

Abstract
Prostate cancer (PCa) and type two diabetes mellitus (T2DM) are both increasing prevalent conditions and often occur concurrently. However, the relationship between the two is more complex than just two prevalent conditions co-existing. This review systematically explores the literature around the interplay between the two conditions. It covers the impact of pre-existing T2DM on PCa incidence, grade and stage, as well as exploring the impact of T2DM on PCa outcomes and mortality and the interaction between T2DM and PCa treatments.
Keywords: type two diabetes, prostate cancer, review
Introduction
Prostate cancer (PCa) remains the commonest cancer in men, affecting one in eight men in the UK [1]. There are also 3.2 million people in the UK who have been diagnosed with type 2 diabetes mellitus (T2DM) and it is estimated that this will rise to 5 million by 2025 [2]. PCa and T2DM thus often occur together in the same individual
T2DM increases the risk of cancer-specific death from several solid malignancies, including colorectal and breast cancer [3], but conflicting evidence exists in the case of PCa. The impact of pre-existing T2DM has also been studied in regard to grade and stage of PCa at presentation, with conflicting results [4]. Moreover, there is emerging evidence that the presence of T2DM and other metabolic abnormalities (dyslipidaemia, hypertension, obesity) is associated with a more rapid progression of PCa [5, 6]. This relationship is further complicated by the fact that standard treatment for advanced PCa, androgen deprivation therapy (ADT), has been suggested to increase incidence of T2DM [7],as well as worsen glycaemic control in those with pre-existing T2DM.
To provide a background on the complex association between PCa and T2DM, this review is set out to explore the following areas:
Impact of pre-existing T2DM on PCa incidence
Impact of pre-existing T2DM on PCa grade and stage
Impact of pre-existing T2DM on PCa outcomes and mortality
Interplay between T2DM and PCa treatments
Impact of pre-existing T2DM on PCa incidence
T2DM that increases the risk of some solid malignancies is now well established, although not all the literature is robust [8]. Some studies suggest that people with T2DM are as much as twice as likely to die from cancer than those without [3]. However, the opposite is seen in PCa, with an inverse association reported in several published meta-analyses [8].
Bonovas et al. published the first meta-analysis examining T2DM and risk of PCa in 2004. They included 14 studies and concluded that T2DM confers a statistically significant 9% decrease in relative risk of developing PCa [9]. This was followed in 2006 by a meta-analysis conducted by Kasper et al. which included 19 studies and reported an inverse relationship of a similar magnitude, relative risk (RR): 0.84 (95%CI: 0.76–0.93) [10]. Following this, Bansal et al. published an updated meta-analysis including 45 studies, involving 8.1 million participants and 132,331 PCa cases, which also reports an inverse association with an RR of 0.86 (95% CI 0.80–0.92) [11]. More recently, Gang et al. published a further updated meta-analysis reviewing the literature up to and including April 2012. This meta-analysis included 56 studies and also reported an inverse association, RR: 0.88 (95%CI: 0.82–0.93) [12]. Here, we perform a further systematic review of the literature up until June 2017.
Evidence acquisition
The systematic review was performed in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guidelines [13], with search terms, inclusion and exclusion criteria all defined a priori.
Search strategy
A computerised literature search of Pubmed to identify full text and abstracts published was performed. The search was done with and without MESH terms (diabetes, diabetes mellitus, PCa, prostate neoplasm, incidence, risk). All references of the selected articles were checked, including hand searches.
Study eligibility
The final articles were chosen based on the following set of inclusion criteria:
examined association of T2DM with PCa incidence/risk
case control or cohort study
English Language
not included in the prior published meta-analyses described above
Articles were excluded if they:
examined association of T2DM with PCa mortality
examined association of T2DM treatments (i.e. drugs) and PCa incidence
were a review article or meta-analysis
Initially, titles were reviewed to assess whether they met inclusion criteria. If, after assessing the abstract, there was any doubt regarding whether it met the relevant criteria, it was kept for more thorough, subsequent assessment. The list of potential articles was further shortened by performing detailed evaluations of the methods and results of each remaining paper. Figure 1 provides more detailed information regarding the exclusion process. The strength of each study was assessed using the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) criteria [14] and is shown in Table 1.
Figure 1. Preferred reporting items for systematic reviews and meta-analysis (PRISMA) flow diagram of article identification, screening, eligibility and inclusion for systematic review on impact of T2DM on PCa incidence.
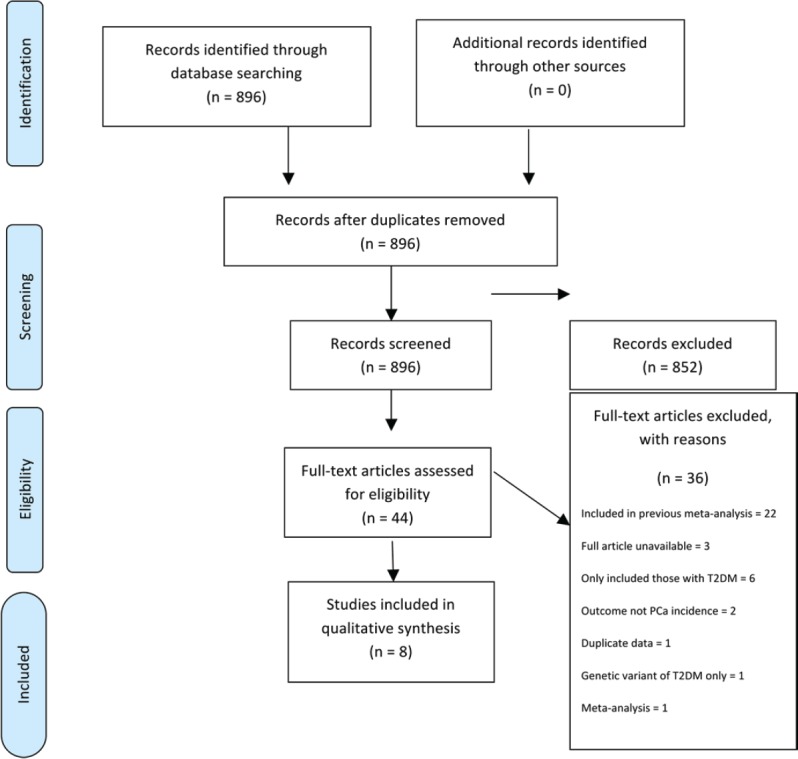
Table 1. Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) checklist for observational studies included in the systematic review on impact of T2DM on PCa incidence.
| Item No. | Recommendation | Dankner 2006 [20] | Tsilidis 2015 [15] | Lai 2013 [16] | Lawrence 2013 [17] | Fall 2013 [18] | Magliano 2012 [21] | Attner 2012 [22] | Moses 2012 [19] | |
|---|---|---|---|---|---|---|---|---|---|---|
| Title and abstract | 1 | (a) Indicate the study’s design with a commonly used term in the title or the abstract | ☒ | ☐ | ☒ | ☐ | ☒ | ☒ | ☒ | ☒ |
| (b) Provide in the abstract an informative and balanced summary of what was done and what was found | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ||
| Introduction | ||||||||||
| Background/rationale | 2 | Explain the scientific background and rationale for the investigation being reported | ☐ | ☒ | ☐ | ☒ | ☒ | ☒ | ☒ | ☒ |
| Objectives | 3 | State specific objectives, including any pre specified hypotheses | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ |
| Methods | ||||||||||
| Study design | 4 | Present key elements of study design early in the paper | ☒ | ☒ | ☒ | ☒ | ☒ | ☐ | ☒ | ☒ |
| Setting | 5 | Describe the setting, locations, and relevant dates, including periods of recruitment, exposure, follow-up, and data collection | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ |
| Participants | 6 | (a) Cohort study—Give the eligibility criteria, and the sources and methods of selection of participants. Describe methods of follow-up | ☒ | ☒ | ☒ | ☒ | n/a | n/a | n/a | ☒ |
| Case-control study—Give the eligibility criteria, and the sources and methods of case ascertainment and control selection. Give the rationale for the choice of cases and controls | n/a | n/a | n/a | n/a | ☒ | ☒ | ☒ | n/a | ||
| (b) Cohort study—For matched studies, give matching criteria and number of exposed and unexposed | ☒ | ☒ | ☒ | ☒ | n/a | n/a | n/a | ☒ | ||
| Case-control study—For matched studies, give matching criteria and the number of controls per case | n/a | n/a | n/a | n/a | ☒ | ☒ | ☒ | n/a | ||
| Variables | 7 | Clearly define all outcomes, exposures, predictors, potential confounders, and effect modifiers. Give diagnostic criteria, if applicable | ☒ | ☒ | ☐ | ☒ | ☒ | ☒ | ☐ | ☒ |
| Data sources/measurement | 8* | For each variable of interest, give sources of data and details of methods of assessment (measurement). Describe comparability of assessment methods if there is more than one group | ☒ | ☒ | ☐ | ☒ | ☒ | ☒ | ☐ | ☒ |
| Bias | 9 | Describe any efforts to address potential sources of bias | ☐ | ☒ | ☒ | ☒ | ☒ | ☒ | ☐ | ☒ |
| Study size | 10 | Explain how the study size was arrived at | ☒ | ☐ | ☐ | ☐ | ☐ | ☐ | ☐ | ☐ |
| Quantitative variables | 11 | Explain how quantitative variables were handled in the analyses. If applicable, describe which groupings were chosen and why | n/a | ☒ | n/a | ☒ | ☒ | ☒ | n/a | ☒ |
| Statistical methods | 12 | (a) Describe all statistical methods, including those used to control for confounding | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ |
| (b) Describe any methods used to examine subgroups and interactions | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ||
| (c) Explain how missing data were addressed | ☐ | ☒ | ☐ | ☐ | ☐ | ☐ | ☐ | ☒ | ||
| (d) Cohort study—If applicable, explain how loss to follow-up was addressed | ☐ | ☒ | ☒ | ☐ | n/a | n/a | n/a | ☒ | ||
| Case-control study—If applicable, explain how matching of cases and controls was addressed | n/a | n/a | n/a | n/a | ☒ | ☒ | ☒ | n/a | ||
| (e) Describe any sensitivity analyses | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ||
| Results | ||||||||||
| Participants | 13* | (a) Report numbers of individuals at each stage of study—e.g., numbers potentially eligible, examined for eligibility, confirmed eligible, included in the study, completing follow-up and analysed | ☒ | ☒ | ☐ | ☒ | ☒ | ☐ | ☐ | ☒ |
| (b) Give reasons for non-participation at each stage | ☐ | ☐ | ☐ | ☐ | ☐ | ☐ | ☐ | ☐ | ||
| (c) Consider the use of a flow diagram | ☒ | ☐ | ☐ | ☐ | ☐ | ☐ | ☐ | ☐ | ||
| Descriptive data | 14* | (a) Give characteristics of study participants (e.g., demographic, clinical, social) and information on exposures and potential confounders | ☒ | ☒ | ☐ | ☒ | ☒ | ☒ | ☐ | ☒ |
| (b) Indicate number of participants with missing data for each variable of interest | ☒ | ☒ | ☐ | ☐ | ☐ | ☐ | ☐ | ☒ | ||
| (c) Cohort study—Summarise follow-up time (e.g., average and total amount) | ☒ | ☒ | ☐ | ☒ | n/a | n/a | n/a | n/a | ||
| Outcome data | 15* | Cohort study—Report numbers of outcome events or summary measures over time | ☒ | ☒ | ☐ | ☒ | n/a | n/a | n/a | ☐ |
| Case-control study—Report numbers in each exposure category, or summary measures of exposure | n/a | n/a | n/a | n/a | ☒ | ☒ | ☒ | n/a | ||
| Main results | 16 | (a) Give unadjusted estimates and, if applicable, confounder-adjusted estimates and their precision (e.g., 95% confidence interval). Make clear which confounders were adjusted for and why they were included | ☐ | ☒ | ☐ | ☐ | ☐ | ☒ | ☒ | ☒ |
| (b) Report category boundaries when continuous variables were categorised | n/a | ☒ | n/a | ☒ | ☒ | ☒ | n/a | ☒ | ||
| (c) If relevant, consider translating estimates of relative risk into absolute risk for a meaningful time period | ☐ | ☐ | ☐ | ☐ | ☐ | ☐ | ☐ | ☐ | ||
| Other analyses | 17 | Report other analyses done—e.g., analyses of subgroups and interactions, and sensitivity analyses | ☒ | ☒ | ☒ | ☐ | ☒ | ☒ | ☐ | ☒ |
| Discussion | ||||||||||
| Key results | 18 | Summarise key results with reference to study objectives | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ |
| Limitations | 19 | Discuss limitations of the study, taking into account sources of potential bias or imprecision. Discuss both direction and magnitude of any potential bias | ☒ | ☐ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ |
| Interpretation | 20 | Give a cautious overall interpretation of results considering objectives, limitations, multiplicity of analyses, results from similar studies, and other relevant evidence | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ |
| Generalisability | 21 | Discuss the generalisability (external validity) of the study results | ☒ | ☐ | ☐ | ☒ | ☐ | ☒ | ☐ | ☐ |
| Other information | ||||||||||
| Funding | 22 | Give the source of funding and the role of the funders for the present study and, if applicable, for the original study on which the present article is based | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☐ |
Data collection
The following details were recorded for each study: author, year of publication, country where study was undertaken, study design, number of patients, population/setting, outcome reported and variables adjusted for in the analysis.
Evidence synthesis
The literature search identified a total of 896 studies, of which 44 were deemed initially relevant. Using the above inclusion and exclusion criteria, 36 were excluded (Figure 1). The reasons for exclusion were included in previously published meta-analysis (n = 22), only T2DM patients included (n = 6), full article not available (n = 3), outcome not PCa incidence (n = 2), meta-analysis (n = 1), duplicate data (n = 1), genetic variant of T2DM examined only (n = 1) (Figure 1). A total of eight studies were included in the systematic review (Table 2).
Table 2. Characteristics of the eight studies included in the systematic review on impact of T2DM on PCa incidence.
| Author, year, country | Study design | No. of patients | Population/setting | Outcome reported | Adjusted for |
|---|---|---|---|---|---|
| Dankner R, 2016, Israel [20] | Retrospective cohort | 2,186,196 | Men aged 21–89 covered by a large healthcare provider | T2DM inversely associated with PCa HR: 0.80; 95%CI: 0.76–0.85 | Age, ethnicity, socioeconomic status |
| Tsilidis K, 2015, Europe [15] | Prospective cohort | 139,131 | Men aged 35–70 from general population | T2DM inversely associated with PCa HR: 0.74; 95%CI: 0.63–0.86 | Education, smoking, BMI, waist circumference, physical activity |
| Lai G, 2013, USA [16] | Prospective cohort | 295,276 | Men aged 50–71 in six US states, general population | T2DM inversely associated with PCa HR: 0.74; 95%CI: 0.70–0.78 | Age, BMI, race, education, marital status, education, family history cancer, diet, smoking |
| Lawrence YR, 2013, Israel [17] | Prospective cohort within RCT | 11,541 | Men aged 36–74 with coronary heart disease enrolled in a secondary prevention trial | T2DM inversely associated with PCa HR: 0.54; 95%CI: 0.40–0.73 | Fasting glucose, triglycerides, HDL, blood pressure, insulin, tobacco, metformin |
| Fall, 2013, Sweden [18] | Nested case control | 44,352 | Men from PCBaSe Sweden | T2DM inversely associated with PCa OR: 0.80; 95%CI: 0.76–0.85 | Socioeconomic status, marital status, comorbidity, age at PCa diagnosis, prevalence of DM in county |
| Magliano DJ, 2012, Australia [21] | Case control | 1,289 cases 5,156 controls |
Cases from Fremantle Diabetes Cohort Study and controls from general population | No significant association reported HR: 0.83; 95%CI: 0.60–1.14 | Age, sex, post code matched controls |
| Attner B, 2012, Sweden [22] | Case control | 3,545 cases 26,654 controls |
Cases from Cancer register Southern Sweden, controls from general population | T2DM inversely associated with PCa RR: 0.81; 95%CI: 0.72–0.93 | Age, sex, county matched controls |
| Moses KA, 2012, USA [19] | Retrospective cohort | 3,162 | Men referred for a prostate biopsy because of abnormal DRE and/or abnormal PSA | T2DM associated with increased odds of positive biopsy OR: 1.26; 95%CI: 1.01–1.55 | Age, race, BMI, prostate volume, family history, PSA, DRE, interaction PSA and DRE |
Of these eight studies, five were cohort studies [15–20] and three case-control studies [21, 22]. Three studies were from European populations, two from the USA, two from Israel and one from Australia.
The studies combined included 2,716,302 subjects. Six reported an inverse association between T2DM and PCa incidence [15–18, 20, 22], one reported no association [21] and in one a positive association was reported [19]. Of the six studies reporting an inverse association, they all reported similar measures of association in the magnitude of a 20% reduction in risk of PCa in those exposed to T2DM as compared to those without T2DM. The one paper which reports no statistically significant association is a case-control study in which cases of T2DM were patients enrolled in the Freemantle Diabetes Study [21]. It is the smallest of the studies, with 1,289 cases and 5,156 controls – which may account for the non- statistical significance of their findings (HR: 0.83, 95%CI: 0.60–1.14) – though the direction and magnitude of the association reported is in line with the other studies. The study which reported a positive association is a retrospective review on 3,162 consecutive men who underwent a prostate biopsy due to either an elevated PSA and/or an abnormal DRE [19]. This design is different from the other studies included here, which were largely based on the general population, not on a selected population attending for a prostate biopsy. This heterogeneity in the design may account for the findings of a 26% increased odds of a positive biopsy in patients with T2DM, compared to those without (OR: 1.26 95%CI: 1.01–1.55).
Discussion
The biological mechanism underlying the inverse association between T2DM and PCa risk is not elucidated. First, several metabolic alterations occur in people with T2DM which may protect from PCa. The Insulin-IGF-1 theory of carcinogenesis suggests that prolonged hyperinsulinaemia results in reduced insulin binding proteins and therefore increased free IGF-1, which results in cellular changes that can lead to carcinogenesis via increased mitosis and decreased apoptosis [23]. There is both laboratory and epidemiological evidence supporting that raised insulin levels are associated with increased PCa risk [24, 25]. Patients with T2DM, though initially may have raised insulin levels, over time develop hypoinsulinaemia. Hence, patients with T2DM who have lower levels of insulin over time would be protected in terms of PCa risk [26]. Several studies have reported a strengthening of the inverse association between T2DM and PCa risks with the duration of T2DM [27–29], which serves to strengthen this hypothesis. Prolonged hypoinsulinaemia may also result in a reduction of leptin, a hormone involved in energy homeostasis [30], raised levels of which have been associated with PCa risk [31, 32]. However, there are no published studies specifically examining this relationship between insulin and leptin levels in T2DM and risk of PCa.
Another metabolic change which occurs in T2DM is a reduction in testosterone levels which has been shown both in animal models and in vitro [33, 34]. PCa is testosterone driven [35]; therefore, a decrease in testosterone is expected to be associated with a decreased risk.
Genetic factors as well as metabolic changes have also been postulated to be involved in the protective effect which T2DM appears to have on PCa risk. The TCF2 gene confers a predisposition to T2DM and has also been shown to have a potential protective effect in PCa. Similarly, other studies have identified different variants in the JAZF1 gene, one associated with T2DM and another associated with PCa [36].
Some cross-sectional studies have shown that men with T2DM have lower PSA levels, compared to those without T2DM [37] and the rate of change over time is also lower [38]. This could result in less screen detected PCa and at least in part account for the difference in the risk seen. This is supported both by studies in which enrolled participants undergo prostate biopsy, which report increased risk of positive biopsies in T2DM [19], and by those which show higher grade PCa detected in those with T2DM [4].
Finally, treatments used for T2DM, including metformin, could be potential confounders in the association between T2DM and PCa risks, which is discussed later in this review.
Conclusion
The updated systematic review of the literature examining the association between T2DM and PCa risks presented here concurs with the previously published findings of several meta-analyses, indicating that T2DM has a protective effect on PCa risk. However, the underlying biological mechanisms are yet to be elucidated.
Impact of pre-existing T2DM on PCa grade and stage
PCa severity can be described in terms of its grade and its clinical or TNM stage. Some studies have examined whether pre-existing T2DM is associated with a particular grade or stage of disease. There is considerable overlap with the literature presented above examining T2DM and PCa incidence and that examining particular stage and grade of PCa. In the systematic review of T2DM and PCa incidence above which includes studies published after 2012, of the eight new studies identified, only two presented subgroup analysis on stage and/or grade [15, 18] (Table 3). These are discussed below but another systematic review was not deemed informative, as one including literature up until 2013 has previously been published [4].
Table 3. Overview of the eight papers included in the systematic review on T2DM and PCa incidence, by subgroup analysis including PCa stage and grade.
| Author, year, country | PCa stage | PCa grade |
|---|---|---|
| Dankner R, 2016, Israel [20] | No | No |
| Tsilidis K, 2015, Europe [15] | Yes | Yes |
| Lai G, 2013, USA [16] | No | No |
| Lawrence YR, 2013, Israel [17] | No | No |
| Fall 2013, Sweden [18] | Yes | Yes |
| Magliano DJ, 2012, Australia [21] | No | No |
| Attner B, 2012, Sweden [22] | No | No |
| Moses KA, 2012, USA [19] | No | No |
Existing literature
In 2013, Xu et al. undertook a meta-analysis of all studies examining the association between T2DM and PCa risks including subgroup analysis by different grade and stage [4]. They included nine studies: five examining stage only, two grade only and two which explored both. They reported findings of an inverse association between T2DM and PCa for both low- and high-grade PCa, defined as Gleason 2–6 and Gleason 7–10 (RR: 0.74, 95%CI: 0.64–0.86 and 0.78, 95%CI: 0.67–0.90). They reported an RR of a similar direction and magnitude for localised and advanced disease (RR: 0.72, 95%CI: 0.68–0.76 and 0.85, 95%CI: 0.75–0.97). This meta-analysis included all studies published up until October 2012.
Updated literature review
Two new studies were identified in the systematic review described above which considered stage and grade of PCa. A nested case-control study by Fall et al. included 44,352 men with PCa in Prostate Cancer data Base Sweden (PCBaSe) Sweden, which is based on the National Prostate Cancer register of Sweden (NPCR) [39]. They showed an inverse association between T2DM and risk of PCa across all risk groups, low, intermediate and high risk/metastatic (OR: 0.71, 95%CI: 0.64–0.80; 0.76, 95%CI: 0.69–0.84; 0.86, 95%CI: 0.80–0.93, respectively). Although they showed a slightly less clear risk pattern for those with high risk and metastatic disease, no significant difference between T2DM and risk category of PCa emerged from this study.
Tsilidis et al. included 139,131 men from the European Prospective Investigation into Cancer and Nutrition (EPIC) prospective cohort, 4,531 of whom went on to develop PCa. They reported no statistical evidence for an inverse association between T2DM and PCa risks. There was no evidence that the association differed by stage (p-heterogeneity, 0.19) or grade (p-heterogeneity, 0.48) of the disease, although the numbers were small in some subgroups and the study may have been under powered to detect differences.
Conclusion
The meta-analysis by Xu et al. [4] concluded that the protective effect of T2DM on PCa risk is seen across different disease grades and stages; however, the number of studies available was too small to confirm this finding. The two new studies identified in the systematic review of T2DM and PCa presented above, which considered grade or stage [15, 18], reported similar findings of no significant difference between different stages and grades of PCa. However, sample sizes remain small and no definitive conclusion can be drawn on the impact of T2DM on risk of PCa of different grades and stages. Larger studies are required to address this question in detail.
Impact of pre-existing T2DM on prostate cancer outcomes and mortality
Introduction
In 2008, a systematic review and meta-analysis examining all-cause mortality in cancer patients reported worse outcomes in cancer patients who had pre-existing diabetes mellitus (DM) [40]. However, the magnitude of the association varied widely between different types of malignancy. This led to a call for research focusing on individual cancer types. Following this, a body of literature has emerged and three published meta-analyses examined the association between pre-existing DM and PCa specific and all-cause mortality [41–43]. The most recent of which was published in 2016. Much of this literature does not distinguish between type 1 and type 2 diabetes; hence in this section refers to DM, encompassing both types, unless otherwise specified.
Existing literature
In 2010, Synder et al. performed a systematic review of the literature (seven papers), but was only able to include four studies in a meta-analysis. They could only investigate all-cause mortality and reported a pooled HR of 1.57 (95%CI: 1.12–2.20) for DM versus no DM. They concluded that more rigorous research was necessary before firm conclusions could be drawn.
Subsequently, a further meta-analysis by Cai et al. [42] was published in 2015, which included 11 cohort studies and looked at outcomes of all-cause mortality, PCa-specific mortality and non-PCa mortality. It reported that DM was positively associated with all three outcomes, with pooled HR of 1.50 (95%CI: 1.25–1.79), 1.26 (95%CI: 1.20–1.33), 1.83 (95%CI: 1.33–2.52), respectively. They concluded that DM was associated with an adverse prognosis in PCa and that clinicians treating patients with both conditions should pay more attention to the dual diagnosis and even consider more aggressive treatment strategies.
The final and most recent meta-analysis by Lee et al. [43] included 17 cohort studies which included 274,677 men. The studies were mainly from the USA (eight) and Europe (six), with two from Taiwan and one from Korea. They reported a 29% increase in PCa-specific mortality (95%CI 1.37–2.96), alongside a 37% increase in all-cause mortality (95%CI: 1.29–1.45). They performed a subgroup analysis of three cohort studies which considered T2DM separately from type 1 DM. This analysis showed a twofold increase in all-cause mortality for those with T2DM as compared to those without DM (95%CI:1.37–2.96), and could not exclude a positive association with PCa-specific mortality (RR:1.17, 95%CI: 0.96–1.42).
Evidence acquisition
As the meta-analysis by Lee was published within the last year, a full systematic review of the literature was not deemed valuable. However, the literature since 2016 was reviewed searching Pubmed using terms (with and without MESH terms): PCa, DM, prognosis and mortality. Only one new paper was identified [44].
Evidence synthesis
The one new paper identified is by Zaorsky et al. It is a retrospective cohort study of 3,217 men with localised PCa undergoing curative radiotherapy. Patients were divided into five groups: 1) No T2DM (n = 2,603); 2) T2DM on oral hypoglycaemic agents (OHA) including metformin (n = 251); 3) T2DM OHA not including metformin (n = 148); 4) T2DM on insulin (n = 89); 5) T2DM – diet controlled (n = 126). They examined several outcomes including OS, freedom for biochemical failure and cancer-specific survival. They showed an increased overall mortality in those on insulin (HR: 2.06, 95%CI: 1.17–3.63) or with diet-controlled T2DM (HR: 2.01, 95%CI: 1.24–3.26), but only an increase in PCa-specific mortality for those on insulin (HR: 3.91, 95%CI: 1.22–11.46). These findings may suggest that OHAs are potentially protective in PCa, the relationship between metformin and PCa is discussed in detail later in this review. Although interesting, this study contained relatively small numbers in each treatment subgroup making it difficult to interpret the results. It also addressed a slightly different research question than the previously discussed meta-analysis focusing in more detail on the treatment for T2DM, rather than just the presence of absence of T2DM.
Discussion
All three meta-analyses showed an increased risk of all-cause mortality for patients with DM compared to those without. The magnitude varied from 37% to 57% increased risk. T2DM increases cardiovascular mortality amongst a multitude of other consequences; increased all-cause mortality for those with T2DM is expected. The evidence for PCa-specific mortality is less clear. Both meta-analyses that examined this reported an increased risk in the order of 25–30%; however, when only studies which included those with T2DM and not type 1 DM were analysed this increased risk was not statistically significant, though sample sizes were small for this subgroup analyses. In the age group affected most commonly with PCa, T2DM is more prevalent than type 1 DM, and so it is probably fair to assume that T2DM is largely contributing to the increases in both all-cause and PCa-specific mortality demonstrated.
A further limitation of the existing literature is that some studies failed to adjust for PCa stage or grade, which is an important co variate associated with PCa mortality [45]. Positive associations reported in these studies could be due to failure to adjust for other important co variates, rather than a true effect of DM. Duration and severity of DM are also important co variates which are often not adjusted for. Bensimon et al. [46] reported a 23% increased risk of PCa-specific mortality and a 25% increased risk of all-cause mortality in those with T2DM. They also examined the effect of duration of T2DM and found a peak increase in PCa risk between 2 and 8 years. They also performed a sensitivity analysis whereby they excluded those people who developed T2DM during follow up, as this could have diluted the risks seen, however this made no difference.
Conclusion
The existing literature indicates that T2DM is associated with increased risk of all-cause and probably also PCa-specific mortality. However, limitations in studies hitherto preclude reliable estimates of what the real sizes of the associations are, especially for PCa-specific mortality.
Interplay between T2DM and PCa treatments
Androgen deprivation therapy and risk of T2DM
ADT is widely used in the management of PCa. It is the recommended first-line treatment in all men with advanced disease, as well as in men with high-risk disease following radical radiotherapy [47]. Even when PCa progresses to a castrate resistant phenotype, it is recommended that treatment with ADT continue, alongside the addition of further therapies. Given the prolonged clinical course of many men with PCa, they can remain on ADT for many years, making any side effects associated with treatment potentially significant.
Common adverse effects of ADT include fatigue, hot flushes and sexual dysfunction [48]. ADT also increases the risk of cardiovascular disease [49, 50], reduced bone mineral density [51] and several North American cohorts have demonstrated an increased risk of diabetes [7, 52–54]. This led the Food and Drug Administration (FDA) in 2010 to require a risk label on all GnRH agonists for increased risk of diabetes and certain cardiovascular diseases (heart attack, sudden cardiac death and stroke) [55].
ADT has been shown to induce a metabolic-like syndrome, in which patients have decreased insulin sensitivity and increased body fat [56]. We have previously undertaken a meta-analysis, including nine published studies, to quantify the association between ADT and MetS [57]. The relative risk of MetS for those on ADT compared to PCa men not on ADT was 1.75 (95%CI 1.27–2.41) and for T2DM alone 1.36 (95%CI 1.17–1.58) (Figure 2). Here we performed an up-to-date systematic review of the literature examining the association between T2DM and ADT.
Figure 2. Forrest plot for association between ADT and risk of diabetes [57].

Evidence acquisition
The systematic review was performed in accordance with the PRISMA guidelines [13] with search terms, inclusion and exclusion criteria all defined a priori.
Search strategy
A computerised literature search of Pubmed to identify full text and abstracts published was performed. The search was done with and without MESH terms (androgen, androgens, deprivation, therapy, therapeutics, diabetes, diabetes mellitus). All references of the selected articles were checked, including hand searches.
Study eligibility
The final articles were chosen based on the following set of inclusion criteria:
Original article
Examined the association of ADT with the risk of developing T2DM
English language article
Excluded if:
Review or meta-analysis
Examined elements of the metabolic syndrome which did not include T2DM (i.e., hyperglycaemia only)
Initially, titles were reviewed to assess whether they met inclusion criteria. If, after assessing the abstract, there was any doubt regarding whether it met the relevant criteria, it was kept for more thorough, subsequent assessment. The list of potential articles was further shortened by performing detailed evaluations of the methods and results of each remaining paper. Figure 3 provides more detailed information regarding the exclusion process. The strength of each study was assessed using the STROBE criteria [14] and is shown in Table 4.
Figure 3. Preferred reporting items for systematic reviews and meta-analysis (PRISMA) flow diagram of article identification, screening, eligibility and inclusion for systematic review on ADT and risk of T2DM.
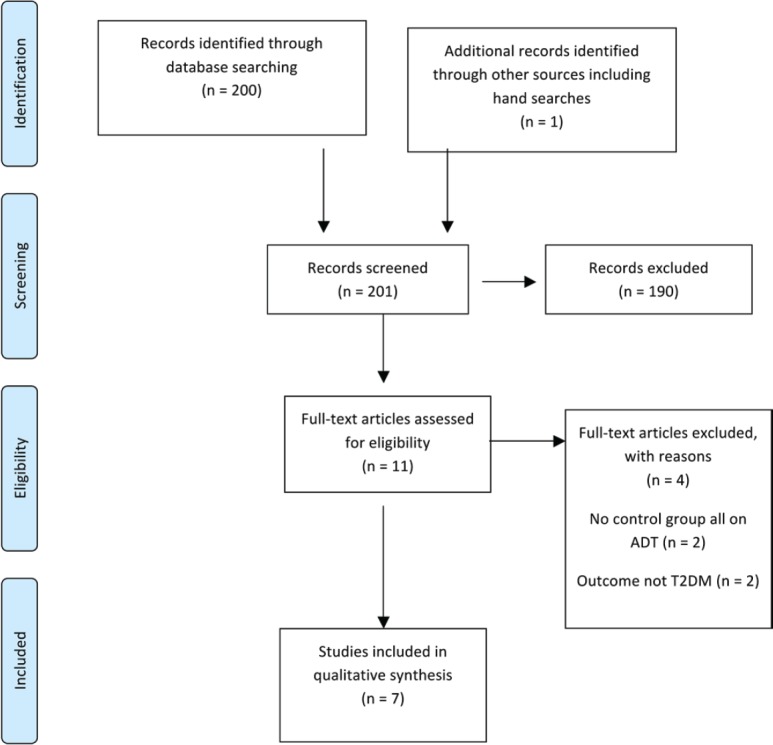
Table 4. Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) checklist for observational studies included in the systematic review of ADT and T2DM.
| Item No. | Recommendation | Keating (2006) [7] | Lage (2007) [53] | Alibhai (2009) [52] | Keating (2010) [54] | Teoh (2015) [60] | Tsai (2015) [58] | Crawley (2016) [59] | |
|---|---|---|---|---|---|---|---|---|---|
| Title and abstract | 1 | (a) Indicate the study’s design with a commonly used term in the title or the abstract | ☒ | ☒ | ☒ | ☒ | ☐ | ☒ | ☒ |
| (b) Provide in the abstract an informative and balanced summary of what was done and what was found | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ||
| Introduction | |||||||||
| Background/rationale | 2 | Explain the scientific background and rationale for the investigation being reported | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ |
| Objectives | 3 | State-specific objectives, including any pre-specified hypotheses | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ |
| Methods | |||||||||
| Study design | 4 | Present key elements of study design early in the paper | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ |
| Setting | 5 | Describe the setting, locations, and relevant dates, including periods of recruitment, exposure, follow-up, and data collection | ☒ | ☒ | ☒ | ☒ | ☐ | ☒ | ☒ |
| Participants | 6 | (a) Cohort study—Give the eligibility criteria, and the sources and methods of selection of participants. Describe methods of follow-up | ☒ | ☒ | ☒ | ☒ | ☐ | ☒ | ☒ |
| Case-control study—Give the eligibility criteria, and the sources and methods of case ascertainment and control selection. Give the rationale for the choice of cases and controls | n/a | n/a | n/a | n/a | n/a | n/a | n/a | ||
| (b) Cohort study—For matched studies, give matching criteria and number of exposed and unexposed | ☒ | ☒ | ☒ | ☒ | ☐ | ☒ | ☒ | ||
| Case-control study—For matched studies, give matching criteria and the number of controls per case | n/a | n/a | n/a | n/a | n/a | n/a | n/a | ||
| Variables | 7 | Clearly define all outcomes, exposures, predictors, potential confounders, and effect modifiers. Give diagnostic criteria, if applicable | ☒ | ☒ | ☒ | ☒ | ☐ | ☒ | ☒ |
| Data sources/measurement | 8* | For each variable of interest, give sources of data and details of methods of assessment (measurement). Describe comparability of assessment methods if there is more than one group | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ |
| Bias | 9 | Describe any efforts to address potential sources of bias | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ |
| Study size | 10 | Explain how the study size was arrived at | ☐ | ☒ | ☐ | ☐ | ☐ | ☒ | ☐ |
| Quantitative variables | 11 | Explain how quantitative variables were handled in the analyses. If applicable, describe which groupings were chosen and why | n/a | n/a | n/a | ☒ | ☒ | n/a | n/a |
| Statistical methods | 12 | (a) Describe all statistical methods, including those used to control for confounding | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ |
| (b) Describe any methods used to examine subgroups and interactions | ☒ | ☐ | ☒ | ☒ | ☐ | ☒ | ☒ | ||
| (c) Explain how missing data were addressed | ☐ | ☐ | ☐ | ☒ | ☐ | ☐ | ☐ | ||
| (d) Cohort study—If applicable, explain how loss to follow-up was addressed | ☐ | ☐ | ☒ | ☐ | ☐ | ☒ | ☐ | ||
| Case-control study—If applicable, explain how matching of cases and controls was addressed | n/a | n/a | n/a | n/a | n/a | n/a | n/a | ||
| (e) Describe any sensitivity analyses | ☒ | ☒ | ☒ | ☒ | ☐ | ☒ | ☒ | ||
| Results | |||||||||
| Participants | 13* | (a) Report numbers of individuals at each stage of study—e.g., numbers potentially eligible, examined for eligibility, confirmed eligible, included in the study, completing follow-up, and analysed | ☒ | ☒ | ☒ | ☒ | ☐ | ☒ | ☒ |
| (b) Give reasons for non-participation at each stage | ☐ | ☐ | ☒ | ☐ | ☐ | ☐ | ☐ | ||
| (c) Consider the use of a flow diagram | ☐ | ☐ | ☒ | ☐ | ☐ | ☒ | ☐ | ||
| Descriptive data | 14* | (a) Give characteristics of study participants (e.g., demographic, clinical, social) and information on exposures and potential confounders | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ |
| (b) Indicate number of participants with missing data for each variable of interest | ☒ | ☐ | ☐ | ☒ | ☐ | ☐ | ☒ | ||
| (c) Cohort study—Summarise follow-up time (e.g., average and total amount) | ☒ | ☐ | ☒ | ☒ | ☒ | ☒ | ☒ | ||
| Outcome data | 15* | Cohort study—Report numbers of outcome events or summary measures over time | ☒ | ☐ | ☐ | ☒ | ☒ | ☒ | ☒ |
| Case-control study—Report numbers in each exposure category, or summary measures of exposure | n/a | n/a | n/a | n/a | n/a | n/a | n/a | ||
| Main results | 16 | (a) Give unadjusted estimates and, if applicable, confounder-adjusted estimates and their precision (e.g., 95% confidence interval). Make clear which confounders were adjusted for and why they were included | ☐ | ☐ | ☐ | ☐ | ☐ | ☐ | ☒ |
| (b) Report category boundaries when continuous variables were categorised | n/a | n/a | n/a | n/a | n/a | n/a | n/a | ||
| (c) If relevant, consider translating estimates of relative risk into absolute risk for a meaningful time period | ☒ | ☐ | ☐ | ☒ | ☐ | ☒ | ☒ | ||
| Other analyses | 17 | Report other analyses done—e.g., analyses of subgroups and interactions, and sensitivity analyses | ☒ | ☒ | ☒ | ☒ | ☐ | ☒ | ☒ |
| Discussion | |||||||||
| Key results | 18 | Summarise key results with reference to study objectives | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ |
| Limitations | 19 | Discuss limitations of the study, taking into account sources of potential bias or imprecision. Discuss both direction and magnitude of any potential bias | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ |
| Interpretation | 20 | Give a cautious overall interpretation of results considering objectives, limitations, multiplicity of analyses, results from similar studies, and other relevant evidence | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ |
| Generalisability | 21 | Discuss the generalisability (external validity) of the study results | ☒ | ☒ | ☒ | ☒ | ☐ | ☒ | ☐ |
| Other information | |||||||||
| Funding | 22 | Give the source of funding and the role of the funders for the present study and, if applicable, for the original study on which the present article is based | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ | ☒ |
Data collection
The following details were recorded for each study: author, year of publication, country where study was undertaken, study design, number of patients, type of ADT, outcome reported and variables adjusted for in the analysis.
Evidence synthesis
The literature search identified a total of 200 studies of which 10 were deemed as initially relevant and a further one study was identified using hand searches. Using the above inclusion and exclusion criteria, four were excluded (Figure 3). The reasons for exclusion were outcomes not T2DM [2] and no control group not on ADT [2]. Seven studies were included in the systematic review (Table 5).
Table 5. Characteristics of the eight Studies included in the systematic review on ADT and T2DM.
| Author, year, country | Study design | No. of patients | ADT type | Main findings | Adjusted for |
|---|---|---|---|---|---|
| Crawley D, 2016, Sweden [59] | Prospective cohort | 34031 ADT vs. 167,205 No ADT | AA, GNRH agonists, Orch | Increased risk GnRH agonists vs. PCa free men HR 1.61 (95%CI: 1.36 – 1.91) No increased risk AA HR 0.74 (95%CI: 0.65 –0.84). |
CCI, PCa risk category, education status |
| Tsai HT, 2015, USA [58] | Retrospective cohort | 2648 ADT vs. 9543 No ADT | GNRH agonist +/- AA | Increased risk with ADT vs. No ADT HR 1.61 (95%CI 1.38–1.88) | Age, race, ethnicity, year of diagnosis, cancer sequence, health plan |
| Teoh JY, 2015, Asia [60] | Retrospective Cohort | 219 ADT vs. 169 No ADT | ?not in abstract? | Increased risk GnRH agonist HR 3.34 (95%CI 1.19–9.39)Orchiectomy HR 6.49 (95%CI 1.48–28.55) vs. No ADT | Age, T Stage, Gleason score, hypertension, dyslipidaemia, ischaemic heart disease, stroke, follow up time, type of ADT, duration of ADT |
| Keating NL, 2010, USA [54] | Retrospective cohort | 14,597 ADT vs. 37,443 No ADT | AA, GNRH agonists, CAB, Orch | Increased risk with GnRH agonist vs No ADT 1.28 (95%CI 1.19–1.38) No increased risk with AA HR 1.02 (95%CI 0.72–1.45) |
Age, race, ethnicity, year of diagnosis, marital status, socioeconomic status, Pca stage and grade, primary treatment, PSA at diagnosis, co morbidities, statin use, finasteride use |
| Alibhai SM, 2009, Canada [52] | Retrospective cohort | 19, 076 ADT vs. 19076 No ADT | LHRH agonists, AA, CAB | Increased risk HR 1.16 (95%CI: 1.11–1.21) | Income and rurality |
| Lage MJ, 2007, USA [53] | Retrospective claims cohort | 1231 ADT vs. 7250 No ADT | Any ADT | Increased risk with ADT HR 1.36 (95%CI 1.07–1.74) | Demographic factors, co morbid conditions, prior statin use |
| Keating NL, 2006, USA [7] | Retrospective Cohort | 26,570 ADT vs. 46,626 No ADT | GNRH agonist, Orch | GnRH agonists HR 1.44 (95%CI 1.34–1.55) vs. No ADT Orch HR 1.34 (95%CI 1.20–1.50) vs. No ADT |
Age, race, Hispanic ethnicity, marital status, residence, SEER region, income and education, tumour grade, comorbidity score, year of diagnosis, primary surgical therapy, prevalent coronary heart disease |
All seven studies were cohort studies. Five were from North American cohorts [7, 52–54, 58], one European [59] and one Asian [60]. The studies combined include 97,893 men on ADT and 287,312 not on ADT. They all report an increased risk of T2DM in those men receiving ADT compared to those men that are not, particularly in those receiving GNRH agonists. The magnitude of this risk varies from a 16% increase reported by Alibhai et al. [52] to 61% [58, 59]. The one outlier is the study by Teoh et al. [60] which reports a much higher increased risk, HR: 3.34 (95%CI: 1.19–9.39). However, this is a much smaller study in comparison with just a few hundred patients compared to the other cohorts which include several thousand patients. This is reflected in the wide confidence intervals surrounding the HR that they present.
Most of the studies in this systematic review included data only on GnRH agonists and orchidectomy or combined all forms of ADT. Only two studies examine anti-androgens (AA) separately [54, 59] and both report no increased risk with those receiving AA alone.
The impact of the duration for which ADT was received and T2DM risk had been examined in two studies [7, 52] but with a relatively short exposure time (25 months). We have previously studied in detail the impact of duration of ADT on risk of T2DM, with exposure times of up to greater than 10 years [59]. This paper showed that the peak risk of T2DM in men receiving GnRH agonists/orchiectomy was within the first three years of exposure [i.e., 1–1.5 years; HR: 1.61 (95%CI: 1.36–1.91)], before the risk off with continued exposure. Thereby showing that the duration of ADT is also important with regards the risk of T2DM.
Discussion
There is good concordance between all studies examining the risk of T2DM with ADT, with all showing an increased risk. The large North American cohort studies, which led to the FDA requiring a risk label on all GnRH agonists for increased risk of T2DM back in 2010, have since been corroborated by further studies in both European [59] and Asian populations [60]. Additionally, the literature demonstrates that both the type and duration of the ADT are important in the risk of T2DM and should be considered by physicians prescribing ADT. The observed temporal changes in risk fit with the physiological and metabolic changes previously described for GnRH agonist treatment [61]. These changes included increased fat mass, reduced lean body mass and increased insulin levels, which all have been demonstrated to occur within three months of commencing ADT [61–63]. Lee et al. measured lean body mass and fat mass in 65 men with PCa on GnRH agonists over a 12-month period. Those with longer prior exposure to treatment with GnRH agonists had less fat accumulation and less loss of lean body mass over the 12-month period [62]. Similarly, GnRH agonists decrease sensitivity to insulin within three months of ADT start [64]. Thus, the adverse metabolic effects of GnRH agonists occur within months of initiation; the consequences of these changes (i.e., developing T2DM) do not peak until several years later.
Conclusion
The literature consistently reports that ADT increases the risk of T2DM and, furthermore, that the type and duration of that ADT is important in determining that risk.
Impact of T2DM treatments on PCa: metformin and PCa
Introduction
Metformin (1,1-dimethylbiguanide hydrochloride) is a biguanide class of OHA and is commonly used for the treatment of T2DM. Metformin inhibits gluconeogenesis and reduces circulating levels of insulin [65]. It is also thought to play a role in lowering triglyceride and LDL cholesterol levels [66]. In addition to its anti-diabetic effect, metformin has also been associated with a reduced risk of various cancers including PCa [67–69]. The literature has reported inconsistent results and several meta-analyses have been undertaken in attempt to clarify results [70–73]. These are summarised below.
Existing literature: metformin and PCa risk
In 2015, Deng et al. [72] reported a decrease in risk of PCa with metformin in a meta-analysis which included seven studies (RR: 0.88; 95%CI: 0.78–0.99). Conversely in the same year, Wu et al. [70] included six cohort and four case-control studies in a meta-analyses and reported no association with PCa risk (RR: 0.92; 95%CI: 0.84–1.02). However, when only cohort studies were considered, a small but statistically significant reduction in risk was reported (RR: 0.92; 95%CI: 0.87–0.96). Similarly in a larger meta-analysis by Gandini et al. in 2014 [69], metformin treatment and PCa risk did not show any association in 12 studies (SRR: 1.06; 95%CI: 0.80–1.41), though a small but statistically significant association was seen when considering just the six prospective studies (SRR: 0.93; 95%CI: 0.89–0.97).
Existing literature: metformin and PCa mortality and outcomes
The meta-analysis by Deng et al. [72] described above also examined how metformin exposure was associated with all-cause mortality (three studies) and biochemical recurrence (BCR) of PCa (four studies). They reported that metformin exposure was not associated with either all-cause mortality (RR: 1.07; 95%CI: 0.86–1.32) or BCR (RR: 0.90; 95%CI: 0.75–1.09). Also in 2015, Raval et al. (73) published a systematic review and meta-analysis of the impact of metformin exposure on clinical outcomes in PCa. They also report no association with all-cause mortality and PCa specific mortality, but reported a marginal association with reduced risk of BCR in five studies (HR: 0.59; 95%CI: 0.67–1.01). In a further meta-analysis by Stopsack et al. [71] in 2016, including nine retrospective cohort studies of 9,186 patients, once again no overall association with PCa-specific mortality was seen, but metformin exposure was associated with improved OS in these studies (HR: 0.88; 95%CI: 0.86–0.90) and with a decreased risk of BCR (HR: 0.79; 95%CI: 0.63–1.00).
Evidence acquisition
As several full meta-analysis and systematic reviews were published in 2015/2016, a full systematic review of the literature was not deemed valuable. However, the literature since 2015 was reviewed searching PubMed using terms (with and without MESH terms): metformin, PCa, risk, mortality and outcomes. Two additional studies were identified [74, 75].
Evidence synthesis
The two new studies identified are summarised in Table 6. Both were large Scandinavian cohort studies. Haggstrom et al. [74] reported that men with more than one-year duration of T2DM had a reduced PCa risk, but that those receiving metformin specifically did not (HR: 0.96; 95%CI: 0.77–1.19). Conversely, the Finnish study by Haring et al. [75] reported that men using antidiabetic drugs had lowered overall PCa risk (HR: 0.85; 95%CI: 0.79–0.92) and among antidiabetic drug users, metformin decreased overall PCa risk (HR: 0.81; 95%CI: 0.69–0.95) in a dose-dependent manner.
Table 6. Two additional studies identified in systematic review of metformin and PCa risk and outcomes.
| Author, year, country | Study design | No of patients | Main findings | Adjusted for |
|---|---|---|---|---|
| Haring, Finland, 2017 [75] | Cohort | 78,615 | Metformin decreased PCa incidence in a dose dependent manner (HR 0.81, 95%CI 0.69–0.95) | Age, trial arm, medications |
| Haggstrom, Sweden, 2017 [74] | Cohort | 612,846 | Metformin did not decrease PCa incidence (HR 0.96 95%CI 0.77–1.19) | Age, education, CCI, county |
Discussion
Despite many studies examining the impact of metformin on PCa risk and outcomes they continue to offer conflicting results. This may be explained by the wide heterogeneity in the design and quality of these studies. Similarly, the meta-analysis, which has been performed to provide clarity, has also provided conflicting results. In their meta-analysis, Stopsack et al. [71] attempted to take into account the differing designs and quality of the literature. In their primary analysis, they only included studies with a clear risk window and in a secondary analysis examined those studies with potential immortal time bias. They showed that an otherwise modest association with reduced PCa risk was magnified in the studies with potential immortal time bias (HR: 0.52; 95%CI: 0.41–0.65). This highlights the need to take into account the quality of studies when performing meta-analysis. Two well-designed and well-powered Canadian studies by Margel et al. [68, 76] reported that an increased cumulative duration of metformin exposure after PC diagnosis was associated with decreases in both all-cause and PC-specific mortality among diabetic men, but was not associated with PCa incidence.
Several biological mechanisms underlying these potential associations have been proposed [77]. One hypothesis is that its anti-neoplastic effect may be via an indirect effect of insulin lowering, which in turn leads to a reduction in IGF-1 levels. Both elevated insulin and IGF-1 levels are known to play a role in PCa development and progression [78]. However, a host of direct molecular mechanisms has also been suggested. Many of these actions are mediated via 5’-AMP- activated protein kinase (AMPK), which is activated under conditions of metabolic stress that leads to intracellular adenosine triphosphate (ATP) being depleted and AMP increasing. Once activated, AMPK inhibits the mammalian target of rapamycin (mTOR) and other protein synthesis. These direct effects can lead to reduced cell proliferation [79] and hence exert an anti-cancer effect. Metformin is a potent activator of AMPK via inhibition of complex I of the respiratory chain, which results in increased AMP [80].
Conclusion
The current epidemiological evidence shows neither a conclusive decrease in PCa risk or improvement in PCa or all-cause mortality with metformin. Further rigorous, well-designed and powered studies are needed to clarify these potential associations.
Impact of PCa on T2DM control and treatments
Introduction
The relationship between PCa and T2DM has been extensively studied with respect to the effects of T2DM on PCa risk and progression, as described above. However, conversely the impact of a PCa diagnosis on the treatment of T2DM has received less attention. In this final section, this review considers the impact of PCa on T2DM control and treatments. In particular, PCa treatments including ADT and corticosteroids given alongside chemotherapy may have an impact on the management of pre-existing T2DM. However, there is little literature in this area. Below we have performed a systematic review of what has been published to date on this subject.
Evidence acquisition
The systematic review was performed in accordance with the PRISMA guidelines [13] with search terms, inclusion and exclusion criteria all defined a priori.
Search strategy
A computerised literature search of Pubmed to identify full text and abstracts published was performed. The search was done with and without MESH terms (PCa, diabetes control). All references of the selected articles were checked, including hand searches.
Study eligibility
The final articles were chosen based on the following set of inclusion criteria:
Original epidemiological study
Examined the impact of PCa diagnosis or treatment on T2DM control or treatment
English language article
The studies were excluded if:
Review or meta-analysis
Initially, titles were reviewed to assess whether they met inclusion criteria. If, after assessing the abstract, there was any doubt regarding whether it met the relevant criteria, it was kept for more thorough, subsequent assessment. The list of potential articles was further shortened by performing detailed evaluations of the methods and results of each remaining paper. Figure 4 shows more detailed information regarding the exclusion process. The strength of each study was assessed using the STROBE criteria [14] and is shown in Table 7.
Figure 4. Preferred reporting items for systematic reviews and meta-analysis (PRISMA) flow diagram of article identification, screening, eligibility and inclusion for systematic review on impact of PCa on T2DM control and treatment.
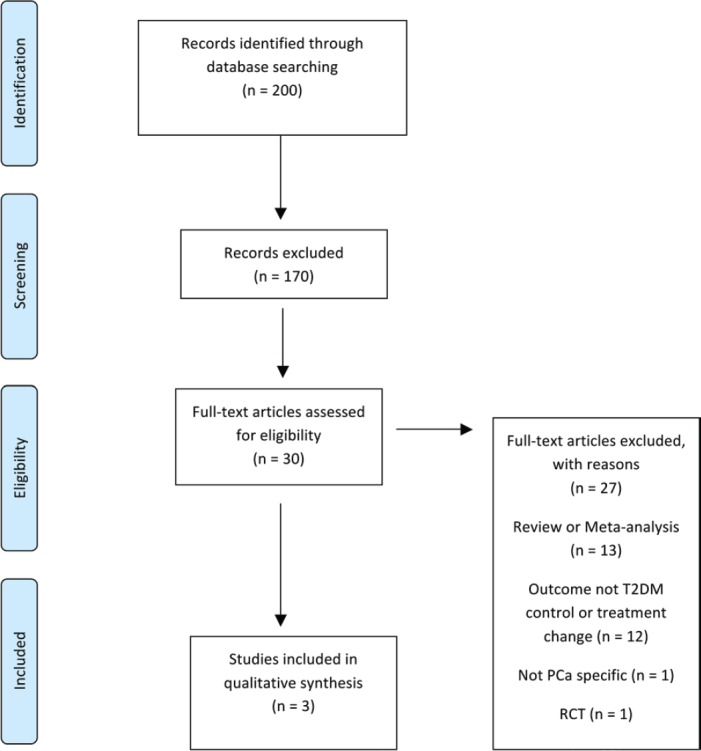
Table 7. Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) checklist for observational studies included in the systematic review of ADT and T2DM.
| Item No. | Recommendation | Rowbottom (2015) [83] | Keating (2014) [81] | Derweesh (2007) [82] | |
|---|---|---|---|---|---|
| Title and abstract | 1 | (a) Indicate the study’s design with a commonly used term in the title or the abstract | ☒ | ☒ | ☒ |
| (b) Provide in the abstract an informative and balanced summary of what was done and what was found | ☒ | ☒ | ☒ | ||
| Introduction | |||||
| Background/rationale | 2 | Explain the scientific background and rationale for the investigation being reported | ☒ | ☒ | ☒ |
| Objectives | 3 | State specific objectives, including any pre specified hypotheses | ☒ | ☒ | ☒ |
| Methods | |||||
| Study design | 4 | Present key elements of study design early in the paper | ☒ | ☒ | ☒ |
| Setting | 5 | Describe the setting, locations, and relevant dates, including periods of recruitment, exposure, follow-up, and data collection | ☒ | ☒ | ☒ |
| Participants | 6 | (a) Cohort study—Give the eligibility criteria, and the sources and methods of selection of participants. Describe methods of follow-up | ☐ | ☒ | ☒ |
| Case-control study—Give the eligibility criteria, and the sources and methods of case ascertainment and control selection. Give the rationale for the choice of cases and controls | n/a | n/a | n/a | ||
| (b) Cohort study—For matched studies, give matching criteria and number of exposed and unexposed | ☐ | ☒ | ☐ | ||
| Case-control study—For matched studies, give matching criteria and the number of controls per case | n/a | n/a | n/a | ||
| Variables | 7 | Clearly define all outcomes, exposures, predictors, potential confounders, and effect modifiers. Give diagnostic criteria, if applicable | ☐ | ☒ | ☒ |
| Data sources/measurement | 8* | For each variable of interest, give sources of data and details of methods of assessment (measurement). Describe comparability of assessment methods if there is more than one group | ☐ | ☒ | ☒ |
| Bias | 9 | Describe any efforts to address potential sources of bias | ☐ | ☒ | ☐ |
| Study size | 10 | Explain how the study size was arrived at | ☐ | ☒ | ☐ |
| Quantitative variables | 11 | Explain how quantitative variables were handled in the analyses. If applicable, describe which groupings were chosen and why | n/a | n/a | n/a |
| Statistical methods | 12 | (a) Describe all statistical methods, including those used to control for confounding | ☐ | ☒ | ☒ |
| (b) Describe any methods used to examine subgroups and interactions | ☐ | ☒ | ☐ | ||
| (c) Explain how missing data were addressed | ☐ | ☐ | ☐ | ||
| (d) Cohort study—If applicable, explain how loss to follow-up was addressed | ☐ | ☒ | ☐ | ||
| Case-control study—If applicable, explain how matching of cases and controls was addressed | n/a | n/a | n/a | ||
| (e) Describe any sensitivity analyses | ☐ | ☒ | ☐ | ||
| Results | |||||
| Participants | 13* | (a) Report numbers of individuals at each stage of study—e.g., numbers potentially eligible, examined for eligibility, confirmed eligible, included in the study, completing follow-up, and analysed | ☒ | ☒ | ☒ |
| (b) Give reasons for non-participation at each stage | ☐ | ☐ | ☐ | ||
| (c) Consider use of a flow diagram | ☐ | ☐ | ☐ | ||
| Descriptive data | 14* | (a) Give characteristics of study participants (e.g., demographic, clinical, social) and information on exposures and potential confounders | ☒ | ☒ | ☒ |
| (b) Indicate number of participants with missing data for each variable of interest | ☐ | ☒ | ☐ | ||
| (c) Cohort study—Summarise follow-up time (e.g., average and total amount) | ☐ | ☒ | ☒ | ||
| Outcome data | 15* | Cohort study—Report numbers of outcome events or summary measures over time | ☒ | ☒ | ☒ |
| Case-control study—Report numbers in each exposure category, or summary measures of exposure | n/a | n/a | n/a | ||
| Main results | 16 | (a) Give unadjusted estimates and, if applicable, confounder-adjusted estimates and their precision (e.g., 95% confidence interval). Make clear which confounders were adjusted for and why they were included | ☐ | ☒ | ☐ |
| (b) Report category boundaries when continuous variables were categorised | n/a | n/a | n/a | ||
| (c) If relevant, consider translating estimates of relative risk into absolute risk for a meaningful time period | ☐ | ☒ | ☐ | ||
| Other analyses | 17 | Report other analyses done—e.g., analyses of subgroups and interactions, and sensitivity analyses | ☐ | ☒ | ☐ |
| Discussion | |||||
| Key results | 18 | Summarise key results with reference to study objectives | ☒ | ☒ | ☒ |
| Limitations | 19 | Discuss limitations of the study, taking into account the sources of potential bias or imprecision. Discuss both direction and magnitude of any potential bias | ☒ | ☒ | ☒ |
| Interpretation | 20 | Give a cautious overall interpretation of results considering objectives, limitations, multiplicity of analyses, results from similar studies, and other relevant evidence | ☒ | ☒ | ☒ |
| Generalisability | 21 | Discuss the generalisability (external validity) of the study results | ☐ | ☒ | ☒ |
| Other information | |||||
| Funding | 22 | Give the source of funding and the role of the funders for the present study and, if applicable, for the original study on which the present article is based | ☒ | ☒ | ☐ |
Data collection
The following details were recorded for each study: author, year of publication, country where study was undertaken, study design, number of patients, main outcome and main findings.
Evidence synthesis
The literature search identified a total of 200 studies of which 30 were deemed as initially relevant. Using the above inclusion and exclusion criteria, 27 were excluded (Figure 4). The reasons for exclusion were review or meta-analysis (n = 13), outcome not T2DM controls or treatment change (n = 12), not PCa specific (n = 1), RCT (n = 1). Three studies were included in the systematic review (Table 8).
Table 8. Characteristics of studies included in the systematic review on the impact of PCa on T2DM control and treatments.
| Author, year, country | Study design | No. of patients | Main outcomes | Main findings |
|---|---|---|---|---|
| Keating, 2014, USA [81] | Cohort with propensity matching | 2237 pairs of propensity matched men with PCa and T2DM who were or were not treated with ADT | The effect of ADT onT2DM control, as measured by HbA1c levels and the intensification of T2DM drug therapy. | HbA1c increased at 1 year for men treated with ADT (7.38 from 7.24 p value 0.04) Receipt of ADT was also associated with an increased risk of addition of T2DM medication (HR 1.20 95% CI: 1.09–1.32) |
| Rowbottom, 2015, Canada [83] | Cohort | 30 GU Cancer patients: 26 PCa 4Bladder Ca | Change in T2DM management or hospitalisation due to T2DM in those receiving corticosteroids with chemotherapy | 40% required a change in their diabetes management (n = 4) 20% (n = 2) required hospitalisations |
| Derweesh, 2007, USA [82] | Cohort | 77 patients | To assess worsening glycaemic control in men with established T2DM after starting ADT for PCa | An increase of ≥ 10% in serum HbA1c in 15 patients (19.5%)An increase of ≥ 10% in FBG in 22 patients (28.6%) |
All three studies identified were North American cohort studies [81–83]. By far the largest of these studies was by Keating et al. [81] which included 2,237 pairs of propensity matched men with PCa and T2DM who did or did not receive ADT. They calculated mean HbA1c at baseline for both the ADT and No ADT groups and then examined the difference in difference at baseline, one and two years between the groups. They reported that HbA1c increased at one year for men treated with ADT to 7.38 and decreased among men not treated with ADT to 7.14, for a difference in differences of + 0.24 (P = 0.008). Results were similar at two years (P = 0.03). They also performed Cox proportional hazards regression model in the propensity matched data to assess if ADT was associated with initiating or adding a new class of anti-diabetes drug. They reported an increased risk of initiating an additional anti-diabetic medication in those men on ADT (HR: 1.20; 95%CI: 1.09–1.32), despite the rise in HbA1c seen in those receiving ADT.
Derweesh et al. [82] also examined glycaemic control in men with pre-existing T2DM starting on ADT. Glycaemic control was defined by comparing mean fasting blood glucose and HbA1c levels before and after treatment. At least three separate values for FBG and HbA1c were averaged to obtain mean values before and after ADT for comparison. Subsets were then analysed to determine the percentage of patients with a ≥ 10% rise in mean FBG or mean HbA1c after starting ADT. They reported an increase of ≥ 10% in serum HbA1c in 15 patients (19.5%) and an increase of ≥ 10% in FBG in 22 patients (28.6%). However, there was no comparison group in this study.
The final study identified in this systematic review is a descriptive cohort study of 30 patients with T2DM and genitourinary cancer, 26 of whom had PCa, who were receiving corticosteroids alongside their chemotherapy [83]. They examined changes in T2DM treatment and hospitalisations due to hyperglycaemia and reported that 40% of patients required a change in their T2DM management (n = 4) and 20% (n = 2) required hospitalisations for hyperglycaemia.
Discussion
This systematic review has highlighted a gap in the existing literature examining the impact of PCa and its treatments on the control and management of pre-existing T2DM. The studies published and described above all suggest that PCa treatments, including ADT and corticosteroids, do impact the management of pre-existing T2DM. However, the existing studies are very limited.
Conclusion
There is a need for further original research into this area, as there is little research evidence available.
Summary of findings
This review began with a systematic review of the impact of pre-existing T2DM on PCa incidence and corroborates previously published findings, indicating that T2DM has a protective effect on PCa risk. Several potential biological mechanisms and possible biases to explain this inverse association were discussed. Secondly, the impact of pre-existing T2DM on stage and grade was explored. Some studies have suggested that the inverse association is seen only in low-risk cancers and that those with T2DM are actually more likely to have higher grade and stage PCa. However, this is not consistent with existing meta-analysis and at present no conclusion can be drawn on the impact of T2DM on the PCa risk of different grades and stages. The existing literature does, however, show that T2DM is consistently associated with increased risk of all-cause and PCa-specific mortality. The relationship between T2DM and PCa is further complicated by the interaction between the two conditions and their treatments. The relationship between ADT and T2DM was examined and there is good concordance between all studies, with all showing an increased risk of T2DM. The epidemiological evidence examining the relationship between metformin exposure and PCa, however, is less convincing. It shows neither a conclusive decrease in the PCa risk or improvement in PCa or all-cause mortality with metformin. Finally, we have highlighted a gap in the existing literature examining the impact of PCa and its treatments on the control and management of pre-existing T2DM. Further research is needed in this area.
References
- 1.UK PC. About Prostate Cancer: Facts and Figures. 2017. [06/17]. [ https://prostatecanceruk.org/prostate-information/about-prostate-cancer]
- 2.UK D. Diabetes Statistics. 2015. [ http://wwwdiabetesorguk/Documents/About%20Us/Statistics/Diabetes-key-stats-guidelines-April2014pdf]
- 3.Seshasai SR, Kaptoge S, Thompson A, et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N Engl J Med. 2011;364(9):829–841. doi: 10.1056/NEJMoa1008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu H, Jiang HW, Ding GX, et al. Diabetes mellitus and prostate cancer risk of different grade or stage: a systematic review and meta-analysis. Diabetes Res Clin Pract. 2013;99(3):241–249. doi: 10.1016/j.diabres.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 5.Flanagan J, Gray PK, Hahn N, et al. Presence of the metabolic syndrome is associated with shorter time to castration-resistant prostate cancer. Ann Oncol. 2011;22(4):801–807. doi: 10.1093/annonc/mdq443. [DOI] [PubMed] [Google Scholar]
- 6.Rudman SM, Gray KP, Batista JL, et al. Risk of prostate cancer-specific death in men with baseline metabolic aberrations treated with androgen deprivation therapy for biochemical recurrence. BJU Int. 2016;118(6):919–926. doi: 10.1111/bju.13428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keating NL, O’Malley AJ, Smith MR. Diabetes and cardiovascular disease during androgen deprivation therapy for prostate cancer. J Clin Oncol. 2006;24(27):4448–4456. doi: 10.1200/JCO.2006.06.2497. [DOI] [PubMed] [Google Scholar]
- 8.Tsilidis KK, Kasimis JC, Lopez DS, et al. Type 2 diabetes and cancer: umbrella review of meta-analyses of observational studies. BMJ. 2015;350:g7607. doi: 10.1136/bmj.g7607. [DOI] [PubMed] [Google Scholar]
- 9.Bonovas S, Filioussi K, Tsantes A. Diabetes mellitus and risk of prostate cancer: a meta-analysis. Diabetologia. 2004;47(6):1071–1078. doi: 10.1007/s00125-004-1415-6. [DOI] [PubMed] [Google Scholar]
- 10.Kasper JS, Giovannucci E. A meta-analysis of diabetes mellitus and the risk of prostate cancer. Cancer Epidemiol Biomarkers Prev. 2006;15(11):2056–2062. doi: 10.1158/1055-9965.EPI-06-0410. [DOI] [PubMed] [Google Scholar]
- 11.Bansal D, Bhansali A, Kapil G, et al. Type 2 diabetes and risk of prostate cancer: a meta-analysis of observational studies. Prostate Cancer Prostatic Dis. 2013;16(2):151–158. doi: 10.1038/pcan.2012.40. (S1) [DOI] [PubMed] [Google Scholar]
- 12.Jian Gang P, Mo L, Lu Y, et al. Diabetes mellitus and the risk of prostate cancer: an update and cumulative meta-analysis. Endocr Res. 2015;40(1):54–61. doi: 10.3109/07435800.2014.934961. [DOI] [PubMed] [Google Scholar]
- 13.Liberati A, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. PLoS Med. 2009;6(7):e1000100. doi: 10.1371/journal.pmed.1000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.von Elm E, Altman DG, Egger M, et al. The strengthening the reporting of observational studies in epidemiology (STROBE) statement: guidelines for reporting observational studies. J Clin Epidemiol. 2008;61(4):344–349. doi: 10.1016/j.jclinepi.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 15.Tsilidis KK, Allen NE, Appleby PN, et al. Diabetes mellitus and risk of prostate cancer in the European prospective investigation into cancer and nutrition. Int J Cancer. 2015;136(2):372–381. doi: 10.1002/ijc.28989. [DOI] [PubMed] [Google Scholar]
- 16.Lai GY, Park Y, Hartge P, et al. The association between self-reported diabetes and cancer incidence in the NIH-AARP diet and health study. J Clin Endocrinol Metab. 2013;98(3):E497–E502. doi: 10.1210/jc.2012-3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawrence YR, Morag O, Benderly M, et al. Association between metabolic syndrome, diabetes mellitus and prostate cancer risk. Prostate Cancer Prostatic Dis. 2013;16(2):181–186. doi: 10.1038/pcan.2012.54. [DOI] [PubMed] [Google Scholar]
- 18.Fall K, Garmo H, Gudbjornsdottir S, et al. Diabetes mellitus and prostate cancer risk; a nationwide case-control study within PCBaSe Sweden. Cancer Epidemiol Biomarkers Prev. 2013;22(6):1102–1109. doi: 10.1158/1055-9965.EPI-12-1046. [DOI] [PubMed] [Google Scholar]
- 19.Moses KA, Utuama OA, Goodman M, et al. The association of diabetes and positive prostate biopsy in a US veteran population. Prostate Cancer Prostatic Dis. 2012;15(1):70–74. doi: 10.1038/pcan.2011.40. [DOI] [PubMed] [Google Scholar]
- 20.Dankner R, Boffetta P, Balicer RD, et al. Time-dependent Risk of cancer after a diabetes diagnosis in a cohort of 2.3 million adults. Am J Epidemiol. 2016;183(12):1098–1106. doi: 10.1093/aje/kwv290. [DOI] [PubMed] [Google Scholar]
- 21.Magliano DJ, Davis WA, Shaw JE, et al. Incidence and predictors of all-cause and site-specific cancer in type 2 diabetes: the Fremantle diabetes study. Eur J Endocrinol. 2012;167(4):589–599. doi: 10.1530/EJE-12-0053. [DOI] [PubMed] [Google Scholar]
- 22.Attner B, Landin-Olsson M, Lithman T, et al. Cancer among patients with diabetes, obesity and abnormal blood lipids: a population-based register study in Sweden. Cancer Causes Control. 2012;23(5):769–777. doi: 10.1007/s10552-012-9946-5. [DOI] [PubMed] [Google Scholar]
- 23.Renehan AG, Frystyk J, Flyvbjerg A. Obesity and cancer risk: the role of the insulin-IGF axis. Trends Endocrinol Metab. 2006;17(8):328–336. doi: 10.1016/j.tem.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Polychronakos C, Janthly U, Lehoux JG, et al. Mitogenic effects of insulin and insulin-like growth factors on PA-III rat prostate adenocarcinoma cells: characterization of the receptors involved. Prostate. 1991;19(4):313–321. doi: 10.1002/pros.2990190405. [DOI] [PubMed] [Google Scholar]
- 25.Hsing AW, Chua S, Jr., Gao YT, et al. Prostate cancer risk and serum levels of insulin and leptin: a population-based study. J Natl Cancer Inst. 2001;93(10):783–789. doi: 10.1093/jnci/93.10.783. [DOI] [PubMed] [Google Scholar]
- 26.Hammarsten J, Hogstedt B. Hyperinsulinaemia: a prospective risk factor for lethal clinical prostate cancer. Eur J Cancer. 2005;41(18):2887–2895. doi: 10.1016/j.ejca.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez C, Patel AV, Mondul AM, et al. Diabetes and risk of prostate cancer in a prospective cohort of US men. Am J Epidemiol. 2005;161(2):147–152. doi: 10.1093/aje/kwh334. [DOI] [PubMed] [Google Scholar]
- 28.Tavani A, Gallus S, Bosetti C, et al. Diabetes and the risk of prostate cancer. Eur J Cancer Prev. 2002;11(2):125–128. doi: 10.1097/00008469-200204000-00003. [DOI] [PubMed] [Google Scholar]
- 29.Giovannucci E, Rimm EB, Stampfer MJ, et al. Diabetes mellitus and risk of prostate cancer (United States) Cancer Causes Control. 1998;9(1):3–9. doi: 10.1023/A:1008822917449. [DOI] [PubMed] [Google Scholar]
- 30.Sandoval DA, Galassetti P, Tate D, et al. Leptin responses to antecedent exercise and hypoglycemia in healthy and type 1 diabetes mellitus men and women. J Diabetes Complications. 2003;17(6):301–306. doi: 10.1016/S1056-8727(02)00243-X. [DOI] [PubMed] [Google Scholar]
- 31.Noda T, Kikugawa T, Tanji N, et al. Longterm exposure to leptin enhances the growth of prostate cancer cells. Int J Oncol. 2015;46(4):1535–1542. doi: 10.3892/ijo.2015.2845. [DOI] [PubMed] [Google Scholar]
- 32.Lopez Fontana CM, Maselli ME, Perez Elizalde RF, et al. Leptin increases prostate cancer aggressiveness. J Physiol Biochem. 2011;67(4):531–538. doi: 10.1007/s13105-011-0098-y. [DOI] [PubMed] [Google Scholar]
- 33.Jackson FL, Hutson JC. Altered responses to androgen in diabetic male rats. Diabetes. 1984;33(9):819–824. doi: 10.2337/diab.33.9.819. [DOI] [PubMed] [Google Scholar]
- 34.Barrett-Connor E. Lower endogenous androgen levels and dyslipidemia in men with non-insulin-dependent diabetes mellitus. Ann Intern Med. 1992;117(10):807–811. doi: 10.7326/0003-4819-117-10-807. [DOI] [PubMed] [Google Scholar]
- 35.Shaneyfelt T, Husein R, Bubley G, et al. Hormonal predictors of prostate cancer: a meta-analysis. J Clin Oncol. 2000;18(4):847–853. doi: 10.1200/JCO.2000.18.4.847. [DOI] [PubMed] [Google Scholar]
- 36.Frayling TM, Colhoun H, Florez JC. A genetic link between type 2 diabetes and prostate cancer. Diabetologia. 2008;51(10):1757–1760. doi: 10.1007/s00125-008-1114-9. [DOI] [PubMed] [Google Scholar]
- 37.Muller H, Raum E, Rothenbacher D, et al. Association of diabetes and body mass index with levels of prostate-specific antigen: implications for correction of prostate-specific antigen cutoff values? Cancer Epidemiol Biomarkers Prev. 2009;18(5):1350–1356. doi: 10.1158/1055-9965.EPI-08-0794. [DOI] [PubMed] [Google Scholar]
- 38.Wallner LP, Morgenstern H, McGree ME, et al. The effects of type 2 diabetes and hypertension on changes in serum prostate specific antigen levels: results from the Olmsted County study. Urology. 2011;77(1):137–141. doi: 10.1016/j.urology.2010.07.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Hemelrijck M, Garmo H, Wigertz A, et al. Cohort profile update: the national prostate cancer register of Sweden and prostate cancer data base-a refined prostate cancer trajectory. Int J Epidemiol. 2016;45(1):73–82. doi: 10.1093/ije/dyv305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barone BB, Yeh HC, Snyder CF, et al. Long-term all-cause mortality in cancer patients with preexisting diabetes mellitus: a systematic review and meta-analysis. Jama. 2008;300(23):2754–2764. doi: 10.1001/jama.2008.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Snyder CF, Stein KB, Barone BB, et al. Does pre-existing diabetes affect prostate cancer prognosis? A systematic review. Prostate Cancer Prostatic Dis. 2010;13(1):58–64. doi: 10.1038/pcan.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cai H, Xu Z, Xu T, et al. Diabetes mellitus is associated with elevated risk of mortality amongst patients with prostate cancer: a meta-analysis of 11 cohort studies. Diabetes Metab Res Rev. 2015;31(4):336–343. doi: 10.1002/dmrr.2582. [DOI] [PubMed] [Google Scholar]
- 43.Lee J, Giovannucci E, Jeon JY. Diabetes and mortality in patients with prostate cancer: a meta-analysis. Springerplus. 2016;5(1):1548. doi: 10.1186/s40064-016-3233-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zaorsky NG, Shaikh T, Ruth K, et al. Prostate cancer patients with unmanaged diabetes or receiving insulin experience inferior outcomes and toxicities after treatment with radiation therapy. Clin Genitourin Cancer. 2017;15(2):326–335. doi: 10.1016/j.clgc.2016.08.020. (e3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Epstein JI, Zelefsky MJ, Sjoberg DD, et al. A contemporary prostate cancer grading system: a validated alternative to the Gleason score. Eur Urol. 2016;69(3):428–435. doi: 10.1016/j.eururo.2015.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bensimon L, Yin H, Suissa S, Pollak MN, et al. Type 2 diabetes and the risk of mortality among patients with prostate cancer. Cancer Causes Control. 2014;25(3):329–338. doi: 10.1007/s10552-013-0334-6. [DOI] [PubMed] [Google Scholar]
- 47.Horwich A, Parker C, de Reijke T, et al. Prostate cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(Suppl 6):vi106–vi114. doi: 10.1093/annonc/mdt208. [DOI] [PubMed] [Google Scholar]
- 48.Rhee H, Gunter JH, Heathcote P, et al. Adverse effects of androgen-deprivation therapy in prostate cancer and their management. BJU Int. 2015;115(Suppl 5):3–13. doi: 10.1111/bju.12964. [DOI] [PubMed] [Google Scholar]
- 49.Van Hemelrijck M, Garmo H, Holmberg L, et al. Absolute and relative risk of cardiovascular disease in men with prostate cancer: results from the population-based PCBaSe Sweden. J Clin Oncol. 2010;28(21):3448–3456. doi: 10.1200/JCO.2010.29.1567. [DOI] [PubMed] [Google Scholar]
- 50.O’Farrell S, Garmo H, Holmberg L, et al. Timing and risk patterns of cardiovascular disease in men with prostate cancer on adrogen deprivation therapy. Eur J Cancer. 2013;49(S3):S16–S17. [Google Scholar]
- 51.Greenspan SL, Coates P, Sereika SM, et al. Bone loss after initiation of androgen deprivation therapy in patients with prostate cancer. J Clin Endocrinol Metab. 2005;90(12):6410–6417. doi: 10.1210/jc.2005-0183. [DOI] [PubMed] [Google Scholar]
- 52.Alibhai SM, Duong-Hua M, Sutradhar R, et al. Impact of androgen deprivation therapy on cardiovascular disease and diabetes. J Clin Oncol. 2009;27(21):3452–3458. doi: 10.1200/JCO.2008.20.0923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lage MJ, Barber BL, Markus RA. Association between androgen-deprivation therapy and incidence of diabetes among males with prostate cancer. Urology. 2007;70(6):1104–1108. doi: 10.1016/j.urology.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 54.Keating NL, O’Malley AJ, Freedland SJ, et al. Diabetes and cardiovascular disease during androgen deprivation therapy: observational study of veterans with prostate cancer. J Natl Cancer Inst. 2010;102(1):39–46. doi: 10.1093/jnci/djp404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. safety sUFdscuto, new roGaantmoGata, cardiovascular sitlrirodac, diseases.
- 56.Smith MR, Lee H, Nathan DM. Insulin sensitivity during combined androgen blockade for prostate cancer. J Clin Endocrinol Metab. 2006;91(4):1305–1308. doi: 10.1210/jc.2005-2507. [DOI] [PubMed] [Google Scholar]
- 57.Bosco C, Crawley D, Adolfsson J, et al. Quantifying the evidence for the risk of metabolic syndrome and its components following androgen deprivation therapy for prostate cancer: a meta-analysis. PLoS One. 2015;10(3):e0117344. doi: 10.1371/journal.pone.0117344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsai HT, Keating NL, Van Den Eeden SK, et al. Risk of diabetes among patients receiving primary androgen deprivation therapy for clinically localized prostate cancer. J Urol. 2015;193(6):1956–1962. doi: 10.1016/j.juro.2014.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Crawley D, Garmo H, Rudman S, et al. Association between duration and type of androgen deprivation therapy and risk of diabetes in men with prostate cancer. Int J Cancer. 2016;139(12):2698–2704. doi: 10.1002/ijc.30403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Teoh JY, Chiu PK, Chan SY, et al. Risk of new-onset diabetes after androgen deprivation therapy for prostate cancer in the Asian population. J Diabetes. 2015;7(5):672–680. doi: 10.1111/1753-0407.12226. [DOI] [PubMed] [Google Scholar]
- 61.Smith MR, Finkelstein JS, McGovern FJ, et al. Changes in body composition during androgen deprivation therapy for prostate cancer. J Clin Endocrinol Metab. 2002;87(2):599–603. doi: 10.1210/jcem.87.2.8299. [DOI] [PubMed] [Google Scholar]
- 62.Lee H, McGovern K, Finkelstein JS, et al. Changes in bone mineral density and body composition during initial and long-term gonadotropin-releasing hormone agonist treatment for prostate carcinoma. Cancer. 2005;104(8):1633–1637. doi: 10.1002/cncr.21381. [DOI] [PubMed] [Google Scholar]
- 63.Smith JC, Bennett S, Evans LM, et al. The effects of induced hypogonadism on arterial stiffness, body composition, and metabolic parameters in males with prostate cancer. J Clin Endocrinol Metab. 2001;86(9):4261–4267. doi: 10.1210/jcem.86.9.7851. [DOI] [PubMed] [Google Scholar]
- 64.Smith MR, Lee H, Fallon MA, et al. Adipocytokines, obesity, and insulin resistance during combined androgen blockade for prostate cancer. Urology. 2008;71(2):318–322. doi: 10.1016/j.urology.2007.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hundal RS, Krssak M, Dufour S, et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000;49(12):2063–2069. doi: 10.2337/diabetes.49.12.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sin HY, Kim JY, Jung KH. Total cholesterol, high density lipoprotein and triglyceride for cardiovascular disease in elderly patients treated with metformin. Arch Pharm Res. 2011;34(1):99–107. doi: 10.1007/s12272-011-0112-5. [DOI] [PubMed] [Google Scholar]
- 67.Wright JL, Stanford JL. Metformin use and prostate cancer in Caucasian men: results from a population-based case-control study. Cancer Causes Control. 2009;20(9):1617–1622. doi: 10.1007/s10552-009-9407-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Margel D, Urbach D, Lipscombe LL, et al. Association between metformin use and risk of prostate cancer and its grade. J Natl Cancer Inst. 2013;105(15):1123–1131. doi: 10.1093/jnci/djt170. [DOI] [PubMed] [Google Scholar]
- 69.Gandini S, Puntoni M, Heckman-Stoddard BM, et al. Metformin and cancer risk and mortality: a systematic review and meta-analysis taking into account biases and confounders. Cancer Prev Res (Phila) 2014;7(9):867–885. doi: 10.1158/1940-6207.CAPR-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu GF, Zhang XL, Luo ZG, et al. Metformin therapy and prostate cancer risk: a meta-analysis of observational studies. Int J Clin Exp Med. 2015;8(8):13089–13098. [PMC free article] [PubMed] [Google Scholar]
- 71.Stopsack KH, Ziehr DR, Rider JR, et al. Metformin and prostate cancer mortality: a meta-analysis. Cancer Causes Control. 2016;27(1):105–113. doi: 10.1007/s10552-015-0687-0. [DOI] [PubMed] [Google Scholar]
- 72.Deng D, Yang Y, Tang X, et al. Association between metformin therapy and incidence, recurrence and mortality of prostate cancer: evidence from a meta-analysis. Diabetes Metab Res Rev. 2015;31(6):595–602. doi: 10.1002/dmrr.2645. [DOI] [PubMed] [Google Scholar]
- 73.Raval AD, Thakker D, Vyas A, et al. Impact of metformin on clinical outcomes among men with prostate cancer: a systematic review and meta-analysis. Prostate Cancer Prostatic Dis. 2015;18(2):110–121. doi: 10.1038/pcan.2014.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Haggstrom C, Van Hemelrijck M, Zethelius B, et al. Prospective study of Type 2 diabetes mellitus, anti-diabetic drugs and risk of prostate cancer. Int J Cancer. 2017;140(3):611–617. doi: 10.1002/ijc.30480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Haring A, Murtola TJ, Talala K, et al. Antidiabetic drug use and prostate cancer risk in the Finnish randomized study of screening for prostate cancer. Scand J Urol. 2017;51(1):5–12. doi: 10.1080/21681805.2016.1271353. [DOI] [PubMed] [Google Scholar]
- 76.Margel D, Urbach DR, Lipscombe LL, et al. Metformin use and all-cause and prostate cancer-specific mortality among men with diabetes. J Clin Oncol. 2013;31(25):3069–3075. doi: 10.1200/JCO.2012.46.7043. [DOI] [PubMed] [Google Scholar]
- 77.Penney KL, Stampfer MJ. The time is ripe for a randomized trial of metformin in clinically localized prostate cancer. J Clin Oncol. 2013;31(25):3054–3055. doi: 10.1200/JCO.2013.50.7715. [DOI] [PubMed] [Google Scholar]
- 78.Zu K, Martin NE, Fiorentino M, et al. Protein expression of PTEN, insulin-like growth factor I receptor (IGF-IR), and lethal prostate cancer: a prospective study. Cancer Epidemiol Biomarkers Prev. 2013;22(11):1984–1993. doi: 10.1158/1055-9965.EPI-13-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Park HU, Suy S, Danner M, et al. AMP-activated protein kinase promotes human prostate cancer cell growth and survival. Mol Cancer Ther. 2009;8(4):733–741. doi: 10.1158/1535-7163.MCT-08-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Keating NL, Liu PH, O’Malley AJ, et al. Androgen-deprivation therapy and diabetes control among diabetic men with prostate cancer. Eur Urol. 2014;65(4):816–824. doi: 10.1016/j.eururo.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Derweesh IH, Diblasio CJ, Kincade MC, et al. Risk of new-onset diabetes mellitus and worsening glycaemic variables for established diabetes in men undergoing androgen-deprivation therapy for prostate cancer. BJU Int. 2007;100(5):1060–1065. doi: 10.1111/j.1464-410X.2007.07184.x. [DOI] [PubMed] [Google Scholar]
- 83.Rowbottom L, Stinson J, McDonald R, et al. Retrospective review of the incidence of monitoring blood glucose levels in patients receiving corticosteroids with systemic anticancer therapy. Ann Palliat Med. 2015;4(2):70–77. doi: 10.3978/j.issn.2224-5820.2015.04.07. [DOI] [PubMed] [Google Scholar]