

Abstract
Within one year, five immunotherapies have been approved for the treatment of patients with locally advanced or metastatic urothelial cancer. The availability of these immunotherapies brings challenges to all stakeholders in the field. Additional research is needed to identify biomarkers that are predictive of individual patient response to different immunotherapies.
Since the accelerated approval of atezolizumab in May 2016 [1], the U.S. Food and Drug Administration (FDA) has approved four additional programmed death receptor‐1 (PD‐1)/programmed death‐ligand 1 (PD‐L1) targeted immunotherapies for the treatment of patients with locally advanced or metastatic urothelial cancer. This includes the treatment of patients who have received prior platinum‐containing chemotherapy as well as patients who are not eligible for cisplatin‐containing chemotherapy. These therapies, approved within 1 year, include atezolizumab, nivolumab, durvalumab, avelumab, and pembrolizumab (Table 1). Atezolizumab, durvalumab, and avelumab are directed against PD‐L1, whereas nivolumab and pembrolizumab are anti‐PD‐1 antibodies. All the approvals received priority review and were granted ahead of the Prescription Drug User Fee Act goal date. This reflects the Agency's efforts, in cooperation with academic investigators and the pharmaceutical community, to make novel effective treatments available to patients earlier. This is especially important in the approval of products to treat those with life‐threatening malignancies, including advanced urothelial cancer.
Table 1. U.S. Food and Drug Administration–approved immunotherapies for advanced urothelial carcinoma.
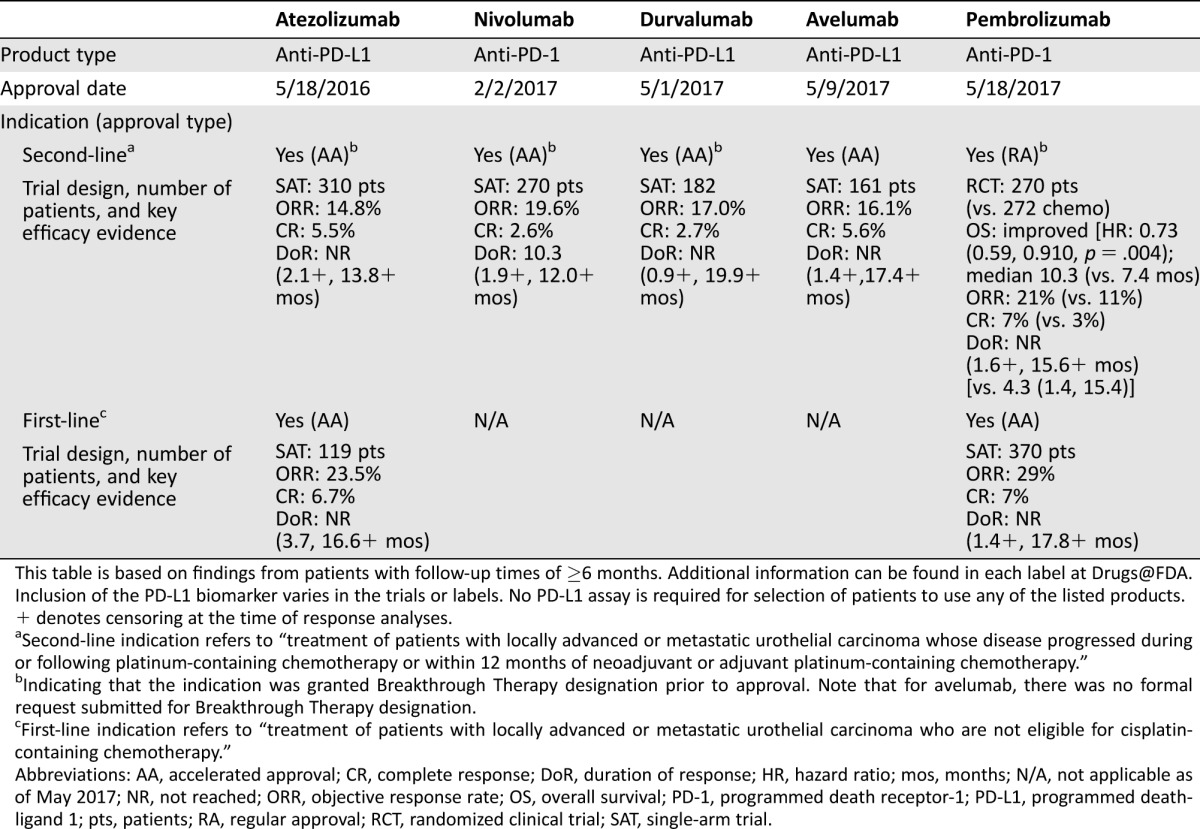
This table is based on findings from patients with follow‐up times of ≥6 months. Additional information can be found in each label at Drugs@FDA. Inclusion of the PD‐L1 biomarker varies in the trials or labels. No PD‐L1 assay is required for selection of patients to use any of the listed products.
+ denotes censoring at the time of response analyses.
Second‐line indication refers to “treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease progressed during or following platinum‐containing chemotherapy or within 12 months of neoadjuvant or adjuvant platinum‐containing chemotherapy.”
Indicating that the indication was granted Breakthrough Therapy designation prior to approval. Note that for avelumab, there was no formal request submitted for Breakthrough Therapy designation.
First‐line indication refers to “treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin‐containing chemotherapy.”
Abbreviations: AA, accelerated approval; CR, complete response; DoR, duration of response; HR, hazard ratio; mos, months; N/A, not applicable as of May 2017; NR, not reached; ORR, objective response rate; OS, overall survival; PD‐1, programmed death receptor‐1; PD‐L1, programmed death‐ligand 1; pts, patients; RA, regular approval; RCT, randomized clinical trial; SAT, single‐arm trial.
Approximately 16,000 patients die from urothelial cancer annually in the U.S. [2]. Standard of care for patients with locally advanced or metastatic disease is platinum‐based chemotherapy (cisplatin or carboplatin) in combination with gemcitabine or other chemotherapeutics. However, most patients experience disease progression during or after platinum‐based chemotherapy and their survival time is limited—approximately 5–9 months [3], [4], [5]. Patients with disease progression during or after platinum‐based chemotherapy have traditionally received taxanes or vinflunine as second‐line therapy. In these trials, objective response rates were low (e.g., 9%–11%) and median response durations were about 5–7 months [3], [4], [5]. The common chemotherapy‐related adverse reactions in these trials include bone marrow toxicities and neuropathy. Until the approval of atezolizumab 1 year ago, no product was FDA approved for second‐line treatment of the disease.
Approximately 25%–50% of patients with advanced urothelial cancer are deemed ineligible or unable to tolerate cisplatin‐containing chemotherapy [6], [7]. This is generally due to comorbidities. Patients ineligible for cisplatin‐based therapy have been defined as those with a poor performance status (e.g., Eastern Cooperative Oncology Group performance status of 2), impairment of renal function (e.g., creatinine clearance of <60 mL/minute), peripheral neuropathy (≥grade 2), or hearing impairment (≥grade 2). For these patients, carboplatin is often substituted for cisplatin in combination with other agents, such as gemcitabine, as first‐line chemotherapy. Data from a randomized trial demonstrated that carboplatin plus gemcitabine was associated with a median overall survival of 9.3 months [8]. Evidence also shows that first‐line use of non–cisplatin combination chemotherapy in cisplatin‐ineligible patients was associated with a response rate of 40%–50% and median response duration of 5–8 months [8], [9]. Clearly, there is an unmet medical need for this cisplatin‐ineligible patient population.
The initial observation of durable responses in 2013–2014 promoted the development of PD‐1/PD‐L1 immunotherapies for advanced urothelial carcinoma. Based on the preliminary reports of response rate and duration of response, in May 2014, the FDA granted atezolizumab Breakthrough Therapy designation for treatment of patients with urothelial cancer who had received prior platinum‐based therapy [10]. Subsequently, nivolumab, durvalumab, and pembrolizumab also received Breakthrough Therapy designation in the same disease setting in 2016. Breakthrough Therapy designation was introduced in Section 902 of the FDA Safety and Innovation Act of 2012. Its goal is to expedite the development and review of investigational products intended to treat serious or life‐threatening diseases. It requires preliminary clinical evidence indicating that a product may demonstrate substantial improvement over available or existing therapies on one or more clinically significant endpoints in patients who have unmet medical needs [11].
Between 2014 and 2016, the review teams at the FDA Office of Hematology and Oncology Products had multiple interactions with relevant sponsors to discuss their proposed plans and trial designs for development of these immunotherapies in advanced urothelial carcinoma. These included discussions of their potential use in second‐line, first‐line, or adjuvant settings. The objective was to enable an efficient development program, ranging from chemistry and manufacturing strategies to clinical investigation, thereby expediting development programs and allowing earlier patient access to therapies. For instance, to expedite the atezolizumab development program, the FDA held more than 10 meetings with the sponsor in 2 years between the time of Breakthrough Therapy designation and accelerated approval [10].
Of the five products approved for the treatment of urothelial cancer following platinum‐based chemotherapy, four (atezolizumab, nivolumab, durvalumab, and avelumab) received accelerated approval and one (pembrolizumab) regular approval. Accelerated approval, a regulatory pathway introduced in 1992, is based on the use of a surrogate endpoint (e.g., response rate, duration of response) that is reasonably likely to predict clinical benefit and must show improvement over available therapies [11], [12]. Regular approval requires substantial evidence of clinical benefit (e.g., improved survival or quality of life). In single‐arm trials, the four products that received accelerated approval demonstrated confirmed objective response rates of 14%–20% with durable durations of response. As noted in Table 1, the median response duration was 10.3 months for nivolumab and was not reached for the other three products. At the time of FDA evaluations, the follow‐up time was short in the remaining three studies and the sponsors have committed to providing additional data concerning the duration of response. The response rates of these four products were similar to or better than the response rates reported for single‐agent chemotherapy (e.g., taxanes or vinflunine) in the same disease setting and carried a distinct safety profile. The duration of response appeared to be much longer with these agents than with conventional chemotherapy. The improvement seen in these two surrogates, response rate and response duration, are reasonably likely to predict clinical benefit in this setting and support accelerated approval.
The regular approval of pembrolizumab was based on a significant improvement in overall survival, as compared with chemotherapy, in a randomized trial of patients with urothelial cancer who had previously received platinum‐based therapy. The chemotherapy control included the investigator's choice of a taxane or vinflunine. As shown in Table 1, pembrolizumab was associated with a 3‐month improvement in median overall survival. Pembrolizumab also elicited durable responses with a confirmed response rate of 21%, similar to those observed in the single‐arm trials of the four products granted accelerated approval.
Atezolizumab and pembrolizumab also received accelerated approval for first‐line use in patients with advanced urothelial cancer who are not eligible for cisplatin‐containing chemotherapy. The approvals were based on confirmed response rates of 24% with atezolizumab and 29% with pembrolizumab. Median response duration was not reached in either trial due to the short follow‐up.
For each of the approved indications, the submitted data and review findings support a favorable benefit‐risk profile. Immune‐related adverse reactions (irARs) that are unique to these products include pneumonitis, hepatitis, colitis, endocrinopathies, and other infrequent irARs such as meningoencephalitis, ocular inflammatory toxicity, and myocarditis. Some irARs were fatal. Approximately 5%–10% of patients in the trials required systemic glucocorticoids (prednisone dose equivalent to ≥40 mg daily) therapy for moderate to severe irARs. These toxicities are different from key toxicities related to chemotherapy used in the disease, which include bone marrow suppression and neuropathy. The differences are reflected in the randomized trial of pembrolizumab compared with chemotherapy discussed previously.
Each of these sponsors independently developed an in vitro PD‐L1 assay. In vitro assays can be approved as a companion or complementary diagnostic. A companion diagnostic assay is an in vitro diagnostic device that provides information essential for the safe and effective use of a corresponding therapeutic product [13]. Companion use of the device is specified in the indication statement for the product (e.g., for identification or selection of patients appropriate for that therapy). A complementary diagnostic is an assay that identifies a biomarker‐defined subset of patients such as those who respond particularly well to a drug and aids in the risk‐benefit assessment. A complementary diagnostic is not deemed essential for the safe and effective use of a therapy.
Three of the five sponsors submitted their assay findings along with clinical data for regulatory evaluation. As summarized in Table 2, these assays vary considerably in the type of cells used to determine PD‐L1 expression (on tumor cells, tumor‐infiltrating immune cells, or both) and have different cutoff points to distinguish patients with high PD‐L1 expression from those with low PD‐L1 expression. With each of the listed products, the response rate is greater in patients with high PD‐L1 expression in tumor specimens (Table 2). However, the differences in response rate between patients with high and low PD‐L1 expression in tumor specimens are not sufficient to support use of any of the assays as a companion diagnostic device, but are appropriate to inform the risk‐benefit decision. With all of these drugs, there were patients with low PD‐L1 expression who had durable responses, indicating that these PD‐L1 assays cannot be considered essential or used as companion diagnostics to select patients who may most benefit from the products. Moreover, the results from single‐arm trials prevent us from assessing whether high or low PD‐L1 expression in tumor specimens is predictive or prognostic. In the same disease setting, the varying methodologies and different cutoff criteria across these reported PD‐L1 assays make clinical interpretation difficult and call for collaborations among sponsors to develop or use the same biomarker assay or harmonize the available assays to ensure equivalent performance characteristics.
Table 2. PD‐L1 assays included in the labels of three products for second‐line use.
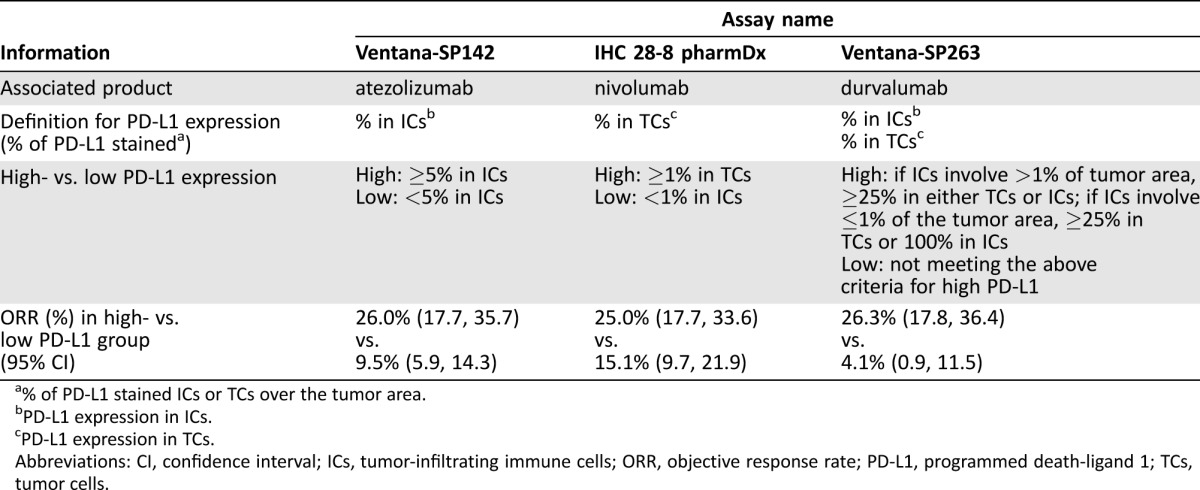
% of PD‐L1 stained ICs or TCs over the tumor area.
PD‐L1 expression in ICs.
PD‐L1 expression in TCs.
Abbreviations: CI, confidence interval; ICs, tumor‐infiltrating immune cells; ORR, objective response rate; PD‐L1, programmed death‐ligand 1; TCs, tumor cells.
Multiple ongoing trials to verify the clinical benefit of these immunotherapies are being conducted as a condition of accelerated approval. Information on these trials can be found at clinicaltrials.gov or in the approval letters at Drugs@FDA. In addition, mature data on the durability of response may provide further evidence to evaluate these therapies. Durable responses, some lasting 1–2 years, have been observed.
The availability of the five immunotherapies for advanced urothelial carcinoma brings challenges to all stakeholders in the field. Additional research is needed to identify biomarkers that are predictive of individual patient response to different immunotherapies. This question may benefit from proactive collaborations among stakeholders. Different strategies should be considered to enhance response (e.g., in combination with other agents used in the disease) or delay the development of resistance to immunotherapy. Furthermore, it is important to assess whether early use of immunotherapy (e.g., in first line or adjuvant setting) is beneficial to patients. In addition to immunotherapy, identification of new drugable targets and efficient development of other novel agents are clearly indicated to expand the effective treatments for patients with urothelial carcinoma.
Footnotes
For Further Reading: Yang‐Min Ning, Daniel Suzman, V. Ellen Maher et al. FDA Approval Summary: Atezolizumab for the Treatment of Patients with Progressive Advanced Urothelial Carcinoma after Platinum‐Containing Chemotherapy. The Oncologist 2017;22:743‐749; first published on April 19, 2017.
Implications for Practice: The accelerated approval of atezolizumab for second‐line use in advanced urothelial carcinoma provides patients with an effective, novel treatment option for the management of their disease. This represents the first immunotherapy approved in this disease setting.
References
- 1. Ning YM, Suzman D, Maher VE et al. FDA Approval Summary: Atezolizumab for the treatment of patients with progressive advanced urothelial carcinoma after platinum‐containing chemotherapy. The Oncologist 2017;22:743–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2016. CA Cancer J Clin 2016;66:7–30. [DOI] [PubMed] [Google Scholar]
- 3. Bellmunt J, Theodore C, Demkov T et al. Phase III trial of vinflunine plus best supportive care compared with best supportive care alone after a platinum‐containing regimen in patients with advanced transitional cell carcinoma of the urothelial tract. J Clin Oncol 2009;27:4454–4461. [DOI] [PubMed] [Google Scholar]
- 4. Choueiri TK, Ross RW, Jacobus S et al. Double‐blind, randomized trial of docetaxel plus vandetanib versus docetaxel plus placebo in platinum‐pretreated metastatic urothelial cancer. J Clin Oncol 2012;30:507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Petrylak DP, Tagawa ST, Kohli M et al. Docetaxel as monotherapy or combined with ramucirumab or icrucumab in second‐line treatment for locally advanced or metastatic urothelial carcinoma: An open‐label, three‐arm, randomized controlled phase II trial. J Clin Oncol 2016;34:1500–1509. [DOI] [PubMed] [Google Scholar]
- 6. Galsky MD, Hahn NM, Rosenberg J et al. Treatment of patients with metastatic urothelial cancer “unfit” for cisplatin‐based chemotherapy. J Clin Oncol 2011;29:2432–2438. [DOI] [PubMed] [Google Scholar]
- 7. Sonpavde G, Galsky MD, Latini D et al. Cisplatin‐ineligible and chemotherapy‐ineligible patients should be the focus of new drug development in patients with advanced bladder cancer. Clin Genitourin Cancer 2014;12:71–73. [DOI] [PubMed] [Google Scholar]
- 8. De Santis M. Bellmunt J, Mead G et al. Randomized phase II/III trial assessing gemcitabine/carboplatin and methotrexate/carboplatin/vinblastine in patients with advanced urothelial cancer who are unfit for cisplatin‐based chemotherapy: EORTC study 30986. J Clin Oncol 2011; 30:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Necchi A. Pond GR, Raggi D et al. Efficacy and safety of gemcitabine plus either taxane or carboplatin in the first‐line setting of metastatic urothelial carcinoma: A systematic review and meta‐analysis. Clin Genitourin Cancer 2016;15:23–30. [DOI] [PubMed] [Google Scholar]
- 10.U.S. Food and Drug Administration . FDA reviews of atezolizumab for second‐line use in advanced urothelial carcinoma. Available at https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/761034Orig1s000TOC.cfm Accessed May 31, 2017.
- 11.U.S. Food and Drug Administration . Guidance for industry, expedited programs for serious conditions—Drugs and biologics. Available at http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm358301.pdf. Accessed May 31, 2017.
- 12. Johnson JR, Ning YM, Farrell A et al. Accelerated approval of oncology products: The Food and Drug Administration experience. J Natl Cancer Inst 2011;103:636–644. [DOI] [PubMed] [Google Scholar]
- 13.U.S. Food and Drug Administration . In vitro companion diagnostic devices: Guidance for industry and Food and Drug Administration staff. Available at https://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM262327.pdf. Accessed May 31, 2017.