

Abstract
Dopamine receptors play an integral role in controlling brain physiology. Importantly, subtype selective agonists and antagonists of dopamine receptors with biased signaling properties have been successful in treating psychiatric disorders with a low incidence of side effects. To this end, we recently designed and developed SK609, a dopamine D3 receptor (D3R) selective agonist that has atypical signaling properties. SK609 has shown efficacy in reversing akinesia and reducing L-dopa-induced dyskinesia in a hemiparkinsonian rats. In the current study, we demonstrate that SK609 has high selectivity for D3R with no binding affinity on D2R high- or low-affinity state when tested at a concentration of 10 μM. In addition, SK609 and its analogues do not induce desensitization of D3R as determined by repeated agonist treatment response in phosphorylation of ERK1/2 functional assay. Most significantly, SK609 and its analogues preferentially signal through the G-protein-dependent pathway and do not recruit β-arrestin-2, suggesting a functional bias toward the G-protein-dependent pathway. Structure–activity relationship (SAR) studies using analogues of SK609 demonstrate that the molecules bind at the orthosteric site by maintaining the conserved salt bridge interactions with aspartate 110 on transmembrane 3 and aryl interactions with histidine 349 on transmembrane 6, in addition to several hydrophobic interactions with residues from transmembranes 5 and 6. The compounds follow a strict SAR with reference to the three pharmacophore elements: substituted phenyl ring, length of the linker connecting phenyl ring and amine group, and orientation and hydrophobic branching groups at the amine among SK609 analogues for efficacy and functional selectivity. These features of SK609 and the analogues suggest that biased signaling is an inherent property of this series of molecules.
Keywords: Biased signaling, β-arrestin, dopamine D3 receptors, desensitization, functional selectivity, G-protein-dependent signaling, hybrid structure-based design, structure–function relationship
Graphical abstract
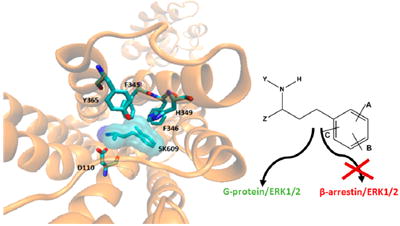
Signaling by G-protein coupled receptors (GPCRs) has been a textbook-example case study to describe the hierarchy and utility of first, second, and third messenger systems in eliciting a functional response. Most of this knowledge was derived based on the effects of endogenous neurotransmitters binding to their cognate receptors and initiating signaling events that lead to the recruitment of various coupling molecules such as G-protein subunits, second messengers, and a cascade of kinases and phosphatases, eventually producing a cellular response.1
Dopamine D2-like receptors consist of the subtypes D2R, D3R, and D4R, which couple to the Gi/o class of G-proteins for neurotransmission. These subtypes are highly homologous to each other and have overlapping distribution and functional roles in the brain.2 Hence, developing subtype selective ligands has been a longstanding challenge in the field of dopamine receptor pharmacology. However, with newer techniques and the use of computational models in understanding molecular determinants of function,3 it is now feasible to design subtype specific molecules. In addition to subtype specificity, recent trends in designing “biased functional” ligands have added further credence to understanding the various molecular signaling pathways.4 Although GPCRs can signal via many pathways, the G-protein-dependent and the G-protein-independent or β-arrestin-dependent pathways have emerged as major players in functional selectivity.5 This suggests that GPCRs must exhibit a range of conformational changes in response to biased ligands, some of which could stabilize and lead to distinct functional effects.1b,6 Several biased signaling beta blockers and opiates have already shown better clinical effects compared with their unbiased counterparts, suggesting that biased signaling molecules could have better utility as clinical agents, with fewer offtarget effects.7
Although several crystal structures of GPCRs are available in apo or ligand bound forms,1b,6b there is little or no structure–function correlation between the orthosteric binding pocket of the GPCRs and the functional response of the different ligand classes, namely, the agonist, antagonist, and partial or inverse agonists.8 These issues are further complicated by the action of biased ligands (agonists or antagonists) that seem to produce distinct conformational changes resulting in functional selectivity. In fact, early references to functional selectivity aptly described it as “functional trafficking”, as it referred not only to the unique conformational changes in the receptor but also to the recruitment of the downstream signaling molecule.9 Hence, it is hypothesized that the signaling molecule determines the chemotypic signature of a given agonist molecule with the functional cellular outcome.10 Studies utilizing fluorescence techniques have provided evidence for the role of functional domains with the receptor that code for G-protein-dependent and -independent mechanisms in vasopressin receptors.11 Accordingly, transmembrane (TM) segment 6 and the third intracellular loop region have been shown to be involved in G-protein-dependent signaling. This has been well validated by crystal structure, nuclear magnetic resonance (NMR) studies, and molecular dynamics studies of the β2-adrenergic receptor.12 Similarly, TM7 and helix-8 regions have been implicated in β-arrestin recruitment and signaling of β2-adrenergic receptor induced by the biased signaling agonist carvedilol.13
The D2-like receptors in their monomeric forms are traditionally known to couple to the Gi/Go class of G-proteins, initiated by a ligand binding event and resulting in activation of either the G-protein-dependent and -independent pathways or, in most cases, both pathways.14 The G-protein-dependent pathway in the case of D2-like receptors leads to inhibition of adenylyl cyclase and reduction of cAMP production, eventually leading to activation of ERK1/2 kinases. The G-protein-independent pathway is initiated by the ligand binding event, followed by phosphorylation of the receptor by the G-protein coupled receptor kinases (GRKs), which further leads to the recruitment of β-arrestin-2 and activation of the glycogen synthase kinase 3β (GSK3β)-mediated pathway.15 The endogenous neurotransmitter dopamine signals through both the G-protein and β-arrestin-mediated/G-dependent pathways, leading to the downstream effects of mitogen-activated protein-independent pathways kinase (MAPK) activation.16 However, synthetic ligands that mimic dopamine’s signaling action of activating multiple pathways cause undesirable side effects, which has led to the design of a new class of dopaminergic agents that have a biased signaling profile through either the G-protein-dependent or β-arrestin-mediated/G-protein-independent pathways. 16,17 Some known chemotypes with D2R biased signaling profile include antipsychotic drugs such as aripiprazole and its analogues, which recruit and signal through the β-arrestin-mediated pathway.18 Similarly, several other clinically used antipsychotic agents have been shown to signal via the β-arrestin-mediated pathway at the D2R, suggesting a common mechanism of action.19 Recently, Free et al.20 have shown a new class of biased D2R agonists that mediate G-protein-dependent signaling and do not recruit β-arrestin, while antagonizing the dopamine stimulated β-arrestin recruitment to D2R. In another interesting study on D2R by Shonberg et al., functional bias between p-ERK1/2 and cAMP pathways was achieved by incorporating fragments of cariprazine to develop structure–activity relationships (SARs) of a novel class of antipsychotic drugs, suggesting that functional bias was directly linked to the chemical signature of the molecule.21
The D3R class of receptors has been overlooked in terms of their pharmacological and biophysical mechanisms of action owing to lack of selective agents that can distinguish the D3R and D2Rs in vivo. However, recent evidence for their differential roles in neurological disorders such as Parkinson’s disease (PD), Huntington’s disease, and L-dopa-induced dyskinesia (LID) has spurred interest in designing selective and biased ligands of D3R.22 Consequently, several new chemotypes that possess selective activity against D3R, with a range of functional features from agonists23 to antagonists,24 have been designed and tested in various disease models. The crystal structure of D3R has further aided in screening and rationally designing novel small molecule modulators of D3R.25 We have recently designed novel atypical D3R agonists with biased signaling properties that differ not only from other known D2-like receptor agonists but also from several well-known D3R agonists.26 Further, the in vivo efficacy of the lead molecule in reversing the akinesias and in blocking LIDs was recently demonstrated using a hemiparkinsonian rodent model of PD.23a In this study, we have further developed the SAR of this novel series of compounds, and importantly demonstrate their functional selectivity to the G-protein-dependent and β-arrestin-independent pathway.
RESULTS AND DISCUSSION
Functional Screening of SK609 and Its Analogues by ERK1/2 Phosphorylation
An in silico screen was performed using the HSB method as discussed previously26 to derive analogues of SK609. Based on the HSB scoring scheme, 17 molecules (including SK609) were chosen for further in vitro screening (Table 1). The molecules were tested for D2R and D3R selectivity using the phosphorylation of ERK1/2 as an end point assay in cells stably expressing either the D2R or D3R. DA and D2R/D3R agonist PD128907 were used as reference compounds.
Table 1.
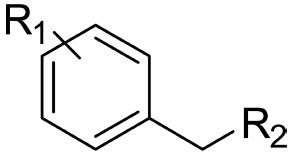
Structure–Activity Relationship Profiles of SK609 and Its Analogues along with Their Docking Scores at D3R
| |||
|---|---|---|---|
| Name | R1 | R2 | Hybrid docking score |
| SK609a | o-Cl |
|
76.8 |
| SK609-R | o-Cl |
|
40.3 |
| SK609-S | o-Cl |
|
81 |
| SK611 | p-OCH3 |
|
57.2 |
| SK610 | p-F |
|
72.6 |
| SK608 | p-Cl |
|
83 |
| SK213 | o-Cl |
|
73.7 |
| SK220 | o-OH |
|
56.2 |
| SK052 | o-I |
|
65 |
| SK058 | o-F |
|
75.9 |
| SK232 | o-Cl |
|
78 |
| SK182 | o-OH |
|
54.8 |
| SK032 | o-Br |
|
64.4 |
| SK041 | o-OCH3 |
|
62 |
| L1 | p-F |
|
28 |
| L2 | p-F |
|
48 |
SK603 (4-(fluran-2-yl)butan-2-amine) was structurally similar to SK609 but had a furan ring instead of an ortho chlorophenyl ring. The hybrid docking score was 64.8.
In CHO-D2R cells (Bmax 1.0 ± 0.15 pmol/mg protein), 1 μM of DA and PD128907 significantly evoked ERK1/2 phosphorylation, but 1 μM of SK609 and its analogues did not have any effect (Figure 1). However, in CHO-D3R cells (Bmax 9.1 ± 0.4 pmol/mg protein), 1 μM of DA, PD128907, SK609, and 9 analogues significantly activated ERK1/2 phosphorylation (Figure 1). As shown in Figure 2A, Supporting Information (SI) Figure 1, and Table 2, DA, SK609, SK608, SK213, and SK232 dose-dependently evoked ERK1/2 phosphorylation with EC50 values of 5.2 ± 1.0, 50.2 ± 5.9, 84.6 ± 21.4, 39.2 ± 4.2, and 859 ± 232 nM, respectively, at CHO-D3R. The Emax values for SK609, SK608, SK213, and SK232 were 81 ± 4.5, 76.1 ± 1.4, 95 ± 5.6, and 106 ± 6.5, respectively. Further, SK609- and analogue-induced ERK1/2 activation in CHO-D3R cells was completely antagonized by pretreatment with 1 μM of D2-like antagonist haloperidol (Figure 2B).
Figure 1.
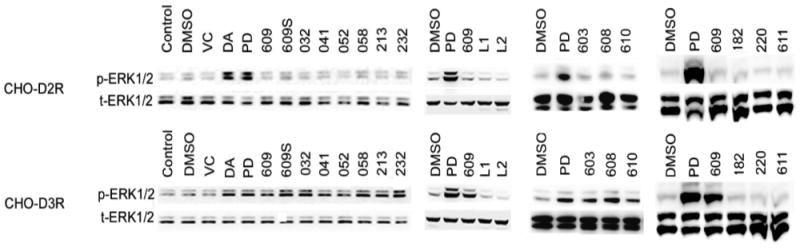
Functional screening of SK609 and its analogues for their ability to selectively phosphorylate ERK1/2. Functional screening of SK609 and all analogues on CHO-D2R or CHO-D3R cells with vehicle or test compounds using ERK1/2 phosphorylation as end point as described in Methods. Representative Western blot (one of three independent experiments) of ERK1/2 phosphorylation (p-ERK1/2) induced by 1 μM of dopamine (DA), PD128907 (PD), SK609, or its analogues for 5 min. SK609 and its analogues are represented by just their numerical part; 609S represents the hydrochloride salt of SK609 and L1 and L2 are the Lichtenberger compounds. DMSO, dimethyl sulfoxide; VC, vitamin C.
Figure 2.
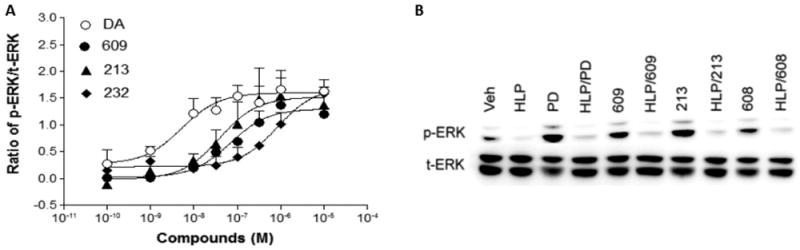
Dose-dependent response of dopamine (DA), SK609, or its analogues and antagonizing effect of antagonist on agonist-induced ERK1/2 phosphorylation. (A) Dose–response curves of ERK1/2 phosphorylation induced by DA, SK609, SK213, and SK232 at CHO-D3R. CHO-D3R cells were pretreated with vehicle, indicated concentrations of DA, SK609, SK213, or SK232 for 5 min, washed once with PBS buffer, and lysed for Western blots. Signals were quantified by densitometry and normalized to total ERK (t-ERK1/2) and the data is presented as ratio of p-ERK/t-ERK minus vehicle control. The graphs represent mean ± SEM of three independent experiments. (B) Haloperidol (HLP) (1 μM) blocked the activation of ERK1/2 induced by PD128907, SK609, and SK608 at CHO-D3R. This image represents one of three independent Western blot experiments of ERK1/2 phosphorylation (p-ERK1/2) induced by 1 μM of PD128907 (PD), SK609, or its analogues in the presence or absence of 1 μM haloperidol for 5 min, which showed consistent results.
Table 2.
Functional Profiles of SK609 and Its Analogues for G-Protein-Dependent and β-Arrestin-Dependent Signaling Mechanisms at D3Ra
| compound | [35S]GTPγS binding
|
ERK1/2 phosphorylation
|
β-arrestin recruitment
|
|||
|---|---|---|---|---|---|---|
| EC50 | Emax | EC50 | Emax | EC50 | Emax | |
| DA | 11.3 ± 2.7 nM | 100 | 5.2 ± 1.0 nM | 100 | ND | ND |
| PD128907 | 1.2 ± 0.18 nM | 96.1 ± 2.1 | 7.1 ± 0.5 nM | 100 | 5.5 ± 0.18 nM | 100 |
| SK609 | 1.1 ± 0.2 μM | 74.3 ± 2.3 | 50.2 ± 5.9 nM | 81 ± 4.5 | 14.4 ± 2.7 μM | 57.3 ± 2.5 |
| SK213 | 1.3 ± 0.3 μM | 98.4 ± 7.9 | 39.2 ± 4.2 nM | 95 ± 5.5 | 6.8 ± 0.5 μM | 66.1 ± 6.5 |
| SK232 | 3.5 ± 0.9 μM | 95.6 ± 8.1 | 859 ± 232 nM | 106 ± 6.5 | ND | ND |
| SK608 | 33.5 ± 8.4 nM | 81.1 ± 5.5 | 84.6 ± 21.4 nM | 76 ± 1.4 | ND | ND |
For ERK1/2 phosphorylation, CHO-D3R cells were treated with DA or test compounds for 5 min at 37 °C. ND, not determined.
Enantiomeric Effect of SK609
The enantiomeric isoforms of SK609 (namely, S-SK609 and R-SK609) were tested for selectivity to D3R and functional effect on inducing ERK1/2 activation in CHO-D2R and CHO-D3R cells. D2R/D3R agonists pramipexole and PD128907, along with the racemate SK609, were used as positive controls. The S-SK609 isoform was found to be selective to D3R and had a functional effect on activation of ERK1/2 identical to that of the racemate SK609, while R-SK609 had neither any effect on either receptor nor any functional effects (Figure 3A). The dose–response curves of S-SK609 and R-SK609, along with pramipexole and racemate SK609, with increasing concentrations (10−10–10−4 M) were assessed. The results confirmed the observation that S-SK609 had a selective and functional D3R effect with an EC50 value of 109 ± 15.4 nM and an Emax value of 93.8 ± 14.5 (Figure 3B), with R-SK609 being virtually inactive. The results suggest that the functional effect of the racemate SK609 is due to the S-enantiomer and that the R-enantiomer has no effect. Indeed, racemic SK609 displayed an EC50 value of 113.9 ± 20.3 nM (with Emax of 88.7 ± 8.6), not appreciably different from what would be expected theoretically with S isomer being the only active component. Based on these results, we utilized the racemate SK609 for all further studies. In this series of experiments, pramipexole did not demonstrate selectivity to D3R (Figure. 3A) but had a potent functional D3R effect, with an EC50 value of 2.85 ± 0.51 and an Emax of 100.
Figure 3.
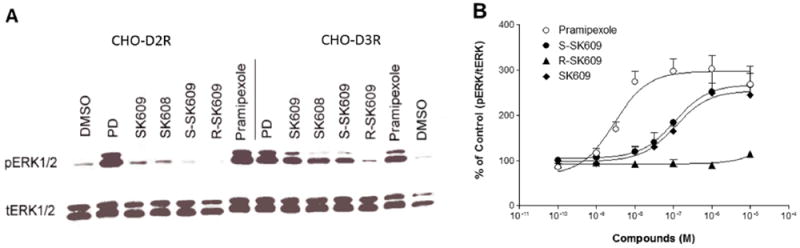
Comparison of functional effects of racemate SK609 and the enantiomeric isoforms (S-SK609, R-SK609) at CHO-D2R and CHO-D3R and dose-dependent response effects at CHO-D3R. (A) Comparison of effects of PD128907, pramipexole, SK609, SK608, S-SK609, and R-SK609 at CHO-D2R or CHO-D3R. Cells were pretreated with vehicle, 1 μM PD128907, pramipexole, SK609, SK608, S-SK609, R-SK609 for 5 min; washed once with PBS buffer; and lysed for Western blots. This represents one of three independent Western blot experiments that showed consistent results. (B) Dose–response curves of ERK1/2 phosphorylation induced by pramipexole, SK609, S-SK609, and R-SK609 at CHO-D3R. CHO-D3R cells were pretreated with vehicle, indicated concentrations of pramipexole, SK609, S-SK609, or R-SK609 for 5 min, washed once with PBS buffer, and lysed for Western blots. Signals were quantified by densitometry and normalized to total ERK (t-ERK1/2). The graphs represent mean ± SEM of three independent experiments.
Structure–Activity Relationship of SK609 and Its Analogues
The structure–activity relationship was determined among the 17 molecules (Table 1) using three pharmacophore elements namely: the phenyl group; length of the linker chain connecting the phenyl and the amine group; and the hydrophobic branching at the amine group (Table 1). Compound SK603 has a furan ring in place of the phenyl ring, and docking studies show that despite binding to the same pocket with the conserved salt bridge with Asp110, it had lower binding scores compared with SK609. This could be due to the loss of aryl interactions with His349 and other hydrophobic groups in TM5 and 6 (Figure 4E). SK603 still maintained selectivity to D3R but had a reduction in functional effect in an ERK1/2 activation assay (Figure 1). Compounds SK058 (F), SK609 (Cl), SK032 (Br), SK182 (OH), SK052 (I), and SK041 (MeO) were used to determine the effects of various electron withdrawing groups when substituted at the ortho position on the phenyl ring. The order of potency was found to be in agreement with the order of electron withdrawing properties of halogen atoms (F > Cl > Br > I) (Figure 1). However, MeO or OH substitutions at either the para or ortho positions led to either weak efficacy (SK041) or a loss of efficacy (SK611, SK182, and SK220) (Figure 1). In contrast, compounds SK609, SK608, SK058, and SK610 have halogen atoms Cl or F substituted at ortho or para positions on the phenyl ring and have the best efficacy. These positions are well-known to be more efficient than the meta position in drawing the electrons away from the pi system and increasing the stability of these compounds. (Figure 4). This feature of halogens has been well demonstrated among all the drug-like molecules, and hence halogens have been labeled as the best electrophilic aromatic substitutions on ring molecules.27
Figure 4.
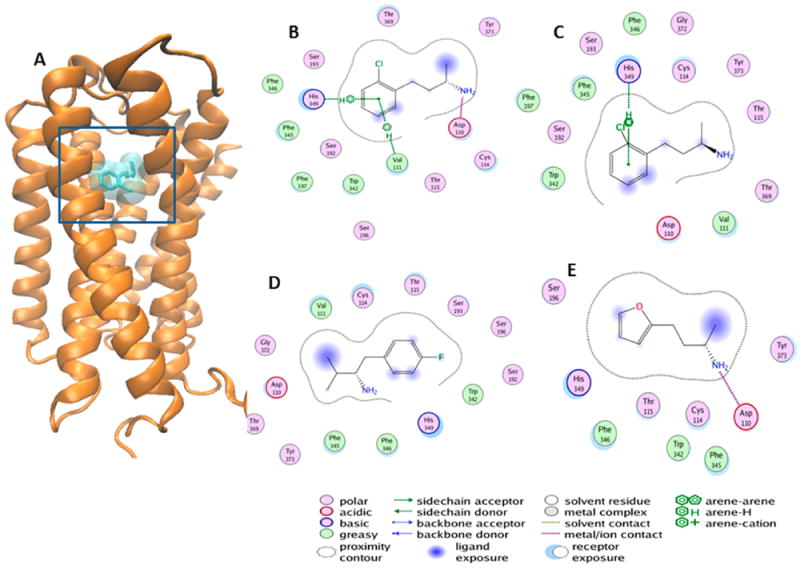
Docked complex of SK609 and its analogues at the orthosteric binding pocket of D3R. (A) SK609 (shown here as licorice sticks and enclosed within a transparent surface) was docked to the binding cavity of D3R. D3R is represented as orange ribbons with important binding pocket residues shown as licorice sticks and colored atom type (carbon, cyan; oxygen, red; nitrogen, blue). (B) 2D representation of the docked S-SK609 at the orthosteric binding pocket. S-SK609 maintains interactions with the aromatic cluster from TM5 and 6 and the salt bridge with ASP110 from TM3, in addition to other favorable interactions. (C) 2D representation of the docked R-SK609 at the orthosteric binding pocket. R-SK609 does not form the salt bridge with Asp110 and hence is a weak binder. (D) 2D representation of the docked L2 compound at the orthosteric binding pocket. Although L2 has the same size as that of SK609, the linker between the primary amine and phenyl group is shorter by one carbon atom, leading to loss of interaction with Asp110 and leading to a low interaction profile. (E) 2D representation of the docked SK603 at the orthosteric binding pocket. SK603 has a furan ring instead of a phenyl ring, which leads to loss favorable aromatic interactions with His349. However, SK603 maintains the salt bridge with Asp110 and a few compensatory hydrophobic interactions with the furan ring. Panels (B)–(E) were generated using the ligX module of the MOE program. The nature of interactions is color coded and explained by the schematic legend.
Effect of Linker Length and Enantiomers
The Lichtenberger compounds28 labeled here as L1 and L2 have a para fluoro substituted phenyl ring and a primary amine group separated by a two-carbon linker (unlike the three-carbon linker found in other analogues of SK609), thus rendering them inactive. This short linker prevents the primary amine in compounds L1 and L2 from forming the salt bridge interaction with the aspartic acid on TM3 (Asp110) and from simultaneously maintaining aromatic pi interactions between the phenyl ring and the aromatic residue cluster from TM5 and TM6 (Figure 4). We also tested the effects of the R- and S-enantiomers in our SAR studies because it is well-known that for most dopaminergic agonists there is a loss of efficacy with the change in orientation of the primary amine group. We found that the S-enantiomer maintained all the same interactions as those of the racemate and in fact had better binding affinity to D3R, with additional arene-cation interaction with Val111 (Figure 4B). However, the R-enantiomer did not form the salt bridge with Asp110 and likely does not bind to D3R (Figure 4C). We have previously demonstrated that atypical agonists such as SK609 induce a specific conformational change that requires simultaneous interactions of the phenyl ring with the aromatic cluster, and a salt bridge between the primary or secondary amines and Asp110 on TM3 of D3R. These interactions are required to induce a signature conformational change that further locks the interactions between extracellular loops 2 and 3 in D3R (Figure 4A).29
Effect of the Hydrophobic Branching Group
Compounds SK220 and SK232 are derived from parent compounds SK182 and SK609, respectively, by increasing the length or the hydrophobicity or hydrophilicity of the branching group. SK220, similar to its parent compound SK182, is moderately active despite gaining additional favorable interactions with Val 111 and Cys114 and the extended hydrophobic tail. However, SK232, with an increased branching with the methanol linker, led to an increase in activity. SK232, despite its increase in activity, could not be used as a lead compound because we observed a loss of activity when it was stored at room temperature, although it was stable when maintained at −20 °C. Compound SK213 was designed to test the effect of converting a primary amine to a secondary amine, which did not impair the efficacy of the molecule. Docking studies showed that the molecule maintained all its interactions with the binding site residues, similar to its parent molecule SK609.
Binding Profiles of SK609 and Its Analogues at Dopamine Receptor Subtypes
To further characterize the binding profiles, we chose a subset of the compounds (SK609, SK608, SK213, SK232, SK032, SK041, SK052, and SK058) and compared them with reference compounds DA or PD128907 and vehicle controls DMSO and/or vitamin C (VC). Binding screening with 10 μM of SK609 and its analogues was conducted using HEK-D1R, CHO-D2R, CHO-D3R, HEK-D4R, and HEK-D5R cell lines (Figure 5). Results from the screening showed that SK609 and its analogues were effective at binding to D3R but none of the test compounds had any significant affinity for D2R (IC50 ≫ 10 μM). A similar profile was observed for D4R with the exception of SK608. However, the reference compounds DA and PD128907 had high binding affinity for D2R, D3R, and D4R in the same assay. None of the test compounds showed binding to D1R or D5R.
Figure 5.
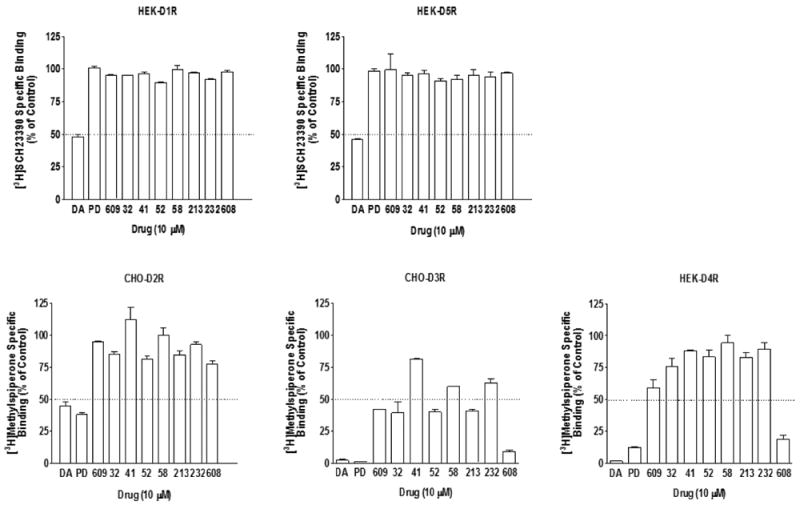
Binding screening of SK609 and its seven analogues at D1R, D5R, D2R, D3R, and D4R. [3H]SCH23390 binding to HEK-D1R or HEK-D5R and [3H]methylspiperone binding to CHO-D2R, CHO-D3R, or HEK-D4R was conducted with vehicle or 10 μM of DA, PD128907 (PD) or test compounds as described in Methods. Each value represents mean ± SEM of three independent experiments performed in duplicate.
Further competitive inhibition (affinity assay) by SK609 and its analogues to D3R was conducted and the Ki values for high affinity (Ki-H) and low affinity (Ki-L) were determined. As shown in Table 3, Ki-H values for the parent compound SK609 and the analogue SK608 were 283 ± 30 nM and 103 ± 25 nM, respectively, compared with reference compounds DA and PD128907, with Ki-H values of 9.9 ± 4.4 and 3.7 ± 1.6 nM, respectively. The Ki-H values of other SK609 analogues varied from 1 to 7 μM. In the radioligand binding assay, SK608 exhibited improvement in binding profile to D3R, with a Ki-H value of 103 nM compared with 283 nM for SK609; however, neither compound had detectable binding to D2R or other DA receptors (Figure 5 and SI Figure 3), except that SK608 also had significant binding to D4R at 10 μM. Molecular docking results suggest these compounds bind at the orthosteric site with specific interactions with residues from TM3, 5, and 6.26
Table 3.
Ki Values (nM) of SK609 and Its Analogue or Reference Compound Binding to the CHO-D3Ra
| compd |
Ki (nM) to CHO-hD3
|
|
|---|---|---|
| Ki-H | Ki-L | |
| DA | 9.9 ± 4.4 | 246 ± 105 |
| PD128907 | 3.7 ± 1.6 | 14.6 ± 3.6 |
| 609 | 283 ± 30 | 4955 ± 1314 |
| 608 | 103 ± 25 | 1029 ± 45 |
| 32 | 1109 ± 310 | 9504 ± 1551 |
| 52 | 1758 ± 901 | 9293 ± 1015 |
| 213 | 3708 ± 1003 | 8082 ± 1185 |
| 232 | 7008 ± 1850 | 7705 ± 1987 |
Competitive inhibition by SK609 and its analogues or reference compounds of [3H]methylspiperone binding to D3R was conducted. The competition curves were fitted by two-site analysis and its high-affinity Ki values (Ki-H) and low-affinity Ki values (Ki-L) were determined using the Prism program. Each value represents mean ± SEM of three experiments performed in duplicate.
In order to confirm the affinity of test compounds to D2R, we reassessed the binding affinity to CHO-D2R in the low- and high-affinity states as described in Methods. Neither DA and PD128907 nor any of the test compounds had affinity for D2R at 10 μM (IC50 ≫ 10 μM) when CHO-D2R was shifted to the low-affinity state in Na+-GTPγS binding buffer (SI Figure 3). However, DA and PD128907 were found to have higher affinity for D2R than in the regular binding buffer (Figure 5), when CHO-D2R was shifted to the high-affinity state with pretreatment of membranes with GDP and Na+ and the binding assay conducted in the presence of Mg2+, a finding in agreement with the studies on D2R and other GPCRs.30 However, SK609 and its analogues had no affinity to D2R even when D2R was in high affinity state. The D2R-like antagonist raclopride had similar high affinity to D2R in both the low- and high-affinity states (SI Figure 3). The results strongly support the conclusion that SK609 and its analogues had no affinity for D2R (IC50 ≫ 10 μM).
It is noteworthy that the binding assay with antagonist [3H]methylspiperone commonly used for D2-like receptors has its limitations in determining agonist binding affinity. The antagonist spiperone can bind both high- and low-affinity sites/states of the D2R-like receptors.31 D2R-like agonists have been reported to show much lower affinity to D2R or D3R using antagonist [3H]methylspiperone than agonist [3H]7-OH-DPAT or other hot agonists.23b,32 In future studies, we will reassess and compare the binding affinities of SK609 and SK608 to D3R using radioligand D2R-like agonists such as [3H]7-OH-DPAT.
Agonist-Induced [35S]GTPγS Binding at Human Dopamine D3R
Next, the compounds were tested for their ability to couple G-proteins. The binding of [35S]GTPγS to the Gα subunit of Gi/o proteins when induced by an agonist is a well-known functional assay to characterize the potency and efficacy of an agonist. Figure 6 showed that 10 μM of SK609 and its analogues increased [35S]GTPγS binding to membranes of CHO-D3R cells. Among these compounds, the values of [35S]GTPγS binding increase induced by compounds SK609, SK052, SK213, and SK232 reached 50% or more of the value of DA, indicating that these compounds are D3R agonists and activate Gi/o proteins (Figure 6). The potencies (EC50) and efficacies (Emax) of SK609 and its analogues and reference compounds DA and PD128907 in stimulating CHO-D3R to enhance [35S]GTPγS binding are shown in Table 2. DA-evoked [35S]GTPγS binding increased, with EC50 values of 11.3 ± 2.7 nM and 319.3 ± 73.1 nM at D3R and D2R, respectively, a finding in agreement with previous studies.23c,33 Similarly, in the same assay PD128907 had EC50 values of 1.19 ± 0.18 and 103.2 ± 29.6 nM at D3R and D2R, respectively, and SK609 had an EC50 value of 1109 ± 273 nM at D3R. Compounds SK608, SK052, SK213, and SK232 had EC50 values of 33.5 ± 8.4, 1324 ± 315, 1285 ± 44, and 3458 ± 868 nM, respectively, at D3R. In comparison with DA (its effect defined as 100%), PD128907 had a full agonist effect on both D3R and D2R, with Emax values of 95.1 ± 2.1 and 92.8 ± 1.7, respectively. In the same assay, SK609 had an EC50 value of ~1 μM, with an Emax of ~75% of DA, suggesting it may be a partial agonist at D3R; however, its analogues SK213 and SK232 had Emax values >95% and were full agonists. In comparison, the analogue SK608 had the strongest potency, with an EC50 value of 33.5 nM and an Emax value 81.1 ± 5.5. Taken together, these results indicate that SK609 and its analogues are D3R agonists to activate Gi/o proteins and downstream ERK1/2 signaling but have no effects on D2R/ERK1/2 signaling.
Figure 6.
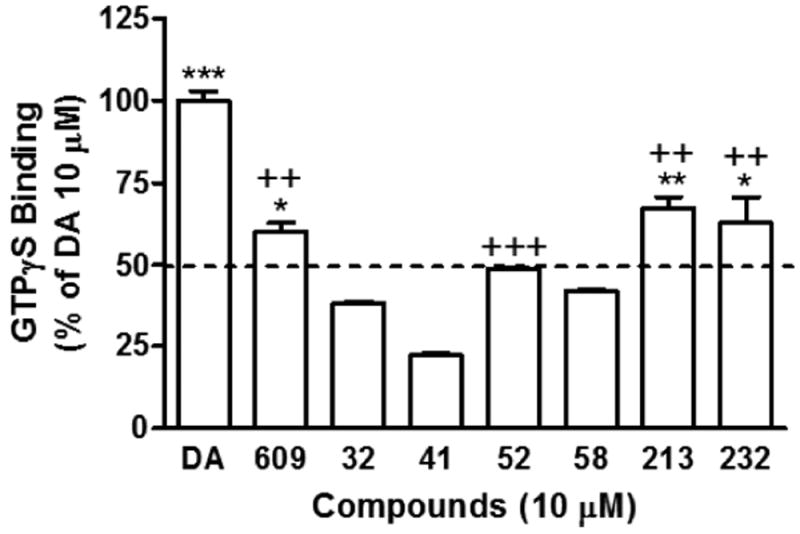
Functional screening of SK609 and its 6 analogues for [35S]GTPγS binding at CHO-D3R. DA-induced [35S]GTPγS binding at CHO-D3R was set as % of vehicle control and the test compounds-induced binding values are presented as % of that of DA. SK032, SK041 and SK058 which induced much lower binding compared with DA were not used for further assays. Each value represents mean ± SEM of three independent experiments performed in duplicate. ++P < 0.01 and +++P < 0.001 when compared with DA or *P < 0.05, **P < 0.01 and ***P < 0.001, compared with SK052, by one-way ANOVA followed by Newman-Keuls multiple-comparison test.
Time Course of SK609 and Its Analogue-Induced ERK1/2 Phosphorylation
In agreement with previous studies from GPCRs,18,34 we observed that DA (1 μM) elicited both early and late phases of phosphorylation of ERK1/2 in CHO-D3R cells. The early phase peaked at about 5 min and the late phase was sustained for nearly 1 h (Figure 7). However, SK609 and its analogue SK213 (1 μM) only induced an early phase (in 10 min) of ERK1/2 phosphorylation, suggesting that SK609 and its analogues may be exhibiting a biased signaling effect to induce ERK1/2 phosphorylation, unlike DA, which is known to signal through both G-protein- and β-arrestin-mediated pathways.
Figure 7.
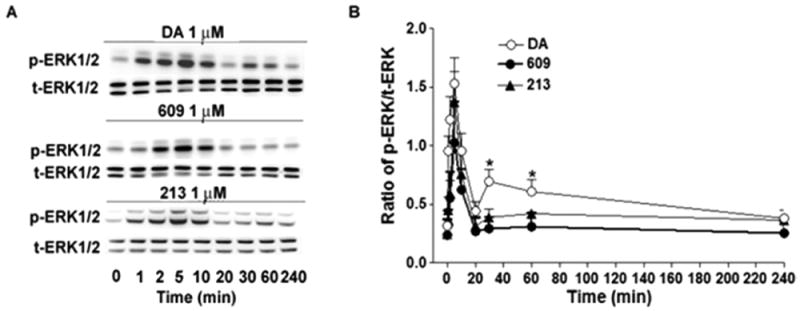
Time course of DA-, SK609-, and SK213-induced ERK1/2 phosphorylation at CHO-D3R. (A) Representative Western blot of ERK1/2 phosphorylation induced by DA, SK609, and SK213 at the indicated times. CHO-D3R cells were pretreated at 0, 1, 2, 5, 10, 20, 30, 60, and 240 min with 1 μM of DA, SK609, or SK213, washed once with PBS buffer and lysed for Western blots as described in Methods. (B) Signals were quantified by densitometry and normalized to total ERK. The graphs represent mean ± SEM of three independent experiments. *P < 0.05 compared with DA by one-way ANOVA followed by Newman–Keuls multiple-comparison test.
SK609 and Its Analogues Do Not Induce Desensitization of the D3R/ERK1/2 Pathway
GPCRs undergo desensitization following prolonged or repeated activation by an agonist. To assess whether SK609 and its analogues induce D3R desensitization compared with reference compounds DA or PD128907, we subjected HEK-D3R cells to two successive treatments with vehicle or 1 μM of PD128907, SK609, and its analogues to repeatedly activate D3R. The response to repeated activation was measured using the phosphorylation signal of ERK1/2. As shown in Figure 8, repeated activation of D3R by PD128907 (PD/PD group) resulted in a significant reduction of ERK1/2 phosphorylation signal compared with the control (DMSO/PD group). However, neither SK609 nor its analogues produced a significant reduction in phosphorylation signal, indicating that these compounds do not induce desensitization of D3R/ERK1/2 signaling. However, other D2R/D3R agonists such as PD128907 (Figure 8) or DA35 significantly induced D3R desensitization. Although p-ERK1/2 is not commonly used to assess GPCR desensitization, we have demonstrated that repeated agonist treatments also affect downstream ERK1/2 pathways for D3R, which was also demonstrated in vivo.36 The mechanisms underlying homologous desensitization of GPCRs are similar among receptors.37 It has been reported that DA-induced desensitization of D3R is associated with pharmacological sequestration in a Gβγ- and β-arrestin-dependent manner.35b
Figure 8.
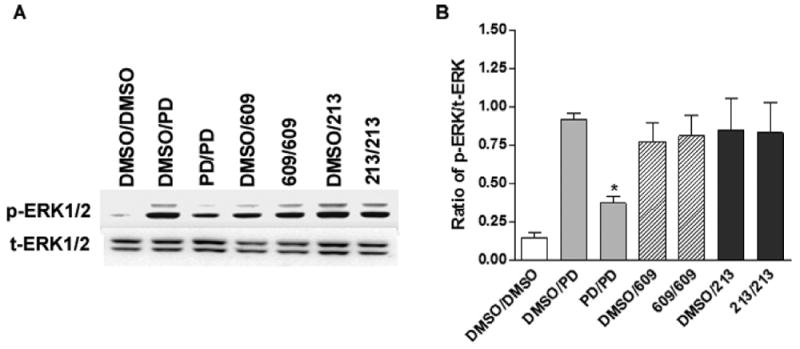
SK609 and SK213 do not induce desensitization of ERK1/2 signaling at HEK-D3R. (A) Representative Western blot of ERK1/2 phosphorylation induced by two successive treatments of vehicle, PD128907 (PD), SK609, and SK213. HEK-D3R cells were pretreated with vehicle, 1 μM of PD128907, SK609, or SK213 for 30 min, washed four times (5 min/per wash), and subsequently treated with vehicle, 1 μM of PD128907, SK609, or SK213 for 2 min, and washed once with PBS buffer and lysed for Western blots as described in Methods. (B) Signals were quantified by densitometry and normalized to total ERK. The graphs represent mean ± SEM of four independent experiments. *P < 0.01, compared with dimethyl sulfoxide (DMSO)/PD by one-way ANOVA followed by a Newman–Keuls multiple-comparison test.
SK609 and Its Analogues are Gi/o/ERK1/2 Biased Signaling D3R Agonists
To determine the biased signaling effects of SK609 and its analogues, we tested their effects on β-arrestin-2 recruitment after D3R activation. We used the PathHunter eXpress (DiscoverX, Fremont, CA) U2OS D3R β-arrestin recruitment assay, which uses a β-galactosidase fragment complementation-based technology to monitor the interaction of β-arrestin with GPCRs. As shown in Figure 9, while PD128907 potently evoked D3R-mediated β-arrestin recruitment, with an EC50 of 5.5 ± 0.18 nM, SK609 and its analogue SK213 had very weak effects, with EC50 values computed as 14.4 ± 2.7 μM and 6.8 ± 0.5 μM, respectively, and Emax values of 57.3 ± 2.5 and 66.1 ± 6.5 (% of PD128907), respectively; the latter values are perhaps underestimates, obtained from incomplete curves that stop at the high practical limit of 100 uM (not reaching the plateau) (Figure 9).
Figure 9.
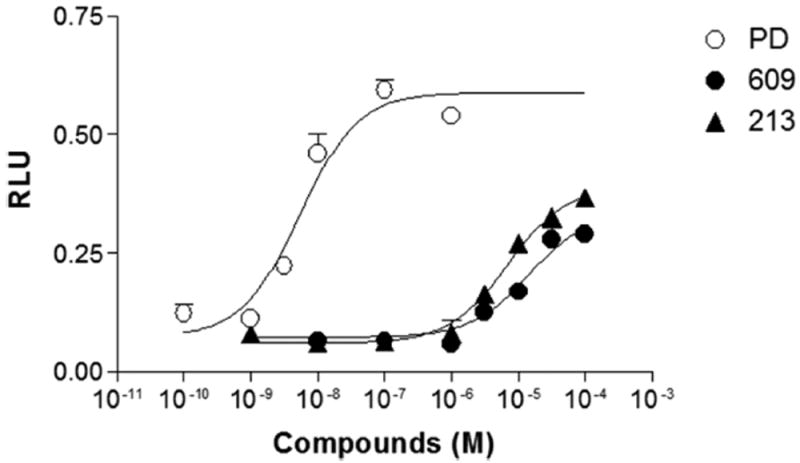
SK609 and SK213 do not recruit β-arrestin-2 for signaling at D3R. Dose–response curve of PD128907, SK609, and SK213 for recruiting β-arrestin-2 at D3R was tested. PathHunter eXpress β-arrestin-2 U2OS cells stably expressing hD3R were plated at 10 000 cells/well in a 96-well plate, incubated for 48 h, and stimulated for 90 min with vehicle and indicated concentrations of compounds PD128907, SK609, and SK213 as described in Methods. Following stimulation, signal was detected using the PathHunter detection reagents following the manufacturer’s protocol. RLU (relative luminescence units) represents a measure of β-arrestin-2 recruitment in this assay, and the data is presented as net change of RLU minus vehicle control (0.027 ± 0.002) of three independent experiments performed in triplicate.
A comparison of the effect of SK609 and its analogues across all the functional pathways is shown in Table 2. The ratios of the EC50 values of SK609 and SK213 at β-arrestin recruitment and G-protein-dependent ERK1/2 phosphorylation are 286 or 173, respectively; the ratios of Emax values of SK609 and SK213 for β-arrestin recruitment and G-protein-induced ERK1/2 phosphorylation are 0.70 or 0.69, respectively. The ratios of EC50 values of SK609 and SK213 for Gi/o coupling and the ERK1/2 pathway are 21 or 33, respectively; the ratios of Emax values of SK609 and SK213 are 0.91 and 1.03, respectively. Taken together, these results indicate that SK609 and SK213 have higher potency for G-protein-induced ERK1/2 phosphorylation, with a low tendency to recruit β-arrestin, and are Gi/o/ERK1/2 biased signaling agonists of D3R.
Screening for Off-Target Effects of SK609 Using β-Arrestin Recruitment Assay
A family of GPCRs (128 receptors) was screened using DiscoverX using their platform β-arrestin recruitment technology in both agonist and antagonist mode. At a high concentration (100 μM), SK609 had significant affinities only to D2R, D3R, CB1, 5HT2A, and β2-adrenergic receptors, and showed agonist activities at D2R and D3R and antagonist activity at β2-adrenergic, cannabinoid, and 5HT2A receptors (Figure 10A). Further testing in a 10-point dose–response study confirmed that neither SK609 nor its analogue SK213 had antagonizing effects on the isoproterenol-induced β2-adrenergic receptor activation, with an IC50 > 10 μM (Figure 10B).
Figure 10.
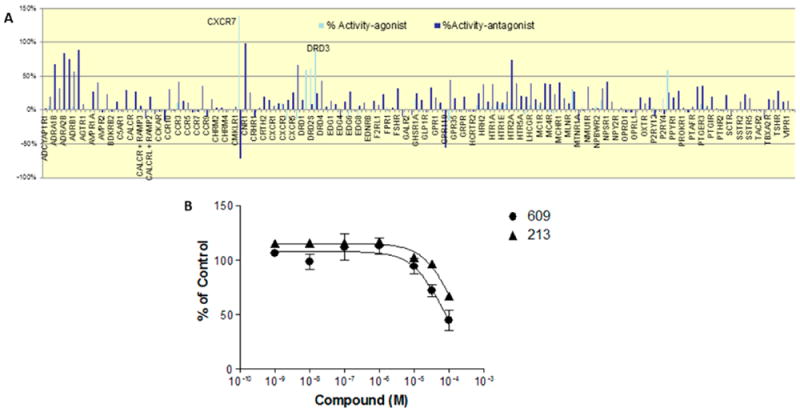
Screening for off-target effects of SK609 at high concentration (100 μM) on a panel of 128 GPCRs using the DiscoveRx β-arrestin recruitment assay. (A) Screening at 100 μM of SK609 in both agonist (light blue bars) and antagonist mode (dark blue bars) resulted in ~70 receptors that had an effect (nonzero value). SK609 has an agonist effect at D2R and D3R and antagonist activity at adrenergic, cannabinoid receptor-1, and 5HT2A receptors at a concentration of 100 μM. Several other hits were found to be false positives. (B) Dose–response curves of SK609 and SK213 for antagonizing isoproterenol-induced β-arrestin-2 recruitment at the β2-adrenergic receptor. CHO-K1 cells stably expressing β2-adrenergic receptor were plated at 10 000 cells/well in a 96-well plate and incubated for 48 h, pretreated with vehicle or indicated concentrations of SK609 or SK213 for 30 min, and subsequently treated with vehicle or 300 nM of isoproterenol (EC80) for 90 min. Following stimulation, signal was detected using the PathHunter detection reagents following the manufacturer’s protocol. Each value represents mean ± SEM of three independent experiments performed in duplicate and represented as percent of control (300 nM of isoproterenol).
In conclusion, the results obtained indicate that SK609 and its analogues are novel biased signaling agonists of D3R. These compounds have no affinity to D2R and do not significantly bind to other GPCRs. They possess unique signaling features in that they do not recruit β-arrestin-2 for downstream signaling and they show specificity toward G-protein-dependent signaling to activate and phosphorylate ERK1/2. In addition to being biased signaling agonists, SK609 and its analogues also possess atypical agonist features that are not commonly known among the D3R agonists. SK609 and its analogues do not induce receptor desensitization as demonstrated with the ERK1/2 phosphorylation assay with repeated agonist treatment. Our results demonstrate that SK609 and its analogues do not significantly recruit β-arrestin and hence do not induce desensitization. These observations are in agreement with our previous reports that SK609 does not induce tolerance at the GIRK channels.26 We have recently demonstrated that SK609 has very potent effects in reversing akinesia in a hemiparkinsonian rodent model.23a The unique signaling features of these molecules (including no tolerance and desensitization) are also reflected in the in vivo effects of SK609, in which chronic treatment with the compound does not lead to loss of efficacy as is seen with most other D3R-selective agents, such as pramipexole.38 In addition to its ability to reverse akinesia, SK609 can be combined with L-dopa synergistically to improve motor deficits without aggravating the dyskinesias. In fact, SK609 can block the dyskinetic effects produced by L-dopa, an action that can be attributed to the partial agonistic property of SK609 at postsynaptic D3R present mostly as D1R-D3R heteromers.39 Our future efforts will be geared toward understanding the functional effects of biased signaling agonists such as SK609 on D1R/D3R cross talk at the membrane and their in vivo effects in naïve and hemiparkinsonian rodent models.
METHODS
Materials
[3H]Methyl spiperone (85.5 Ci/mmol), [3H]SCH23390 (73.1 Ci/mmol), and [35S]GTPγS (1250 Ci/mmol) were purchased from PerkinElmer (Boston, MA). Tetracycline, hygromycin, blasticidin, dopamine (DA), (+) butaclamol, fluphenazine, phenylmethylsulfonyl fluoride (PMSF), GDP, GTPγS, and ascorbic acid were obtained from Sigma-Aldrich (St. Louis, MO). G418 was obtained from Gemini Bio-Products (West Sacramento, CA); cell culture reagents were obtained from Invitrogen (Carlsbad, CA); and protease inhibitor cocktail, phosphatase inhibitor cocktail, and lysis buffer were obtained from Thermo Scientific (Rockford, IL). All test molecules (Table 1) were either synthesized in-house as described below or purchased from vendors. Asinex Corp. (Winston-Salem, NC): SK603, SK609, SK610, SK611; Enamine, Ltd. (Kiev, Ukraine): Compounds described in Lichtenberger patent28 L1 and L2; Asiba Pharmatech, Inc. (Milltown, NJ): R- and S-enantiomers of SK609. DA and D2R/D3R agonists PD128907 and pramipexole were purchased from Tocris Bioscience (Bio-Techne, Inc., CA).
Synthesis of SK609 and Its Analogues
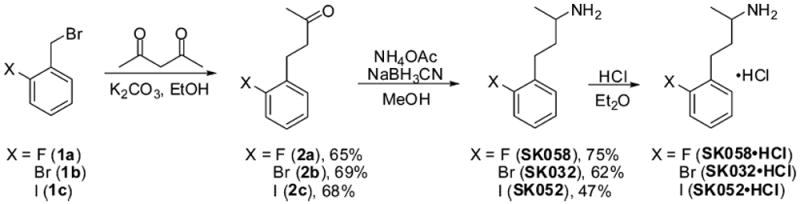
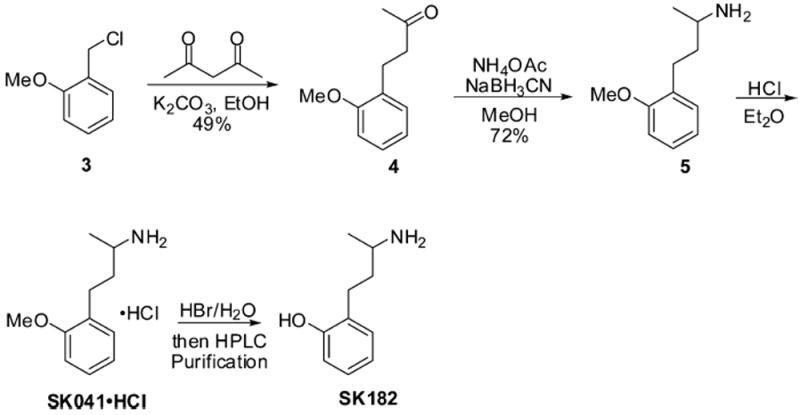
The preparation of a series of SK609 analogues was achieved through exploration of different synthetic routes (as outlined in detail in Supporting Information). For example, synthesis of SK032·HCl began with an alkylation/deacylation cascade, using starting material benzyl bromide 1b (Scheme 1). Reductive amination was carried out using NaBH3CNas reducing agent, to afford the desired product SK032, which was then converted to its HCl salt. Compounds SK058·HCl and SK052·HCl, both as HCl salts, were prepared following the same synthetic sequence. This route can also be applied to deliver compound SK041·HCl, which was then treated with HBr in H2O to generate the phenol analogue SK182 (Scheme 2).
Scheme 1.
Syntheses of Compounds SK058·HCl, SK032·HCl, and SK052·HCl
Scheme 2.
Syntheses of Compounds SK041·HCl and SK182
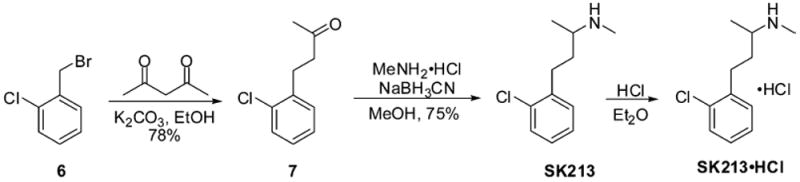
To prepare compound SK213·HCl, the alkylation/deacylation sequence was performed on benzyl chloride starting material 6 to afford ketone 7 (Scheme 3). Reductive amination reaction employing MeNH2·HCl then produced a secondary amine SK213 in good yield (75%), which again was converted to the HCl salt using HCl in Et2O solution.
Scheme 3.
Synthesis of Compound SK213·HCl
To access compound SK220, benzyl chloride 8 was first alkylated with β-keto ester 9, with NaH as base, and in the presence of tetrabutylammonium iodide (TBAI) (Scheme 4). The resulting ester 10 was then heated in EtOH and H2O under basic conditions (KOH) to hydrolyze the ethyl ester, followed by decarboxylation to deliver ketone intermediate 11 in 51% yield over two steps. The carbonyl moiety in 11 was then converted to the corresponding primary amine using the method as described above (NH4OAc and NaBH3CN). Finally, demethylation of compound 12 with HBr in H2O was achieved, followed by high-performance liquid chromatography (HPLC) purification, to afford phenol analogue SK220.
Scheme 4.
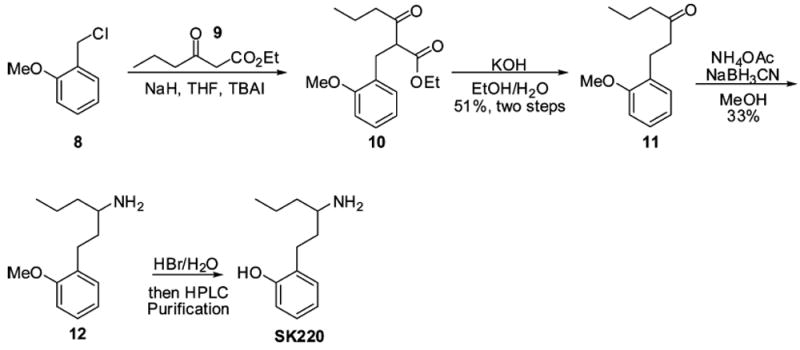
Synthesis of Compound SK220
The synthesis of primary hydroxyl analogue SK232·HCl utilized the bromo starting material 13, which was first alkylated with diester 14 (Scheme 5). The diester 15 that was thus generated was then treated with LiBr/H2O in DMF to produce intermediate 16. The ester functionality in 16 was reduced to alcohol with LiAlH4, and the Boc protecting group was subsequently removed to deliver the desired product SK232·HCl.
Scheme 5.
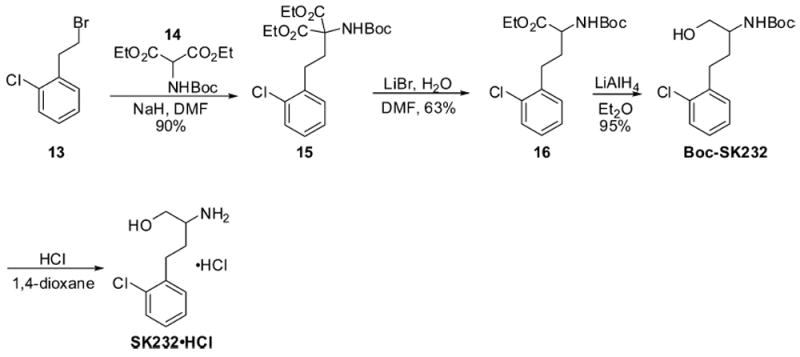
Synthesis of Compound SK232·HCl
Finally, compound SK609 was commercially purchased as a racemate and converted to its HCl salt using the previously described procedure, without complications (Scheme 6). Detailed experimental procedures and characterization data (1H NMR, 13C NMR, and high-resolution mass spectrometry [HRMS]) of all the above-mentioned compounds has been described in the Supporting Information.
Scheme 6.
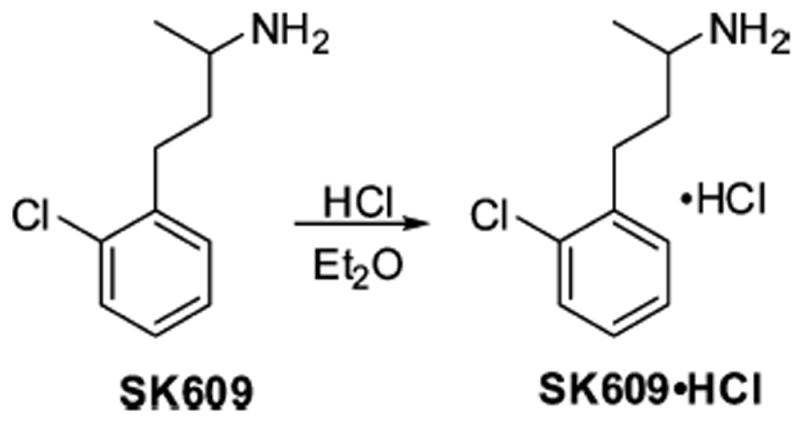
Synthesis of Compound SK609·HCl
Enantiomeric Forms
The enantiomeric forms of SK609 were purchased from Asiba Pharmatech and were synthesized in an asymmetric fashion with an enantiomeric enrichment of ~98%. The enantiomer enrichment was performed using chiral HPLC as analytical method and diastereomeric salt formation or isomer separation as an enrichment method. The enantiomeric forms and purity were confirmed using liquid chromatography–mass spectrometry (LC-MS) (SI Figure 2).
In Silico Screening and Molecular Docking
We have previously reported on differences in receptor conformations induced by tolerance producing and nontolerance producing agonists at the D3R.26,29 In addition, using iterative rounds of site-directed mutagenesis and modeling studies, we have demonstrated the role of the extracellular loop-2 (EL2) and TM3-IL2 regions in contributing to a tolerance phenotype in quinpirole and PD128907.29,40 Using this information and the pharmacophore of cis-8-OH-PBZI (a nontolerance producing D3R agonist) as template to the hybrid structure based (HSB) method of in silico screening, we identified SK609 as the best-ranking molecule that displayed high selectivity for D3R.26 The crystal structure of D3R25a was prepared by adding hydrogen atoms and setup in a POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) membrane environment. Further the membrane bound D3R was refined with 10 ns of molecular dynamics simulation using the NAMD (Nanoscale Molecular Dynamics) program and Amber99 (Assisted Model Building with Energy Refinement) charges and force field.41 We utilized the three major pharmacophore features of SK609 to derive analogues using the HSB method. The features included an aromatic ring with electrophilic substituents, a secondary or tertiary amine group, and a three-carbon linker between the aromatic ring and the amine group. The best 50 hits that conformed with respect to at least two features (aromatic ring and amine group) and ranked by Tanimoto similarity score (similarity to SK609) were chosen for docking studies. The molecules were docked to the previously characterized26 binding site consisting of residues from TM3, 5, and 6 and EL2 using GOLD program (ver 4.1).42 The complexes were scored using a hybrid scoring scheme,43 consisting of weighting the GOLD score with a custom score that was derived by scoring ring interactions with residues from TM5 and 6 and amine interactions with Asp110 and penalizing reversed poses.43 The best-ranking molecules and a few worst-ranking molecules (docked with the correct pose but without interactions with Asp110, such as R-SK609, L1, and L2 molecules) were chosen for further in vitro validation.
Cell Culture
HEK-293 cells stably expressing human DA D1 receptor (HEK-D1R), D2 receptor (HEK-D2R), D3 receptor (HEK-D3R), D4 receptor (HEK-D4R), or D5 receptor (HEK-D5R) and CHO cells stably expressing human D2 receptor (CHO-D2R) or human D3 receptor (CHO-D3R) were grown in 100 mm culture dishes in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal calf serum, 100 units/mL penicillin, and 100 μg/mL streptomycin in a humidified atmosphere consisting of 5% CO2 and 95% air at 37 °C. HEK-D1R, HEK-D2R, or HEK-D3R cells, which have the CMV promoter controlled by a Tet-on mechanism, were grown in the presence of 50 μg/mL hygromycin and 5 μg/mL blasticidin to maintain T-rex and receptor selection, respectively. The HEK cells were induced to express D1R, D2R, or D3R with 1 μg/mL tetracycline for 12 to 24 h before binding or functional assays. CHO-D2R, CHO-D3R, HEK-D4R, or HEK-D5R cells were grown in the presence of 0.1 mg/mL G418 or 5 μg/mL blasticidin to maintain high receptor expression levels.
Receptor Binding
Dopamine receptor binding was performed according to our published procedure.33,33 For D1R and D5R, [3H]SCH23390 was used as the radiolabeled ligand and fluphenazine (10 μM) was used to define nonspecific binding, whereas [3H]-methylspiperone and (+)-butaclamol (4 μM) were used for radiolabeling and nonspecific binding, respectively for D2R, D3R, and D4R. Membranes were prepared from transfected CHO-D2R and CHO-D3R cells or HEK-D1R, HEK-D5R, HEK-D2R, HEK-D3R, and HEK-D4R cells as described previously.33 Saturation binding of [3H]-methylspiperone to D2R and D3R was performed with at least 8 concentrations of [3H]methylspiperone (ranging from 20 pM to 10 nM). Binding was carried out in 50 mM Tris-HCl buffer containing 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, and 1 mM MgCl2 (pH 7.4) at room temperature for 1 h in duplicate in a volume of 250 μL with 10 to 100 μg membrane protein depending on receptor expression level. Incubations were terminated by filtration through Whatman GF/B filters (GE Healthcare Lifesciences) under vacuum, and radioactivity on filters was measured. Competitive inhibition of [3H]methylspiperone (1 nM) binding to D2R or D3R by test compounds and positive controls DA and PD128907 was performed with varying concentrations (10−11 to 10−5 M) of test compounds. Binding data was analyzed with the Prism program (GraphPad, San Diego, CA), Kd and Bmax values were determined, and further Ki values were determined using the one-site or two-site analysis. A similar analysis could not be performed for D2R because the compounds had no affinity to D2R at 10 μM. To further distinguish the effect of compounds on D2Rs’ high- and low-affinity states,44 two additional binding buffers were used to determine the affinity of test compounds in competing for the binding of [3H]-methylspiperone to CHO-D2R. The Na+-GTPγS binding buffer (50 mM Tris-HCl, pH 7.4, with 154 mM NaCl, 0.025% ascorbic acid, 10 μM GTPγS, and 0.001% bovine serum albumin), which was used to convert the receptors to a low-affinity state for agonists30a and the Mg2+ binding buffer (50 mM Tris-HCl, pH 7.4, 6 mM MgCl2, 0.025% ascorbic acid, and 0.001% bovine serum albumin) was used for high-affinity state agonist binding. In addition to using the Mg2+ buffer, the membranes were preincubated with 100 μM GDP and 100 mM NaCl for 30 min at room temperature and washed and centrifuged at 40,000 × g for 15 min to remove excess Na+ and GDP from membranes, which was used to promote the receptors to a high-affinity state for agonists in the presence of Mg2+.30b,45 [3H]methylspiperone (about 0.25 nM) binding to D2R was conducted with vehicle or 10 μM of DA and PD128907 and D2R-like antagonist raclopride (RAC) or test compounds for 1 h at 30 °C in the two binding buffers, respectively. Haloperidol 10 μM was used to define nonspecific binding.
[35S]GTPγS Binding Assay
Determination of [35S]GTPγS binding to G proteins was carried out using a modification of our published procedure.33,46 Briefly, membranes of CHO-D2R or CHO-D3R cells were prepared using published procedures.47 Membranes (20 μg protein) were incubated in reaction buffer containing [35S]GTPγS (80–100 pM) and 3 or 6 μM GDP, 10 or 3 mM MgCl2, and 150 or 100 mM NaCl with various concentrations of each test compound (10−10 to 10−4 M) in a total volume of 0.5 mL for 60 min at 30 °C. Nonspecific binding was determined in the presence of 10 μM GTPγS. Bound and free [35S]GTPγS were separated by filtration. Data was analyzed and EC50 and Emax values were determined with the Prism program.
Immunoblot Analysis of Phosphorylated ERK1/2 MAPK
CHO-D3R and CHO-D2R cells (0.3 × 106) or HEK-D2R and HEK-D3R cells (0.6 × 106) were seeded in 6-well plates and cultured in 3 mL of DMEM complete growth medium for 48 h. Cells were washed with 1 mL of phosphate-buffered saline (PBS) and incubated with 3 mL of prewarmed DMEM without serum for 2 h. Reference compound DA (1 μM with 10 μg/mL ascorbic acid) or D2R-like agonist PD128907 (1 μM in 0.01% dimethyl sulfoxide [DMSO]) and test compounds (1 μM in 0.01% DMSO) were added to the cells and incubated for the indicated times at 37 °C. The reaction was stopped by aspirating the medium and washing the cells with 1 mL PBS followed by the addition of 100 μL/well lysis buffer with 1× Protease Inhibitor Cocktail, 1× Phosphatase Inhibitor Cocktail, and 1 mM PMSF to solubilize cells. Cell lysates were assayed for protein content using the DC Protein Assay Kit II (Bio-Rad). Loading buffer was added to protein samples and boiled for 5 min at 100 °C in a water bath, and spun at 2500g. Five μg total protein/lane was loaded to Novex 4–20% Tris-Glycine Mini Gel for separation (Invitrogen) for 2 h and transferred to a PVDF membrane (Thermo Scientific). Immunoblotting was performed with rabbit anti-phosphop44/ 42 MAPK polyclonal antibody (1:5000, Cell Signaling Technology) and peroxidase-conjugated goat anti-rabbit IgG (H+L) (1:5000, Jackson Immuno Research Laboratories, Inc.,). Total p44/42 MAPK levels in the same blots were also determined with rabbit antip44/42 MAPK monoclonal or polyclonal antibody (1:5000, Cell Signaling Technology) after stripping the blots of the same PVDF membrane. Chemiluminescence detection was performed using the SuperSignal West Dura Extended Duration Substrate detection kit (Thermo Scientific). Phosphorylated or total ERK1/2 MAPK immunoblots were quantified by densitometry with ImageQuant LAS4000 (GE Healthcare Bio-Sciences, Pittsburgh, PA) and normalized to vehicle treatment. Data was analyzed and EC50 and Emax values were determined with the Prism program.
For desensitization experiments, 0.6 × 106 HEK-D3R cells were seeded in 6-well plates and cultured in 3 mL of DMEM complete growth medium for 48 h. Cells were washed with 1 mL of PBS and incubated with 3 mL of prewarmed DMEM without serum for 2 h. Vehicle, 1 μM of PD128907 or test compounds was added to the cells and incubated for 30 min (first time pretreatment) at 37 °C in CO2 incubator. The reaction was stopped by aspirating the medium and the cells were washed four times (5 min/per wash in CO2 incubator) with 3 mL of DMEM without serum and subsequently treated a second time with vehicle or 1 μM of PD128907, test compounds for 2 min at 37 °C. The reaction was stopped by aspirating the medium and the cells were washed once with 1 mL PBS followed by the addition of 100 μL/well lysis buffer with 1× Protease Inhibitor Cocktail and 1× Phosphatase Inhibitor Cocktail and 1 mM PMSF to solubilize cells for immunoblot assays.
β-Arrestin Recruitment DiscoveRx Assay
The measurement of β-arrestin recruitment stimulated by D3R or β2-adrenergic receptor activation was conducted using D3R U2OS or ADRB2-CHO-K1 PathHunter express cells purchased from DiscoveRx (Fremont, CA) according to the manufacturer’s protocol. In brief, cells were thawed, resuspended in the supplied medium, plated at 10 000 cells/well in a 96-well plate, and incubated for 48 h at 37 °C. After stimulation with vehicle or various concentrations (10−10–10−4 M) of test compounds in triplicate for 90 min at 37 °C, the detection reagent solution was added and incubated for 60 min at room temperature. For an antagonizing effect of test compounds on isoproterenol-induced β-arrestin recruitment, ADRB2-CHO-K1 cells were pretreated with vehicle or various concentrations (10−9–10−4 M) of test compounds for 30 min at 37 °C. Further, vehicle or 300 nM of isoproterenol (EC80) was added and incubated for 90 min at 37 °C, and the detection reagent solution was then added. Chemiluminescence was recorded using Fluoroskan Ascent FL (Thermo Fisher Scientific). Data was analyzed and EC50 or IC50 and Emax values were determined using the Prism program. The β-arrestin recruitment assay was also used to assess the effect of SK609 in a GPCR panel assay consisting of 124 receptors in both agonist and antagonist mode at a high concentration of 100 μM.
Determination of the Protein Content
Protein content in the membrane was determined using the DC Protein Assay Kit II with bovine serum albumin as the standard (Bio-Rad, Hercules, CA), following the manufacturer’s protocol.
Statistical Analysis
All data is presented as the mean ± SEM unless specified otherwise. For comparison of multiple groups, data was analyzed using one-way ANOVA followed by a Newman–Keuls multiple-comparison test. For comparison of two groups, Student’s t test was performed. P < 0.05 was defined as a statistically significant difference.
Supplementary Material
Acknowledgments
Funding
S.K. acknowledges funding support from Drexel University Innovation fund, Drexel-Coulter translational research grants and faculty development funds from Department of Microbiology, Drexel University. A.M.T. was funded by a Drexel University undergraduate summer research program fellowship. L.-Y. L.-C. acknowledges funding from NIH P30 DA013429.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro. 6b00221.
Additional synthesis and 1H NMR profiles of all the compounds synthesized in our laboratory and described under Schemes 1-6; LC-MS profiles of R, S, and racemate forms of SK609; Western blot image associated with Figure 2A; comparison of binding affinity of SK609 and its analogues to CHO-D2R in the low affinity state and the high affinity state (PDF)
Notes
The authors declare the following competing financial interest(s): The compounds described in the Article are part of portfolio licensed by Drexel University to PolyCore Therapeutics LLC. Dr. Sandhya Kortagere is a co-founder and owns equity in Polycore Therapeutics LLC. However, no part of this study was funded by PolyCore Therapeutics LLC.
References
- 1.(a) Rohrer DK, Kobilka BK. G protein-coupled receptors: functional and mechanistic insights through altered gene expression. Physiol Rev. 1998;78(1):35–52. doi: 10.1152/physrev.1998.78.1.35. [DOI] [PubMed] [Google Scholar]; (b) Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459(7245):356–63. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Beaulieu JM, Espinoza S, Gainetdinov RR. Dopamine receptors - IUPHAR Review 13. British journal of pharmacology. 2015;172(1):1–23. doi: 10.1111/bph.12906. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jackson DM, Westlind-Danielsson A. Dopamine receptors: molecular biology, biochemistry and behavioural aspects. Pharmacol Ther. 1994;64(2):291–370. doi: 10.1016/0163-7258(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 3.Kortagere S, Gmeiner P, Weinstein H, Schetz JA. Certain 1,4-disubstituted aromatic piperidines and piperazines with extreme selectivity for the dopamine D4 receptor interact with a common receptor microdomain. Mol Pharmacol. 2004;66(6):1491–1499. doi: 10.1124/mol.104.001321. [DOI] [PubMed] [Google Scholar]
- 4.(a) Luttrell LM, Maudsley S, Bohn LM. Fulfilling the Promise of “Biased” G Protein-Coupled Receptor Agonism. Mol Pharmacol. 2015;88(3):579–88. doi: 10.1124/mol.115.099630. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stott LA, Hall DA, Holliday ND. Unravelling intrinsic efficacy and ligand bias at G protein coupled receptors: A practical guide to assessing functional data. Biochem Pharmacol. 2016;101:1–12. doi: 10.1016/j.bcp.2015.10.011. [DOI] [PubMed] [Google Scholar]; (c) Wisler JW, Xiao K, Thomsen AR, Lefkowitz RJ. Recent developments in biased agonism. Curr Opin Cell Biol. 2014;27:18–24. doi: 10.1016/j.ceb.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Jin M, Min C, Zheng M, Cho DI, Cheong SJ, Kurose H, Kim KM. Multiple signaling routes involved in the regulation of adenylyl cyclase and extracellular regulated kinase by dopamine D(2) and D(3) receptors. Pharmacol Res. 2013;67(1):31–41. doi: 10.1016/j.phrs.2012.09.012. [DOI] [PubMed] [Google Scholar]; (b) Kurko D, Kapui Z, Nagy J, Lendvai B, Kolok S. Analysis of functional selectivity through G protein-dependent and -independent signaling pathways at the adrenergic alpha(2C) receptor. Brain Res Bull. 2014;107:89–101. doi: 10.1016/j.brainresbull.2014.07.005. [DOI] [PubMed] [Google Scholar]; (c) Shukla AK, Xiao K, Lefkowitz RJ. Emerging paradigms of beta-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem Sci. 2011;36(9):457–69. doi: 10.1016/j.tibs.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Kahsai AW, Xiao K, Rajagopal S, Ahn S, Shukla AK, Sun J, Oas TG, Lefkowitz RJ. Multiple ligand-specific conformations of the beta2-adrenergic receptor. Nat Chem Biol. 2011;7(10):692–700. doi: 10.1038/nchembio.634. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shukla AK, Singh G, Ghosh E. Emerging structural insights into biased GPCR signaling. Trends Biochem Sci. 2014;39(12):594–602. doi: 10.1016/j.tibs.2014.10.001. [DOI] [PubMed] [Google Scholar]; (c) Tikhonova IG, Selvam B, Ivetac A, Wereszczynski J, McCammon JA. Simulations of biased agonists in the beta(2) adrenergic receptor with accelerated molecular dynamics. Biochemistry. 2013;52(33):5593–603. doi: 10.1021/bi400499n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Woo AY, Song Y, Zhu W, Xiao RP. Advances in receptor conformation research: the quest for functionally selective conformations focusing on the beta2-adrenoceptor. British journal of pharmacology. 2015;172(23):5477–88. doi: 10.1111/bph.13049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) DeWire SM, Violin JD. Biased ligands for better cardiovascular drugs: dissecting G-protein-coupled receptor pharmacology. Circ Res. 2011;109(2):205–16. doi: 10.1161/CIRCRESAHA.110.231308. [DOI] [PubMed] [Google Scholar]; (b) Violin JD, Soergel DG, Boerrigter G, Burnett JC, Jr, Lark MW. GPCR biased ligands as novel heart failure therapeutics. Trends Cardiovasc Med. 2013;23(7):242–9. doi: 10.1016/j.tcm.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 8.Chini B, Parenti M, Poyner DR, Wheatley M. G-protein-coupled receptors: from structural insights to functional mechanisms. Biochem Soc Trans. 2013;41(1):135–6. doi: 10.1042/BST20120344. [DOI] [PubMed] [Google Scholar]
- 9.(a) Mistry SN, Shonberg J, Draper-Joyce CJ, Klein Herenbrink C, Michino M, Shi L, Christopoulos A, Capuano B, Scammells PJ, Lane JR. Discovery of a Novel Class of Negative Allosteric Modulator of the Dopamine D2 Receptor Through Fragmentation of a Bitopic Ligand. J Med Chem. 2015;58(17):6819–43. doi: 10.1021/acs.jmedchem.5b00585. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kenakin T. Functional selectivity through protean and biased agonism: who steers the ship? Molecular pharmacology. 2007;72(6):1393–401. doi: 10.1124/mol.107.040352. [DOI] [PubMed] [Google Scholar]; (c) Kenakin T. Collateral efficacy in drug discovery: taking advantage of the good (allosteric) nature of 7TM receptors. Trends Pharmacol Sci. 2007;28(8):407–15. doi: 10.1016/j.tips.2007.06.009. [DOI] [PubMed] [Google Scholar]; (d) Watson C, Chen G, Irving P, Way J, Chen WJ, Kenakin T. The use of stimulusbiased assay systems to detect agonist-specific receptor active states: implications for the trafficking of receptor stimulus by agonists. Mol Pharmacol. 2000;58(6):1230–1238. doi: 10.1124/mol.58.6.1230. [DOI] [PubMed] [Google Scholar]
- 10.Szabo M, Klein Herenbrink C, Christopoulos A, Lane JR, Capuano B. Structure-activity relationships of privileged structures lead to the discovery of novel biased ligands at the dopamine D(2) receptor. J Med Chem. 2014;57(11):4924–39. doi: 10.1021/jm500457x. [DOI] [PubMed] [Google Scholar]
- 11.(a) Mouillac B, Mendre C. Vasopressin receptors and pharmacological chaperones: from functional rescue to promising therapeutic strategies. Pharmacol Res. 2014;83:74–8. doi: 10.1016/j.phrs.2013.10.007. [DOI] [PubMed] [Google Scholar]; (b) Rahmeh R, Damian M, Cottet M, Orcel H, Mendre C, Durroux T, Sharma KS, Durand G, Pucci B, Trinquet E, Zwier JM, Deupi X, Bron P, Baneres JL, Mouillac B, Granier S. Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc Natl Acad Sci U S A. 2012;109(17):6733–8. doi: 10.1073/pnas.1201093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science. 2012;335(6072):1106–10. doi: 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Warne T, Edwards PC, Leslie AG, Tate CG. Crystal structures of a stabilized beta1-adrenoceptor bound to the biased agonists bucindolol and carvedilol. Structure. 2012;20(5):841–9. doi: 10.1016/j.str.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huff RM. Signal transduction pathways modulated by the D2 subfamily of dopamine receptors. Cell Signalling. 1996;8(6):453–9. doi: 10.1016/s0898-6568(96)00074-5. [DOI] [PubMed] [Google Scholar]
- 15.Min C, Cho DI, Kwon KJ, Kim KS, Shin CY, Kim KM. Novel regulatory mechanism of canonical Wnt signaling by dopamine D2 receptor through direct interaction with beta-catenin. Molecular pharmacology. 2011;80(1):68–78. doi: 10.1124/mol.111.071340. [DOI] [PubMed] [Google Scholar]
- 16.Urs NM, Caron MG. The physiological relevance of functional selectivity in dopamine signalling. Int J Obes. 2014;4(Suppl 1):S5–8. doi: 10.1038/ijosup.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Peterson SM, Pack TF, Wilkins AD, Urs NM, Urban DJ, Bass CE, Lichtarge O, Caron MG. Elucidation of G-protein and beta-arrestin functional selectivity at the dopamine D2 receptor. Proc Natl Acad Sci U S A. 2015;112(22):7097–102. doi: 10.1073/pnas.1502742112. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Urs NM, Bido S, Peterson SM, Daigle TL, Bass CE, Gainetdinov RR, Bezard E, Caron MG. Targeting beta-arrestin2 in the treatment of L-DOPA-induced dyskinesia in Parkinson’s disease. Proc Natl Acad Sci U S A. 2015;112(19):E2517–26. doi: 10.1073/pnas.1502740112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allen JA, Yost JM, Setola V, Chen X, Sassano MF, Chen M, Peterson S, Yadav PN, Huang XP, Feng B, Jensen NH, Che X, Bai X, Frye SV, Wetsel WC, Caron MG, Javitch JA, Roth BL, Jin J. Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc Natl Acad Sci U S A. 2011;108(45):18488–93. doi: 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Chen X, Sassano MF, Zheng L, Setola V, Chen M, Bai X, Frye SV, Wetsel WC, Roth BL, Jin J. Structurefunctional selectivity relationship studies of beta-arrestin-biased dopamine D(2) receptor agonists. J Med Chem. 2012;55(16):7141–53. doi: 10.1021/jm300603y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Masri B, Salahpour A, Didriksen M, Ghisi V, Beaulieu JM, Gainetdinov RR, Caron MG. Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc Natl Acad Sci U S A. 2008;105(36):13656–61. doi: 10.1073/pnas.0803522105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Free RB, Chun LS, Moritz AE, Miller BN, Doyle TB, Conroy JL, Padron A, Meade JA, Xiao J, Hu X, Dulcey AE, Han Y, Duan L, Titus S, Bryant-Genevier M, Barnaeva E, Ferrer M, Javitch JA, Beuming T, Shi L, Southall NT, Marugan JJ, Sibley DR. Discovery and characterization of a G proteinbiased agonist that inhibits beta-arrestin recruitment to the D2 dopamine receptor. Mol Pharmacol. 2014;86(1):96–105. doi: 10.1124/mol.113.090563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Shonberg J, Herenbrink CK, Lopez L, Christopoulos A, Scammells PJ, Capuano B, Lane JR. A structure-activity analysis of biased agonism at the dopamine D2 receptor. J Med Chem. 2013;56(22):9199–221. doi: 10.1021/jm401318w. [DOI] [PubMed] [Google Scholar]; (b) Shonberg J, Lopez L, Scammells PJ, Christopoulos A, Capuano B, Lane JR. Biased agonism at G protein-coupled receptors: the promise and the challenges–a medicinal chemistry perspective. Med Res Rev. 2014;34(6):1286–330. doi: 10.1002/med.21318. [DOI] [PubMed] [Google Scholar]
- 22.(a) Fiorentini C, Savoia P, Bono F, Tallarico P, Missale C. The D3 dopamine receptor: From structural interactions to function. Eur Neuropsychopharmacol. 2015;25(9):1462–9. doi: 10.1016/j.euroneuro.2014.11.021. [DOI] [PubMed] [Google Scholar]; (b) Bezard E, Ferry S, Mach U, Stark H, Leriche L, Boraud T, Gross C, Sokoloff P. Attenuation of levodopa-induced dyskinesia by normalizing dopamine D3 receptor function. Nat Med. 2003;9(6):762–7. doi: 10.1038/nm875. [DOI] [PubMed] [Google Scholar]; (c) Rangel-Barajas C, Coronel I, Floran B. Dopamine Receptors and Neurodegeneration. Aging and disease. 2015;6(5):349–68. doi: 10.14336/AD.2015.0330. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sokoloff P, Diaz J, Le Foll B, Guillin O, Leriche L, Bezard E, Gross C. The dopamine D3 receptor: a therapeutic target for the treatment of neuropsychiatric disorders. CNS Neurol Disord : Drug Targets. 2006;5(1):25–43. doi: 10.2174/187152706784111551. [DOI] [PubMed] [Google Scholar]
- 23.(a) Simms SL, Huettner DP, Kortagere S. In vivo characterization of a novel dopamine D3 receptor agonist to treat motor symptoms of Parkinson’s disease. Neuropharmacology. 2016;100:106–15. doi: 10.1016/j.neuropharm.2015.04.004. [DOI] [PubMed] [Google Scholar]; (b) Zou MF, Keck TM, Kumar V, Donthamsetti P, Michino M, Burzynski C, Schweppe C, Bonifazi A, Free RB, Sibley DR, Janowsky A, Shi L, Javitch JA, Newman AH. Novel Analogues of (R)-5-(Methylamino)-5,6-dihydro-4H-imidazo[4,5,1-ij]- quinolin-2(1H)-one (Sumanirole) Provide Clues to Dopamine D2/D3 Receptor Agonist Selectivity. J Med Chem. 2016;59(7):2973–88. doi: 10.1021/acs.jmedchem.5b01612. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Das B, Vedachalam S, Luo D, Antonio T, Reith ME, Dutta AK. Development of a Highly Potent D2/D3 Agonist and a Partial Agonist from Structure-Activity Relationship Study of N(6)-(2-(4-(1H-Indol-5-yl)piperazin-1-yl)ethyl)-N(6)-propyl-4,5,6,7-tetrahydroben zo- [d]thiazole-2,6-diamine Analogues: Implication in the Treatment of Parkinson’s Disease. J Med Chem. 2015;58(23):9179–95. doi: 10.1021/acs.jmedchem.5b01031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(a) Keck TM, John WS, Czoty PW, Nader MA, Newman AH. Identifying Medication Targets for Psychostimulant Addiction: Unraveling the Dopamine D3 Receptor Hypothesis. J Med Chem. 2015;58(14):5361–80. doi: 10.1021/jm501512b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sabioni P, Di Ciano P, Le Foll B. Effect of a D3 receptor antagonist on context-induced reinstatement of nicotine seeking. Prog Neuro-Psychopharmacol Biol Psychiatry. 2016;64:149–54. doi: 10.1016/j.pnpbp.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 25.(a) Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330(6007):1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Michino M, Beuming T, Donthamsetti P, Newman AH, Javitch JA, Shi L. What can crystal structures of aminergic receptors tell us about designing subtype-selective ligands? Pharmacol Rev. 2015;67(1):198–213. doi: 10.1124/pr.114.009944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuzhikandathil EV, Kortagere S. Identification and characterization of a novel class of atypical dopamine receptor agonists. Pharm Res. 2012;29(8):2264–75. doi: 10.1007/s11095-012-0754-0. [DOI] [PubMed] [Google Scholar]
- 27.Kortagere S, Ekins S, Welsh WJ. Halogenated ligands and their interactions with amino acids: implications for structure-activity and structure-toxicity relationships. J Mol Graphics Modell. 2008;27(2):170–7. doi: 10.1016/j.jmgm.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Erfinder, W. S. G. W. L. Olaf New phenylethyl amine derivatives useful in the treatment of psychiatric and/or neurological disorders. DE102011015842 A1 2012
- 29.Westrich L, Gil-Mast S, Kortagere S, Kuzhikandathil EV. Development of tolerance in D3 dopamine receptor signaling is accompanied by distinct changes in receptor conformation. Biochem Pharmacol. 2010;79(6):897–907. doi: 10.1016/j.bcp.2009.10.016. [DOI] [PubMed] [Google Scholar]
- 30.(a) Zhen J, Antonio T, Ali S, Neve KA, Dutta AK, Reith ME. Use of radiolabeled antagonist assays for assessing agonism at D2 and D3 dopamine receptors: comparison with functional GTPgammaS assays. J Neurosci Methods. 2015;248:7–15. doi: 10.1016/j.jneumeth.2015.03.028. [DOI] [PubMed] [Google Scholar]; (b) Liu-Chen LY, Yang HH, Li S, Adams JU. Effect of intracerebroventricular beta-funaltrexamine on mu opioid receptors in the rat brain: consideration of binding condition. J Pharmacol Exp Ther. 1995;273(3):1047–1056. [PubMed] [Google Scholar]
- 31.Roberts DJ, Lin H, Strange PG. Investigation of the mechanism of agonist and inverse agonist action at D2 dopamine receptors. Biochem Pharmacol. 2004;67(9):1657–65. doi: 10.1016/j.bcp.2003.12.030. [DOI] [PubMed] [Google Scholar]
- 32.Lahti RA, Evans DL, Figur LM, Carrigan KJ, Moon MW, Hsi RS. Dopamine D2 receptor binding properties of [3H]U-86170, a dopamine receptor agonist. Eur J Pharmacol. 1991;202(2):289–91. doi: 10.1016/0014-2999(91)90308-d. [DOI] [PubMed] [Google Scholar]
- 33.Xu W, Wang Y, Ma Z, Chiu YT, Huang P, Rasakham K, Unterwald E, Lee DY, Liu-Chen LY. L-isocorypalmine reduces behavioral sensitization and rewarding effects of cocaine in mice by acting on dopamine receptors. Drug Alcohol Depend. 2013;133(2):693–703. doi: 10.1016/j.drugalcdep.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.(a) Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, Eckhardt AE, Cowan CL, Spurney RF, Luttrell LM, Lefkowitz RJ. Distinct beta-arrestin- and G proteindependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J Biol Chem. 2006;281(16):10856–64. doi: 10.1074/jbc.M513380200. [DOI] [PubMed] [Google Scholar]; (b) McLennan GP, Kiss A, Miyatake M, Belcheva MM, Chambers KT, Pozek JJ, Mohabbat Y, Moyer RA, Bohn LM, Coscia CJ. Kappa opioids promote the proliferation of astrocytes via Gbetagamma and beta-arrestin 2-dependent MAPK-mediated pathways. J Neurochem. 2008;107(6):1753–65. doi: 10.1111/j.1471-4159.2008.05745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sneddon WB, Friedman PA. Beta-arrestin-dependent parathyroid hormone-stimulated extracellular signal-regulated kinase activation and parathyroid hormone type 1 receptor internalization. Endocrinology. 2007;148(8):4073–9. doi: 10.1210/en.2007-0343. [DOI] [PubMed] [Google Scholar]
- 35.(a) Kim KM, Gainetdinov RR, Laporte SA, Caron MG, Barak LS. G protein-coupled receptor kinase regulates dopamine D3 receptor signaling by modulating the stability of a receptor-filamin-beta-arrestin complex. A case of autoreceptor regulation. J Biol Chem. 2005;280(13):12774–80. doi: 10.1074/jbc.M408901200. [DOI] [PubMed] [Google Scholar]; (b) Min C, Zheng M, Zhang X, Caron MG, Kim KM. Novel roles for betaarrestins in the regulation of pharmacological sequestration to predict agonist-induced desensitization of dopamine D3 receptors. British journal of pharmacology. 2013;170(5):1112–29. doi: 10.1111/bph.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cote SR, Kuzhikandathil EV. In vitro and in vivo characterization of the agonist-dependent D3 dopamine receptor tolerance property. Neuropharmacology. 2014;79:359–67. doi: 10.1016/j.neuropharm.2013.11.023. [DOI] [PubMed] [Google Scholar]
- 37.Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–44. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- 38.(a) Rokosik SL, Napier TC. Pramipexole-induced increased probabilistic discounting: comparison between a rodent model of Parkinson’s disease and controls. Neuropsychopharmacology. 2012;37(6):1397–408. doi: 10.1038/npp.2011.325. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dardou D, Chassain C, Durif F. Chronic pramipexole treatment increases tolerance for sucrose in normal and ventral tegmental lesioned rats. Fronti Neurosci. 2014;8:437. doi: 10.3389/fnins.2014.00437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.(a) Fiorentini C, Savoia P, Savoldi D, Missale C. Receptor heteromers in Parkinson’s disease and L-DOPA-induced dyskinesia. CNS Neurol Disord : Drug Targets. 2013;12(8):1101–1113. [PubMed] [Google Scholar]; (b) Guitart X, Navarro G, Moreno E, Yano H, Cai NS, Sanchez-Soto M, Kumar-Barodia S, Naidu YT, Mallol J, Cortes A, Lluis C, Canela EI, Casado V, McCormick PJ, Ferre S. Functional selectivity of allosteric interactions within G protein-coupled receptor oligomers: the dopamine D1-D3 receptor heterotetramer. Mol Pharmacol. 2014;86(4):417–29. doi: 10.1124/mol.114.093096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gil-Mast S, Kortagere S, Kota K, Kuzhikandathil EV. An amino acid residue in the second extracellular loop determines the agonist-dependent tolerance property of the human D3 dopamine receptor. ACS Chem Neurosci. 2013;4(6):940–51. doi: 10.1021/cn3002202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267(3):727–48. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 43.Kortagere S, Welsh WJ. Development and application of hybrid structure based method for efficient screening of ligands binding to G-protein coupled receptors. J Comput -Aided Mol Des. 2007;20(12):789–802. doi: 10.1007/s10822-006-9077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grigoriadis D, Seeman P. Complete conversion of brain D2 dopamine receptors from the high- to the low-affinity state for dopamine agonists, using sodium ions and guanine nucleotide. J Neurochem. 1985;44(6):1925–35. doi: 10.1111/j.1471-4159.1985.tb07189.x. [DOI] [PubMed] [Google Scholar]
- 45.Wong CS, Su YF, Watkins WD, Chang KJ. Continuous intrathecal opioid treatment abolishes the regulatory effects of magnesium and guanine nucleotides on mu opioid receptor binding in rat spinal membranes. J Pharmacol Exp Ther. 1992;262(1):317–326. [PubMed] [Google Scholar]
- 46.Xu W, Chen C, Li JG, Dimattio K, Wang Y, Unterwald E, Liu-Chen LY. PKA and ERK1/2 are involved in dopamine D(1) receptor-induced heterologous desensitization of the delta opioid receptor. Life Sci. 2013;92(23):1101–9. doi: 10.1016/j.lfs.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu J, Luo LY, Li JG, Chen C, Liu-Chen LY. Activation of the cloned human kappa opioid receptor by agonists enhances [35S]GTPgammaS binding to membranes: determination of potencies and efficacies of ligands. J Pharmacol Exp Ther. 1997;282(2):676–684. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.