

Abstract
The sirtuin enzymes are important regulatory deacylases in a variety of biochemical contexts and may therefore be potential therapeutic targets through either activation or inhibition by small molecules. Here, we describe the discovery of the most potent inhibitor of sirtuin 5 (SIRT5) reported to date. We provide rationalization of the mode of binding by solving co-crystal structures of selected inhibitors in complex with both human and zebrafish SIRT5, which provide insight for future optimization of inhibitors with more “drug-like” properties. Importantly, enzyme kinetic evaluation revealed a slow, tight-binding mechanism of inhibition, which is unprecedented for SIRT5. This is important information when applying inhibitors to probe mechanisms in biology.
Keywords: enzyme inhibitors, sirtuins, posttranslational modifications, deacylases, drug discovery
Table of Contents
SAR study of mechanism-based inhibitors, combined with structural insight from X-ray co-crystal structures provide potent inhibitors of the sirtuin 5 hydrolase. Kinetic investigations furthermore reveal unprecedented slow, tight-binding behaviour of several compounds.
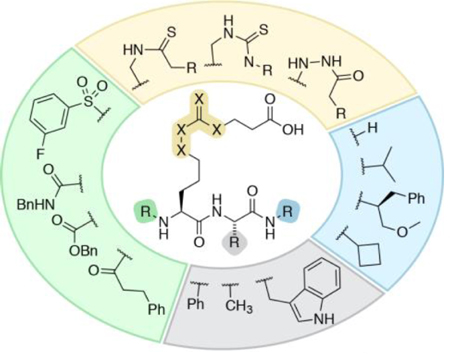
Sirtuins are a family of NAD+-dependent silent information regulator 2 (Sir2) enzymes that catalyze the removal of acyl groups from ε-N-amino groups of lysine residues in the proteome.[1] The human genome codes for seven different sirtuin isoforms (SIRT1–7), which are classified according to sequence similarity and localize to different cellular compartments.[2] Recently, it has become evident that different enzyme isoforms exhibit preference for different ε-N-acyllysine posttranslational modifications (PTMs).[1c, 3] Thus, ε-N-acetyllysine (Kac) functionalities are targeted primarily by SIRT1 and 6 in the nucleus, SIRT2 in the cytoplasm, and SIRT3 in the mitochondria.[3a] In addition, long chain acyl groups, such as ε-N-myristoyllysine (Kmyr), are also cleaved by SIRT1–3 and 6.[4] SIRT5 has been shown to selectively cleave ε-N-carboxyacyllysine derivatives based on malonate (Kmal),[5] succinate (Ksuc),[5b] and glutarate (Kglu).[6] Recently, the ability of SIRT4 to cleave the negatively charged ε-N-(3-methylglutaryl)lysine (Kmg) and ε-N-(3-methylglutaconyl)lysine (Kmgc) has also been demonstrated.[7]
Although the role of SIRT5 is not fully understood, it has been shown to regulate several metabolic enzymes, e.g., carbamoyl phosphate synthetase 1 (CPS1), succinate dehydrogenase (SDH), and 3-hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2).[8] Additionally, SIRT5 is involved in detoxification of reactive oxygen species, by deacylating proteins such as SOD1[9], IDH2, and G6PD.[10] Furthermore, SIRT5 has been implicated in tumor growth in non-small cell lung cancer[10b, 11] and has an anti-apoptotic effect in neuroblastoma cells,[12] highlighting the potential of SIRT5 as a therapeutic target.
Insight into the NAD+-dependent hydrolytic mechanism of the sirtuins has been exploited for design of highly potent substrate-mimicking inhibitors that contain thioamide or thiourea functionalities, forming stalled intermediates with ADPR in the sirtuin active sites (Scheme 1A).[13]
Scheme 1.

(A) Sirtuin hydrolytic mechanism. (B) Previous mechanism-based inhibitor. (C) Inhibitor optimization in this study. X = O or S, NAD+ = reduced nicotinamide adenine dinucleotide, ADP = adenosine diphosphate, Cbz = carboxybenzyl.
Taking advantage of the acyl-substrate specificity of SIRT5, this strategy has been successfully adapted to selectively inhibit this isozyme (Scheme 1B).[6b, 15] Here, we performed an extensive iterative structure–activity relationship (SAR) study, evaluating more than 70 compounds, which furnished SIRT5-selective inhibitors exhibiting nanomolar affinity via a slow, tight-binding mechanism. These are the most potent SIRT5 inhibitors reported to date; however, our study also highlights the necessity for thorough assessment of inhibitor mechanism and calls into question the application of IC50 values as the sole measure of potency for these chemotypes.
As starting point, we chose ε-N-thioglutaryllysine over ε-N-thiosuccinyllysine due to the lower KM value of glutarylated substrates.[6a] The α-amino group was kept Cbz protected to address the C-terminal, first by introduction of a series of amines (1–6 Scheme 2 and Supporting Scheme S1). Inspired by examination of co-crystal structures of SIRT5 with a peptide substrate (PDB 3RIY and 4GIC), we then extended the series to di- and tripeptides (7–17), addressing the importance of side chain bulkiness, stereochemistry, and presence of backbone secondary amide (Scheme 2, gray area). The two latter proved important with a preference for l-configuration at position i+1 (10 vs 11), while the steric bulk of the side chain had minor effect (12 vs 14).
Scheme 2.

Subset of the structure–activity relationship study, measuring compound potency against recombinant SIRT5 as previously described.[6a, 14] Potencies are given as IC50 values or %-inhibition at the highest tested concentration (see Figure S1 for dose-response curves). Please consult the Supporting Information for a list of additional compounds and their potencies (Scheme S1) as well as synthetic Schemes S2–S25.
Furthermore, alkylation of the C-terminal amide was beneficial for potency and extending the structure with an i+2 amino acid resulted in a slight increase in potency in one case (15). However, to limit the peptidic nature of the ligand, we chose 10 for further SAR. Next, we explored modifications of the PTM and the ε-amide bond (18–29Scheme 2, yellow area). Inspired by work on Kac surrogates by Cole and Denu, introducing hydrazide[16] and urea[17] functionalities, respectively, we designed compounds 20 and 21, as well as extended the series with semicarbazide 22 and carbamate 23. Compound 24 was inspired by work on fluorinated acetamides,[17–18] and inverted amide (25) as well as Glu(Cbz) 26 have been introduced as side chains previously.[6b, 15c] Finally, 3-methylglutaryl-mimicking[7] analogues (27 and 28) were prepared along with compound 29, the thiourea analogue of 10. Collectively, this exercise showed that thioamide- and thiourea-based compounds were the most potent and thus, compound 29 was chosen for individual optimization of the N-terminal (30–44Scheme 2, green area) and modifications to the C-terminal N-alkyl group (45–47Scheme 2, cyan area).
Co-crystal structures of NAD+-derived intermediates of both lead compounds 10 and 29 in complex with SIRT5 from either zebrafish (zSIRT5) or man (hSIRT5) were solved (Supporting Table S1). These structures revealed detailed insight into the binding modes of the two compounds (Figure 1A–D). Both structures of zSIRT5 contained the compound (10 or 29) bound as bicyclic intermediate with ADP-ribose, similar to structure III in Scheme 1A (mixed with a fraction bound as intermediate II in Scheme 1A for compound 29), and with expected interactions of the carboxylate with Y98 and R101 (Figure 1A,B and Supporting Figure S2). Slight structural deviations between the complexes were observed in a Zn-domain loop, the co-factor binding loop, and helix α3, presumably due to the subtle differences in the ligand acyl groups. Interestingly, the structure of 29 in complex with hSIRT5 revealed only the ADP-ribose-1′-thioimidate intermediate II of Scheme 1 and no bicyclic intermediate (Figure 1C). However, the protein chains of the hSIRT5 and zSIRT5 complexes with 29 were almost identical (rmsd 0.31 Å for 225 Cα atoms), and the reason for partially stalling at different intermediate states remains to be elucidated. Whereas thioamides have been co-crystallized with sirtuins previously,[19] these are the first structures with thiourea-based inhibitors. It is reassuring to observe examples for both intermediates II and III, confirming that this functionality behaves similarly to thioamides. Important interactions of the compound with hSIRT5 were again the glutaryl carboxylate with Y102 and R105 as well as the ε-NH to backbone carbonyl of V221 and additional backbone–backbone amide interactions (Figure 1C). Four SIRT5 chains with variations in the rotation of the Cbz and indole moieties of the ligand were resolved (Figure 1D). These varying end group conformations are influenced by crystal packing, and the indole positions indicate a flexibility in the SIRT5 complex that is in agreement with the minor effect of this group observed in the SAR.
Figure 1.

Co-crystal structures resulting from incubation of zSIRT5 with NAD+ and inhibitors 10 or 29 as well as hSIRT5 with NAD+ and inhibitor 29. (A) Superposition of co-crystal structures of zSIRT5 with either bicyclic intermediate III (compound 10 shown in pale blue) or an indistinguishable mixture of bicyclic intermediate III and ADP-ribose-1′-thioimidate intermediate II (compound 29 shown in pale green). (B) Active site zoom of the zSirt5 complexes in panel A with interactions between protein and ligand represented as dashed lines. (C) The active site of co-crystal structure of hSIRT5 and ADP-ribose-1′-thioimidate intermediate II with compound 29. Hydrogen bonding interactions between protein and ligand are shown as dashed lines. (D) Surface view of the hSIRT5 complex containing the ADPR29 intermediate, showing the different positions of the Cbz and indole moieties, while the rest of the ligand is tightly bound.
Furthermore, the observed flexibility of the Cbz group as well as its lack of specific interactions with the protein surface (Figure 1D) indicated that a variety of functionalities could be tested in the continued SAR. Thus, for the ease of synthesis, we decided to investigate the N-terminal relative to compound 30, which is devoid of the C-terminal i-propyl group, allowing for ready preparation of the series 30–44 by solid-phase synthesis. We included amide, urea, and sulphonamide analogues of the Cbz group, including various lengths (30–37). Due to the potency of 37 and the high abundance of sulphonamides in approved drugs, we prepared analogues 38–44 as well.
In parallel, we briefly re-investigated the importance of steric bulk at the C-terminus, now in the context of compound 29 (45–47Scheme 2, cyan area). Combining the results of these two series, we prepared compounds 48–50 in a final iteration, providing compound 49 as the most potent inhibitor with an improvement in IC50 value of >100-fold from compound 1. Not surprisingly based on the well-documented substrate specificity of SIRT5, selected compounds (29 and 48–50) exhibited excellent selectivity for SIRT5 over SIRT1–3 and 6 (Figure 2A). We were then interested in gaining insight into the kinetic behaviour of our most potent compound (49) along with intermediate lead compounds 10 and 29 as well as compound 1 and patented compound V (Scheme 1) as a control. To achieve this, we first performed a continuous assay[14] to establish whether the inhibition occurred at steady-state kinetics. Not surprisingly, since this has been reported previously for mechanism-based inhibitors of SIRT1,[20] compounds 10, 29 and 49 exhibited slow, tight-binding kinetics (Figure 2B and Supporting Figure S3). Interestingly, the less potent compounds 1 and V behaved like standard fast-on–fast-off inhibitors (Figure 2B and Supporting Figure S3), indicating that the change in mechanism is not associated with the thioamide or thiourea amide bond surrogate, but rather developing as the backbone-interacting part of the molecule gains affinity.
Figure 2.

Biochemical evaluation in vitro. (A) Selectivity of compounds 29 and 48–50, measured at an inhibitor concentration of 10 µM. Acyllysines of substrates used are indicated for individual sirtuins. (B) Progression curves and data fitting for inhibition of recombinant SIRT5 by compounds 1, 29, and 49. (C) Preincubation experiments of compounds 10, 19–25, 29, and 49.
Nevertheless, slow binding could be associated with interaction with NAD+ and enzyme or enzyme alone, so we performed pre-incubation experiments of selected inhibitors and SIRT5 with or without NAD+ to address these scenarios and to evaluate whether compounds 19–25 exhibited slow-binding as well (Figure 2C). Not surprisingly, the slow-binding behaviour of compounds 10, 19, 29 and 49 was depending on the presence of NAD+, indicating that it involves formation of the stalled intermediate. However, it is intriguing that optimization of the scaffold’s contribution to affinity imposes a change in mechanism of inhibition. Interestingly, the urea-containing compound 21 also exhibited NAD+-dependent slow-binding, which may revive the use of this functionality for future inhibitor design. The remaining compounds did not exhibit slow-binding within the time-frame of the pre-incubation experiments (Figure 2C).
Finally, we tested an ethylester prodrug version of compound 29 (Et-29) for its ability to affect the degree of lysine glutarylation in cells. However, it is unfortunately not trivial to detect changes in either lysine malonylation, succinylation, or glutarylation even compared to the control CRISPR-Cas9 SIRT5 knockout cell line (Supporting Figures S4 and S5). Further optimization of the experimental design and subsequent evaluation of these compounds in a cellular context will be of immediate future interest.
In summary, we describe mechanism-based inhibitors of sirtuin 5 that exhibit up to a 100-fold increase in IC50 values compared to a patented reference compound included in our assays. Importantly, we show that kinetic analyses of inhibitors of these enzymes is important for appropriate comparison of potencies as we disclose the first examples of slow, tight-binding behaviour for SIRT5 inhibitors. This calls for more thorough investigation mechanism-based inhibitors of all sirtuins. We also describe structural information for the binding mode of thiourea-based sirtuin inhibitors for the first time, which provides important insight for future inhibitor design.
Supplementary Material
Acknowledgments
We thank Ms Alessia Lucidi and Dr. Jonas S. Harild for donation of building blocks. We thank BESSY (operated by HZI Berlin, Germany) and SLS (operated by Paul Scherrer Institut, Villigen, Switzerland) for synchrotron beam time and experimental support. This work was supported by the University of Copenhagen (PhD fellowship to N.R.), the Oberfrankenstiftung (04115; C.S.), the Lundbeck Foundation (Group Leader Fellowship R52-2010-5054; C.A.O.), the Carlsberg Foundation (2011-01-0169, 2013-01-0333, and CF15-011; C.A.O.), the Novo Nordisk Foundation (NNF15OC0017334; C.A.O.), and the European Research Council (ERC-CoG-725172 – SIRFUNCT; C.A.O.). We thank the COST action CM1406 (EPICHEMBIO) for support.
Footnotes
Supporting information for this article is given via a link at the end of the document. X-ray: diffraction data and coordinates have been deposited with the wwPDB (www.wwpdb.org) under accession numbers 6ENX (zSIRT5/10), 6EO0 (zSIRT5/29), 6EQS (hSIRT5/29).
Conflict of interest
The authors declare no conflict of interest.
References
- 1.a) Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R. Proc. Natl. Acad. Sci. U. S. A. 2000;97:5807–5811. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Imai S, Armstrong CM, Kaeberlein M, Guarente L. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]; c) Bheda P, Jing H, Wolberger C, Lin H. Annu. Rev. Biochem. 2016;85:405–429. doi: 10.1146/annurev-biochem-060815-014537. [DOI] [PubMed] [Google Scholar]
- 2.a) Frye RA. Biochem. Biophys. Res. Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]; b) Marmorstein R. Structure. 2001;9:1127–1133. doi: 10.1016/s0969-2126(01)00690-6. [DOI] [PubMed] [Google Scholar]; c) Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Mol. Biol. Cell. 2005;16:4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Chen B, Zang W, Wang J, Huang Y, He Y, Yan L, Liu J, Zheng W. Chem. Soc. Rev. 2015;44:5246–5264. doi: 10.1039/c4cs00373j. [DOI] [PubMed] [Google Scholar]; b) Sabari BR, Zhang D, Allis CD, Zhao Y. Nat. Rev. Mol. Cell Biol. 2017;18:90–101. doi: 10.1038/nrm.2016.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Jiang H, et al. Nature. 2013;496:110–113. doi: 10.1038/nature12038. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Feldman JL, Baeza J, Denu JM. J. Biol. Chem. 2013;288:31350–31356. doi: 10.1074/jbc.C113.511261. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Galleano I, Schiedel M, Jung M, Madsen AS, Olsen CA. J. Med. Chem. 2016;59:1021–1031. doi: 10.1021/acs.jmedchem.5b01532. [DOI] [PubMed] [Google Scholar]; d) Madsen AS, et al. J. Biol. Chem. 2016;291:7128–7141. doi: 10.1074/jbc.M115.668699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Peng C, et al. Mol. Cell. Proteomics. 2011;10 doi: 10.1074/mcp.M111.012658. M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Du J, et al. Science. 2011;334:806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Tan M, et al. Cell Metab. 2014;19:605–617. doi: 10.1016/j.cmet.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Roessler C, et al. Angew. Chem. Int. Ed. 2014;53:10728–10732. doi: 10.1002/anie.201402679. [DOI] [PubMed] [Google Scholar]
- 7.Anderson KA, et al. Cell Metab. 2017;25:838–855. doi: 10.1016/j.cmet.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gertz M, Steegborn C. Cell. Mol. Life Sci. 2016;73:2871–2896. doi: 10.1007/s00018-016-2180-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin ZF, Xu HB, Wang JY, Lin Q, Ruan Z, Liu FB, Jin W, Huang HH, Chen X. Biochem. Biophys. Res. Commun. 2013;441:191–195. doi: 10.1016/j.bbrc.2013.10.033. [DOI] [PubMed] [Google Scholar]
- 10.a) Zhou L, et al. EMBO Rep. 2016;17:811–822. doi: 10.15252/embr.201541643. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xiangyun Y, Xiaomin N, Linping G, Yunhua X, Ziming L, Yongfeng Y, Zhiwei C, Shun L. Oncotarget. 2017;8:6984–6993. doi: 10.18632/oncotarget.14346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu W, Zuo Y, Feng Y, Zhang M. Tumour Biol. 2014;35:10699–10705. doi: 10.1007/s13277-014-2372-4. [DOI] [PubMed] [Google Scholar]
- 12.Liang F, Wang X, Ow SH, Chen W, Ong WC. Neurotox. Res. 2017;31:63–76. doi: 10.1007/s12640-016-9664-y. [DOI] [PubMed] [Google Scholar]
- 13.Jiang Y, Liu J, Chen D, Yan L, Zheng W. Trends Pharmacol. Sci. 2017;38:459–472. doi: 10.1016/j.tips.2017.01.009. [DOI] [PubMed] [Google Scholar]
- 14.Madsen AS, Olsen CA. J. Med. Chem. 2012;55:5582–5590. doi: 10.1021/jm300526r. [DOI] [PubMed] [Google Scholar]
- 15.a) He B, Du J, Lin H. J. Am. Chem. Soc. 2012;134:1922–1925. doi: 10.1021/ja2090417. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lin H. (Cornell University), WO 2014/197775 A1. 2014 [Google Scholar]; c) He Y, Yan L, Zang W, Zheng W. Org. Biomol. Chem. 2015;13:10442–10450. doi: 10.1039/c5ob01721a. [DOI] [PubMed] [Google Scholar]; d) Zang W, Hao Y, Wang Z, Zheng W. Bioorg. Med. Chem. Lett. 2015;25:3319–3324. doi: 10.1016/j.bmcl.2015.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Polletta L, et al. Autophagy. 2015;11:253–270. doi: 10.1080/15548627.2015.1009778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dancy BCR, et al. J. Am. Chem. Soc. 2012;134:5138–5148. doi: 10.1021/ja209574z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith BC, Denu JM. J. Biol. Chem. 2007;282:37256–37265. doi: 10.1074/jbc.M707878200. [DOI] [PubMed] [Google Scholar]
- 18.a) Smith BC, Denu JM. Biochemistry. 2007;46:14478–14486. doi: 10.1021/bi7013294. [DOI] [PubMed] [Google Scholar]; b) Smith BC, Denu JM. J. Am. Chem. Soc. 2007;129:5802–5803. doi: 10.1021/ja070162w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gertz M, Fischer F, Nguyen GT, Lakshminarasimhan M, Schutkowski M, Weyand M, Steegborn C. Proc. Natl. Acad. Sci. U. S. A. 2013;110:E2772–E2781. doi: 10.1073/pnas.1303628110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Hirsch BM, Du Z, Li X, Sylvester JA, Wesdemiotis C, Wang Z, Zheng W. MedChemComm. 2011;2:291. [Google Scholar]; b) Asaba T, Suzuki T, Ueda R, Tsumoto H, Nakagawa H, Miyata N. J. Am. Chem. Soc. 2009;131:6989–6996. doi: 10.1021/ja807083y. [DOI] [PubMed] [Google Scholar]; c) Suzuki T, Asaba T, Imai E, Tsumoto H, Nakagawa H, Miyata N. Bioorg. Med. Chem. Lett. 2009;19:5670–5672. doi: 10.1016/j.bmcl.2009.08.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.