

Abstract
Localization microscopy techniques – such as photoactivation localization microscopy (PALM), fluorescent PALM (FPALM), ground state depletion (GSD), and stochastic optical reconstruction microscopy (STORM) – provide the highest precision for single molecule localization currently available. However, localization microscopy has been largely limited to cell cultures due to the difficulties that arise in imaging thicker tissue sections. Sample fixation and antibody staining, background fluorescence, fluorophore photoinstability, light scattering in thick sections, and sample movement create significant challenges for imaging intact tissue. We have developed a sample preparation and image acquisition protocol to address these challenges in rat brain slices. The sample preparation combined multiple fixation steps, saponin permeabilization, and tissue clarification. Together, these preserve intracellular structures, promote antibody penetration, reduce background fluorescence and light scattering, and allow acquisition of images deep in a 30 μm thick slice. Image acquisition challenges were resolved by overlaying samples with a permeable agarose pad and custom-built stainless steel imaging adapter, and sealing the imaging chamber. This approach kept slices flat, immobile, bathed in imaging buffer, and prevented buffer oxidation during imaging. Using this protocol, we consistently obtained single molecule localizations of synaptic vesicle and active zone proteins in three-dimensions within individual synaptic terminals of the striatum in rat brain slices. These techniques may be easily adapted to the preparation and imaging of other tissues, substantially broadening the application of super-resolution imaging.
Keywords: Super-resolution microscopy, single molecule localization, brain slice, tissue imaging
1. Introduction
Single-molecule localization techniques, including PALM, FPALM, STORM and GSD, currently provide the highest precision for protein localization in micrographs, with 20 nm precision in the X and Y axes and 50 nm precision in the Z axis (1). Despite the successful application of super-resolution imaging to thin, sparse samples such as primary cell cultures, the widespread adoption of localization microscopy to thicker samples, such as tissues or organs, is limited by substantial obstacles presented during sample preparation and imaging. As a result, only a few studies have been published using tissue slices for single-molecule super-resolution imaging (2–4) (See Note 1).
Imaging brain slices using single-molecule localization microscopy poses five fundamental challenges that this protocol addresses. First, fine synaptic terminal structures must be preserved, but thick sections must be sufficiently permeabilized to allow antibody penetrance deep into the tissue. This can be accomplished through cardiac perfusion and extensive fixation with paraformaldehyde followed by saponin permeabilization during staining. Second, prolonged fixation with aldehydes and thick tissues create background autofluorescence that must be reduced to ensure precise single-molecule localization and allow imaging at depth, which can be achieved through sodium borohydride incubation prior to antibody staining and tissue clarification prior to imaging. Third, light scattering must be reduced to allow deep imaging (~ 5 – 10 μm) within a tissue slice and maximize the precision of particle localization, which can be achieved by clarifying slices with Scale U2 buffer prior to imaging accomplished. Fourth, the tissue must be immersed within a reducing buffer to allow fluorophore blinking and maintenance of the dark state (5), but remain completely immobile on the coverslip while imaging. Fifth, oxygen promotes dye photobleaching (6), so the sample chamber must be sealed for the duration of the imaging session and/or the imaging buffer must be replaced regularly. To solve these two problems, this protocol utilizes an agarose pad and stainless steel adapter to prevent sample movement during imaging, yet allow imaging buffer to access the tissue and be changed repeatedly. To prevent imaging buffer oxygenation during imaging, the chamber is sealed with a parafilm-lined culture plate lid. Using the methods outlined within this protocol, three-dimensional super-resolution images can be obtained deep within tissue slices. The effectiveness of this protocol in imaging thick brain slices is demonstrated herein through staining and imaging of the vesicular monoamine transporter-2 (VMAT2), a marker of dopaminergic synaptic vesicles, and Bassoon, a structural protein of presynaptic active zones, within individual synaptic terminals of the striatum from 30 μm thick rat brain slices. The sample preparation, staining, and imaging steps of this protocol can be adapted to thick tissue slices of other complex organ systems.
2. Materials
2.1 Equipment
Perfusion pump
Rodent and small animal guillotine
Freezing stage microtome
- Single-molecule localization super-resolution microscope (Vutara 350; Bruker Nanosurfaces, Salt Lake City, UT, USA):
- Irradiation lasers at 405 nm, 488 nm, 561 nm, and 647 nm (15 kW/cm2)
- Water-immersion objective (60×, 1.2NA)
- EM-CCD camera
2.2 Brain Collection and Slicing
Male Sprague-Dawley rats: 300 – 400 g in weight (see Note 2)
Pentobarbital sodium salt: 250 mg/kg in 0.9% sterile saline solution. Prepare in small batches, vortex into solution, and use immediately.
Handheld wire cutters
Straight operating scissors; 4.5 – 5.5 inches
Small dressing forceps
Razor blades
Phosphate-buffered saline (PBS): 137 mM NaCl, 2.7 mM KCl, 10.1 mM Na2HPO4, and 1.8 mM KH2PO4 in ddH2O, pH 7.5. Store at 4° C.
Four percent paraformaldehyde (4% PFA): 4% (wt/vol) paraformaldehyde in PBS, pH 7.5, store at 4 °C.
Thirty percent sucrose (wt/vol) in 4% PFA, store at 4° C
Dry ice
Shandon cyromatrix freezing medium
90% ethanol or methanol
Paint brush
2.3 Slice Staining
Blocking buffer: mix 5% (vol/vol) normal donkey serum (species from which secondary antibody is derived), 1% bovine serum albumin (wt/vol), and 0.1% saponin (wt/vol) in PBS and pH to 7.5
PBS with 0.1% saponin (PBS-saponin)
- Primary antibodies
- Rabbit anti-vesicular monoamine transporter-2 polyclonal antibody (VMAT2; made by Bethyl Laboratories, Montgomery, TX, USA; see Note 3)
- Guinea pig anti-Bassoon polyclonal antibody (Synaptic Systems, Göttingen, Germany)
- 4. Secondary antibodies
- Goat anti-rabbit Alexa Fluor 647 IgG (Invitrogen, ThermoFisher Scientific, Pittsburgh, PA, USA)
- Donkey anti-guinea pig Cy3B IgG (conjugated in house, see Note 4; IgG antibody from Jackson Immunoresearch, West Grove, PA, USA; Cy3B NHS-ester from GE Healthcare Life Sciences, Pittsburgh, PA, USA)
5. Parafilm M
6. Scale U2 buffer: mix 4M urea, 30% glycerol (vol/vol), 0.1% triton X-100 (vol/vol), and bring to volume in ddH2O.
2.4 Super-Resolution Imaging
Two percent agarose stock: mix in ddH2O, microwave until the agarose goes into solution (45 seconds to 2 minutes), stir briefly, and aliquot into vials or test tubes. Store at 4 °C until use.
10 cm petri dish
6 cm petri dish cover
25 mm #1 circular coverslips (0.12 mm thick)
Imaging buffer: mix 20 mM cysteamine, 1% 2-mercaptoethanol (vol/vol), 168.8 active units glucose oxidase per ml of buffer, 1404 active units catalase per ml of buffer, 10 mM NaCl, 10% (wt/vol) glucose, 50 mM Tris-HCl, bring to volume in ddH2O and pH to 8.0.
Stainless steel imaging chamber (Life Sciences A-7816)
Stainless steel imaging insert (machined in-house; see Fig. 1D for schematics): the bottom of the insert was painted with Rust-Oleum Painter’s Touch Ultra Cover Semi-gloss Black to prevent light backscattering during laser excitation
Figure 1.

Brain slice mounting for super-resolution imaging. (A) A brain slice is floated, mounted on circular glass coverslips and then allowed to completely dry. (B, C) Liquid 2% agarose is pipetted along the edges of the brain slice and then across the entire surface to form an immobile pad. (D) Schematics of the stainless steel imaging chamber insert with all dimensions in mm and radii with a curvature measurement of 2 mm. (E) The stainless steel insert is placed in the imaging chamber along with imaging buffer and (F) sealed with a parafilm-lined lid of a 35 mm culture plate for imaging.
3. Method
3.1 Brain Collection and Slicing
Deeply anaesthetize a rat with an intraperitoneal 250 mg/kg sodium pentobarbital injection.
Once the animal no longer responds to both a tail/foot pinch and an audible startle (usually within 5 min of injection), clean the chest fur and skin with ethanol, open the chest cavity to expose the heart. Cut a small incision into the right ventricle of the heart and perfuse 100 ml of ice-cold 4% PFA at a rate of 25 ml/min into the left ventricle of the heart (see Note 5).
Decapitate the rat by guillotine following perfusion. To dissect the brain, open the skull by placing the wire cutter shears in the optic foramen and fracturing the nasal bridge of the frontal bone. Next, place the wire snip shears in the auditory canal and the foramen magnum and fracture the bones within the masseter region. Starting in the foramen magnum, press the bottom shear of the operating scissors against the parietal bone of the skull and slowly cut along the sagittal fissure until the snipped region of nasal bridge, shortly past bregma, is reached. Gently pry away the parietal and frontal bones to expose the brain. Scoop the brain out the skull with the forceps and place it into ice-cold 4% PFA.
Incubate brains in 4% PFA for at least 24 hours at 4 °C. Brains can be stored for prolonged periods (months to years) at 4 °C provided they are kept submerged 4% PFA.
Cryoprotect the brain by transferring it to a 50 ml conical tube containing 30% sucrose in PFA and incubating until the brain sinks to the bottom of the tube (approximately 72 h).
Using a razor blade, remove the cerebellum and olfactory bulbs prior to slicing the brain.
Cool the microtome stage with dry ice placed into the bath containing 90% ethanol and place a sufficient amount of Shandon cryomatrix freezing medium on the stage to embed at least 3 mm of the caudal brain region.
Place the brain, rostral end up, into the freezing medium just as it begins to freeze. Quickly apply a few mm of medium to the base of the brain to prevent separation from the stage during slicing.
Flash freeze the brain by covering it with crushed dry ice for 1.5 minutes.
Cut 30 μm coronal slices through the striatum (or brain region of interest), remove them from the microtome blade with the paintbrush, and place them into 24-well plate wells containing PBS. Store slices at 4 °C until use.
3.2 Slice Staining
Incubate sections with 0.1% NaBH4 in PBS while gently rotating for 15 min at room temperature. See Note 6.
Wash sections once for 10 min then twice for 5 min with PBS while gently rotating at room temperature.
Incubate sections in blocking buffer for at least 2 h while gently rotating at room temperature. 250 μl of buffer is sufficient per each well in a 24-well plate.
Remove blocking solution and incubate with primary antibody mixture in PBS-saponin (200 μl total volume; See Note 7) while gently rotating overnight at 4 °C. To prevent antibody evaporation during this long incubation stage, a sheet of
Wash sections once for 10 minutes then twice for 5 min with PBS-saponin while gently rotating at room temperature.
Incubate sections in secondary antibody mixture (200 μl; PBS-saponin) for 1.5 h in the dark (or covered) while rotating gently at room temperature,
Wash sections twice for 5 min with PBS-saponin and three times for 5 min with PBS while rotating gently at room temperature.
Post-fix sections in 4% PFA for 1 h while rotating gently at room temperature (See Note 8).
Wash sections three times for 5 min with PBS while rotating gently at room temperature.
Incubate samples in Scale U2 buffer overnight (12 h minimum) and store covered at 4 °C until imaging (see Note 9).
3.3 Super-Resolution Imaging
Remove 2% agarose stock from 4 °C storage, liquefy in a microwave for 45 sec, and incubate at 55 °C until use.
Submerge a coverslip and brain slice in a PBS-filled 10 cm petri dish and use the paint brush to gently float the brain slice onto the coverslip.
While still wet, gently move the brain slice to the center of the coverslip and flatten. Allow the brain slice to dry completely on the coverslip before proceeding (Fig. 1A; see Note 10).
Place the coverslip into the imaging chamber and rapidly pipette liquid (55 °C; approximately 150 μl) 2% agarose onto the slice, starting at the edges and working in a spiral pattern (Fig. 1B, C). This step must be performed rapidly or the agarose will solidify within the pipette.
Allow the agarose to cool to room temperature for 5 min. Since rapid temperature reduction deforms the agarose pad, failure to perform this step leads to agarose dissociation from the coverslip when ice-cold imaging buffer is applied.
Place the stainless steel insert (Fig. 1D; see Note 11) over the sample (Fig. 1E), fill the chamber with imaging buffer (approximately 750 μl), and seal the chamber with a parafilm-lined 30 mm culture plate lid (Fig. 1F; see Note 12).
Image the brain slice, replacing imaging buffer every 1 h. See Figure 2 for a representative image of VMAT2 and Bassoon within striatal terminals, and Note 13 for further discussion. To store slices for future imaging sessions, replace the imaging buffer with PBS and incubating at 4 °C.
Figure 2.
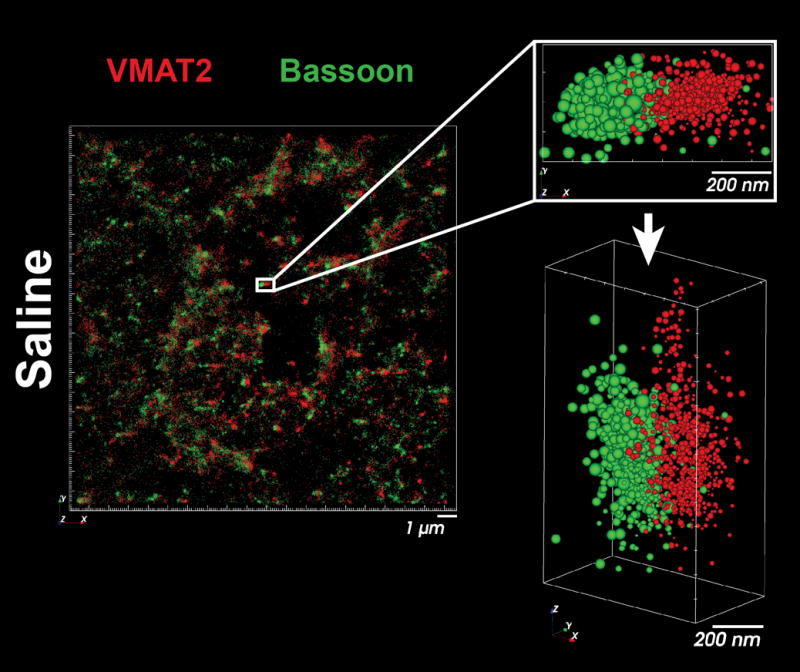
The distribution of VMAT2 (red) and Bassoon (green) within individual presynaptic terminals of the striatum.
Acknowledgments
We thank Sam Feinman for his assistance in creating the imaging adapter blueprints in SolidWorks. E.M. Jorgensen is a Howard Hughes Medical Institute (HHMI) investigator. This work was funded by the NIH grants R01NS034307 (EMJ and MVG), DA019447 (AEF), DA13367 (CLG and AEF) and DA11389 (CLG and AEF).
Footnotes
Conflicts of Interest
M.V. Gudheti is an employee of Bruker Nano Surfaces.
4. Notes
Other approaches have imaged thinner tissue sections by sandwiching them between the slide and coverslip with a small amount of imaging buffer and sealing the edges of the coverslip with nailpolish (3). We found this approach problematic for at least two reasons. First, prolonged imaging sessions (greater than an hour) required frequent replacement of the imaging buffer to ensure fluorophore stability and blinking, which wasn’t possible when the coverslip was sealed. Second, the coverslip alone did not provide sufficient weight to flatten the slice along the surface of the slide, which led to slight tissue folding, flotation, and movement of the tissue. The tissue folding and flotation led to z-axis inconsistency, while slight tissue movement led to dramatic reduction in the accuracy of particle localization in all axes.
Rats were housed with food and water ad libitum in a temperature controlled environment and all experiments were approved by the University of Utah Institutional Animal Care and Use Committee.
The VMAT2 rabbit polycolonal antibody was produced by Bethyl Labs and raised against a 20-amino acid c-terminal sequence (CTQNNIQSYPIGEDEESESD) of human VMAT2. Proper immunoreactivity of this antibody against VMAT2 was validated through western blotting, immunoprecipitation and tandem mass spectrometry identification of immunoreactive bands (data not shown).
Cy3b donkey anti-guinea pig antibody was created by crosslinking Cy3b NHS-ester to donkey anti-guinea pig antibody which was then purified over NAP-5 columns per manufacturer’s direction.
The integrity of fine central nervous system structures, such as pre- and post-synaptic terminals, was accomplished through thorough fixation, cryoprotection, and gentle detergent use (saponin) during staining. 4% PFA perfusion and the prolonged, 24 h incubation that follows ensure structures deep within the brain are well-fixed, and sucrose cryoprotection is a critical step that prevents ice crystal formation and membrane lysis during flash freezing.
Free aldehyde fluorescence can be quite pronounced in super-resolution images because of the prolonged fixation and thickness of sections; thus, autofluorescence was quenched with NaBH4 treatment prior to staining.
Antibody penetration into thick tissue samples requires tissue permeabilization using a detergent. Saponin, unlike Triton X-100, is a mild and reversible non-ionic detergent that improves staining efficiency but does not damage the structure of organelles and vesicles in presynaptic terminals (7), and was included in all blocking, staining, and wash steps.
The post-fixation step is critical for preventing tissue destruction and loss of signal when clarifying slices with the Scale U2 steps that follow.
Tissue clarification using Scale U2 buffer dramatically reduced both light scattering and tissue autofluorescence, and was necessary for imaging deeper than a few microns from the slice surface. See (8) for further discussion of Scale buffer formulation and use.
Allowing the brain slice to dry to the coverslip is important for two reasons. First, it completely flattens the tissue to the surface of the coverslip, which aids in z-plane imaging. Second, it prevents tissue folding when the warm agarose is subsequently added.
Keeping brain slices flat and immobile on the coverslip while immersed in buffer during image acquisition is a significant challenge for localization microscopy of tissues. To solve these problems, slices were overlayed with a 2% agarose pad dense enough to immobilize the tissue but porous enough to allow buffer penetrance. Since the agarose pad can occasionally detach and/or move during imaging, a stainless steel insert was designed and fabricated to sit on top of the agarose pad, provide sufficient weight to prevent movement, and still allow imaging buffer to access the tissue slice.
Throughout the course of imaging, the glucose oxidase and catalase necessary for fluorophore stability converts oxygen and glucose into gluconic acid (6). The progressive acidification of the sample impairs fluorophore blinking, necessitating the replacement of imaging buffer every 45 minutes. Covering the imaging chamber with Parafilm M and sealing it with the petri dish cover reduces oxygen exposure during imaging, prolonging imaging time before the buffer must be changed.
VMAT2 is clustered adjacent to Bassoon throughout the striatum in pairs approximately 500 nm in width, consistent with the scale of dopaminergic terminals identified by electron microscopy (9). Prior to these data, the capacity to resolve the distribution of active zone components within individual terminals of complex tissues, such as brain slices, has largely been limited to immuno-electron microscopy.
References
- 1.Maglione M, Sigrist SJ. Seeing the forest tree by tree: super-resolution light microscopy meets the neurosciences. Nat Neurosci. 2013;16(7):790–7. doi: 10.1038/nn.3403. [DOI] [PubMed] [Google Scholar]
- 2.Baddeley D, et al. 4D super-resolution microscopy with conventional fluorophores and single wavelength excitation in optically thick cells and tissues. PLoS One. 2011;6(5):e20645. doi: 10.1371/journal.pone.0020645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dani A, et al. Superresolution imaging of chemical synapses in the brain. Neuron. 2010;68(5):843–56. doi: 10.1016/j.neuron.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nanguneri S, et al. Three-dimensional, tomographic super-resolution fluorescence imaging of serially sectioned thick samples. PLoS One. 2012;7(5):e38098. doi: 10.1371/journal.pone.0038098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Endesfelder U, et al. Chemically induced photoswitching of fluorescent probes—a general concept for super-resolution microscopy. Molecules. 2011;16(4):3106–18. doi: 10.3390/molecules16043106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi X, Lim J, Ha T. Acidification of the oxygen scavenging system in single-molecule fluorescence studies: in situ sensing with a ratiometric dual-emission probe. Anal Chem. 2010;82(14):6132–8. doi: 10.1021/ac1008749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldenthal KL, et al. Postfixation detergent treatment for immunofluorescence suppresses localization of some integral membrane proteins. J Histochem Cytochem. 1985;33(8):813–20. doi: 10.1177/33.8.3894499. [DOI] [PubMed] [Google Scholar]
- 8.Hama H, et al. Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nat Neurosci. 2011;14(11):1481–8. doi: 10.1038/nn.2928. [DOI] [PubMed] [Google Scholar]
- 9.Nirenberg MJ, et al. Vesicular monoamine transporter-2: immunogold localization in striatal axons and terminals. Synapse. 1997;26(2):194–8. doi: 10.1002/(SICI)1098-2396(199706)26:2<194::AID-SYN10>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]