

Mice with the actin Glu99Lys hypertrophic cardiomyopathy mutation (ACTC E99K) are prone to sudden cardiac death around 40 days, associated with increased Ca2+ transients, spark mass, and fibrosis. However, adult survivors have normal Ca2+ transients and spark density accompanied by hypertrophy. Penetrance of the sudden cardiac death phenotype depends on the genetic background of the mouse.
Keywords: calcium sparks, calcium waves, sarcoplasmic reticulum, myofilament sensitivity
Abstract
Patients with hypertrophic cardiomyopathy, particularly young adults, can die from arrhythmia, but the mechanism underlying abnormal rhythm formation remains unknown. C57Bl6 × CBA/Ca mice carrying a cardiac actin (ACTC) E99K (Glu99Lys) mutation reproduce many aspects of human hypertrophic cardiomyopathy, including increased myofilament Ca2+ sensitivity and sudden death in a proportion (up to 40%) of young (28−40 day old) animals. We studied the hearts of transgenic (TG; ACTC E99K) mice and their non-TG (NTG) littermates when they were in their vulnerable period (28–40 days old) and when they were adult (8–12 wk old). Ventricular myocytes were isolated from the hearts of TG and NTG mice at these two time points. We also examined the hearts of mice that died suddenly (SCD). SCD animals had approximately four times more collagen compared with age-matched NTG mice, yet myocyte cell size was normal. Young TG mice had double the collagen content of NTG mice. Contraction and Ca2+ transients were greater in cells from young TG mice compared with their NTG littermates but not in cells from adult mice (TG or NTG). Cells from young TG mice had a greater propensity for Ca2+ waves than NTG littermates, and, despite similar sarcoplasmic reticulum Ca2+ content, a proportion of these cells had larger Ca2+ spark mass. We found that the probability of SCD in young TG mice was increased when the mutation was expressed in animals with a CBA/Ca2+ background and almost eliminated in mice bred on a C57Bl6 background. The latter TG mice had normal cellular Ca2+ homeostasis.
NEW & NOTEWORTHY Mice with the actin Glu99Lys hypertrophic cardiomyopathy mutation (ACTC E99K) are prone to sudden cardiac death around 40 days, associated with increased Ca2+ transients, spark mass, and fibrosis. However, adult survivors have normal Ca2+ transients and spark density accompanied by hypertrophy. Penetrance of the sudden cardiac death phenotype depends on the genetic background of the mouse.
Listen to this article’s corresponding podcast at http://ajpheart.podbean.com/e/calcium-regulation-in-e99k-mouse-heart/.
INTRODUCTION
Inherited heart diseases are responsible for a significant proportion of cardiac-related death and morbidity and are currently difficult to treat. Hypertrophic cardiomyopathy (HCM) is a primary disease of cardiac muscle clinically defined by a hypertrophied nondilated left ventricle in the absence of any other etiology. It is a common disorder affecting 1 in 500 of the population and is the leading cause of sudden cardiac death (SCD) in young individuals (27). HCM is largely caused by monogenic mutations in genes encoding sarcomeric proteins, commonly myosin heavy chain 7 (MYH7) and myosin-binding protein C3 (MYBPC3), although ~12% are in genes encoding proteins of the thin filament (1). HCM presents with a variable sequence of clinical events. Some patients can remain stable over long periods of time, whereas others suffer adverse events, including SCD, embolic stroke, and heart failure. Sudden death (not associated with heart failure) is the most devastating of the adverse events and most frequently occurs in adolescents and young adults, although it can also extend into later life (28).
The molecular basis of HCM has been extensively studied, and the most likely primary mechanism involves derangement of Ca2+ regulation of the sarcomere (30). A recent literature search yielded 71 independent measurements of increased Ca2+ sensitivity due to HCM mutations, ranging from a 1.15- to 3.8-fold increase (mean: 1.87 ± 0.07) (29). Thus, the cardiac muscle in patients with HCM has a hypercontractile phenotype that causes incomplete relaxation and diastolic dysfunction. Moreover, the increased Ca2+ sensitivity has been proposed to predispose the heart to certain forms of arrhythmia that may provoke SCD (18, 46, 47).
We have developed a transgenic (TG) mouse with a cardiac actin Glu99Lys (ACTC E99K) mutation (34) that enables us to investigate the proposed mechanism of SCD. TG mice express the mutation in 50% of total sarcomeric actin. This level is achieved by 21 days and is maintained throughout life. The mutation causes an increase in myofilament Ca2+ sensitivity observed in single myofilaments using in vitro motility assays, myofibrils, and isolated papillary muscle (46, 47). Not only do these mice reproduce the apical hypertrophy, myocyte disarray, interstitial fibrosis, arrhythmias, and subsequent development of heart failure noted in human HCM but also they have a striking predisposition to SCD between 4 and 6 wk of age, which is more pronounced in female mice (46, 47). Up to 40% of mice die during this early vulnerable period, whereas survivors have a normal lifespan despite less efficient cardiac work and in later life experienced progressive cardiac hypertrophy, fibrosis, and diastolic dysfunction. The close recapitulation of the human condition by this mouse model offers an opportunity to investigate the mechanism underlying SCD and account for the observation that only a proportion of mice die early in life. The underlying cause of the SCD in patients with HCM is usually arrhythmia, and Baudenbacher et al. (3) demonstrated that Ca2+ sensitization per se can be proarrhythmic in the absence of typical anatomical abnormalities produced by HCM. The mechanism underlying arrhythmia generation when the primary disturbance in cell biology is a sarcomeric mutation remains unknown.
We studied the hearts of TG mice and their non-TG (NTG) littermates in their vulnerable period (28–40 days old), as adults (8–12 wk old), and when they died suddenly. We investigated if the development of hypertrophy and fibrosis correlated with SCD and if the higher Ca2+ sensitivity in TG mice disturbed intracellular Ca2+ homeostasis, causing changes to cellular Ca2+ handling and electrophysiological stability. Here, we show that the underlying proarrhythmic mechanism is that some young TG mice have an increased propensity for spontaneous Ca2+ release that could lead to the formation of afterdepolarizations, predisposing their hearts to potentially fatal arrhythmias. Moreover, we found that the probability that young TG mice have this aberrant cellular Ca2+ homeostasis is dependent on the background strain of the mice.
METHODS
ACTC E99K Mice
The generation of ACTC E99K TG mice has been previously described by Song et al. (46). Male ACTC E99K mice were bred with female C57Bl6 × CBA/Ca F1 hybrid mice, and the resulting phenotype was described in detail by Song et al. (46). Mice were housed at 21 ± 1°C on a 12:12-h light-dark cycle and provided with standard feed and water ad libitum. All procedures on mice were performed in accordance with the United Kingdom Animals (Scientific Procedures) Act of 1986, EU Directive 2010/63/EU for animal experiments and with university ethical approval. To obtain heart tissue, mice were deeply anesthetized using 5% isoflurane in 100% oxygen and subsequently killed by cervical dislocation. Tissues were then removed from the animal.
Genotyping TG Mice
Ear notch samples produced ~5 µg DNA. DNA extraction was performed using a Qiagen DNeasy blood and tissue kit (Qiagen) according to the manufacturer’s instructions, and the extracted DNA was stored at 4°C until required for PCR. PCR primers were obtained from Invitrogen. The forward primer was located in the promoter element of the transgene sequence (5′-TGACAGACAGATCCCTCCTATCTCC-3′), and the reverse primer was located in the mutant actin (5′-GGCATCTTAGAAGCATTTGCGG-3′).
RNA Extraction and Gene Expression Analysis
Heart tissue (20 mg) was homogenized and lysed in 350 μl TRI Reagent buffer (Sigma). Primers used for gene expression analysis are listed below. RNA was extracted with the RNeasy Mini Kit (Qiagen). NanoDrop 2000 (ThermoScientific) was used for the quantification of RNA. RT-PCR was performed with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) based on the manufacturer instructions. Real-time PCR were performed with TaqMan gene expression assays (Applied Biosystems). All samples were run in triplicate with the housekeeping gene GAPDH. Quantitative PCR data were analyzed by the comparative threshold cycle (Ct) method ( method) using the average of three technical replicates.
Western Blot Analysis
Heart tissues were manually homogenized in T-PER Tissue Protein Extraction Reagent (Thermo Scientific) containing 2 μg/ml each of the protease inhibitors chymostatin, leupeptin, and pepstatin A. Six times of the volume of the T-PER solution to the weight of frozen heart tissue were added to dissolve the pulverzied tissue. The same amount of 2× SDS buffer was added (20 mM Tris·HCl, 5% SDS, and 10% β-mercaptoethanol). Proteins were transferred to a 0.2-μm nitrocellulose membrane using the TransBlot Turbo Transfer System (Bio-Rad). Proteins were visualized using the Pierce Reversible Protein Stain Kit (Thermo Scientific) and imaged using the G:Box Chemi XT Imager (Syngene). Membranes were blocked with 20% SeaBlock blocking buffer (Pirece Chemical) and 0.1% Tween 20 in PBS (pH 7.2−7.4) for 1 h. The custom-made rabbit polyclonal antibody specific to the E99K mutation was diluted at 5,000:1 in 20% blocking solution and incubated at 4°C overnight with agitation. Membranes were washed with PBS containing 0.1% Tween 20 three times for 5 min each followed by incubation with the secondary antibody for 1 h at room temperature with agitation. Wash steps in PBS were repeated three times. Immunoreactivity bands were visualized using Amersham ECL Select Western Blotting Detection Reagents (GE Healthcare) and detected in the imager. GeneTools (Syngene) was used for the quantification of band density. The density of antibody relative to the density of α-actinin on the protein-stained membrane was used to control for loading variations between samples.
Histology and Immunohistochemistry
Freshly dissected hearts were stored in liquid nitrogen. They were gradually thawed, fixed in 4% paraformaldehyde overnight at 4°C, and then cryoprotected with 30% sucrose. Fixed hearts were frozen in OCT compound vertically, and transverse cryosections were cut at 7-μm intervals.
Wheat germ agglutinin staining for cell size.
For wheat germ agglutinin (WGA) staining, sections were rehydrated with PBS. Slides were blocked with 20% goat serum diluted in PBS for 30 min at room temperature. Next, slides were incubated with the Alexa Fluor 488 conjugate of WGA (10 μg/ml, diluted in PBS, Invitrogen) for 30 min with protection from light. Slides were washed two times with PBS, mounted in Vectashield with DAPI (Vector Laboratories), and covered with coverslips. Images were recorded with fluorescence microscopy (Zeiss Axio Observer Inverted Widefield Microscope with LED illumination) at excitation and emission wavelengths of 495 and 519 nm, respectively. For each section, four images were captured using ×20 magnification, and in each image 50–60 cells were calculated in ImageJ software.
Picrosirius red staining for collagen.
Sections were rehydrated with distilled water and stained in a solution of 0.1% sirius red F3BA in saturated aqueous picric acid (Sigma-Aldrich) for 35 min at room temperature followed by four washing steps in 100% methanol, as modified from another study (24). Subsequently, slides were rinsed two times in xylene for 10 min each. Slides were mounted with Vectashield with DAPI (Vector Laboratories), covered with coverslips, and left to dry overnight at room temperature. A series of images of one section was acquired at ×10 magnification and automatically stitched together using bright-field mode of the LSM-780 inverted confocal laser scanning microscope (Zeiss). Collagen content was quantified in ImageJ software. Total collagen content was defined as the proportion of positive pixels to total pixels of stained area.
Isolation of Cardiac Myocytes and Loading with Ca2+-Sensitive Dyes
Cardiac myocytes were isolated by enzymatic digestion and loaded with Ca2+-sensitive dyes using established techniques (24, 25, 45, 50).
Ca2+ Transients and Cell Length Measurements
Fura 2 was excited at 360 and 380 nm, and fluorescence was collected at 510 nm using an Ionoptix system (Ionoptix, Milton, MA) with a photomultiplier tube connected to a personal computer running IonWizard software. Cell contraction was measured using a charge-coupled device video camera (MyoCam-S, IonOptix) also connected to the personal computer running IonWizard software.
Ca2+ Sparks and Waves
Ca2+ sparks were recorded by previously described methods, and analysis of the line scan images was performed using ImageJ (National Institutes of Health) with the SparkMaster plugin (40) and custom macros (45). Detection criteria for Ca2+ sparks were set at 3.8 times the SD above the mean background value. Spark mass was calculated using the formulas derived by Hollingworth et al. (16). To assess wave frequency and characteristics, cells were stimulated at up to 5 Hz for 30 s. Field stimulation was then stopped, and waves were recorded during a subsequent 20-s period.
Determination of Sarcoplasmic Reticulum Ca2+ Content
Determinations of sarcoplasmic reticulum (SR) Ca2+ content were made using the change in the fura 2 ratio as a qualitative index of load after rapid application of 10 mM caffeine in Na+-free/Ca2+-free solution. The caffeine evokes SR Ca2+ release, whereas the 0 Na+/0 Ca2+ solution prevents Na+/Ca2+ exchange from effluxing Ca2+ and attenuating the size of the induced transient. Assessments of Na+/Ca2+ exchange and SR Ca2+-ATPase function were made by comparing the decay times of caffeine-induced Ca2+ transients either in the continued presence of caffeine alone (for Na+/Ca2+ exchange function) or in Na+-free/Ca2+-free solution after caffeine removal (for SR Ca2+-ATPase function) (see Fig. 4) (2).
Fig. 4.

A: typical traces of spontaneous contractile waves (denoted by arrows) occurring in nontransgenic (NTG) and transgenic (TG) cardiac myocytes isolated from young animals. These spontaneous contractions occurred during a quiescent 30-s period after 1-Hz stimulation. B: pooled data of wave frequency in the quiescent period in NTG and TG cells presented as means ± SE; n = 80 cells from 8 NTG animals and 80 cells from 9 TG animals. ***P < 0.001. C: Ca2+ waves in young cells recorded using fura 2. Typical responses are shown for NTG (top) and TG (bottom) cells. Cells were paced at a particular frequency for 1 min, the stimulation train was stopped, and spontaneous Ca2+ waves were recorded over the next 30 s. D: effect of the stimulation train frequency on the occurrence of Ca2+ waves in young NTG and TG cardiac myocytes. Data are presented as means ± SE; n = 20 cells from 5 NTG animals and 20 cells from 4 TG animals.
Statistical Analyses
Data are presented as means ± SE, and statistical significance between means was taken when P < 0.05. Data sets were normally distributed except for Ca2+ spark parameters. Logarithmic transformations of these data were carried out before the Student’s t-test or ANOVA was applied.
RESULTS
Timeline for the Development of the HCM Phenotype
Using an antibody specific to the ACTC E99K mutation, we found that expression of the mutation was switched on shortly after birth and reached adult levels between postnatal day 14 (P14) and P21, well before the SCD window. Postmortem analysis of SCD mice showed that ACTC E99K expression levels were the same as TG adult survivors (Fig. 1).
Fig. 1.
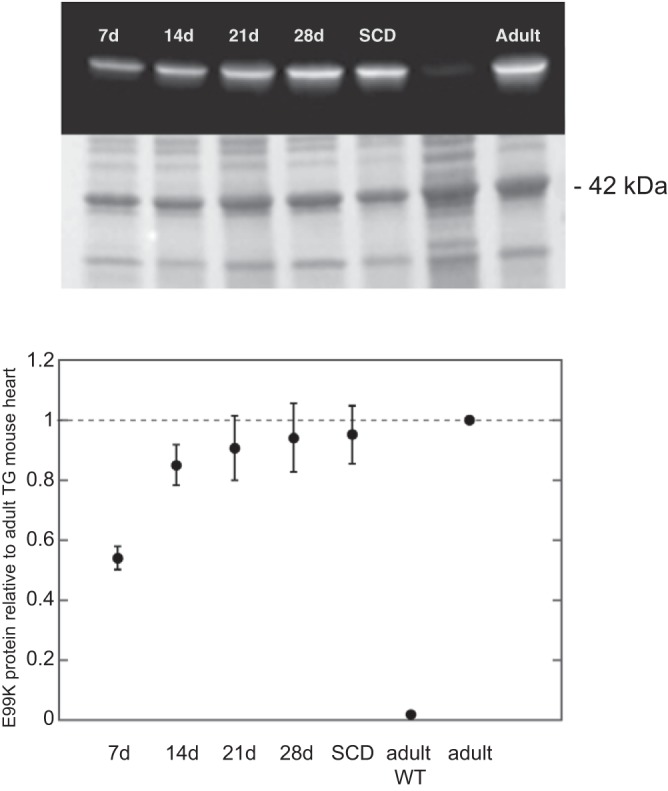
Quantification of transgene expression by Western blots with antibody specific to the E99K mutation. Top: Western blot and corresponding whole protein-stained membrane. Bottom: time dependence of level of E99K expressed relative to the adult. d, Day; SCD, sudden cardiac death; TG, transgenic; WT, wild type.
We measured ventricular muscle cell sizes in two ways. Cross sections of ventricular wall were stained with WGA, and cell cross-sectional area was measured using planimetry. Figure 2, A–C, right, shows representative histological WGA-stained sections of hearts from NTG (A), ACTC E99K TG (B), and ACTC E99K TG (C) mice that suffered SCD. Cell areas of young TG mice and TG mice that died suddenly were not significantly different from NTG mice (Fig. 2D). Cell hypertrophy was observed in older TG mice, but, in NTG hearts, cell areas were the same in young and adult mice. Cell areas and perimeter lengths of enzymatically dissociated ventricular myocytes were measured after image capture and analysis by ImageJ. Young TG mice had smaller perimeters than their NTG counterparts (173 ± 3 vs. 297 ± 4 µm, P < 0.001) and reduced cell areas (1,711 ± 44 vs. 2,201 ± 66 μm2, 178 cells from 15 NTG animals and 125 cells from 10 TG animals, P < 0.001). These combined approaches of cell size measurement indicate that cell hypertrophy does not contribute to the SCD phenotype.
Fig. 2.

A–C: representative histological sections of hearts from nontransgenic mice (NTG; A), cardiac actin (ACTC) mutation E99K transgenic (TG) mice (B), and ACTC E99K TG mice that suffered sudden cardiac death (SCD; C). Left, picrosirus-stained heart sections (for collagen determination); right, wheat germ agglutinin sections (for cell cross-sectional area measurements). D: mean ± SE cross-sectional area of myocytes from sections similar to those shown in A−C. More than 690 myocytes were analyzed from each group. E: mean ± SE percentage of the field of view that stained positively for collagen with picrosirus red. Data were acquired from 3 NTG hearts, 4 TG hearts, 2 hearts from TG mice that suffered SCD, and 3 NTG and 3 TG hearts from mice on a pure C57Bl6 background. F and G: differences in COL1A1 (F) and COL3A1 (G) mRNA levels in NTG, TG, and SCD hearts. COL1A1 encodes the pro-α1 chains of collagen type I, and COL3A1 encodes the pro-α1 chains of collagen type III. F: means ± SE; n > 6. ANOVA with Tukey's multiple-comparison test: *P < 0.05 and **P < 0.01. G: means ± SE; n = 3.
Figure 2, A–C, left, shows representative histological sections of hearts stained with picrosirus red for collagen determination. We found a strong association between the extent of cardiac fibrosis and SCD in TG mice. These data (Fig. 2E) suggest there is approximately four times more collagen in SCD mice compared with age-matched NTG mice. Young TG mice have approximately double the collagen content of NTG mice, and, notably, NTG and TG mice bred on pure C57Bl6 backgrounds have similar collagen content to NTG hybrid mice. These findings were supported by measurements of collagen type 1a1 and collagen type 3a1 gene expression (Fig. 2, F and G).
Contraction and Ca2+ Transients
Figure 3 shows that ventricular myocytes isolated from young TG mice (between 20 and 45 days old) have a hypercontractile phenotype (A) and larger Ca2+ transients (B) compared with their NTG littermates and with adult mice of both genotypes (B and D). The larger contractions can be explained by higher myofilament Ca2+ sensitivity in TG mice found in previous work (46, 47). The larger contractions in young TG animals are observed over a range of stimulation frequencies. In contrast, ventricular myocytes isolated from adult ACTC E99K mice (between 56 and 84 days old) do not have a hypercontractile phenotype and had smaller Ca2+ transients compared with their NTG littermates (Fig. 3, B and D). Ca2+ transient amplitudes recorded from TG cardiac myocytes isolated from younger animals had a larger spread of datapoints extending above the range seen in the control NTG group (Fig. 3E). Almost a quarter of young TG cells exhibited Ca2+ transients above the largest fura 2 ratio observed in young NTG cells (~0.9 change in fura 2 ratio, dashed lines in Fig. 3E). In contrast, in the adult group of mice (8–12 wk), the Ca2+ transient amplitude range was not significantly different between NTG and TG cells (Fig. 3F). Mean Ca2+ transient amplitudes recorded in myocytes isolated from adult and young NTG hearts were not different. TG animals (both young and adult) had lower diastolic fura 2 ratios (Table 1), but there was no difference in decay times of caffeine-induced Ca2+ transients between NTG or TG cells either in the continued presence of caffeine or in Na+-free/Ca2+-free solution, suggesting unchanged Na+/Ca2+ exchange and SR Ca2+-ATPase functions.
Fig. 3.

Contraction and Ca2+ transients in young (25–45 days; A) and adult (8–12 wk; B) mice. Left, typical traces of contraction profiles (expressed in terms of sarcomere length changes) of nontransgenic (NTG; dashed line) and transgenic (TG; solid line) cells. Right, corresponding pooled sarcomere shortening data (means ± SE). C and D: typical traces of Ca2+ transients of NTG (dashed line) and TG (solid line) cells, with the corresponding pooled data (means ± SE) on the right for young and adult mice, respectively. Mean data were calculated from 92 cells isolated from 10 NTG young animals, 77 cells from 8 TG young animals, 64 cells isolated from 10 NTG adult animals, and 41 cells from 8 TG adult animals. ***P < 0.001 and **P < 0.01, t-test. E and F: scatter graphs of Ca2+ transient amplitudes recorded from young (E) and adult (F) TG and NTG cardiac myocytes. The dashed lines indicate the greatest transient amplitude recorded from young NTG mice. Almost one-quarter of tyoung TG cells had Ca2+ transient amplitudes greater than the largest transients observed in NTG cells.
Table 1.
Resting (diastolic) fura 2 ratios
| Young |
Adult |
|||||
|---|---|---|---|---|---|---|
| Means ± SE | n | P value | Means ± SE | n | P value | |
| Nontransgenic | 1.49 ± 0.02 | 109 | <0.05 | 1.41 ± 0.01 | 93 | <0.01 |
| Transgenic | 1.44 ± 0.02 | 69 | 1.29 ± 0.01 | 65 | ||
Values shown are resting Ca2+ concentration (fura 2 ratio); n, no. of cells.
Ca2+ Waves
The next experiments were designed to assess if young TG animals exhibited more spontaneous contractions (SCs). Cells were paced at 0.5–5.0 Hz for 1 min to load the SR. The stimulation train was followed by a 30-s pause during which SCs were recorded (Fig. 4A). Figure 4B shows that, after stimulation at 1.0 Hz, the frequency of SCs was greater in young TG cells compared with their NTG counterparts. Similar relationships were found with Ca2+ waves measured using fura 2 (Fig. 4C). TG cells showed more spontaneous Ca2+ wave events compared with cells isolated from NTG littermates, a relationship that remained at all SR loading stimulation frequencies (Fig. 4D).
The time to the first SC was measured as the time from the last electrically stimulated contraction (at 1 Hz) to the first SC. This was represented in Kaplan-Meier survival curves, as shown in Fig. 5. The SC-free survival period was reduced in TG cells (P < 0.001, log-rank curve comparison test). This relationship remained at all stimulation frequencies.
Fig. 5.
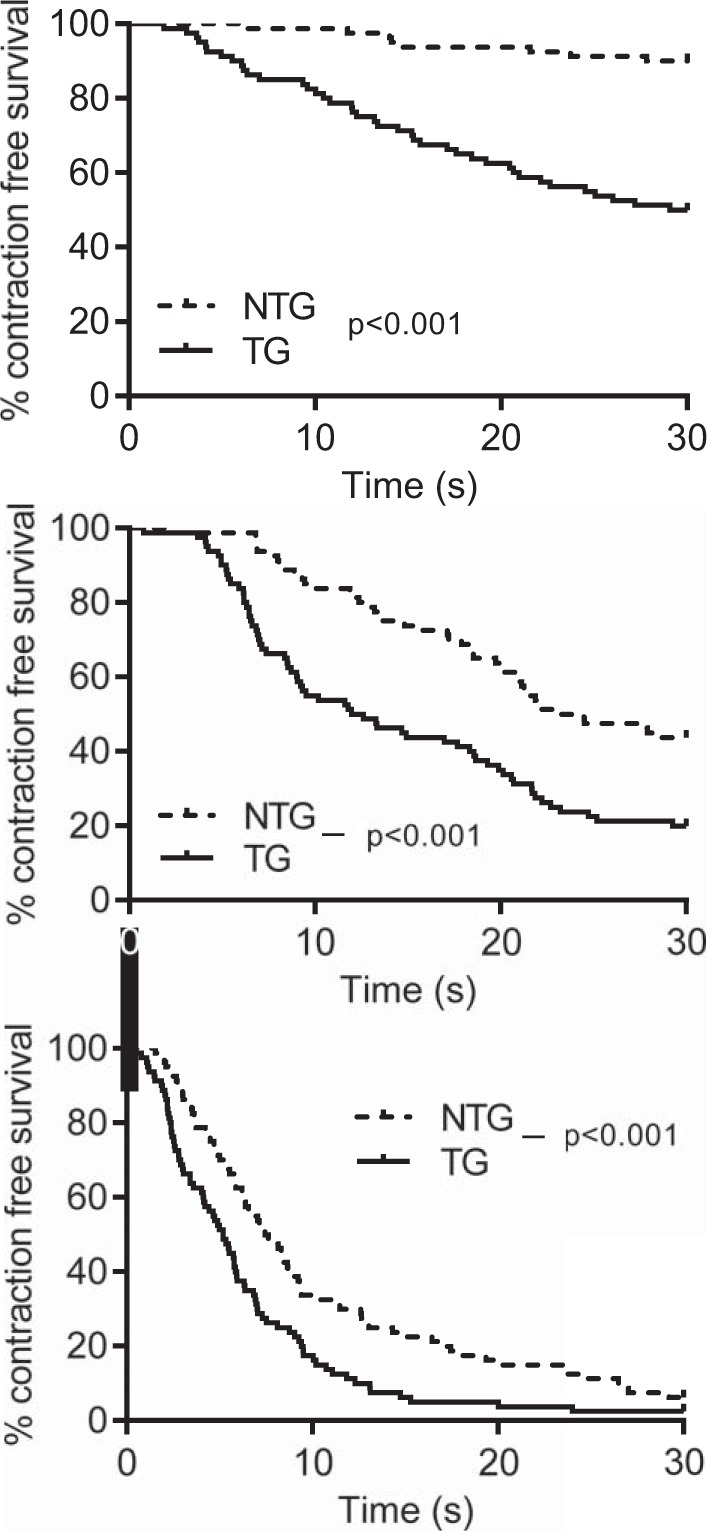
Wave-free survival of young transgenic (TG) and nontransgenic (NTG) myocytes after different pacing rates. The time to the first spontaneous contraction was measured as the time from the last electrically stimulated contraction to the first spontaneous contraction. This was represented in a Kaplan-Meier survival curve. Top: survival after a 0.5-Hz train; middle: survival after a 2.0-Hz train; bottom, survival after a 5.0-Hz train. Points are means calculated from 80 TG cells from 9 animals and 80 NTG cells from 8 animals.
The increased occurrence of waves could be a result of an increase in SR Ca2+ content (8, 42), so we assessed this using rapid application of 10 mM caffeine in Na+-free/Ca2+-free solution. Caffeine evoked SR Ca2+ release, whereas Na+-free/Ca2+-free solution prevented the Na+/Ca2+ exchange from effluxing Ca2+ and reduced the size of the induced transient (Fig. 6A). The SR Ca2+ contents of myocytes isolated from young TG mice were not different from their NTG counterparts (Fig. 6B), suggesting the increased occurrence of waves in these mice is dependent on some other factor. The SR Ca2+ contents of myocytes isolated from adult surviving TG mice were less than their NTG counterparts (Fig. 6C). Because Ca2+ release depends on SR Ca2+ content, the reduction in transient size observed in the adult surviving mice may be explained by a reduction in stored Ca2+.
Fig. 6.

A: determinations of sarcoplasmic reticulum Ca2+ content in young and adult myocytes assessed using rapid application of 10 mM caffeine in Na+-free/Ca2+-free solution. B and C: pooled data showing the differences in sarcoplasmic reticulum Ca2+ content of myocytes isolated from young (B) and adult (C) mice. Data are presented as means ± SE; n = 56 cells from 10 young nontransgenic (NTG) animals and 25 cells from 8 young transgenic (TG) animals and n = 34 cells from 10 adult NTG animals and 28 cells from 6 adult TG animals. **P < 0.01, t-test.
Ca2+ Regulation in Male and Female TG Mice
In previous work, we reported that the prevalence of SCD in young ACTC E99K mice was more frequent in female than male mice (40% of female mice vs. 25% male mice die when they are 30–45 days old). Sex differences in the onset and progression of cardiovascular disease have been noted (31, 37, 39) as well as some variations in cardiomyocyte intracellular Ca2+ regulation (11, 54). We examined if there were any alterations in intracellular Ca2+ regulation between cardiomyocytes isolated from surviving female and male ACTC E99K mice. We found two significant differences; these are shown in Fig. 7. Although there were no differences in Ca2+ transient decay times between the pooled TG and NTG data, the decay of the electrically stimulated Ca2+ transient in female TG myocytes was slower than the transient decay in male myocytes. Separation of the data into male and female groups also revealed that the decay times of caffeine-induced Ca2+ transients (in the continued presence of caffeine, thus allowing the assessment of Ca2+ efflux mediated by Na+/Ca2+ exchange and slow systems) were slower in TG female myocytes compared with cells from their NTG littermates. A similar difference was not evident in male myocytes.
Fig. 7.

Sex-based alterations in intracellular Ca2+ regulation between cardiomyocytes isolated from surviving female (F) and male (M) actin (ACTC) E99K mutant mice. The time to 50% decay of Ca2+ transients evoked electrically (left) and by rapid application of caffeine (right) is shown. n = 46 cells from male nontransgenic (NTG) animals, 37 cells from male transgenic (TG) animals, 47 cells from female NTG animals, and 42 cells from female TG animals. *P < 0.05 and **P < 0.01, ANOVA.
Ca2+ Sparks
Cells from TG animals (both young and adult) had greater spark frequencies compared with cells from NTG animals (Fig. 8, B, C, F, and G). However, spark amplitudes were greater in young TG animals compared with NTG littermates (Fig. 8, D and E). Spark amplitudes measured in TG and NTG cells from older surviving mice were not different (Fig. 8, H and I). These data were skewed, so log transformations were used to produce approximately Gaussian distributions (Fig. 8, C, E, G, and I), and a t-test with Welch’s correction was applied.
Fig. 8.

Spark properties of cardiac myocytes isolated from young and adult transgenic (TG) and nontransgenic (NTG) hearts. A: typical line scan recordings of Ca2+ sparks in young NTG and TG cells. Horizontal scale bar = 50 μm, vertical scale bar = 500 ms. Scatter graphs of data for spark frequency and spark amplitude for young cells are shown in B and D with their corresponding log transforms in C and E. Bars on the scatter graphs indicate the means of these data. The log transforms are plotted with boxes (25th–75th percentiles) and whiskers (entire range of data points). The line in the box signifies the median. B–E: data from young hearts. NTG: 68 myocytes isolated from 5 hearts; TG: 60 myocytes isolated from 4 hearts. ***P < 0.001 and **P < 0.01, t-test. F–I: data from adult hearts. The graphs are laid out in an identical way. Scatter graphs of data for spark frequency and spark amplitude for adult cells are shown in F and G with their corresponding log transforms in H and I. NTG: 70 myocytes isolated from 5 hearts; TG: 49 myocytes isolated from 3 hearts. ***P < 0.001, t-test.
Spark mass [an index of the volume integral of the change in fluo 4 fluorescence and calculated using the formula derived by Hollingworth et al. (16)] was greater in young TG animals compared with NTG littermates (Fig. 9, A and B). Figure 9A shows that the range of spark mass was also much larger in young TG cells. Again, these data were skewed, so log transformations were used to produce approximately Gaussian distributions (Fig. 9B). ANOVA with Bonferroni correction indicated highly significant differences (P < 0.001) between the two groups. The dashed line in Fig. 9A indicates the largest spark mass in the young NTG population for ease of comparison. Notably, spark masses measured in cells isolated from older surviving TG mice were not different from NTG littermates (Fig. 9, C and D). Only one spark measured from the TG group had a mass greater than the largest mass measured in the young NTG group, as indicated by the dashed line (Fig. 9C).
Fig. 9.

Spark masses measured in cardiac myocytes isolated from young (A) and adult (C) transgenic (TG) and non-TG (NTG) hearts with a mixed (hybrid) C57/Bl6 × CBA/Ca background. E: similar measurements on young NTG and E99K TG mice bred on a pure C57Bl6 background. The corresponding log transformations in box and whisker form are shown in B, D, and F. The boxes represent the 25th–75th percentiles, and the whiskers represent the entire range of data points. The line in the box signifies the median. ***P < 0.001, ANOVA with Bonferroni. For A and B, NTG: 68 myocytes isolated from 5 hearts and TG: 60 myocytes isolated from 4 hearts. For C and D, NTG: 70 myocytes isolated from 5 hearts and TG: 49 myocytes isolated from 3 hearts. For E and F, NTG: 75 myocyes isolated from 6 hearts and TG: 67 myocytes isolated from 8 hearts.
Effect of the ACTC E99K Mutation Depends on Strain of Mouse
It is evident that some of the young ACTC E99K mice are phenotypically hypercontractile and have increased Ca2+ transients and spark mass, whereas TG survivors are not different from their NTG littermates. Because we were studying the ACTC E99K mutation on a hybrid background (C57/Bl6 × CBA/Ca), a simple explanation would be that mice segregate into two populations, some with more C57/Bl6 characteristics and some with more CBA/Ca2+ characteristics. To test this idea, we bred our TG mice on pure C57Bl6 backgrounds. ACTC E99K mice backcrossed with pure C57/Bl6 female mice for at least eight generations lost the SCD trait (see Fig. 10), and young ACTC E99K mice on this pure C57Bl6 background showed low spark masses comparable with NTG littermates (Fig. 9, E and F).
Fig. 10.

Left: Kaplan-Meier survival plots of each transgenic (TG) mouse line. The solid line shows survival of the mixed hybrid TG mice (i.e., those with the C57/Bl6 × CBA/Ca background), the dashed line shows the survival of mice bred on a pure C57Bl6 background, and the dotted line shows the survival of first-generation TG mice bred on a pure CBA/Ca2+ background, which showed the shortest survival duration. Right: distribution of age at death in hybrid TG mice (n = 35).
When we bred our hybrid ACTC E99K male mice with CBA/Ca2+ female mice, the first generation showed that the TG mice had a greater probability of SCD (Fig. 10), but, unfortunately, a second backcrossing yielded no TG pups. Kaplan-Meier survival plots of each of these mouse lines are shown in Fig. 10. TG mice bred on a pure C57Bl6 background lost the sharp SCD time window, whereas first-generation TG mice bred on a CBA/Ca2+ background showed the lowest survival duration. The hybrid TG mice (on the C57/Bl6 × CBA/Ca background) had an intermediate survival duration with the now well-described window of SCD around 40 days.
DISCUSSION
HCM is the most common monogenic cardiovascular disease and carries a significant risk of SCD due to arrhythmias. Adult ACTC E99K TG mice express the mutation at 50% of total sarcomeric actin and reproduce many aspects of HCM observed in humans with this mutation (34, 46), such as apical and septal hypertrophy, trabeculation, myocyte disarray, and interstitial fibrosis.
The most striking characteristic of the ACTC E99K TG mouse population is the high probability of SCD in a proportion of young mice; 40% of female mice and 25% of male mice were reported to die at 30–45 days old (47). To understand this, we investigated the changes that take place in young mice near the vulnerable window (25–40 days) and compared these with the mice that died suddenly and the adult survivors. The results show that young TG mice have a different Ca2+ regulatory phenotype from adult TG survivors and that this is strongly dependent on the genetic background of the mice studied. There are reports of developmental changes in intracellular Ca2+ regulation, transverse tubule organization, and excitation-contraction (EC) coupling, particularly in the rat (4, 10, 43, 49), just after birth. However, such changes are an unlikely explanation for the present findings because the integrity of the transverse tubules systems, synchronization of triggered Ca2+ release, and EC coupling mechanisms are fully developed by day 20 in mice (41).
In common with most sarcomeric mutations that cause HCM, the primary effect of the ACTC E99K mutation is enhanced myofilament Ca2+ sensitivity and an uncoupling of the relationship between Ca2+ sensitivity and troponin I phosphorylation by PKA. Thus, the mutation leads to a hypercontractile phenotype (47). The mechanisms by which this leads to hypertrophy and to fibrosis are not clear, but Baudenbacher et al. (3) demonstrated that increased Ca2+ sensitivity per se was sufficient to increase the probability of arrhythmia. The major secondary characteristics of HCM seen in patients are hypertrophy and interstitial fibrosis. We could not find evidence for cellular hypertrophy, particularly in young mice, so the former could not contribute to the SCD phenotype that we observed. On the other hand, young ACTC E99K mice that had died suddenly showed a high level of interstitial fibrosis similar to that found in other mouse models with SCD at this age (21). Although interstitial fibrosis is unlikely to be an initiating cause of the events leading up to SCD, since potentially fatal arrhythmias can be induced by acute Ca2+ sensitization in the absence of fibrosis (3), it is recognized that fibrosis is associated with persistent atrial fibrillation, increased anisotropy in ventricular tissue, and the separation of muscle bundles and so provides a substrate for multiple reentrant circuits, perhaps allowing spontaneous depolarizations to proliferate. This is supported by examination of the hearts from mice with the ACTC E99K transgene that have been bred on a pure C57Bl6 background, which have low levels of fibrosis (38). We noted that there were 15% fewer myocytes per square millimeter in SCD mice compared with wild-type mice, suggesting that cell death and replacement fibrosis are associated with the processes leading to SCD.
There may also be an interaction between fibrosis and Ca2+ sensitivity and fibrosis and cell-cell coupling, which may create a potentially arrhythmogenic substrate. For example, a study by Lu et al. (23) suggests that collagen can directly modulate Ca2+ dynamics and electrical activities of atrial cardiomyocytes. Electrical and/or mechanical coupling can be disrupted if the extracellular matrix and cytoskeletal links are altered by increased integrin signaling (52), and this may also contribute to the HCM phenotype.
Ca2+ Transients
When the young ACTC E99K mice were compared with NTG mice, the most striking observation was that the hypercontractility due to the intrinsically higher Ca2+ sensitivity in mutant mice was accompanied by an increased Ca2+ transient that would magnify the hypercontractile state of the TG mice.
It is interesting to note that the values for the Ca2+ transient magnitude were distributed over a wide range of values and appeared to consist of two populations, one with a range similar to the wild type plus 20–30% with values considerably higher that may correspond to the mice destined for SCD.
The observations that SR load is no different and diastolic Ca2+ concentration less in TG hearts, yet the Ca2+ transient amplitude is greater, implies there is an increase in trigger Ca2+ or that EC coupling is more sensitive because of a lowering of the threshold for Ca2+ activation either on the cytoplasmic (33) or luminal (13) sides of the ryanodine receptor (RyR). In HCMs, it is not known if RyR Ca2+ activation thresholds change, but such alterations in RyR properties may arise from abnormal expression of associated proteins, e.g., calsequestrin, junction, and triadin (14), or changes in phosphorylation status (51). Increased RyR-mediated Ca2+ leak from the SR is observed in heart failure in humans (58) and various animal models (22, 24, 44), although the underlying pathogenesis remains highly controversial (17).
In contrast, cardiac myocytes from older 8- to 12-wk-old TG animals showed no differences in maximum sarcomere shortening compared with their NTG counterparts, but the Ca2+ transients were smaller. The attainment of similar shortening amplitude with less Ca2+ delivery derives from the hypercontractile nature of ACTC E99K myocytes. This is a similar finding to most studies using TG mice with HCM mutations, which reported results from adult mice [e.g., 4–6 mo old (15, 19) or more elderly (48)]. The evidence for abnormal Ca2+ transients associated with HCM mutations is equivocal and appears to be mutation specific. Cardiac myocytes isolated from TG mice with a troponin T (TnT) mutation that is associated with a high risk of SCD in patients (ΔE160) had a decreased resting Ca2+ concentration and larger and faster Ca2+ transients (19), but other TnT mutations (TnT R92L, which is not linked with SCD, and TnT I79N, which is associated with a high incidence of SCD) did not show these changes. The decreased Ca2+ transients in our older ACTC E99K TG mouse hearts appear to provide compensation to normalize contraction and hence preserve energy reserves in the face of the hypercontractile myofilaments and compromised ATP supply present at all ages (15, 19).
In previous work, we reported that SCD in young ACTC E99K mice is more frequent in female mice than in male mice (46). Many studies have recognized that there can be sex differences both in the onset and progression of cardiovascular diseases (31, 37, 39) and in cardiomyocyte intracellular Ca2+ regulation (11, 54). Although we found very few sex differences in Ca2+ regulation, we did find that the decay of the electrically stimulated Ca2+ transient in female TG myocytes was slower than the transient decay in male TG myocytes, whereas there were no differences in decay time in NTG cells. Differences in decay between males and females have also been noted in rats by Leblanc et al. (20) and Curl et al. (6). We could not determine any sex differences in TG mice in spark frequency or mass, yet others have suggested possible sex-dependent alterations in RyR regulation (11). Therefore, we can only state at this stage that sex differences in TG mice are not large, but they may contribute to the complex interactions occurring to produce the final phenotype.
Ca2+ Waves and Arrhythmia Provocation
The consequence of the sensitized release process found in young TG mice is an alteration to the normal gating behavior of RyRs that cause aberrant activation of the channels as evidenced by the increased frequency and larger amplitudes and masses of Ca2+ sparks recorded in myocytes isolated from young TG hearts. As with the Ca2+ transient amplitude, there is a wide dispersal of values and evidence of two populations, whereas Ca2+ sparks recorded in myocytes isolated from NTG and adult TG hearts do not appear to have different characteristics. With maintained SR Ca2+ content (53), the larger spontaneous Ca2+ release events in young TG hearts translate into the increased appearance of Ca2+ waves (5, 9, 55).
It is well described that the appearance of waves will activate electrogenic Na+/Ca2+ exchange (12, 32), producing an inward current that can depolarize the cell. If a large fraction of the total SR Ca2+ content is released in producing the wave, the resultant delayed afterdepolarization can be large enough to initiate an ectopic triggered action potential (32) that could generate arrhythmias and provoke spatially discordant alternans (56). The factors affecting source-sink mismatches that tend to cause afterdepolarizations to be suppressed are unknown, but such mismatches may be overcome by microfibrosis and structural remodeling (57), allowing conduction spread. We hypothesize that, while the disturbed single myocyte Ca2+ handling may initiate potentially arrhythmic depolarizations, changes in the passive properties of the heart tissue could allow such triggering events to develop and propagate (7), slow conduction, and promote reentry (36).
Age and Mouse Strain Determine SCD
The occurrence of SCD in a time window centered on 36–40 days in ACTC E99K and troponin I3 R193H TG mouse studied by Li et al. (21) indicates that important changes occur at this stage in development. This age corresponds to the end of a growth spurt whereby the heart and body weight increase 3.8- and 3.5-fold, respectively, between P10 and P35, reaching 80% of final body weight but only 70% of final heart weight and is immediately followed by puberty (35). Although evidence suggests that transverse tubule systems and EC coupling mechanisms are fully developed by P20 in mice (41), there may not yet be appropriate adaptation to the intrinsically high myofibrillar Ca2+ sensitivity, leaving the TG mouse heart particularly vulnerable at this age. Studies grouped around this time should be more informative than those involving older animals in which complex secondary changes may have occurred that could mask initial triggers for HCM.
It is evident that some of the young mice have a hypercontractile phenotype associated with increased Ca2+ transients and spark volumes, whereas TG survivors are not very different from their NTG littermates. Does this indicate that compensatory changes develop in adult mice, or are there two populations of mice in our study? The following observations would suggest that the latter possibility seems likely. The Kaplan-Meier plots of TG mouse survival show that not all animals die suddenly, and, moreover, there is a striking dependence on the genetic background of the mice studied. There is very little SCD on a pure C57Bl6 background and more SCD on a CBA/Ca2+-enriched background compared with hybrid C56Bl6 × CBA/Ca mice. In every generation of hybrid mice, two populations of mice are consistently observed, those that die suddenly and those that do not, despite the same level of expression of the mutant actin protein in both populations (see Fig. 1 and Ref. 46). The survivors have a similar lifespan to NTG animals. The SCD phenotype correlates with an obvious fraction of cells isolated from the hearts of young TG animals that have larger Ca2+ transients, larger spark masses, and more Ca2+ waves, suggesting segregation of phenotypes when TG mice are bred on a hybrid C57Bl6 × CBA/Ca genetic background. These are properties not shared by the survivors or NTG animals, suggesting that the animals with high spark mass are destined for SCD.
This is supported by results with the ACTC E99K transgene bred on a pure C57Bl6 background, where both young and old TG mice showed the same spark mass as NTG littermates, corresponding to the absence of SCD; thus, it is the genetically determined secondary response to the initial mutation-induced abnormality that is crucial for provoking SCD. The C57Bl6 haplotype is known to be resistant to arrhythmia provocation and angiotensin-induced myopathy, whereas CBA/Ca2+ is genetically closer to mouse strains that show much more susceptibility to arrhythmias, e.g., BalbC (26, 38).
The underlying mechanism is not known. This may take place because of subtleties in dyad architecture, energy, or phosphorylation status. Alternatively, the primed proarrhythmic substrate orchestrated by Ca2+ dysregulation in TG mice may be activated by structural changes, such as fibrosis, that could occur in parallel and be genetically determined, for example, by T helper cell genotypes 1 and 2 and their role in cardiac remodeling (38).
It is particularly interesting that the influence of genetic background on the manifestation of the SCD trait due to the ACTC E99K mutation parallels the well-known variability of penetrance in families with the same HCM-linked mutation. Indeed, a study of 76 individuals from 11 families with the ACTC E99K mutation revealed SCD in 8 cases, but 5 of these were in the same family (34, 46). The ability to reproduce variable penetrance of SCD in a mouse model could enable us to determine the background factors that determine pathogenicity.
GRANTS
This work was supported by British Heart Foundation Program Grant PG11/020/29266 and a British Heart Foundation PhD studentship (Grant FS10/021/20244).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.R., T.O., S.L., S.C., S.P., H.-Y.Y., W.S., R.W., A.A.-L., and K.G. performed experiments; C.R., T.O., S.L., S.C., S.P., H.-Y.Y., R.W., A.A.-L., S.M., and K.T.M. analyzed data; C.R., T.O., S.L., S.C., S.P., H.-Y.Y., W.S., R.W., A.A.-L., K.G., S.M., and K.T.M. interpreted results of experiments; C.R., T.O., H.-Y.Y., and K.T.M. prepared figures; C.R., T.O., S.L., S.C., H.-Y.Y., W.S., R.W., A.A.-L., K.G., S.M., and K.T.M. approved final version of manuscript; K.G., S.M., and K.T.M. conceived and designed research; K.G., S.M., and K.T.M. edited and revised manuscript; S.M. and K.T.M. drafted manuscript.
ACKNOWLEDGMENTS
We acknowledge the technical assistance of O’Neal Copeland for producing the Western blot data shown in Fig. 1.
REFERENCES
- 1.Alfares AA, Kelly MA, McDermott G, Funke BH, Lebo MS, Baxter SB, Shen J, McLaughlin HM, Clark EH, Babb LJ, Cox SW, DePalma SR, Ho CY, Seidman JG, Seidman CE, Rehm HL. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med 17: 880–888, 2015. doi: 10.1038/gim.2014.205. [DOI] [PubMed] [Google Scholar]
- 2.Bassani JWM, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J Physiol 476: 279–293, 1994. doi: 10.1113/jphysiol.1994.sp020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, Potter JD, Knollmann BC. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest 118: 3893–3903, 2008. doi: 10.1172/JCI36642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Dordrecht, The Netherlands: Kluwer Academic, 2001. doi: 10.1007/978-94-010-0658-3. [DOI] [Google Scholar]
- 5.Cheng H, Lederer MR, Lederer WJ, Cannell MB. Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am J Physiol Cell Physiol 270: C148–C159, 1996. [DOI] [PubMed] [Google Scholar]
- 6.Curl CL, Wendt IR, Kotsanas G. Effects of gender on intracellular. Pflugers Arch 441: 709–716, 2001. doi: 10.1007/s004240000473. [DOI] [PubMed] [Google Scholar]
- 7.de Jong S, van Veen TAB, van Rijen HVM, de Bakker JMT. Fibrosis and cardiac arrhythmias. J Cardiovasc Pharmacol 57: 630–638, 2011. doi: 10.1097/FJC.0b013e318207a35f. [DOI] [PubMed] [Google Scholar]
- 8.Díaz ME, Trafford AW, O’Neill SC, Eisner DA. Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. J Physiol 501: 3–16, 1997. doi: 10.1111/j.1469-7793.1997.003bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eisner DA, Valdeolmillos M. A study of intracellular calcium oscillations in sheep cardiac Purkinje fibres measured at the single cell level. J Physiol 372: 539–556, 1986. doi: 10.1113/jphysiol.1986.sp016024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Escobar AL, Ribeiro-Costa R, Villalba-Galea C, Zoghbi ME, Pérez CG, Mejía-Alvarez R. Developmental changes of intracellular Ca2+ transients in beating rat hearts. Am J Physiol Heart Circ Physiol 286: H971–H978, 2004. doi: 10.1152/ajpheart.00308.2003. [DOI] [PubMed] [Google Scholar]
- 11.Farrell SR, Ross JL, Howlett SE. Sex differences in mechanisms of cardiac excitation-contraction coupling in rat ventricular myocytes. Am J Physiol Heart Circ Physiol 299: H36–H45, 2010. doi: 10.1152/ajpheart.00299.2010. [DOI] [PubMed] [Google Scholar]
- 12.Fedida D, Noble D, Rankin AC, Spindler AJ. The arrhythmogenic transient inward current iTI and related contraction in isolated guinea-pig ventricular myocytes. J Physiol 392: 523–542, 1987. doi: 10.1113/jphysiol.1987.sp016795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Györke I, Györke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J 75: 2801–2810, 1998. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Györke S, Terentyev D. Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc Res 77: 245–255, 2008. doi: 10.1093/cvr/cvm038. [DOI] [PubMed] [Google Scholar]
- 15.Haim TE, Dowell C, Diamanti T, Scheuer J, Tardiff JC. Independent FHC-related cardiac troponin T mutations exhibit specific alterations in myocellular contractility and calcium kinetics. J Mol Cell Cardiol 42: 1098–1110, 2007. doi: 10.1016/j.yjmcc.2007.03.906. [DOI] [PubMed] [Google Scholar]
- 16.Hollingworth S, Peet J, Chandler WK, Baylor SM. Calcium sparks in intact skeletal muscle fibers of the frog. J Gen Physiol 118: 653–678, 2001. doi: 10.1085/jgp.118.6.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Houser SR. Controversies in cardiovascular research: role of protein kinase A mediated hyperphosphorylation of the ryanodine receptor at serine 2808 in heart failure and arrhythmias. Circ Res 114: 1320–1327, 2014. doi: 10.1161/CIRCRESAHA.114.300569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huke S, Knollmann BC. Increased myofilament Ca2+-sensitivity and arrhythmia susceptibility. J Mol Cell Cardiol 48: 824–833, 2010. doi: 10.1016/j.yjmcc.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knollmann BC, Kirchhof P, Sirenko SG, Degen H, Greene AE, Schober T, Mackow JC, Fabritz L, Potter JD, Morad M. Familial hypertrophic cardiomyopathy-linked mutant troponin T causes stress-induced ventricular tachycardia and Ca2+-dependent action potential remodeling. Circ Res 92: 428–436, 2003. doi: 10.1161/01.RES.0000059562.91384.1A. [DOI] [PubMed] [Google Scholar]
- 20.Leblanc N, Chartier D, Gosselin H, Rouleau JL. Age and gender differences in excitation-contraction coupling of the rat ventricle. J Physiol 511: 533–548, 1998. doi: 10.1111/j.1469-7793.1998.533bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Zhang L, Jean-Charles PY, Nan C, Chen G, Tian J, Jin JP, Gelb IJ, Huang X. Dose-dependent diastolic dysfunction and early death in a mouse model with cardiac troponin mutations. J Mol Cell Cardiol 62: 227–236, 2013. doi: 10.1016/j.yjmcc.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Louch WE, Hake J, Mørk HK, Hougen K, Skrbic B, Ursu D, Tønnessen T, Sjaastad I, Sejersted OM. Slow Ca2+ sparks de-synchronize Ca2+ release in failing cardiomyocytes: evidence for altered configuration of Ca2+ release units? J Mol Cell Cardiol 58: 41–52, 2013. doi: 10.1016/j.yjmcc.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 23.Lu YY, Chen YC, Kao YH, Wu TJ, Chen SA, Chen YJ. Extracellular matrix of collagen modulates intracellular calcium handling and electrophysiological characteristics of HL-1 cardiomyocytes with activation of angiotensin II type 1 receptor. J Card Fail 17: 82–90, 2011. doi: 10.1016/j.cardfail.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 24.Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, Korchev YE, Harding SE, Gorelik J. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci USA 106: 6854–6859, 2009. doi: 10.1073/pnas.0809777106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacLeod KT, Harding SE. Effects of phorbol ester on contraction, intracellular pH and intracellular Ca2+ in isolated mammalian ventricular myocytes. J Physiol 444: 481–498, 1991. doi: 10.1113/jphysiol.1991.sp018889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maguire CT, Wakimoto H, Patel VV, Hammer PE, Gauvreau K, Berul CI. Implications of ventricular arrhythmia vulnerability during murine electrophysiology studies. Physiol Genomics 15: 84–91, 2003. doi: 10.1152/physiolgenomics.00034.2003. [DOI] [PubMed] [Google Scholar]
- 27.Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA 287: 1308–1320, 2002. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 28.Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, Shah PM, Spencer WH III, Spirito P, Ten Cate FJ, Wigle ED, Vogel RA, Abrams J, Bates ER, Brodie BR, Danias PG, Gregoratos G, Hlatky MA, Hochman JS, Kaul S, Lichtenberg RC, Lindner JR, O’Rourke RA, Pohost GM, Schofield RS, Tracy CM, Winters WL, Klein WW, Priori SG, Alonso-Garcia A, Blomström-Lundqvist C, De Backer G, Deckers J, Flather M, Hradec J, Oto A, Parkhomenko A, Silber S, Torbicki A; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents; European Society of Cardiology Committee for Practice Guidelines . American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. Eur Heart J 24: 1965–1991, 2003. doi: 10.1016/S0195-668X(03)00479-2. [DOI] [PubMed] [Google Scholar]
- 29.Marston SB. Why is there a limit to the changes in myofilament Ca2+-sensitivity associated with myopathy causing mutations? Front Physiol 7: 415, 2016. doi: 10.3389/fphys.2016.00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marston SB. How do mutations in contractile proteins cause the primary familial cardiomyopathies? J Cardiovasc Transl Res 4: 245–255, 2011. doi: 10.1007/s12265-011-9266-2. [DOI] [PubMed] [Google Scholar]
- 31.Martínez-Sellés M, Doughty RN, Poppe K, Whalley GA, Earle N, Tribouilloy C, McMurray JJV, Swedberg K, Køber L, Berry C, Squire I; Meta-Analysis Global Group In Chronic Heart Failure (MAGGIC) . Gender and survival in patients with heart failure: interactions with diabetes and aetiology. Results from the MAGGIC individual patient meta-analysis. Eur J Heart Fail 14: 473–479, 2012. doi: 10.1093/eurjhf/hfs026. [DOI] [PubMed] [Google Scholar]
- 32.Mechmann S, Pott L. Identification of Na-Ca exchange current in single cardiac myocytes. Nature 319: 597–599, 1986. doi: 10.1038/319597a0. [DOI] [PubMed] [Google Scholar]
- 33.Meissner G, Henderson JS. Rapid calcium release from cardiac sarcoplasmic reticulum vesicles is dependent on Ca2+ and is modulated by Mg2+, adenine nucleotide, and calmodulin. J Biol Chem 262: 3065–3073, 1987. [PubMed] [Google Scholar]
- 34.Monserrat L, Hermida-Prieto M, Fernandez X, Rodríguez I, Dumont C, Cazón L, Cuesta MG, Gonzalez-Juanatey C, Peteiro J, Álvarez N, Penas-Lado M, Castro-Beiras A. Mutation in the alpha-cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular non-compaction, and septal defects. Eur Heart J 28: 1953–1961, 2007. doi: 10.1093/eurheartj/ehm239. [DOI] [PubMed] [Google Scholar]
- 35.Naqvi N, Li M, Calvert JW, Tejada T, Lambert JP, Wu J, Kesteven SH, Holman SR, Matsuda T, Lovelock JD, Howard WW, Iismaa SE, Chan AY, Crawford BH, Wagner MB, Martin DI, Lefer DJ, Graham RM, Husain A. A proliferative burst during preadolescence establishes the final cardiomyocyte number. Cell 157: 795–807, 2014. doi: 10.1016/j.cell.2014.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen TP, Qu Z, Weiss JN. Cardiac fibrosis and arrhythmogenesis: the road to repair is paved with perils. J Mol Cell Cardiol 70: 83–91, 2014. doi: 10.1016/j.yjmcc.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nieminen MS, Harjola VP, Hochadel M, Drexler H, Komajda M, Brutsaert D, Dickstein K, Ponikowski P, Tavazzi L, Follath F, Lopez-Sendon JL. Gender related differences in patients presenting with acute heart failure. Results from EuroHeart Failure Survey II. Eur J Heart Fail 10: 140–148, 2008. doi: 10.1016/j.ejheart.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 38.Peng H, Yang XP, Carretero OA, Nakagawa P, D’Ambrosio M, Leung P, Xu J, Peterson EL, González GE, Harding P, Rhaleb NE. Angiotensin II-induced dilated cardiomyopathy in Balb/c but not C57BL/6J mice. Exp Physiol 96: 756–764, 2011. doi: 10.1113/expphysiol.2011.057612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petrie MC, Dawson NF, Murdoch DR, Davie AP, McMurray JJV. Failure of women’s hearts. Circulation 99: 2334–2341, 1999. doi: 10.1161/01.CIR.99.17.2334. [DOI] [PubMed] [Google Scholar]
- 40.Picht E, Zima AV, Blatter LA, Bers DM. SparkMaster: automated calcium spark analysis with ImageJ. Am J Physiol Cell Physiol 293: C1073–C1081, 2007. doi: 10.1152/ajpcell.00586.2006. [DOI] [PubMed] [Google Scholar]
- 41.Reynolds JO, Chiang DY, Wang W, Beavers DL, Dixit SS, Skapura DG, Landstrom AP, Song LS, Ackerman MJ, Wehrens XH. Junctophilin-2 is necessary for T-tubule maturation during mouse heart development. Cardiovasc Res 100: 44–53, 2013. doi: 10.1093/cvr/cvt133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Satoh H, Blatter LA, Bers DM. Effects of [Ca2+]i, SR Ca2+ load, and rest on Ca2+ spark frequency in ventricular myocytes. Am J Physiol Heart Circ Physiol 272: H657–H668, 1997. [DOI] [PubMed] [Google Scholar]
- 43.Seki S, Nagashima M, Yamada Y, Tsutsuura M, Kobayashi T, Namiki A, Tohse N. Fetal and postnatal development of Ca2+ transients and Ca2+ sparks in rat cardiomyocytes. Cardiovasc Res 58: 535–548, 2003. doi: 10.1016/S0008-6363(03)00255-4. [DOI] [PubMed] [Google Scholar]
- 44.Shannon TR, Pogwizd SM, Bers DM. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res 93: 592–594, 2003. doi: 10.1161/01.RES.0000093399.11734.B3. [DOI] [PubMed] [Google Scholar]
- 45.Sikkel MB, Collins TP, Rowlands C, Shah M, O’Gara P, Williams AJ, Harding SE, Lyon AR, MacLeod KT. Flecainide reduces Ca2+ spark and wave frequency via inhibition of the sarcolemmal sodium current. Cardiovasc Res 98: 286–296, 2013. doi: 10.1093/cvr/cvt012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song W, Dyer E, Stuckey DJ, Copeland O, Leung MC, Bayliss C, Messer A, Wilkinson R, Tremoleda JL, Schneider MD, Harding SE, Redwood CS, Clarke K, Nowak K, Monserrat L, Wells D, Marston SB. Molecular mechanism of the E99K mutation in cardiac actin (ACTC gene) that causes apical hypertrophy in man and mouse. J Biol Chem 286: 27582–27593, 2011. doi: 10.1074/jbc.M111.252320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song W, Vikhorev PG, Kashyap MN, Rowlands C, Ferenczi MA, Woledge RC, MacLeod K, Marston S, Curtin NA. Mechanical and energetic properties of papillary muscle from ACTC E99K transgenic mouse models of hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol 304: H1513–H1524, 2013. doi: 10.1152/ajpheart.00951.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spindler M, Saupe KW, Christe ME, Sweeney HL, Seidman CE, Seidman JG, Ingwall JS. Diastolic dysfunction and altered energetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. J Clin Invest 101: 1775–1783, 1998. doi: 10.1172/JCI1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tanaka H, Shigenobu K. Effect of ryanodine on neonatal and adult rat heart: developmental increase in sarcoplasmic reticulum function. J Mol Cell Cardiol 21: 1305–1313, 1989. doi: 10.1016/0022-2828(89)90676-7. [DOI] [PubMed] [Google Scholar]
- 50.Terracciano CMN, Souza AI, Philipson KD, MacLeod KT. Na+-Ca2+ exchange and sarcoplasmic reticular Ca2+ regulation in ventricular myocytes from transgenic mice overexpressing the Na+-Ca2+ exchanger. J Physiol 512: 651–667, 1998. doi: 10.1111/j.1469-7793.1998.651bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ullrich ND, Valdivia HH, Niggli E. PKA phosphorylation of cardiac ryanodine receptor modulates SR luminal Ca2+ sensitivity. J Mol Cell Cardiol 53: 33–42, 2012. doi: 10.1016/j.yjmcc.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Valencik ML, Zhang D, Punske B, Hu P, McDonald JA, Litwin SE. Integrin activation in the heart: a link between electrical and contractile dysfunction? Circ Res 99: 1403–1410, 2006. doi: 10.1161/01.RES.0000252291.88540.ac. [DOI] [PubMed] [Google Scholar]
- 53.Venetucci LA, Trafford AW, Eisner DA. Increasing ryanodine receptor open probability alone does not produce arrhythmogenic calcium waves: threshold sarcoplasmic reticulum calcium content is required. Circ Res 100: 105–111, 2007. doi: 10.1161/01.RES.0000252828.17939.00. [DOI] [PubMed] [Google Scholar]
- 54.Wasserstrom JA, Kapur S, Jones S, Faruque T, Sharma R, Kelly JE, Pappas A, Ho W, Kadish AH, Aistrup GL. Characteristics of intracellular Ca2+ cycling in intact rat heart: a comparison of sex differences. Am J Physiol Heart Circ Physiol 295: H1895–H1904, 2008. doi: 10.1152/ajpheart.00469.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wier WG, Cannell MB, Berlin JR, Marban E, Lederer WJ. Cellular and subcellular heterogeneity of [Ca2+]i in single heart cells revealed by fura-2. Science 235: 325–328, 1987. doi: 10.1126/science.3798114. [DOI] [PubMed] [Google Scholar]
- 56.Xie LH, Weiss JN. Arrhythmogenic consequences of intracellular calcium waves. Am J Physiol Heart Circ Physiol 297: H997–H1002, 2009. doi: 10.1152/ajpheart.00390.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xie Y, Sato D, Garfinkel A, Qu Z, Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J 99: 1408–1415, 2010. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang C, Chen B, Guo A, Zhu Y, Miller JD, Gao S, Yuan C, Kutschke W, Zimmerman K, Weiss RM, Wehrens XH, Hong J, Johnson FL, Santana LF, Anderson ME, Song LS. Microtubule-mediated defects in junctophilin-2 trafficking contribute to myocyte transverse-tubule remodeling and Ca2+ handling dysfunction in heart failure. Circulation 129: 1742–1750, 2014. doi: 10.1161/CIRCULATIONAHA.113.008452. [DOI] [PMC free article] [PubMed] [Google Scholar]