

A Rare Tumor Clinic, emphasizing precision medicine, was recently initiated. This article describes preliminary experiences at the clinic and reports a case highlighting the successful use of ErbB2‐targeting therapy in an ultrarare tumor.
Keywords: Precision therapy, Targeted therapy, Genomics, Protein analyses, Rare tumors
Abstract
Background.
Patients with rare tumors may lack approved treatments and clinical trial access. Although each rare tumor is uncommon, cumulatively they account for approximately 25% of cancers. We recently initiated a Rare Tumor Clinic that emphasized a precision medicine strategy.
Materials and Methods.
We investigated the first 40 patients presenting at the Rare Tumor Clinic. Next‐generation sequencing (NGS) of tissue and plasma‐derived, circulating‐tumor DNA (ctDNA), and protein markers were assessed.
Results.
Median age was 58 years (range, 31–78 years); 70% (28/40) were women; median number of previous systemic therapies was 2 (range 0–7). The most common diagnoses were sarcoma (n = 7) for solid tumors and Erdheim‐Chester disease (n = 5) for hematologic malignancies. Twenty distinct diagnoses were seen. Examples of ultrarare tumors included ameloblastoma, yolk sac liver tumor, ampullary cancer, and Castleman's disease. Altogether, 32 of 33 patients (97%) with tissue NGS and 15 of 33 (45%) with ctDNA sequencing harbored ≥1 alteration. Overall, 92.5% of patients (37/40) had ≥1 actionable target based on either genomic (n = 32) or protein (n = 27) markers. In total, 52.5% (21/40) received matched therapy; 52.4% (11/21) achieved stable disease (SD) ≥6 months (n = 3), partial remission (PR; n = 6), or complete remission (CR; n = 2). Matched therapy resulted in significantly longer progression‐free survival compared with last prior unmatched therapy (hazard ratio 0.26, 95% confidence interval 0.10–0.71, p = .008).
Conclusion.
Identifying genomic and protein markers in patients with rare/ultrarare tumors was feasible. When therapies were matched, >50% of patients attained SD ≥6 months, PR, or CR. Further precision medicine clinical investigations focusing on rare and ultrarare tumors are urgently needed.
Implications for Practice.
Although rare tumors are infrequent by definition, when all subtypes of rare cancers are combined, they account for approximately 25% of adult malignancies. However, patients with rare tumors may lack approved treatments and clinical trial access. This paper describes an institutional a Rare Tumor Clinic focused on a precision medicine strategy. Performing genomics and protein analyses was feasible amongst patients with rare cancers. Over 50% of patients attained SD ≥6 months, PR, or CR when they received matched therapy (genomically targeted and/or immunotherapy). Further studies investigating the efficacy of the precision therapy approach among rare tumors are warranted.
Introduction
In 2012, there were 14.1 million new cases of cancer and 8.2 million cancer‐related deaths worldwide, making cancer one of the most common causes of death [1]. Among diverse cancer subtypes, certain cancers fall into the category of rare tumors. The definition of rare tumors differs depending on the country. For example, in the U.S., rare tumors are defined as those with an incidence of fewer than 15 cases per 100,000 per year; in Europe and Japan, 6 cases per 100,000 per year; however, incidence of fewer than 15 cases per 100,000 per year is a widely accepted definition [2], [3], [4], [5]. Among rare tumors, cancers with prevalence of fewer than 2,000 or incidence of fewer than 2 cases per 100,000 are referred to as ultrarare tumors [6], [7].
Although each type of rare tumor is uncommon by definition, when all subtypes of rare cancers are combined, they account for 22%–25% of all adult tumors [2], [3]. Hence the overall burden of rare tumors is significant. Clinical management of rare malignancies can be challenging due to lack of information, which can lead to difficulty making the diagnosis, as well as a shortage of therapeutic options that are approved by the U.S. Food and Drug Administration (FDA) and experimental options in clinical trials. Rare cancers can also be scientifically challenging to study, and substantial parts of the literature regarding rare cancers are case reports, single‐institution case series, or smaller multicenter case series rather than phase III randomized trials [3]. Thus, patients with rare cancers tend to lack novel therapeutic approaches such as those with a targeted therapy. Conceivably due to these limitations, patients with rare tumors are reported to have lower 5‐year overall survival when compared with those with common tumors (47% vs. 66%) [2]. To overcome these challenges, in‐depth understanding of the biology of rare tumors is required.
Rapid technological advancements have revolutionized the way we evaluate and diagnose patients’ cancer. The standard evaluation is based on the light microscope, but more recently the “molecular microscope,” which includes comprehensive genomic interrogation by techniques such as next‐generation sequencing (NGS), transcriptomics, and protein analyses, has been exploited [8]. Consequently, the way clinical trials are conducted is also transforming from a “one‐size‐fits‐all” approach to a more customized, precision strategy that incorporates basket or umbrella trials, each of which are applicable even in patients with rare cancers. (A basket trial focuses on a specific mutation across multiple cancer types; an umbrella trial focuses on a specific type of cancer with treatment based on any one of multiple molecular alterations as assessed by genomic profiling.) One example is the basket/umbrella study using imatinib among diverse cancers known to express imatinib‐sensitive tyrosine kinases [9]. This study showed responses among multiple rare malignancies, including dermatofibrosarcoma protuberans (response rate [RR] 83%, targeting PDGFRB), hypereosinophilic syndrome (RR 43%, targeting PDGFRA/KIT), myeloproliferative disorders (RR 57%, targeting PDGFRB), and systemic mastocytosis (RR 20%, targeting PDGFRA/KIT), which facilitated FDA approval of imatinib for these rare and ultrarare disease conditions [9]. Furthermore, a basket trial with vemurafenib in BRAF V600 mutation‐positive cancers also demonstrated clinical benefit among patients with rare cancers (RR of 43% for Erdheim‐Chester disease or Langerhans’ cell histiocytosis and RR of 29% for anaplastic thyroid cancer) [10]. Moreover, accumulating evidence from clinical trials and large‐scale meta‐analyses suggests that the matched targeted therapy approach (biomarker‐based) can achieve better outcomes when compared with a non‐biomarker‐based approach [11], [12], [13], [14].
Based on the unmet need for novel treatments for patients with rare cancers, we have initiated a Rare Tumor Clinic that emphasized a precision medicine strategy utilizing genomic and protein analysis to guide individualized therapy. Herein, we report our preliminary experience across patients as well as an illustrative case highlighting the successful use of ErbB2‐targeting therapy in an ultrarare tumor.
Materials and Methods
Patients
We investigated clinical characteristics and treatment outcomes among patients presenting at the Rare Tumor Clinic at the University of California, San Diego (UCSD) Moores Cancer Center (n = 40). This study was performed in accordance with the guidelines of the UCSD Internal Review Board (PREDICT [Profile Related Evidence Determining Individualized Cancer Therapy] protocol; NCT02478931) an investigational studies for which the patients gave consent.
Target Identification Through Next‐Generation Sequencing and Protein Analysis
When available, we performed NGS on both tissue and plasma (circulating‐tumor DNA [ctDNA]) to seek actionable genomic alterations. Protein markers were also analyzed as appropriate.
The majority of tissue NGS were performed at Foundation Medicine with assay panels of 236 or 315 genes according to previously reported methods in a laboratory certified by Clinical Laboratory Improvement Amendments (CLIA; n = 31; Cambridge, MA, www.foundationmedicine.com) [15], [16], [17]. This method of sequencing allows for detection of copy number alterations, gene rearrangements, and somatic mutations with 99% specificity and >99% sensitivity for base substitutions at ≥5 mutant allele frequency and >95% sensitivity for copy number alterations. A threshold of ≥8 copies for gene amplification was used. A smaller subset of patients had tissue NGS done using other platforms, including UCSD (n = 2, 397 genes), NantOmics (n = 2, 434 genes; Culver City, CA, http://www.nantomics.com/), and Washington University (n = 1; St. Louis, MO, http://gps.wustl.edu/patient-care/sequencing-tests/).
Blood‐derived ctDNA analysis was performed by Guardant Health (n = 33; Redwood City, CA, www.guardanthealth.com), a CLIA‐certified laboratory, with assay panels of 54, 68, or 70 genes, as previously described [18]. All ctDNA was sequenced, including somatic ctDNA and the germline ctDNA derived from leukocyte lysis. Germline alterations were filtered out and not reported. The assay reports single nucleotide variants in all genes and selected copy number amplifications, fusions, and indel events [18].
Most protein analyses with immunohistochemistry (IHC) were performed at Caris Life Sciences (n = 25; Irving, TX, www.carismolecularintelligence.com). The IHC markers to be tested were selected by the treating physician. Selected protein death‐ligand 1 (PD‐L1) testing was performed through Pathline (n = 5) or Emerge (n = 11; Ramsey, NJ, www.pathline-emerge.com/).
Endpoints and Statistical Methods
Descriptive statistics were used to summarize the patient characteristics. Progression‐free survival (PFS) was measured using the method of Kaplan and Meier [19] and defined as the time interval between the start of therapy and the date of disease progression or removal from therapy for any reason, whichever occurred first. Patients who were progression‐free (for PFS) at the time of last follow‐up were censored on that date. Response to therapy was evaluated using the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 [20].
Results
Patient Characteristics
Among 40 patients who presented to the Rare Tumor Clinic, the median age was 58 years (range 31–78 years). Seventy percent of patients were women (28/40). The median number of previous systemic therapies was 2 (range 0–7). The most common diagnosis was sarcoma (n = 7, 17.5%) followed by Erdheim‐Chester disease (12.5% [5/40]), Castleman's disease (10% [4/40]) and high‐grade serous ovarian cancer (10% [4/40]). Overall, 20 distinct diagnoses were seen. Thirty patients had ultrarare tumors, including ameloblastoma, yolk sac liver tumor, ampullary cancer, Castleman's disease, and desmoid tumor (Table 1 and supplemental online Table 1).
Table 1. Patient characteristics (n = 40).
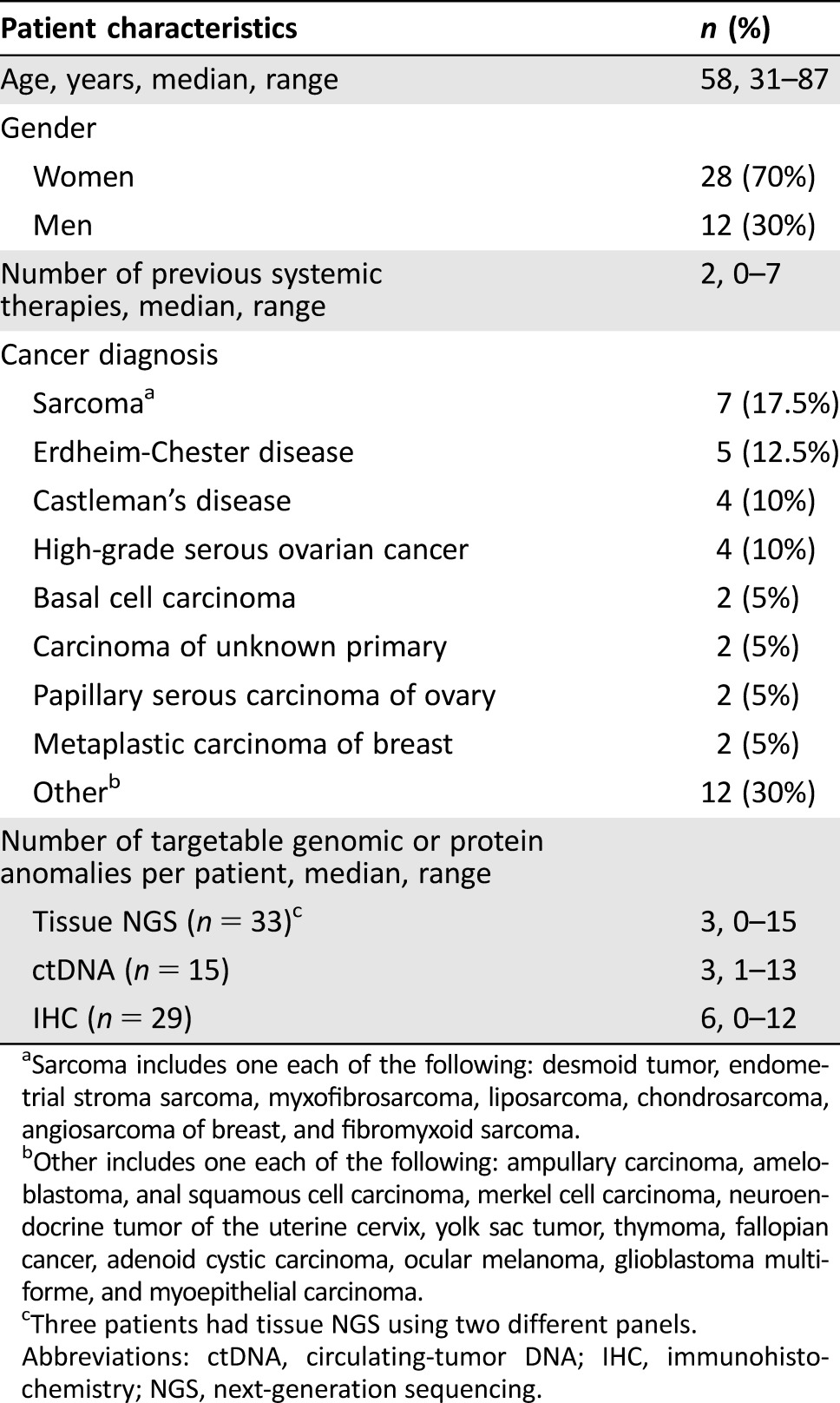
Sarcoma includes one each of the following: desmoid tumor, endometrial stroma sarcoma, myxofibrosarcoma, liposarcoma, chondrosarcoma, angiosarcoma of breast, and fibromyxoid sarcoma.
Other includes one each of the following: ampullary carcinoma, ameloblastoma, anal squamous cell carcinoma, merkel cell carcinoma, neuroendocrine tumor of the uterine cervix, yolk sac tumor, thymoma, fallopian cancer, adenoid cystic carcinoma, ocular melanoma, glioblastoma multiforme, and myoepithelial carcinoma.
Three patients had tissue NGS using two different panels.
Abbreviations: ctDNA, circulating‐tumor DNA; IHC, immunohistochemistry; NGS, next‐generation sequencing.
Target Identification with Tissue NGS, ctDNA, and IHC Among Patients with Rare Tumors
Among 40 patients who presented to the Rare Tumor Clinic, 37 patients (92.5%) had at least one theoretically actionable target (by either an FDA‐approved or an investigational agent) detected by either tissue NGS, ctDNA, or IHC or similar test (supplemental online Tables 1, 2, and 3).
Tissue NGS.
Among 40 patients with rare tumors, 33 patients underwent tissue NGS. Most patients had tissue NGS through Foundation Medicine (n = 31); others had NGS through the laboratory at NantOmics (n = 2), UCSD (n = 2), and Washington University (n = 1). Three patients had NGS using two different panels. (See Materials and Methods section for assay details.) Among those 33 patients, the most common alteration was in the TP53 gene (45.5% [15/33]), followed by CDKN2A/B loss (12.1% [4/33]), FRS2 amplification (12.1% [4/33]), MDM2 amplification (12.1% [4/33]), RB1 alteration (12.1% [4/33]), and KRAS alteration (12.1% [4/33]; Fig. 1A and supplemental online Table 4). The median number of alterations detected per patient was 3 (range 0–24; Table 1 and supplemental online Table 1). The median number of alterations that were potentially targetable with either an FDA‐approved or an investigational agent was 3 per patient (range 0–15; supplemental online Tables 1 and 3). Among 33 patients who had tissue NGS, 32 patients had theoretically actionable aberrations. One patient, ID13 (supplemental online Tables 1 and 3), had BRAF V600E mutation detected by polymerase chain reaction, but tissue NGS failed to reveal the same mutation. Of the 33 patients, 32 (97%) had an alteration targetable by an FDA‐approved drug (supplemental online Tables 1 and 3).
Figure 1.

Genomic and protein analyses among patients presented at Rare Tumor Clinic (n = 40). (A): Frequency of genomic alterations detected by tissue next‐generation sequencing (236 to 434 genes) in the Rare Tumor Clinic (n = 33). Included gene alterations with n ≥2. Among 33 patients who had tissue next‐generation sequencing, the most common alteration was TP53 alteration (45.5% [15/33]), followed by CDKN2A/B loss (12.1% [4/33]), FRS2 amplification (12.1% [4/33]), MDM2 amplification (12.1% [4/33]), RB1 alteration (12.1% [4/33]), and KRAS alteration (12.1% [4/33]; supplemental online Table 4). (B): Frequency of characterized genomic alterations detected by circulating‐tumor DNA (ctDNA) in the Rare Tumor Clinic (Guardant Health; 54 to 70 genes; n = 33). Among 33 patients who had ctDNA evaluation, the most common alteration was TP53 alteration (21.2% [7/33]), followed by BRAF amplification (18.2% [6/33]), MYC amplification (15.2% [5/33]), and MET amplification (12.1% [4/33]; supplemental online Table 5). (C): Frequency of protein aberrations detected via immunohistochemistry in the Rare Tumor Clinic (n = 29). Among 29 patients who had immunohistochemistry, the most common potentially actionable marker was RRM1 negative (81.0% [17/21]), followed by ERCC1 negative (70.8% [17/24]), TLE3 positive (70.6% [12/17]), and TOPO1 positive (66.7% [16/24]). Supplemental online Table 2 shows abbreviations and implications of each protein marker.
Blood‐Derived ctDNA.
ctDNA was evaluated in 33 patients using panels of 54 to 70 genes (see Materials and Methods). Among those 33 patients, 15 had detectable, nonsynonymous, characterized alterations. The most common alteration was in the TP53 gene (21.2% [7/33]), followed by BRAF amplification (18.2% [6/33]), MYC amplification (15.2% [5/33]), and MET amplification (12.1% [4/33]; Fig. 1B and supplemental online Table 5). Among 15 patients with detectable ctDNA alterations, the median number of alterations detected per patient was 3 (range 1–14; Table 1 and supplemental online Table 1). The median number of alterations that were potentially targetable with either an FDA‐approved or an investigational agent was 3 per patient (range 1–13; Table 1 and supplemental online Tables 1 and 3). Of the 15 patients who were found to have alterations detected by ctDNA, all 15 had ≥1 alterations potentially targetable by an FDA‐approved drug. Of the 33 patients evaluated for ctDNA, 27 patients had both ctDNA and tissue NGS analyses. Concordances between ctDNA and tissue NGS for commonly altered genes were 66.7% (18/27) for TP53, 74.1% (20/27) for BRAF, 88.9% (24/27) for MYC, and 85.2% (23/27) for MET alterations (supplemental online Table 6).
Protein Immunohistochemistry.
Among patients who had IHC testing, most had IHC performed by Caris Life Sciences (n = 25). Occasionally, PD‐L1 testing was performed through Pathline (22C3 antibody; n = 5) or Emerge (SP142 antibody; n = 11). Altogether, 29 patients had IHC testing, 27 of whom were found to have potentially actionable IHC results (Table 1). Two patients (IDs 20 and 28, Table 1) who did not have actionable IHC only had PD‐L1 testing, which was negative. The most common potentially actionable IHC result was RRM1 negative (81.0% [17/21]), followed by ERCC1 negative (70.8% [17/24]), TLE3 positive (70.6% [12/17]), and TOPO1 positive (66.7% [16/24]; Fig. 1C and supplemental online Table 2 show implications of test results). Among 29 patients, the median number of hypothetically druggable IHC results per patient was 6 (range 0–12; Table 1, supplemental online Tables 1 and 2). All IHC results were potentially targetable with FDA‐approved agents (supplemental online Table 2).
Clinical Outcome Among Patients Who Presented at Rare Tumor Clinic
Among patients who presented to the Rare Tumor Clinic (n = 40), 21 received matched targeted therapy and were assessable for response (Fig. 2, supplemental online Table 3, and supplemental online Fig. 1). Other patients (n = 19) were not included in response assessment due to the following reasons: treatment had not yet been started or it was too early for first response assessment (n = 6), underlying disease was stable or in remission with prior systemic therapy (n = 6), the patient had undergone surgical management and was on surveillance (n = 4), or the patient had poor performance status that obviated being treated (n = 3).
Figure 2.

Waterfall (A) and swimmer plot (B) among patients who received matched therapy in the Rare Tumor Clinic (n = 21). Patient ID corresponds to supplemental online Tables 1 and 3, which describe the genomic/protein markers and matched targeted therapy patients received. Supplemental online Figure 1 shows 3D‐waterfall plot corresponding to waterfall and swimmer plot.
Abbreviation: ID, identification.
Among 21 patients who underwent matched targeted therapy, 14.3% (3/21) attained SD (stable disease) ≥6 months, 28.6% (6/21) had partial response (PR), and 9.5% (2/21) achieved complete response (CR), for a total of 52.4% of patients with SD ≥6 months, PR, or CR. Median PFS with matched therapy was 19.6 months (range 0.99+ to 26.1+ months) (Fig. 2, supplemental online Table 3, and supplemental online Fig. 1).
Twelve of the 21 patients were evaluable to compare the therapeutic outcome of matched therapy with last prior unmatched therapy. (Nine were not evaluable for the following reasons: matched therapy was the first‐line therapy [n = 5], prior therapy was matched therapy [n = 3], and prior therapy was given in an adjuvant setting [n = 1].) Among those evaluable patients who could be assessed for matched versus last prior unmatched therapy, matched therapy had statistically significant improvement in PFS when compared with last prior unmatched therapy (hazard ratio [HR] 0.26, 95% confidence interval [CI] 0.10–0.71, p = .008, median PFS 19.7 vs. 3.5 months; Fig. 3). Eight of 12 patients (66.7%) had PFS ratio of ≥1.3 (range 0.23–5.60; PFS of matched therapy divided by PFS of last prior unmatched therapy) [21] (supplemental online Fig. 2). On the other hand, no patient achieved PFS ratio of ≥1.3 when subsequent unmatched therapy was administered (range 0.17–1.19; n = 6 were evaluable).
Figure 3.

Progression‐free survival (PFS) from matched therapy in the Rare Tumor Clinic versus last prior unmatched therapy (n = 12). Twelve individuals had available data for the comparison between matched therapy and last prior unmatched therapy. Median PFS was 19.7 months for matched therapy and 3.5 months for last prior unmatched therapy (HR 0.26, 95% CI 0.10–0.71, p = .008).
Abbreviations: CI, confidence interval; HR, hazard ratio.
Patient with Ampullary Carcinoma Managed with Matched Therapy Based on Tissue DNA
A 68‐year‐old woman with ampullary carcinoma presented with recurrent disease of the lung 5 years after completing perioperative management with neoadjuvant and adjuvant chemotherapy. Biopsy of the lung mass confirmed metastatic ampullary carcinoma. Further analysis with tissue NGS revealed multiple alterations, including ERBB2 amplification (supplemental online Table 1, ID #1). Matched therapy with a combination of trastuzumab and pertuzumab was initiated after consent (protocol: My Pathway, NCT02091141). The patient achieved a partial response (59% reduction per RECIST 1.1). Treatment is ongoing after 15+ months of therapy (Fig. 4). There was no significant toxicity.
Figure 4.

Patient with ampullary carcinoma and ERBB2 amplification treated with anti‐human epidermal growth receptor 2 therapy (trastuzumab and pertuzumab). A 68‐year‐old woman with ampullary carcinoma presented with recurrent disease to lung 5 years after the completion of perioperative therapy with neoadjuvant chemotherapy (5‐fluorouracil, irinotecan, and oxaliplatin) followed by Whipple procedure and adjuvant 5‐ fluorouracil. Biopsy of lung mass confirmed metastatic ampullary carcinoma. Tissue next‐generation sequencing revealed alterations, including ERBB2 amplification. Patient received trastuzumab and pertuzumab, demonstrating partial response. Left: Computerized axial tomography (CAT) scan of chest before treatment. Right: CAT scan 14 months after the treatment, showing about 59% reduction in size of lung mass (per Response Evaluation Criteria in Solid Tumors, version 1.1). Progression‐free survival = 15.2+ months (supplemental online Table 1, ID #1).
Discussion
Here we report our preliminary experience in the Rare Tumor Clinic at the UCSD Center for Personalized Cancer Therapy (n = 40 patients). When available, genomics and protein analyses were performed to guide a precision therapy strategy (supplemental online Table 1). Overall, 37 patients (92.5%) had at least one potentially actionable target (by either an FDA‐approved or an investigational agent) by genomics (from tissue and/or blood) as well as protein analyses, indicating that this approach is feasible among patients with rare tumors (supplemental online Tables 1, 2, and 3).
As mentioned, each case of rare cancer is rare by definition; however, when all the subtypes of rare cancers are included, they account for one‐fourth of all adult tumors, making “rare” tumors a rather common condition [2], [3]. Moreover, in the current era of genomic typing [8], common tumors are being subgrouped into rare subsets or even n‐of‐one conditions. For example, patients with adenocarcinoma of the lung are now subdivided based on their underlying molecular characteristics, including KRAS (30%), EGFR (10–15%), BRAF (7%), MET (7%), ROS1 (2%), ALK (1%), and RET (<1%) mutations [22]. Although adenocarcinoma of the lung diagnosed by standard microscopic exam is categorized as one of the most common cancers (incidence of 62 per 100,000 per year [23]), patients with ROS1, ALK, and RET alterations comprise infrequent subgroups of this malignancy. Furthermore, through the lens of the “molecular microscope,” each individual can have a distinct and complex genomic makeup [24], [25], [26]. Understanding that each individual has a unique molecular portfolio is important because it suggests that most patients may need an individualized precision therapy approach rather than the canonical strategy based on histological commonalities [11], [12], [13], [14].
We have utilized the personalized matched targeted therapy approach at the Rare Tumor Clinic (Fig. 2, supplemental online Tables 1, 2, and 3). Overall, among 21 patients who received a matched therapy, 52.4% (11/21) attained SD ≥6 months, PR, or CR (14.3% [3/21], SD ≥6 months; 28.6% [6/21], PR; and 9.5% [2/21], CR) with a median PFS of 19.6 months (range 0.99+ to 26.1+ months). Median overall survival from initiation of matched therapy has not reached (range 1.21 to 29.21 months) (Fig. 2 and supplemental online Table 3). Moreover, the matched therapy approach had a statistically significant improvement in PFS when compared with last prior unmatched therapy (HR: 0.26, 95% CI: 0.10–0.71, p = .008; median PFS 19.7 vs. 3.5 months [n = 12 evaluable patients]); Fig. 3). Our pilot experience with the matched targeted therapy approach in rare cancers suggests feasibility in a Rare Tumor Clinic.
Among our patients with exceptional responses was an individual diagnosed with ampullary carcinoma. Her tumor harbored an ERBB2 amplification, and her disease was successfully managed with anti‐human epidermal growth receptor 2 therapy (trastuzumab and pertuzumab; PR, 59% decrease, with PFS of 15.2+ months). Examples of a successful matched targeted therapy approach also include patients treated with immunotherapy. Recent literature suggests that high tumor mutation burden (TMB) status, high microsatellite instability (MSI‐high) and the expression/amplification of PD‐L1 can be predictive of response to immune checkpoint inhibitors [27], [28]. We have treated three patients with aggressive, advanced/metastatic ultrarare malignancies (two with advanced/metastatic basal cell cancers and one with high‐grade, large‐cell neuroendocrine gynecologic cancer) with PD‐1 inhibitors based on the genomic information (n = 3 patients with high TMB, one of whom also had PDL1 amplification; n = 1 with both high TMB and MSI‐high); all had remarkable responses (n = 1 with CR and n = 2 with PR; Fig. 2, supplemental online Tables 1 and 3, ID #4, 16, and 21) [29], [30].
At the Rare Tumor Clinic, although more than half of the cases demonstrated SD ≥6 months, PR, or CR with the matching approach, it is important to note that not all patients had favorable clinical outcomes despite rationally selected treatments (Fig. 2 and supplemental online Table 3). Interestingly, we have previously shown that a high Matching Score (number of alterations matched with targeted therapies divided by total number of alterations identified) is independently associated with improvement in all outcome parameters [31], [32]. The number of patients in the current pilot study in our Rare Tumor Clinic is still too small to calculate the impact of a Matching Score, but this is planned for future reports. There are several other limitations to the current report. This study was performed retrospectively, and 20 different cancer types were included in this study, precluding a more in‐depth analysis of any one histology. However, the diversity of tumors could suggest that the conclusions are generalizable across different rare tumors.
Conclusion
We have demonstrated that genomic and protein analysis to direct therapy is feasible among patients with rare and ultrarare tumors. Most patients (37/40 [92.5%]) had at least one actionable target detected by either genomics or protein analyses (supplemental online Tables 1, 2, and 3). Among patients who received matched targeted therapy, >50% of patients had SD ≥6 months, PR, or CR (Fig. 2 and supplemental online Table 3). Moreover, matched targeted therapy had a statistically significant improvement in PFS when compared with last prior unmatched therapy (HR: 0.26, 95% CI: 0.10–0.71, p = .008; Fig. 3). These pilot study results may be important because patients with rare and ultrarare tumors often have no FDA‐approved treatments and may be ineligible for clinical trials. For this reason, we (Southwest Oncology Group/National Cancer Institute) recently initiated the national Dual Anti‐CTLA‐4 and anti‐PD‐1 blockade in Rare Tumors (DART) immunotherapy trial for rare cancers (NCT02834013). Further studies investigating the efficacy of an individualized precision therapy approach in patients with rare/ultrarare neoplasms are needed.
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Material
Acknowledgments
This work was supported in part by the Joan and Irwin Jacobs Fund and by National Cancer Institute grant P30 CA016672 (R.K.).
Footnotes
For Further Reading: Todd C. Knepper, Gillian C. Bell, J. Kevin Hicks et al. Key Lessons Learned from Moffitt's Molecular Tumor Board: The Clinical Genomics Action Committee Experience. The Oncologist 2017;22:144‐151; first published on February 8, 2017.
Implications for Practice: It is clear that the increasing practicality of genetic tumor sequencing technology has led to its incorporation as part of routine clinical practice. Subsequently, many cancer centers are seeking to develop a personalized medicine services and/or molecular tumor board to shepherd precision medicine into clinical practice. This article discusses the key lessons learned through the establishment and development of a molecular tumor board and personalized medicine clinical service. This article highlights practical issues and can serve as an important guide to other centers as they conceive and develop their own personalized medicine services and molecular tumor boards.
Editor's Note: See the related commentary, “Trailblazing Precision Oncology for Rare Tumor Subtypes,” by Kevin Shee and Todd W. Miller on page 143 of this issue.
Author Contributions
Conception/design: Shumei Kato, Razelle Kurzrock
Provision of study material or patients: Shumei Kato, Kellie Kurasaki, Sadakatsu Ikeda
Collection and/or assembly of data: Shumei Kato, Kellie Kurasaki
Data analysis and interpretation: Shumei Kato, Razelle Kurzrock
Manuscript writing: Shumei Kato, Kellie Kurasaki, Sadakatsu Ikeda, Razelle Kurzrock
Final approval of manuscript: Shumei Kato, Kellie Kurasaka, Sadakatsu Ikeda, Razelle Kurzrock
Disclosures
Razelle Kurzrock: X‐Biotech, Actuate Therapeutics (C/A), Genentech, Pfizer, Sequenom, Guardant, Foundation Medicine, Merck Serono (RF), CureMatch Inc (OI). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1. Ferlay J, Soerjomataram I, Dikshit R et al. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015;136:E359–E386. [DOI] [PubMed] [Google Scholar]
- 2. Gatta G, van der Zwan JM, Casali PG et al. Rare cancers are not so rare: The rare cancer burden in Europe. Eur J Cancer 2011;47:2493–2511. [DOI] [PubMed] [Google Scholar]
- 3. Greenlee RT, Goodman MT, Lynch CF et al. The occurrence of rare cancers in U.S. adults, 1995–2004. Public Health Rep 2010;125:28–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Munoz J and Kurzrock R. Targeted therapy in rare cancers–Adopting the orphans. Nat Rev Clin Oncol 2012;9:631–642. [DOI] [PubMed] [Google Scholar]
- 5. Tamaki T, Dong Y, Ohno Y et al. The burden of rare cancer in Japan: Application of the RARECARE definition. Cancer Epidemiol 2014;38:490–495. [DOI] [PubMed] [Google Scholar]
- 6. Beck M. Rare and ultra rare diseases? J Dev Drugs 2012;1:e107. [Google Scholar]
- 7. Hughes DA, Tunnage B, Yeo ST. Drugs for exceptionally rare diseases: Do they deserve special status for funding? QJM 2005;98:829–836. [DOI] [PubMed] [Google Scholar]
- 8. Subbiah V, Kurzrock R. Universal genomic testing needed to win the war against cancer: Genomics IS the diagnosis. JAMA Oncol 2016;2:719–720. [DOI] [PubMed] [Google Scholar]
- 9. Heinrich MC, Joensuu H, Demetri GD et al. Phase II, open‐label study evaluating the activity of imatinib in treating life‐threatening malignancies known to be associated with imatinib‐sensitive tyrosine kinases. Clin Cancer Res 2008;14:2717–2725. [DOI] [PubMed] [Google Scholar]
- 10. Hyman DM, Puzanov I, Subbiah V et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jardim DL, Schwaederle M, Wei C et al. Impact of a biomarker‐based strategy on oncology drug development: A meta‐analysis of clinical trials leading to FDA approval J Natl Cancer Inst 2015;107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schwaederle M, Zhao M, Lee JJ et al. Impact of precision medicine in diverse cancers: A meta‐analysis of phase II clinical trials. J Clin Oncol 2015;33:3817–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schwaederle M, Zhao M, Lee JJ et al. Association of biomarker‐based treatment strategies with response rates and progression‐free survival in refractory malignant neoplasms: A meta‐analysis. JAMA Oncol 2016;2:1452–1459. [DOI] [PubMed] [Google Scholar]
- 14. Tsimberidou AM, Iskander NG, Hong DS et al. Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center initiative. Clin Cancer Res 2012;18:6373–6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frampton GM, Fichtenholtz A, Otto GA et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thomas RK, Nickerson E, Simons JF et al. Sensitive mutation detection in heterogeneous cancer specimens by massively parallel picoliter reactor sequencing. Nat Med 2006;12:852–855. [DOI] [PubMed] [Google Scholar]
- 17. Wagle N, Berger MF, Davis MJ et al. High‐throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov 2012;2:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lanman RB, Mortimer SA, Zill OA et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell‐free circulating tumor DNA. PLoS One 2015;10:e0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc 1958;53:457–481. [Google Scholar]
- 20. Eisenhauer EA, Therasse P, Bogaerts J et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–247. [DOI] [PubMed] [Google Scholar]
- 21. Von Hoff DD, Stephenson JJ Jr., Rosen P et al. Pilot study using molecular profiling of patients' tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol 2010;28:4877–4883. [DOI] [PubMed] [Google Scholar]
- 22. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dela Cruz CS, Tanoue LT, Matthay RA. Lung cancer: Epidemiology, etiology, and prevention. Clin Chest Med 2011;32:605–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kato S, Elkin SK, Schwaederle M et al. Genomic landscape of salivary gland tumors. Oncotarget 2015;6:25631–25645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kurzrock R, Giles FJ. Precision oncology for patients with advanced cancer: The challenges of malignant snowflakes. Cell Cycle 2015;14:2219–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wheler JJ, Parker BA, Lee JJ et al. Unique molecular signatures as a hallmark of patients with metastatic breast cancer: Implications for current treatment paradigms. Oncotarget 2014;5:2349–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Patel SP, Kurzrock R. PD‐L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther 2015;14:847–856. [DOI] [PubMed] [Google Scholar]
- 28. Topalian SL, Taube JM, Anders RA et al. Mechanism‐driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 2016;16:275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. FalchoIt is clear that the increasing practicality of genetic tumor sequencing technology has led to its incorporation as part of routine clinical practice. Subsequently, many cancer centers are seeking to develop a personalized medicine services and/or molecular tumor board to shepherd precision medicine into clinical practice. This article discusses the key lessons learned through the establishment and development of a molecular tumor board and personalized medicine clinical service. This article highlights practical issues and can serve as an important guide to other centers as they conceive and develop their own personalized medicine services and molecular tumor boards.ok GS, Leidner R, Stankevich E et al. Responses of metastatic basal cell and cutaneous squamous cell carcinomas to anti‐PD1 monoclonal antibody REGN2810. J Immunother Cancer 2016;4:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ikeda S, Goodman AM, Cohen PR et al. Metastatic basal cell carcinoma with amplification of PD‐L1: Exceptional response to anti‐PD1 therapy. NPJ Genom Med 2016;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schwaederle M, Parker BA, Schwab RB et al. Precision oncology: The UC San Diego Moores Cancer Center PREDICT experience. Mol Cancer Ther 2016;15:743–752. [DOI] [PubMed] [Google Scholar]
- 32. Wheler JJ, Janku F, Naing A et al. Cancer therapy directed by comprehensive genomic profiling: A single center study. Cancer Res 2016;76:3690–3701. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.