed by t test (**p < 0.01). n = number of cells analyzed per condition. Error bars for FRAP indicate 95% confidence interval. Results shown are representative of at least two experiments. See also Figure S4.
Finally, if TCR and CD4 bind pMHCII independently in a V-like arch, then CD4 t1/2 should not be impacted by the quality of peptides presented therein, but the peptide sequence would impact the CD4 t1/2 in a compact macrocomplex. For 5c.c7 cells, a decrease in CD4TmCh t1/2 was observed on surfaces in which MCC:I-Ek was diluted from 50% to 12.5% to 4% into Hb:I-Ek that was not mirrored by CD4TΔbind.mCh t1/2 on such surfaces (Figures 4I, 4J, and S4H–S4J). The most dramatic difference was observed from 50% to 12.5% MCC:I-Ek, so we tested 2B4 cells under these conditions and obtained similar results (Figures 4K, 4L, S4K, and S4L). These data further establish that TCR engagement impacts CD4 dwell time.
TCR-CD3 Mobility on pMHCII Surfaces Is Impeded by CD4-MHCII Interactions
We next asked whether CD4-MHCII interactions impact TCR-CD3 dwell time by measuring TCR-CD3TG mobility on TCR+CD3T+CD4T+ or CD4TΔbind+ M12 cells. CD4T minimally impacted TCR-CD3TG recovery on MCC surfaces, compared with CD4TΔbind, for either 5c.c7 or 2B4 cells (Figures S5A and S5B). As our initial analysis suggested that TCR rebinding on the high density MCC surface impeded TCR-CD3TG mobility, we also analyzed TCR-CD3TG mobility on surfaces with 12.5% MCC:I-Ek diluted into Hb:I-Ek. Here, the presence of CD4T slightly reduced the mobile fraction but dramatically increased the TCR-CD3TG t1/2 compared to CD4TΔbind for both the 5c.c7 and 2B4 TCRs (Figures 5A–5D, S5C, and S5D), showing the CD4T can increase TCR dwell time on low ligand densities of agonist pMHCII. To ask whether CD4T impacts TCR dwell times on lower-affinity ligands we monitored TCR-CD3TG mobility on T102S surfaces. Again, CD4T reduced the mobile fraction and increased TCR-CD3TG t1/2 compared to CD4TΔbind for both the 5c.c7 and 2B4 TCRs (Figures 5E–5H, S5E, and S5F). These data are most consistent with a compact macrocomplex.
Figure 5. CD4 Increases TCR-CD3 Confine-ment on pMHCII.

FRAP of TCR-CD3TG complexes for TCR+ CD4T+ or CD4TΔbind+ M12 cells.
(A–D) FRAP of TCR-CD3TG on surfaces coated with 12.5% MCC:I-Ek in Hb:I-Ek. (A) Mobile fraction (%) and (B) half-life (t1/2) for 5c.c7 TCR + CD3δTG. (C) Mobile fraction (%) and (D) half-life (t1/2) for 2B4 TCR (2B4βG+).
(E–H) FRAP of TCR-CD3TG on T102S:I-Ek-coated surfaces. (E) Mobile fraction (%) and (F) half-life (t1/2) for 5c.c7 TCR + CD3δTG. (G) Mobile fraction (%) and (H) half-life (t1/2) for 2B4 TCR (2B4βG+).
n = number of cells analyzed per condition. Error bars are 95% confidence interval. Results shown are representative of at least three experiments. See also Figure S5.
Mutations in the CD4 D3 Domain Impair Function
A final difference between the V-like and compact macrocomplex models is that the former dictates that CD4 only binds MHCII via the D1 domain, while the latter predicts that CD4 contacts the TCR-CD3-pMHCII axis along its length. Our CD4Δbind mutant provides evidence for the importance of the D1 domain, but to further test these models required mutants in an MHCII-distal region of CD4.
Prior studies proposed that the D3 domain surface interacts with the TCR-CD3 complex, as mutating F208 and F227 (using UniProt convention) that extend into the CD4 D3 domain hydrophobic core impair signaling (Figure S6A) (Vignali and Vignali, 1999). Since the cavity created by F208A and F227A could destabilize the D3 domain surface, we examined the surface above F208 and F227 and found that P228 and F231, as well as P281 on an adjacent loop, constitute a conserved nonpolar patch that borders a hydrophobic pocket (Figures S6A and S6B). Such patches can mediate protein:protein interactions (DeLano et al., 2000), leading us to ask whether changing the nature of this surface by mutagenesis (CD4TP228E+F231E and CD4TP281E) would reduce its contribution to TCR signaling, proximity to the TCR-CD3 complex, and CD4 mobility upon pMHCII engagement. Glutamate was used to impart a negative charge on this surface, while mutating the rigid pro-lines was also likely to impact the shape, mobility, or surface stability (Wang et al., 2009). Indeed, for P281 that facilitates a turn on one side of the nonpolar patch, the CD4TP281E mutant had slightly reduced cell-surface expression and increased CD4TP281E::CD4TP281E Förster resonance energy transfer (FRET) compared with the WT or CD4TP228E+F231E mutant, which may indicate instability and increased aggregation (Figures S6C–S6E).
58α−β− cell lines expressing the 5c.c7 TCR and wild-type or mutant CD4T molecules were generated to assess the functional impact of these mutations. CD4TP228E+F231E cells had reduced IL-2 production, TCR downregulation, and CD69 expression in response to tethered MCC:I-Ek, T102S:I-Ek, or T102G:I-Ek pMHCII expressed on M12 cells compared with CD4T cells (Figures 6A–6C). After sorting CD4T and CD4TP281E cells for matched surface expression (Figure S6F), CD4TP281E cells also made less IL-2 upon TCR engagement than the CD4T cells (Figure 6D). Since sorting requires coating cells with mAbs to CD4, which could deliver a signal and impact the responsiveness of cells that expanded thereafter, we used unsorted cells for our analysis of TCR downregulation and CD69 expression as flow cytometry allowed us to directly compare matched subsets by gating on equivalent CD4 expression (Figure S6G). Here, again we observed reduced responses by the CD4TP281E cells relative to the CD4T cells for multiple independently generated sets of lines (Figures 6E and 6F).
Figure 6. The CD4 D3 Domain Contributes to TCR-CD3 Signaling.

(A–C) 5c.c7+ CD4T+ or CD4TP228E+F231E+ 58α−β− cells were cultured with M12 cells expressing tethered pMHCII for 16 hr. (A) Relative IL-2 production, (B) TCR downregulation, and (C) CD69 upregulation were assessed. Each data point represents an experiment with an independently generated cell line. Bars represent mean.
(D–F) 5c.c7+ CD4T+ or CD4TP281E+ 58α−β− cells were cultured with M12 cells expressing tethered pMHCII for 16 hr. (D) Relative IL-2 production for fluorescence-activated cell sorted (FACS) CD4T and CD4TP281E cells. Each point represents an independent experiment from one set of sorted cell lines. Bars represent mean. (E) TCR downregulation and (F) CD69 upregulation were assessed on unsorted cells that were CD4 expression-matched by flow cytometry. Each point represents an experiment with an independently generated cell line. Bars represent mean.
Relative IL-2 values were normalized to CD4T controls for each condition. CD69 upregulation and TCR downregulation data are presented relative to the Hb condition for each cell line. Data in (A)–(F) were from 3 experiments and analyzed using a paired Student’s t test (*p < 0.05, **p < 0.01, ***p < 0.001).
(G and H) 5c.c7+ CD4-Lck+, CD4P228E+F231E-Lck+, or CD4TΔbind-Lck+ 58α−β− cells were cultured with M12 cells expressing (G) MCC4A:I-Ek or (H) 2W4A:I-Ab for 16 hr.
IL-2 was measured by ELISA. Data are mean ± SEM of triplicate wells. Results shown are representative of four independent experiments. See also Figure S6.
As a final functional assay, we made 5c.c7+ 58α−β− cells expressing a CD4-Lck fusion, CD4TP228E+F231E-Lck, or CD4Δbind-Lck since we have reported that 5c.c7+ CD4-Lck+ 58α−β− cells make IL-2 in response to high expression levels of MHCII presenting “shaved” peptides with alanine substitutions in the TCR contact residues (Parrish et al., 2016). Our prior data suggest that the TCR and CD4 constantly work together to scan the peptide content of MHCII, so we reasoned that the CD4P228E+F231E should have a profound impact on TCR scanning if the macrocomplex model has merit. Indeed, for CD4-Lck and CD4P228E+F231E-Lck cells with equivalent expression, the mutation nearly ablated IL-2 production in response to the shaved MCC4A peptide in I-Ek or the shaved 2W4A peptide in I-Ab, while the CD4Δbind-Lck cells had no response (Figures 6G, 6H, and S6H).
D3 Domain Mutants Impair the Formation and Stability of the Macrocomplex
Next we asked whether the CD4TP228E+F231E and CD4TP281E mutants impact TCR-CD3 accumulation on immobile T102S: I-Ek surfaces. Both the CD4TP228E+F231E and CD4TP281E cells had reduced CD3δTG intensity compared to CD4T cells (Figure 7A).
Figure 7. Mutations in the CD4 D3 Domain Impact MacrocomplexAssembly and Stability.

(A–C) 5c.c7+ CD3T+ CD4T+, CD4TP228E+F231E+, CD4TP281E+, or CD4TΔbind+ M12 cells were analyzed for (A) CD3δTG intensity on T102S:I-Ek-coated surfaces in TIRF as in Figure 2, (B) CD3δTG::CD4TmCh FRETE on immobile MCC:I-Ek surfaces, and (C) FRAP of the indicated CD4T molecules adhered to MCC:I-Ek-coated surfaces as in Figure 3. Each dot represents an individual cell; black bars represent the mean value. n = number of cells analyzed per condition. Intensity and FRETE data were assessed for normality using a D’Agostino and Pearson omnibus normality test followed by a one-way analysis of variance (ANOVA) and Dunnet’s multiple comparison test (*p < 0.05, ****p < 0.0001). Results shown are representative of at least two experiments. See also Figure S7.
Since CD4 is closer to the CD3 subunits than predicted by a V-like arch (Glassman et al., 2016), we also used donor recovery after acceptor photobleaching to calculate the efficiency of FRETE between CD3δTG and CD4TmCh, CD4TP228E+F231E.mCh or CD4TP281E.mCh. M12 cells expressing the 5c.c7 TCR, CD3T subunits (including CD3δTG), and CD4TmCh or the mutants were used to reduce our analysis to interactions mediated by the ectodomains of CD4 and the TCR-CD3 complex in situ in the absence of signaling or other TCR engagement-associated events. Immobile MCC:I-Ek surfaces were used to maximize the formation of putative CD4 docking sites along the TCR-CD3-pMHC axis (Figures S7A and S7B). The lower level of CD4TP281E expression compared with CD4T or CD4TP228E+F231E cells was accounted for by analyzing subsets of cells matched for the intensity of CD3δTG and CD4TmCh (Figures S7D–S7G), as described previously (Glassman et al., 2016). Without pMHCII, CD3δTG::CD4TmCh FRETE was indistinguishable between the cell lines (Figure S7C), but on immobile MCC surfaces the CD4TP228E+F231E, CD4TP281E and CD4TΔbind cells showed reductions in FRETE compared with the CD4T cells (Figure 7B). The CD4TP228E+F231E cells indicate that the D3 domain is important for increasing the frequency of CD4 and TCR-CD3 complexes that come into close association upon interactions with agonist pMHCII. The CD4TP281E cells support this conclusion, although their tendency to aggregate (Figure S6C) means that fewer CD4TP281E molecules may be available to interact with the TCR-CD3 complex.
Finally, we used FRAP to determine how the CD4 mutants impact the kinetics of macrocomplex formation on immobile MCC:I-Ek surfaces since the higher-affinity TCR-pMHCII interactions should provide the highest resolving power. While the D3 domain mutations did not impact the mobile fraction, the hierarchy of t1/2 (CD4T > CD4TP228E+F231E > CD4TP281E > CD4TΔbind) mirrored the FRETE results (Figures 7C, S7H, and S7I).
These data further indicate that the D3 domain of CD4 is involved in the formation and stabilization of the TCR-CD3-pMHC-CD4 macrocomplex, albeit the same caveat holds for the CD4TP281E result here as above. Whether the CD4TP228E+F231E mutations disrupted interactions between CD4 and the TCR-CD3-pMHC axis mediated by the native residues, or induced allosteric changes in residues that mediate interactions cannot be known from the data provided here. The D3 hydrophobic core was not altered, so any allosteric changes should be localized to adjacent residues rather than propagated to a distant site. Altogether, the results suggest that the conserved nonpolar patch or proximal residues are involved in macrocomplex assembly and function.
DISCUSSION
CD4 has long been enigmatic. Originally, it was viewed as an accessory molecule until antibody-crosslinking experiments were interpreted to mean that the TCR and CD4 physically interact; the term coreceptor was then offered to define CD4 as a component of a multi-subunit receptor complex for pMHCII in order to distinguish it from a subsidiary function such as adhesion (Janeway, 1988). Mutagenesis data were subsequently taken as evidence for TCR-CD4 interactions (Vignali et al., 1996; Vignali and Vignali, 1999). However, the V-like crystal structures altered this thinking since the TCR and CD4 would not directly interact (Wang et al., 2001a; Yin et al., 2012). A model that could reconcile these discordant results was proposed whereby the TCR-CD3 complex pre-associates with the CD4 D3 and D4 domains and crosslinks one TCR-pMHCII assembly to another (Irvine et al., 2002; Krogsgaard et al., 2005). But recent FRET and super-resolution data show that the TCR-CD3 complex and CD4 do not pre-associate in the absence of coincident pMHCII engagement or adopt the spatial relationship predicted by that model (Glassman et al., 2016; Roh et al., 2015). Thus, fundamental questions persist regarding how CD4 fits and works within this multi-subunit molecular machine.
The data presented here do not conform with expectations regarding how the TCR and CD4 should behave if they were to bind pMHCII in a V-like arch, but they are consistent with pMHCII receptor signaling having converged on a mechanism akin to cytokine signaling (Kuhns and Badgandi, 2012; Madrenas et al., 1997). For example, binding of the cytokine-specific IL-4Rα subunit to IL-4 forms a composite docking interface to which the degenerate γc subunit binds with a higher affinity than to IL-4 alone; only then are the IL-4Rα and γc-associated Janus Kinases positioned in a spatial and temporal relationship that facilitates signaling (Wang et al., 2009). By analogy, our data are consistent with the TCR-CD3 complex serving as a specific receptor subunit for pMHCII, while CD4 represents a degenerate subunit with no appreciable affinity for pMHCII on its own. In this scenario, the constrained docking polarity observed for TCRs on pMHCII (Rossjohn et al., 2015) results in a highly reproducible composite surface along the TCR-CD3-pMHCII axis to which CD4 would bind with higher affinity than to MHCII alone. Docking would then position Lck in the proper spatial and temporal relationship with the CD3 ITAMs to allow for phosphorylation.
The most straightforward extracellular docking topology would follow an apparent shape complementarity between CD4 and the TCR-CD3-pMHCII axis (Figure S1A) since (1) structural data indicate that large-scale conformational changes in the TCR or CD3 ectodomains do not occur upon pMHCII or mitogenic mAb engagement; (2) experimental data point to a stable TCR-CD3 unit bound by continuous interactions between the ecto and transmembrane domains; and (3) FRET analysis indicate that CD4 is proximal to CD3δ (Call et al., 2002; Ding et al., 1999; Fernandes et al., 2012; Garcia et al., 1999; Glassman et al., 2016; Kuhns and Badgandi, 2012; Kuhns and Davis, 2007; Kuhns et al., 2010; Sun et al., 2001; Xu et al., 2006). An alternative hypothesis, akin to the permissive geometry model, is that TCR engagement moves the CD3 ectodomains to expose a CD4 contact site on the TCR (Minguet et al., 2007). Either way, initial CD4-MHCII interactions could allow a pivot from a V-like arch to a lower energy docked position via a ball-and-socket movement akin to the VH-CH elbow of antibodies (Lesk and Chothia, 1988). Or, docking may occur straight away. Importantly, once docked, an additional anchor could form via intracellular interactions between Lck and the CD3 subunits (Li et al., 2017; Mingueneau et al., 2008; Xu and Littman, 1993).
Prior studies suggest that macrocomplexes form constantly, regardless of the peptide content of an MHCII, can signal in response to very weak ligands on thymocytes and that both the TCR-pMHCII dwell time and the frequency of CD4-Lck interactions influence the signaling outcome (Kao and Allen, 2005; Parrish et al., 2016; Stepanek et al., 2014; Wang et al., 2001b). Here and elsewhere, CD4 minimally impacted agonist TCR-pMHCII interactions if only agonist pMHCII is present, indicating there is a TCR dwell time above which CD4 is more likely to cycle through these macrocomplexes than enhance TCR-pMHCII dwell time (Crawford et al., 1998; Hong et al., 2015; Huppa et al., 2010; O’Donoghue et al., 2013; Stepanek et al., 2014; Wooldridge et al., 2006). However, CD4 did influence TCR dwell time on agonist ligands diluted among null ligands—conditions that were not explored in prior studies— suggesting that CD4 can make a relevant contribution to TCR dwell time on agonist pMHCII. Our data also demonstrate that CD4 increases TCR dwell time on low-affinity pMHCII. These results are consistent with biomembrane force probe (BFP) 2D affinity measurements of the 3.L2 TCR that showed no contribution of CD4 to the agonist Hb:I-Ek pMHCII, but a significant contribution to binding of an antagonist (I72) pMHCII and a trend toward significance with a weak agonist ligand (T72; p = 0.06) (Hong et al., 2015). Importantly, we found the CD4 dwell time on pMHCII to be proportional to that of the TCR in a bulk assay, yet the CD4 t1/2 was always faster than the TCR; therefore, the kinetics of TCR-pMHCII interactions would influence the duration that a docking interface remains intact for CD4 molecules to cycle through in a processive-like manner (Stepanek et al., 2014). By increasing TCR-CD3 dwell time on lower-affinity pMHCII, CD4 might help recruit a greater breadth of clonotypes from the CD4+ T cell repertoire that differentiate to distinct effector phenotypes (Gottschalk et al., 2010; Tubo et al., 2013). This also appears likely to be key for signaling in response to the weak TCR-pMHCII interactions that drive CD4+ T cell development and homeostasis (Kao and Allen, 2005; Parrish et al., 2016; Stepanek et al., 2014; Wang et al., 2001b).
A final implication of this macrocomplex design is that it provides an explanation for how distinct constituents of the TCR repertoire can reproducibly direct the appropriate response to an immunological challenge. At issue is that if TCRs bind pMHCII in a noncanonical orientation, then the docking interface for CD4 would not be formed and any recruitment of Lck to the ITAMs by CD4 would be subject to the problems outlined above for a V-like arch with extraordinarily weak CD4-MHCII interactions. So, if two TCRs interact with the same pMHCII via the same kinetic properties but different docking modalities, then they would likely produce very different outcomes due to differences in both the spatial relationship between Lck and the ITAMs, and the duration with which they are held there. But it is now clear that there is a reliable relationship between the kinetics of TCR-pMHCII interactions and CD4+ T cell signaling, gene expression, and effector functions (Allison et al., 2016; Corse et al., 2010; Gottschalk et al., 2010; Govern et al., 2010; Kersh et al., 1998; Stepanek et al., 2014; Tubo et al., 2013; van Panhuys et al., 2014; Vanguri et al., 2013). Since the highly ordered macrocomplex imposes uniform requirements for assembly on a diverse collection of TCRs within the repertoire, the spatial relationship between Lck and each of the ten ITAMs will always be the same for any TCR that docks appropriately, as supported by our recent FRET analysis (Glassman et al., 2016). Consequently, the quantity and quality of ITAM phosphorylation by CD4-associated Lck would be determined strictly by the kinetic factors that influence the stability of the macrocomplex, and thus the duration of Lck proximity to any given ITAM, as suggested previously (Kersh et al., 1998). We thus postulate that the precise assembly of this molecular machine has evolved as an equalizer that ensures a reproducible translation of information, regarding specificity at the TCR-pMHCII interface, into scalable signaling at the ITAMs; in turn, the extent of ITAM phosphorylation would determine CD4+ T cell responses (Guy et al., 2013; Holst et al., 2008; Hwang et al., 2015). Future experiments will be required to interrogate how these molecular mechanisms influence cell-fate decisions in vivo.
EXPERIMENTAL PROCEDURES
Cell lines, constructs, standard experimental procedures, and previously described image acquisition and analysis can be found in Supplemental Experimental Procedures.
Mice
6- to 8-week-old male and female 5c.c7 TCR Tg mice were used for spleenocyte and thymocyte co-cultures. Mice were maintained under specific pathogen-free conditions in the University of Arizona animal facility. Experiments were conducted under the guidelines and approval of the University of Arizona Institutional Animal Care and Use Committee.
Image Analysis
For subunit accumulation, cells were bisected with a line scan ROI in bright field, and the median mEGFP intensity was exported using SlideBook6 (3I). Data were imported into Prism (GraphPad) and displayed as fluorescence intensity.
FRAP analysis was performed as described (Klammt et al., 2015). Background subtracted median fluorescence intensity in the photobleached regions and a 6.45-μm2 photobleach control region were exported for analysis in MATLAB (MathWorks). Photoablation was calculated as the ratio of post-bleach to prebleach fluorescence intensity, Abl = It = 0/It = −1 where t = −1 is the time point prior to bleaching and t = 0 is first frame postbleach. Analyzed events had photobleaching to <20% of prebleach intensity. Recovery (%) was computed for each time point (t = x) as Recovery = (It = x−It = 0)/(It = −1−It = 0)*100 where t = −1 is the time point prior to bleaching and t = 0 is first frame postbleach. FRAP data were fitted with a single-term exponential function, F = A(1-e−kt) where F is fluorescence intensity, A is the mobile fraction, t is the elapsed time following photobleaching and k is related to the half-life such that t1/2 = ln(2)/k.
Statistical Analysis
Statistical analyses were performed using Prism 6 (GraphPad software) and are indicated in the figure legends along with sample size (n). Data were assessed for normality using a D’Agostino and Pearson omnibus normality test. Data that were normally distributed were further analyzed using a one-way analysis of variance (ANOVA) and Dunnet’s multiple comparison test or a t test, as indicated in the figure legends. Nonparametric data were analyzed using a Mann-Whitney test. For FRAP analysis, fit and 95% confidence intervals (CIs) were generated using MATLAB nonlinear fitting (Klammt et al., 2015).
Supplementary Material
Highlights.
The affinity of TCR-pMHCII interactions impacts CD4 confinement to MHCII
CD4 increases the duration of TCR-pMHCII interactions in the absence of signaling
The CD4 D1 and D3 domains contribute to macrocomplex stability and functionality
Acknowledgments
We thank Morgan Huse, John Purdy, Lonnie Lybarger, Sam Campos, and Janko Nikolich-Zugich for critical feedback on the manuscript as well as members of the Frelinger, Schenten, and Wu labs for thoughtful comments. Jessica Seng, Karen Hernandez, Katrina Lichauco, and Joseph Wagner provided technical assistance. The UACC/ARL Cytometry Core Facility and the Cancer Center Support Grant (CCSG-CA 023074) supported our flow cytometry. M.S.K. is a Pew Scholar in the Biomedical Sciences, supported by The Pew Charitable Trusts. This work was also supported by the University of Arizona College of Medicine (M.S.K.), the Bio5 Institute (M.S.K.), and NIH/NIAID R01AI101053 (M.S.K.).
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and two movies and can be found with this article online at https://doi.org/10.1016/j.celrep.2017.12.104.
AUTHOR CONTRIBUTIONS
M.S.K. directed the research. C.R.G., H.L.P., and M.S.K. designed the experiments. C.R.G., H.L.P., and M.S.L. generated key reagents, analytical tools, and conducted the experiments. All authors analyzed the data, wrote the manuscript, and edited the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Allison KA, Sajti E, Collier JG, Gosselin D, Troutman TD, Stone EL, Hedrick SM, Glass CK. Affinity and dose of TCR engagement yield proportional enhancer and gene activity in CD4+ T cells. eLife. 2016;5 doi: 10.7554/eLife.10134. Published online July 4, 2016. https://doi.org/10.7554/eLife.10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: A further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Call ME, Pyrdol J, Wiedmann M, Wucherpfennig KW. The organizing principle in the formation of the T cell receptor-CD3 complex. Cell. 2002;111:967–979. doi: 10.1016/s0092-8674(02)01194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly JM, Hansen TH, Ingold AL, Potter TA. Recognition by CD8 on cytotoxic T lymphocytes is ablated by several substitutions in the class I alpha 3 domain: CD8 and the T-cell receptor recognize the same class I molecule. Proc Natl Acad Sci USA. 1990;87:2137–2141. doi: 10.1073/pnas.87.6.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corse E, Gottschalk RA, Krogsgaard M, Allison JP. Attenuated T cell responses to a high-potency ligand in vivo. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000481. Published online September 14, 2010. https://doi.org/10.1371/journal.pbio.1000481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford F, Kozono H, White J, Marrack P, Kappler J. Detection of antigen-specific T cells with multivalent soluble class II MHC covalent peptide complexes. Immunity. 1998;8:675–682. doi: 10.1016/s1074-7613(00)80572-5. [DOI] [PubMed] [Google Scholar]
- DeLano WL, Ultsch MH, de Vos AM, Wells JA. Convergent solutions to binding at a protein-protein interface. Science. 2000;287:1279–1283. doi: 10.1126/science.287.5456.1279. [DOI] [PubMed] [Google Scholar]
- Ding YH, Baker BM, Garboczi DN, Biddison WE, Wiley DC. Four A6-TCR/peptide/HLA-A2 structures that generate very different T cell signals are nearly identical. Immunity. 1999;11:45–56. doi: 10.1016/s1074-7613(00)80080-1. [DOI] [PubMed] [Google Scholar]
- Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol. 2009;10:375–384. doi: 10.1038/ni.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes RA, Shore DA, Vuong MT, Yu C, Zhu X, Pereira-Lopes S, Brouwer H, Fennelly JA, Jessup CM, Evans EJ, et al. T cell receptors are structures capable of initiating signaling in the absence of large conformational rearrangements. J Biol Chem. 2012;287:13324–13335. doi: 10.1074/jbc.M111.332783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia KC, Teyton L, Wilson IA. Structural basis of T cell recognition. Annu Rev Immunol. 1999;17:369–397. doi: 10.1146/annurev.immunol.17.1.369. [DOI] [PubMed] [Google Scholar]
- Gil D, Schamel WW, Montoya M, Sánchez-Madrid F, Alarcón B. Recruitment of Nck by CD3 epsilon reveals a ligand-induced conformational change essential for T cell receptor signaling and synapse formation. Cell. 2002;109:901–912. doi: 10.1016/s0092-8674(02)00799-7. [DOI] [PubMed] [Google Scholar]
- Glaichenhaus N, Shastri N, Littman DR, Turner JM. Requirement for association of p56lck with CD4 in antigen-specific signal transduction in T cells. Cell. 1991;64:511–520. doi: 10.1016/0092-8674(91)90235-q. [DOI] [PubMed] [Google Scholar]
- Glassman CR, Parrish HL, Deshpande NR, Kuhns MS. The CD4 and CD3δε cytosolic juxtamembrane regions are proximal within a compact TCR-CD3-pMHC-CD4 macrocomplex. J Immunol. 2016;196:4713–4722. doi: 10.4049/jimmunol.1502110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk RA, Corse E, Allison JP. TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. J Exp Med. 2010;207:1701–1711. doi: 10.1084/jem.20091999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govern CC, Paczosa MK, Chakraborty AK, Huseby ES. Fast on-rates allow short dwell time ligands to activate T cells. Proc Natl Acad Sci USA. 2010;107:8724–8729. doi: 10.1073/pnas.1000966107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy CS, Vignali KM, Temirov J, Bettini ML, Overacre AE, Smeltzer M, Zhang H, Huppa JB, Tsai YH, Lobry C, et al. Distinct TCR signaling pathways drive proliferation and cytokine production in T cells. Nat Immunol. 2013;14:262–270. doi: 10.1038/ni.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst J, Wang H, Eder KD, Workman CJ, Boyd KL, Baquet Z, Singh H, Forbes K, Chruscinski A, Smeyne R, et al. Scalable signaling mediated by T cell antigen receptor-CD3 ITAMs ensures effective negative selection and prevents autoimmunity. Nat Immunol. 2008;9:658–666. doi: 10.1038/ni.1611. [DOI] [PubMed] [Google Scholar]
- Hong J, Persaud SP, Horvath S, Allen PM, Evavold BD, Zhu C. Force-regulated in situ TCR-peptide-bound MHC class II kinetics determine functions of CD4+ T cells. J Immunol. 2015;195:3557–3564. doi: 10.4049/jimmunol.1501407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppa JB, Axmann M, Mörtelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, Klein LO, Schütz GJ, Davis MM. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963–967. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, Palin AC, Li L, Song KD, Lee J, Herz J, Tubo N, Chu H, Pepper M, Lesourne R, et al. TCR ITAM multiplicity is required for the generation of follicular helper T-cells. Nat Commun. 2015;6:6982. doi: 10.1038/ncomms7982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine DJ, Purbhoo MA, Krogsgaard M, Davis MM. Direct observation of ligand recognition by T cells. Nature. 2002;419:845–849. doi: 10.1038/nature01076. [DOI] [PubMed] [Google Scholar]
- Janeway CA., Jr T-cell development. Accessories or coreceptors? Nature. 1988;335:208–210. doi: 10.1038/335208a0. [DOI] [PubMed] [Google Scholar]
- Jönsson P, Southcombe JH, Santos AM, Huo J, Fernandes RA, McColl J, Lever M, Evans EJ, Hudson A, Chang VT, et al. Remarkably low affinity of CD4/peptide-major histocompatibility complex class II protein interactions. Proc Natl Acad Sci USA. 2016;113:5682–5687. doi: 10.1073/pnas.1513918113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao H, Allen PM. An antagonist peptide mediates positive selection and CD4 lineage commitment of MHC class II-restricted T cells in the absence of CD4. J Exp Med. 2005;201:149–158. doi: 10.1084/jem.20041574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersh GJ, Kersh EN, Fremont DH, Allen PM. High- and low-potency ligands with similar affinities for the TCR: The importance of kinetics in TCR signaling. Immunity. 1998;9:817–826. doi: 10.1016/s1074-7613(00)80647-0. [DOI] [PubMed] [Google Scholar]
- Killeen N, Littman DR. Helper T-cell development in the absence of CD4-p56lck association. Nature. 1993;364:729–732. doi: 10.1038/364729a0. [DOI] [PubMed] [Google Scholar]
- Klammt C, Novotná L, Li DT, Wolf M, Blount A, Zhang K, Fitchett JR, Lillemeier BF. T cell receptor dwell times control the kinase activity of Zap70. Nat Immunol. 2015;16:961–969. doi: 10.1038/ni.3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König R, Huang LY, Germain RN. MHC class II interaction with CD4 mediated by a region analogous to the MHC class I binding site for CD8. Nature. 1992;356:796–798. doi: 10.1038/356796a0. [DOI] [PubMed] [Google Scholar]
- Krogsgaard M, Prado N, Adams EJ, He XL, Chow DC, Wilson DB, Garcia KC, Davis MM. Evidence that structural rearrangements and/or flexibility during TCR binding can contribute to T cell activation. Mol Cell. 2003;12:1367–1378. doi: 10.1016/s1097-2765(03)00474-x. [DOI] [PubMed] [Google Scholar]
- Krogsgaard M, Li QJ, Sumen C, Huppa JB, Huse M, Davis MM. Agonist/endogenous peptide-MHC heterodimers drive T cell activation and sensitivity. Nature. 2005;434:238–243. doi: 10.1038/nature03391. [DOI] [PubMed] [Google Scholar]
- Kuhns MS, Badgandi HB. Piecing together the family portrait of TCR-CD3 complexes. Immunol Rev. 2012;250:120–143. doi: 10.1111/imr.12000. [DOI] [PubMed] [Google Scholar]
- Kuhns MS, Davis MM. Disruption of extracellular interactions impairs T cell receptor-CD3 complex stability and signaling. Immunity. 2007;26:357–369. doi: 10.1016/j.immuni.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Kuhns MS, Girvin AT, Klein LO, Chen R, Jensen KD, Newell EW, Huppa JB, Lillemeier BF, Huse M, Chien YH, et al. Evidence for a functional sidedness to the alphabetaTCR. Proc Natl Acad Sci USA. 2010;107:5094–5099. doi: 10.1073/pnas.1000925107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Glassman CR, Deshpande NR, Badgandi HB, Parrish HL, Uttamapinant C, Stawski PS, Ting AY, Kuhns MS. A mechanical switch couples T cell receptor triggering to the cytoplasmic juxta-membrane regions of CD3ζζ. Immunity. 2015;43:227–239. doi: 10.1016/j.immuni.2015.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesk AM, Chothia C. Elbow motion in the immunoglobulins involves a molecular ball-and-socket joint. Nature. 1988;335:188–190. doi: 10.1038/335188a0. [DOI] [PubMed] [Google Scholar]
- Li L, Guo X, Shi X, Li C, Wu W, Yan C, Wang H, Li H, Xu C. Ionic CD3-Lck interaction regulates the initiation of T-cell receptor signaling. Proc Natl Acad Sci USA. 2017;114:E5891–E5899. doi: 10.1073/pnas.1701990114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrenas J, Chau LA, Smith J, Bluestone JA, Germain RN. The efficiency of CD4 recruitment to ligand-engaged TCR controls the agonist/partial agonist properties of peptide-MHC molecule ligands. J Exp Med. 1997;185:219–229. doi: 10.1084/jem.185.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malissen B, Bongrand P. Early T cell activation: Integrating biochemical, structural, and biophysical cues. Annu Rev Immunol. 2015;33:539–561. doi: 10.1146/annurev-immunol-032414-112158. [DOI] [PubMed] [Google Scholar]
- Mingueneau M, Sansoni A, Grégoire C, Roncagalli R, Aguado E, Weiss A, Malissen M, Malissen B. The proline-rich sequence of CD3epsilon controls T cell antigen receptor expression on and signaling potency in preselection CD4+CD8+ thymocytes. Nat Immunol. 2008;9:522–532. doi: 10.1038/ni.1608. [DOI] [PubMed] [Google Scholar]
- Minguet S, Swamy M, Alarcón B, Luescher IF, Schamel WW. Full activation of the T cell receptor requires both clustering and conformational changes at CD3. Immunity. 2007;26:43–54. doi: 10.1016/j.immuni.2006.10.019. [DOI] [PubMed] [Google Scholar]
- Newell EW, Ely LK, Kruse AC, Reay PA, Rodriguez SN, Lin AE, Kuhns MS, Garcia KC, Davis MM. Structural basis of specificity and cross-reactivity in T cell receptors specific for cytochrome c-I-E(k) J Immunol. 2011;186:5823–5832. doi: 10.4049/jimmunol.1100197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donoghue GP, Pielak RM, Smoligovets AA, Lin JJ, Groves JT. Direct single molecule measurement of TCR triggering by agonist pMHC in living primary T cells. eLife. 2013;2:e00778. doi: 10.7554/eLife.00778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish HL, Glassman CR, Keenen MM, Deshpande NR, Bronnimann MP, Kuhns MS. A transmembrane domain GGxxG motif in CD4 contributes to its Lck-independent function but does not mediate CD4 dimerization. PLoS ONE. 2015;10:e0132333. doi: 10.1371/journal.pone.0132333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish HL, Deshpande NR, Vasic J, Kuhns MS. Functional evidence for TCR-intrinsic specificity for MHCII. Proc Natl Acad Sci USA. 2016;113:3000–3005. doi: 10.1073/pnas.1518499113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh KH, Lillemeier BF, Wang F, Davis MM. The coreceptor CD4 is expressed in distinct nanoclusters and does not colocalize with T-cell receptor and active protein tyrosine kinase p56lck. Proc Natl Acad Sci USA. 2015;112:E1604–E1613. doi: 10.1073/pnas.1503532112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossjohn J, Gras S, Miles JJ, Turner SJ, Godfrey DI, McCluskey J. T cell antigen receptor recognition of antigen-presenting molecules. Annu Rev Immunol. 2015;33:169–200. doi: 10.1146/annurev-immunol-032414-112334. [DOI] [PubMed] [Google Scholar]
- Savage PA, Boniface JJ, Davis MM. A kinetic basis for T cell receptor repertoire selection during an immune response. Immunity. 1999;10:485–492. doi: 10.1016/s1074-7613(00)80048-5. [DOI] [PubMed] [Google Scholar]
- Spangler JB, Moraga I, Mendoza JL, Garcia KC. Insights into cytokine-receptor interactions from cytokine engineering. Annu Rev Immunol. 2015;33:139–167. doi: 10.1146/annurev-immunol-032713-120211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanek O, Prabhakar AS, Osswald C, King CG, Bulek A, Naeher D, Beaufils-Hugot M, Abanto ML, Galati V, Hausmann B, et al. Coreceptor scanning by the T cell receptor provides a mechanism for T cell tolerance. Cell. 2014;159:333–345. doi: 10.1016/j.cell.2014.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun ZJ, Kim KS, Wagner G, Reinherz EL. Mechanisms contributing to T cell receptor signaling and assembly revealed by the solution structure of an ectodomain fragment of the CD3 epsilon gamma heterodimer. Cell. 2001;105:913–923. doi: 10.1016/s0092-8674(01)00395-6. [DOI] [PubMed] [Google Scholar]
- Tubo NJ, Pagán AJ, Taylor JJ, Nelson RW, Linehan JL, Ertelt JM, Huseby ES, Way SS, Jenkins MK. Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell. 2013;153:785–796. doi: 10.1016/j.cell.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Panhuys N, Klauschen F, Germain RN. T-cell-receptor-dependent signal intensity dominantly controls CD4(+) T cell polarization In Vivo. Immunity. 2014;41:63–74. doi: 10.1016/j.immuni.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanguri V, Govern CC, Smith R, Huseby ES. Viral antigen density and confinement time regulate the reactivity pattern of CD4 T-cell responses to vaccinia virus infection. Proc Natl Acad Sci USA. 2013;110:288–293. doi: 10.1073/pnas.1208328110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal K, Daniel C, Hill M, Littman DR, Allen PM. Differential requirements for CD4 in TCR-ligand interactions. J Immunol. 1999;163:4811–4818. [PubMed] [Google Scholar]
- Vignali DA, Vignali KM. Profound enhancement of T cell activation mediated by the interaction between the TCR and the D3 domain of CD4. J Immunol. 1999;162:1431–1439. [PubMed] [Google Scholar]
- Vignali DA, Carson RT, Chang B, Mittler RS, Strominger JL. The two membrane proximal domains of CD4 interact with the T cell receptor. J Exp Med. 1996;183:2097–2107. doi: 10.1084/jem.183.5.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH, Meijers R, Xiong Y, Liu JH, Sakihama T, Zhang R, Joachimiak A, Reinherz EL. Crystal structure of the human CD4 N-terminal two-domain fragment complexed to a class II MHC molecule. Proc Natl Acad Sci USA. 2001a;98:10799–10804. doi: 10.1073/pnas.191124098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Strong J, Killeen N. Homeostatic competition among T cells revealed by conditional inactivation of the mouse Cd4 gene. J Exp Med. 2001b;194:1721–1730. doi: 10.1084/jem.194.12.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lupardus P, Laporte SL, Garcia KC. Structural biology of shared cytokine receptors. Annu Rev Immunol. 2009;27:29–60. doi: 10.1146/annurev.immunol.24.021605.090616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooldridge L, Scriba TJ, Milicic A, Laugel B, Gostick E, Price DA, Phillips RE, Sewell AK. Anti-coreceptor antibodies profoundly affect staining with peptide-MHC class I and class II tetramers. Eur J Immunol. 2006;36:1847–1855. doi: 10.1002/eji.200635886. [DOI] [PubMed] [Google Scholar]
- Wu LC, Tuot DS, Lyons DS, Garcia KC, Davis MM. Two-step binding mechanism for T-cell receptor recognition of peptide MHC. Nature. 2002;418:552–556. doi: 10.1038/nature00920. [DOI] [PubMed] [Google Scholar]
- Xu H, Littman DR. A kinase-independent function of Lck in potentiating antigen-specific T cell activation. Cell. 1993;74:633–643. doi: 10.1016/0092-8674(93)90511-n. [DOI] [PubMed] [Google Scholar]
- Xu C, Call ME, Wucherpfennig KW. A membrane-proximal tetracysteine motif contributes to assembly of CD3deltaepsilon and CD3gammaepsilon dimers with the T cell receptor. J Biol Chem. 2006;281:36977–36984. doi: 10.1074/jbc.M607164200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Wang XX, Mariuzza RA. Crystal structure of a complete ternary complex of T-cell receptor, peptide-MHC, and CD4. Proc Natl Acad Sci USA. 2012;109:5405–5410. doi: 10.1073/pnas.1118801109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuñiga-Pflücker JC, McCarthy SA, Weston M, Longo DL, Singer A, Kruisbeek AM. Role of CD4 in thymocyte selection and maturation. J Exp Med. 1989;169:2085–2096. doi: 10.1084/jem.169.6.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.