

Right ventricular (RV) failure is a serious disease with a poor prognosis and no effective treatments. In the mouse bleomycin model of RV failure, we tested the efficacy of a treatment using the α1A-adrenergic receptor subtype agonist A61603. Chronic A61603 treatment improved RV contraction and reduced multiple indexes of RV injury, suggesting that the α1A-subtype is a therapeutic target to treat RV failure.
Keywords: α1-adrenergic therapy, right ventricle, reactive oxygen species, oxidative stress
Abstract
Failure of the right ventricle (RV) is a serious disease with a poor prognosis and limited treatment options. Signaling by α1-adrenergic receptors (α1-ARs), in particular the α1A-subtype, mediate cardioprotective effects in multiple heart failure models. Recent studies have shown that chronic treatment with the α1A-subtype agonist A61603 improves function and survival in a model of left ventricular failure. The goal of the present study was to determine if chronic A61603 treatment is beneficial in a RV failure model. We used tracheal instillation of the fibrogenic antibiotic bleomycin in mice to induce pulmonary fibrosis, pulmonary hypertension, and RV failure within 2 wk. Some mice were chronically treated with a low dose of A61603 (10 ng·kg−1·day−1). In the bleomycin model of RV failure, chronic A61603 treatment was associated with improved RV fractional shortening and greater in vitro force development by RV muscle preparations. Cell injury markers were reduced with A61603 treatment (serum cardiac troponin I, RV fibrosis, and expression of matrix metalloproteinase-2). RV oxidative stress was reduced (using the probes dihydroethidium and 4-hydroxynonenal). Consistent with lowered RV oxidative stress, A61603 was associated with an increased level of the cellular antioxidant superoxide dismutase 1 and a lower level of the prooxidant NAD(P)H oxidase isoform NOX4. In summary, in the bleomycin model of RV failure, chronic A61603 treatment reduced RV oxidative stress, RV myocyte necrosis, and RV fibrosis and increased both RV function and in vitro force development. These findings suggest that in the context of pulmonary fibrosis, the α1A-subtype is a potential therapeutic target to treat the failing RV.
NEW & NOTEWORTHY Right ventricular (RV) failure is a serious disease with a poor prognosis and no effective treatments. In the mouse bleomycin model of RV failure, we tested the efficacy of a treatment using the α1A-adrenergic receptor subtype agonist A61603. Chronic A61603 treatment improved RV contraction and reduced multiple indexes of RV injury, suggesting that the α1A-subtype is a therapeutic target to treat RV failure.
INTRODUCTION
Failure of the right ventricle (RV) is a serious clinical problem with a poor prognosis (11, 25). RV failure, defined as the inability of the RV to provide adequate blood flow through the pulmonary circulation at a normal preload (11), is the leading determinant of symptoms and survival in patients with pulmonary arterial hypertension (10, 32).
Unfortunately, treatment options for patients with RV failure are limited (24). Standard therapies that have been developed to treat patients with failure of the left ventricle (LV) are ineffective for improving function or survival of patients with RV failure (25). There is a need for new therapies to treat RV failure.
Clinical and experimental evidence suggests that in heart failure involving the LV, α1-adrenergic receptors (α1-ARs) mediate multiple cardioprotective effects, in vivo and in vitro (3, 14, 23). Consistent with a beneficial effect mediated by α1-ARs, recent studies have suggested that chronic stimulation of a single α1-AR subtype (α1A) is beneficial in a doxorubicin toxicity model of LV failure (4, 20). However, it is not known if this novel α1A-subtype therapy is effective for RV failure models.
In a model of RV failure due to bleomycin-induced pulmonary fibrosis, we reported that α1-AR inotropic responses were increased (33) and that the α1A-subtype was solely responsible for the increased α1-AR inotropy (8). We hypothesized that increased α1A-subtype signaling would be beneficial in RV failure and a potential therapeutic strategy.
The goal of this project was to evaluate the α1A-subtype as a therapeutic target for treating RV failure. We used the bleomycin mouse model of RV failure. The fibrogenic antibiotic bleomycin instilled into the trachea of anesthetized mice causes pulmonary fibrosis leading to pulmonary arterial hypertension, RV hypertrophy, and RV failure within 2 wk (12). Histologically, this model resembles human idiopathic pulmonary fibrosis (IPF) (12). For patients with IPF, the presence of pulmonary hypertension has a significant adverse impact on survival (21). Using the bleomycin model, we chronically treated animals with a low dose of the α1A-subtype agonist A61603. Chronic A61603 treatment in the bleomycin RV failure model had a beneficial effect on RV contraction and on multiple markers of RV injury. This study suggests that in the context of pulmonary fibrosis, the α1A-subtype is a novel therapeutic target to treat RV failure by improving RV function and decreasing RV injury.
METHODS
This institution is accredited by the American Association for the Accreditation of Laboratory Animal Care (Institutional PHS Assurance Number: A3476-01). The study was approved by the Animal Care and Use Subcommittee of the San Francisco Veterans Affairs Medical Center and conformed with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (Revised 2011).
RV injury model and A61603 therapy.
Adult male C57BL/6J mice (Jackson Laboratory) were used (age: 10–14 wk, body weight: ≈28 g at the beginning of the experiment). We used a RV-specific heart failure model. The fibrogenic antibiotic bleomycin (Sigma-Aldrich, St. Louis, MO) was introduced into the lungs by endotracheal instillation (0.075 units bleomycin in 100 μl saline), which leads to pulmonary fibrosis, pulmonary hypertension, and RV failure in ~2 wk (12). Animals were harvested after 10–14 days. Control animals were instilled with saline. Concurrently, mice were chronically treated for 2 wk with A61603 (Tocris Bioscience, Bristol, UK), a potent and highly specific agonist for the α1A-subtype that does not stimulate the other two α1-AR subtypes (α1B or α1D). A subhypertensive dose of A61603 (10 ng·kg−1·day−1) (20) or saline was given by continuous subcutaneous infusion with an osmotic mini-pump (model 1002, Alzet, Durect) that was implanted between the scapulae under isoflurane anesthesia. This is a disease prevention study design.
In vivo hemodynamics.
Echocardiography was performed on conscious, gently restrained mice using an Acuson S2000 (Siemens) with a 5- to 14-MHz multidimensional matrix transducer. RV dimensions were measured using two-dimensional guided M-mode, acquired from five consecutive cardiac cycles in the long-axis view. Measurements were taken at midway along the heart. RV fractional shortening was calculated from the diastolic minus systolic chamber dimension expressed as a percentage of the diastolic dimension. An estimate of the RV cardiac output was computed from the difference between the diastolic versus systolic dimension multiplied by the heart rate. The echocardiographer was blinded to the treatments of all mice.
Serum cardiac troponin I.
Mice were deeply anesthetized with pentobarbital sodium (100 mg/kg ip) and heparinized (100 U). A midline thoracotomy was performed, and blood was collected by apical LV puncture with a 20-gauge needle. Whole blood, 1–2 drops, was assayed immediately for cardiac troponin I (cTnI) concentration (in ng/ml) using VetScan i-STAT cTnI cartridges (Abaxis).
Fibrosis.
Freshly isolated RV samples were fixed in phosphate-buffered 4% paraformaldehyde (Fisher Scientific, Fair Lawn, NJ) at 4°C for at least 24 h. Paraffin-embedded 5-µm-thick sections were stained with hematoxylin and eosin and picrosirius red as previously described (7). Images of stained cross sections (3 images/animal) were analyzed using ImageJ (National Institutes of Health, Bethesda, MD). Collagen content in picrosirius red-stained sections was quantified using ImageJ thresholding analysis for collagen-positive areas.
In vitro contraction of demembranated RV cardiac muscle preparations.
After removal, hearts were immersed in 4°C arrest solution containing (in mM) 120 NaCl, 30 KCl, and 0.1 CaCl2 and then perfused through the aorta with modified Krebs-Henseleit solution containing (in mM) 137 NaCl, 10 KCl, 1.2 MgSO4, 1.2 NaH2PO4, 10 glucose, 20 NaHCO3, 0.2 CaCl2, and 30 2,3-butanedione monoxime. The perfusate was oxygenated with 95% O2-5% CO2 to give a pH of 7.4 at 22°C. The RV free wall was removed, pinned onto a silicone substrate, and immersed for 24 h at 4°C in a demembranating solution that consisted of relaxing solution (see below) containing 1% Triton X-100 (Sigma-Aldrich) (34). Free-running unbranched muscle preparations were dissected from the tricuspid valve region on the endocardial RV surface. Demembranated muscle preparations were attached with aluminum T-clips to a force transducer and anchor (model 1400A, Permeabilized Fiber Test System, Aurora Scientific) and bathed in relaxing solution containing (in mM) 20 EGTA, 7.05 MgCl2, 6.31 Na2ATP, 10 creatine phosphate, and 80 N,N-bis(2-hydroxyethyl)2-aminoethane sulfonic acid with pH adjusted to 7.1 with KOH and ionic strength adjusted to 200 mM with KCl (29). Muscle width and thickness were measured using a dissection scope (×40), and muscle cross-sectional area was estimated assuming an elliptical cross section. Muscle sarcomeres were observed using a ×40 objective, and sarcomere lengths were assessed using a video-based system (model 900B, Aurora Scientific). Muscle length was adjusted to set the muscle sarcomere length to 2.1 µm. Demembranated muscle preparations were briefly transitioned to preactivating solution in which Ca2+ buffering was reduced by replacing 19.5 mM EGTA with hexamethylenediamine-N,N,N´,N´-tetraacetate (Fluka). We used activating solution containing 20 mM Ca2+ EGTA, and activated muscles by exposure to solutions of various Ca2+ concentrations ([Ca2+]) (achieved by mixing relaxing solution and activating solution to obtain intermediate Ca2+ levels) (34). All solutions contained 1% (vol/vol) protease inhibitor cocktail P-8340 and 10 IU/ml creatine kinase (Sigma-Aldrich).
Measurements of muscle force were normalized to muscle cross-sectional area. The relationship between force development (F) versus [Ca2+] was fit to the following Hill equation: , where Fmax is the maximum Ca2+-activated force, EC50 is the [Ca2+] at which F is 50% of Fmax, and nH is the Hill coefficient reflecting the slope of the Ca2+-force relationship at EC50.
Quantitative RT-PCR.
Total RNA from mouse heart samples was isolated using TRIzol (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Isolated RNA was further purified using a DNA-free RNA kit (Zymo Research, Irvine, CA), and 1 μg RNA was used to synthesize cDNA using a Transcriptor First Strand cDNA Synthesis Kit (Roche Applied Bioscience, Indianapolis, IN). Quantitative RT-PCR was performed to quantify the expression of full-length and NH2-terminus truncated (NTT) matrix metalloproteinase (MMP)-2 isoforms using a LightCycler 480 SYBR Green I Master Kit (Roche Applied Bioscience). Each sample was plated in triplicate in a 384-well PCR plate (ThermoFisher, Waltham, MA). Primer sequences are shown in Table 1. Amplification reactions were performed with 40 cycles of 95°C for 15 s, 58°C for 45 s, and 72°C for 1 min and normalized to β2-microglobulin. Melt curves were used to verify the absence of primer dimers and other nonspecific products in the amplification reactions. Fold changes in mRNA expression were calculated using ∆∆CT (where CT is the threshold cycle).
Table 1.
Quantitative PCR primer sequences
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Full-length MMP-2 | 5′-GACCTCTGCGGGTTCTCTGC-3′ | 5′-TTGCAACTCTCCTTGGGGCAGC-3′ |
| NTT-MMP-2 | 5′-GGCTCTGGAGCATGACCGCTT-3′ | 5′-TTGCAACTCTCCTTGGGGCAGC-3′ |
| β2-Microglobulin | 5′-TAAGCATGCCAGTATGGCCG-3′ | 5′-AGAAGTAGCCACAGGGTTGG-3′ |
MMP-2, matrix metalloproteinases-2; NTT, NH2-terminus truncated.
Dihydroethidium fluorescence to assess ROS.
Freshly isolated hearts were exsanguinated by flushing via the aorta with ice-cold PBS. The RV free wall was removed, embedded in optimal cutting temperature medium, and frozen in isopentane that was cooled by liquid nitrogen. RV cross sections (8 μm) were incubated in a light-protected chamber for 15 min at 37°C with 20 μM dihydroethidium (DHE; Santa Cruz Biotechology) in PBS. Slides were washed three times in PBS and mounted with VECTASHIELD mounting medium for fluorescence. Slides were imaged at ×40 using a Zeiss LSM 510 confocal microscope. Oxidized DHE was detected at excitation/emission wavelengths of 405 and 570 nm, respectively (22). Oxidized DHE fluorescence was quantified using ImageJ thresholding analysis. Three to four images were measured per section, and the integrated density was averaged for each sample.
Western blot analysis.
RV free wall samples (10–20 mg) were homogenized 1:10 (wt/vol) in ice-cold RIPA buffer (50 mM Tris, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS) with protease and phosphatase inhibitors (Cell Signaling) using Dounce tissue grinders (Thomas Scientific). Samples were centrifuged for 10 min at 10,000 g at 4°C, and the supernatant was collected and stored at −80°C. Quantification of protein concentration was performed using the BCA protein assay (Thermo Scientific). Samples were prepared for immunoblot analysis by diluting equal amounts of protein in Laemmli sample buffer (Bio-Rad Laboratories) followed by boiling the samples for 5 min. Equal amounts of protein were separated on 4–20% Criterion TGX SDS-PAGE gels and then transferred to polyvinylidine difluoride membranes. Membranes were blocked in 5% nonfat milk in PBS with 0.05% Tween 20 (PBS-T) for 1 h and then incubated overnight at 4°C with primary antibody for 4-hydroxynonenal (4-HNE; 1:1,000, ab46545), superoxide dismutase 1 (SOD1; 1:2,500, sc-25778), or NAD(P)H oxidase 4 (NOX4; 1:1,000, NB11-58849). The membrane was then washed with PBS-T and incubated with horseradish peroxidase-conjugated secondary antibody (sc-2054) in 5% nonfat skim milk for 1 h. Immunoreactive bands were developed using Clarity Western ECL substrate (Bio-Rad Laboratories), and membranes were exposed to light-sensitive film. The film was scanned, and the optical density of protein bands was quantified using ImageJ. To ensure equal protein loading and transfer, membranes were stained with India ink. Briefly, the membrane was treated with 1% (wt/vol) KOH for 5 min, washed in PBS-T at 37°C for 30 min, and then washed at room temperature with PBS-T for 30 min before the membrane was stained with 0.2% India ink solution in PBS (Alfa Aesar) overnight at room temperature (26). The membrane was then washed in PBS-T for 30 min before it was air dried and scanned, and the individual protein bands were quantified using ImageJ. The optical density of protein bands per lane was summed. The presence of 4-HNE motifs and the expression of SOD1 and NOX4 were expressed relative to total membrane protein (30).
Statistical analysis.
Data are presented as means ± SE. Statistical tests (ANOVA with post hoc analysis using Bonferroni multiple comparisons and paired t-test) were performed using Prism 6 software (GraphPad Software, La Jolla, CA) with a significance level set at P < 0.05.
RESULTS
Chronic A61603 treatment improves in vivo function in the bleomycin model of RV failure.
In the bleomycin model, the presence of RV failure was suggested by reduced RV fractional shortening, reduced estimated RV cardiac output, and increased RV weight relative to body weight. Other signs common to both RV failure and lung injury were evident: increased lung weight, dyspnea, and lethargy. These findings are consistent with a previous study (12) that found that the bleomycin model resulted in pulmonary hypertension, decreased RV function, and death after ∼2 wk (Table 2).
Table 2.
Effect of tracheal instillation of bleomycin or saline on body weight and RV weight
| Number of Test Animals | Body Weight, g | RV Weight, mg | RV Weight/Body Weight, mg/g | |
|---|---|---|---|---|
| Saline | 9 | 27.6 ± 1 | 28.4 ± 1.3 | 1.03 ± 0.04 |
| Bleomycin | 12 | 20.2 ± 0.5§ | 23.6 ± 0.6† | 1.18 ± 0.04* |
| Bleomycin + A61603 | 10 | 20.8 ± 0.3§ | 22.4 ± 1‡ | 1.08 ± 0.03 |
Values are presented as means ± SE. A single instillation of bleomycin (0.075 units) or saline was given. After 2 wk, bleomycin resulted in decreases in body weight and right ventricular (RV) free wall weight. RV weight relative to body weight was increased by bleomycin. Significant differences compared with saline are shown:
P < 0.05,
P < 0.01,
P < 0.001, and
P < 0.0001.
We measured RV function in vivo noninvasively using echocardiography in conscious mice (Fig. 1A). Two weeks after bleomycin instillation, there was appreciably reduced RV fractional shortening compared with the nonfailing group (saline instillation). Moreover, 2 wk after bleomycin instillation, estimated RV cardiac output was reduced 44 ± 8% (P < 0.01) relative to the nonfailing group. Figure 1A also shows that RV fractional shortening was not reduced by bleomycin in mice that received concurrent A61603 infusion. Thus, chronic A61603 treatment prevented the fall in RV fractional shortening due to bleomycin-induced pulmonary fibrosis. Bleomycin-induced lung injury was associated with a doubling of lung weight relative to body weight (Fig. 1B) and severe pulmonary fibrosis (Fig. 1C). Moreover, A61603 treatment did not affect the increased lung weight or lung fibrosis due to bleomycin, suggesting that the beneficial effect of A61603 treatment on RV fractional shortening was not due to a reduction of lung injury. A61603 treatment in the absence of bleomycin did not affect lung weight, lung fibrosis, or RV fractional shortening (not shown).
Fig. 1.

Chronic A61603 treatment improved failing right ventricular (RV) function without affecting lung injury. Values from individual animals are shown with group means ± SE superimposed. A: echocardiographic assessment of RV fractional shortening (FS) 2 wk after tracheal instillation of bleomycin (Bleo) or saline (nonfailing) both with or without treatment with A61603. After bleomycin instillation, RV fractional shortening was reduced versus the nonfailing RV (***P < 0.001). After bleomycin instillation, RV fractional shortening was higher with A61603 treatment (**P < 0.01). B: lung weight (relative to body weight) was increased by bleomycin instillation (****P < 0.0001) but was not affected by A61603 treatment (ns, not significant; P > 0.999), suggesting lung injury was not reduced by A61603 treatment. C: lung histology showed that relative to saline instillation, bleomycin instillation resulted in severe lung fibrosis that was not affected by A61603 treatment (magnification ×40).
Chronic A61603 treatment reduces RV injury in the bleomycin model of RV failure.
Figure 2A shows the levels of serum cTnI measured in serum for mice 2 wk after tracheal instillation of saline (nonfailing), bleomycin, or bleomycin plus 2-wk concurrent treatment with A60103. Bleomycin instillation caused increased serum cTnI, consistent with RV cardiomyocyte injury and necrosis. However, chronic A61603 treatment resulted in significantly lower cTnI after bleomycin instillation, suggesting a protective effect of chronic α1A-subtype signaling. We assessed myocardial fibrosis with histological staining using picrosirius red. Figure 2B shows that the bleomycin model induced RV fibrosis, consistent with RV cardiomyocyte necrosis and replacement fibrosis. Figure 2B shows that for the bleomycin model, RV fibrosis was appreciably lower with chronic A61603 treatment.
Fig. 2.

Chronic A61603 treatment reduced right ventricular (RV) injury due to bleomycin (Bleo). A: serum levels of cardiac troponin I (cTnI) 2 wk after tracheal instillation of bleomycin or saline (nonfailing) both with or without treatment with A61603. Compared with the nonfailing RV, after bleomycin instillation cTnI was increased (****P < 0.0001) but was appreciably lower with concurrent A61603 treatment (****P < 0.0001). B: compared with the nonfailing RV, after bleomycin instillation RV fibrosis was increased versus the nonfailing RV (***P < 0.001) but was lower with concurrent A61603 treatment (*P < 0.05).
Chronic A61603 treatment improves myofilament force in the bleomycin model of RV failure.
Force development by cardiac myofilaments was assessed using demembranated RV muscle samples placed in activating solutions with various [Ca2+] (Fig. 3A). Fmax of demembranated RV myocardium obtained at saturating [Ca2+] was lower in the bleomycin-treated group compared with nonfailing group (Fig. 3B). However, chronic treatment with A61603 improved Fmax. There were no differences among groups in the sensitivity to [Ca2+] estimated from the EC50 for each curve (not shown).
Fig. 3.

Chronic A61603 treatment improved myofilament force development. A: myofilament force development in demembranated right ventricular (RV) muscle samples at various levels of activator Ca2+ concentration ([Ca2+]), expressed as pCa (−log [Ca2+]). Compared with the nonfailing RV, force development was impaired in the bleomycin (Bleo) model. Force development was higher with concurrent A61603 treatment. B: maximum Ca2+-activated force (Fmax) determined by fitting data to the Hill equation (35). The reduction of Fmax in the bleomycin model (****P < 0.0001) was attenuated by A61603 (***P < 0.001).
For the failing RV, the increased Fmax with A61603 treatment paralleled the greater fractional shortening with A61603 treatment (Fig. 1). Figure 4 shows the relationships between cardiomyocyte necrosis assessed from serum levels of cTnI and both RV fractional shortening and Fmax. Figure 4 shows that compared with the nonfailing RV, bleomycin resulted in elevated serum cTnI and reduced fractional shortening and Fmax. The changes due to bleomycin were markedly attenuated by concurrent treatment with A61603.
Fig. 4.
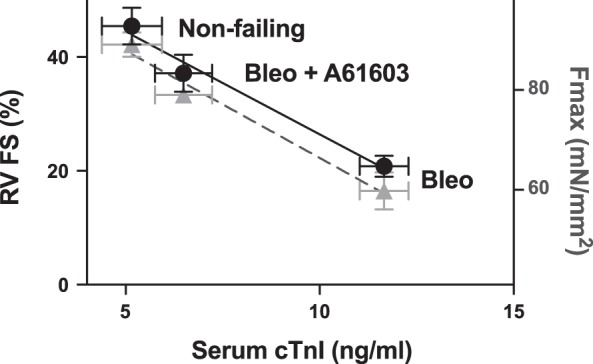
Chronic A61603 treatment improved contraction and reduced cardiomyocyte necrosis. Shown is a summary of averaged data for right ventricular (RV) fractional shortening (FS; black circles) and maximum myofilament force (Fmax; gray triangles) relative to the serum level of cardiac troponin I (cTnI; from data shown in Figs. 1–3). Bleomycin (Bleo) increased cTnI and caused proportional reductions in both RV FS and Fmax. These changes were attenuated by concurrent treatment with A61603.
Chronic A61603 treatment reduces expression of MMP-2 in the bleomycin model of RV failure.
MMP-2 is involved in remodeling of both the extracellular and intracellular compartments of the heart. Intracellular MMP-2 can cleave multiple contractile proteins and cause decreased myofilament force (9). We studied two MMP-2 isoforms: the canonical full-length MMP-2 (FL-MMP-2) and a novel NTT isoform (NTT-MMP-2) (16, 17). Figure 5 shows that in the bleomycin model, RV samples had appreciably elevated levels of both MMP-2 isoforms. Chronic treatment with A61603 resulted in lower levels of both MMP-2 isoforms. The A61603-mediated reduction of both MMP-2 isoforms may improve RV function by reducing MMP-2-mediated myofilament proteolysis.
Fig. 5.

Chronic A61603 treatment reduced expression of two isoforms of matrix metalloproteinase-2 (MMP-2). A and B: summary of quantitative RT-PCR measurements of the expression of two MMP-2 isoforms: the canonical full-length isoform (FL-MMP-2; A) and the NH2-terminal truncated isoform (NTT-MMP-2; B). *P < 0.05. Bleo, bleomycin.
Chronic A61603 treatment reduces oxidative stress in the bleomycin model of RV failure.
We assessed the presence of ROS using the fluorescent ROS probe DHE. Figure 6 shows that compared with the nonfailing RV, DHE fluorescence was increased in the RV from mice with bleomycin-induced RV injury. Moreover, for mice with bleomycin-induced RV injury, DHE fluorescence was appreciably lower for mice that were chronically treated with A61603.
Fig. 6.

Chronic A61603 treatment reduced reactive oxygen species (ROS)-dependent dihydroethidium (DHE) fluorescence. A: representative images of DHE fluorescence from frozen sections of right ventricular myocardium. DHE fluorescence was low in nonfailing hearts and markedly increased in the bleomycin (Bleo) model. This increase was prevented with concurrent treatment with A61603. B: pooled data. *P < 0.05; **P < 0.01.
Oxidative stress is associated with the generation of the lipid peroxidation product 4-HNE. 4-HNE reacts with numerous cellular proteins, and the presence of 4-HNE protein adducts is used as an indicator of oxidative stress (1). We found that levels of 4-HNE were increased in the RV from mice with bleomycin-induced injury but that 4-HNE levels were not increased in mice that received chronic treatment with A61603 (Fig. 7). This finding is consistent with the DHE data (Fig. 6) and indicates that chronic A61603 treatment reduced the level of oxidative stress in the RV failure model.
Fig. 7.
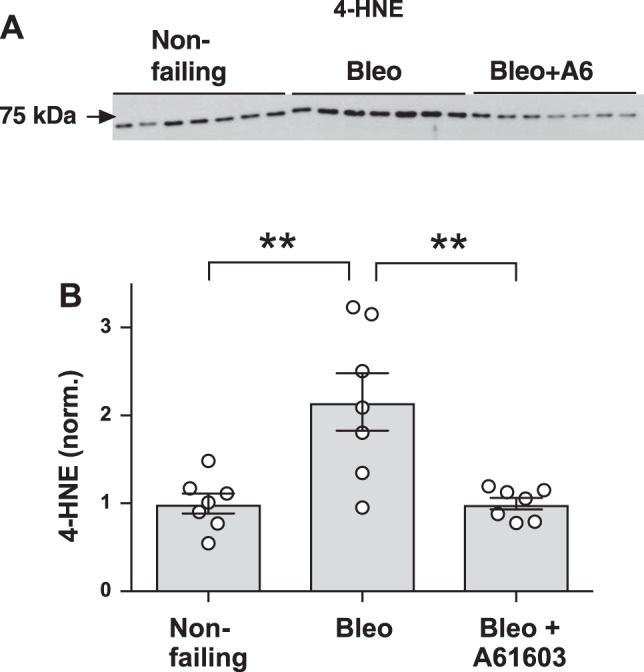
Chronic A61603 treatment reduced reactive oxygen species-dependent 4-hydroxynonenal (4-HNE). A: Western blot for detection of 4-HNE in right ventricular homogenates. B: pooled data showing that 4-HNE was increased in the bleomycin (Bleo) model and that this increase was prevented with chronic A61603 treatment. **P < 0.01.
To investigate potential mechanisms for oxidative stress reduction, we measured the levels of the cellular antioxidant SOD1 and levels of the oxidant-producing enzyme isoform NOX4 (15). Figure 8 shows that in the bleomycin model of RV injury, SOD1 was increased with chronic A61603 treatment. Increased SOD1 might have contributed to the finding that chronic A61603 treatment reduced oxidative stress, as evidenced by reduced levels of DHE and 4-HNE. Figure 9 shows that for the bleomycin model, chronic treatment with A61603 reduced the level of NOX4. Thus, treatment with A61603 reduces RV oxidant stress by improving the balance between antioxidant (SOD1) and oxidant (NOX4) enzymes.
Fig. 8.
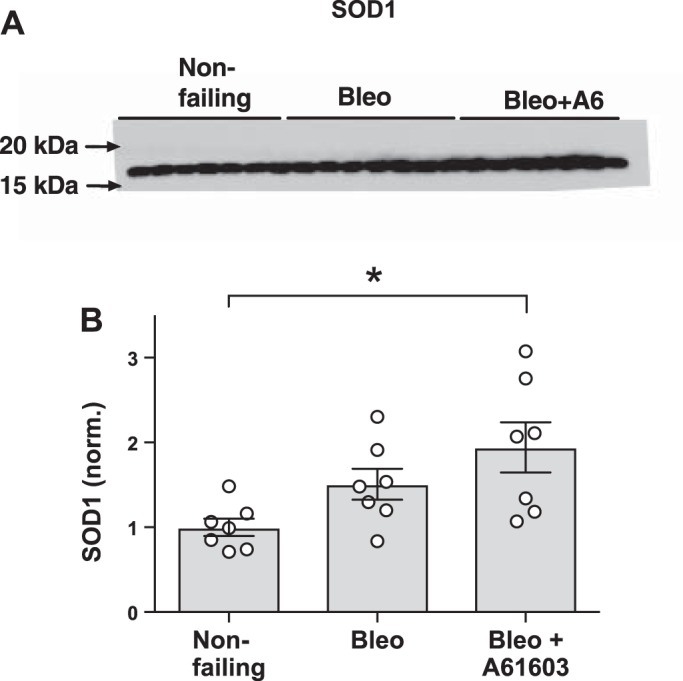
Chronic A61603 treatment increased superoxide dismutase 1 (SOD1). A: Western blot for detection of SOD1 in right ventricular homogenates. B: pooled data showing that SOD1 was increased in the bleomycin (Bleo) model with chronic A61603 treatment. *P < 0.05.
Fig. 9.

Chronic A61603 treatment reduced NAD(P)H oxidase 4 (NOX4). A: Western blot for detection of NOX4 in right ventricular homogenates. B: pooled data showing that NOX4 was lower in the bleomycin (Bleo) model with chronic A61603 treatment. *P < 0.05.
DISCUSSION
The major finding of this study is that in the mouse bleomycin model of RV failure, chronic treatment with the α1A-subtype agonist A61603 had a beneficial effect on RV function in vivo and beneficial effects on multiple measures of RV injury. Thus, the primary significance of this study is the finding that the α1A-subtype may be a novel therapeutic target to treat RV failure.
We found that in the bleomycin model of RV failure, chronic A61603 treatment resulted in lowered serum cTnI levels, consistent with reduced RV cardiomyocyte necrosis. Consistent with this, chronic A61603 treatment resulted in lowered RV replacement fibrosis. These findings are consistent with a previous report showing that α1A-subtype signaling has a prosurvival effect in cardiomyocytes (13).
In the bleomycin model of RV failure, chronic A61603 treatment resulted in significantly higher in vitro Fmax per unit area of RV myocardium. A component of this beneficial effect of A61603 might be due to the lowered RV fibrosis obtained with chronic A61603 treatment (and thus a greater area fraction of RV myocardium occupied by cells vs. fibrosis). However, the effect of chronic A61603 treatment to reduce RV fibrosis is quantitatively much smaller (≈3%) than the effect to cause higher Fmax (≈32%). This suggests that in the bleomycin model of RV failure, the beneficial effect of chronic A61603 treatment is the result primarily of greater absolute RV myofilament contraction force.
The relationship between RV fractional shortening versus cardiac injury assessed with cTnI was similar to the relationship between Fmax and cTnI. The data suggest that Fmax is a key determinant of RV fractional shortening and that the beneficial effect of chronic A61603 treatment is mediated in part by a higher Fmax. Thus, the beneficial effect of A61603 treatment on myofilament function might be central to the therapeutic effect in this model of RV failure.
Chronic A61603 treatment also resulted in lowered oxidative stress, as evidenced by lowered ROS-dependent DHE fluorescence and lowered abundance of the ROS-dependent protein adduct 4-HNE. The reduced oxidative stress due to A61603 treatment was associated with a higher level of the antioxidant SOD1 and a reduced abundance of the oxidant producer NOX4. Together, these changes could contribute to overall lowered oxidative stress.
ROS directly reduces contractile protein function (19). Thus, lowered oxidative stress with A61603 treatment could improve myofilament contraction by reducing myofilament damage induced by ROS. Oxidative stress also results in increased transcription and activity of MMP-2, which negatively affects both intracellular and extracellular cardiomyocyte function (6, 31). Intracellular MMP-2 causes intracellular damage to the myofilaments in cardiomyocytes (17, 18, 27, 28, 36) and cleaves multiple myofilament-associated proteins, including troponin I, titin, α-actinin, and myosin essential light chain, an activity attributed to intracellular FL-MMP-2 (2). We have previously reported that cardiac-specific transgenic expression of the NTT-MMP-2 isoform reduces cardiomyocyte contractility due to defective Ca2+ handling (16). Given that A61603 reduced expression of both MMP-2 isoforms, the beneficial effect of A61603 on RV function may involve improved Ca2+ handling and a reduction in myofilament injury.
These findings suggest a hypothesis for the beneficial effect of chronic A61603 treatment in the failing RV (Fig. 10). In the progression to RV failure, RV pressure overload involves increased ROS production that directly damages myofilaments and induces synthesis of both MMP-2 isoforms. These events lead to decreased myofilament contraction and resultant RV failure. Chronic A61603 treatment may be beneficial by intervening is this pathway by increasing SOD1 and reducing NOX4, which together lower ROS and thus minimize ROS-mediated reductions of myofilament contraction. In addition, Fig. 10 shows a potential direct inotropic effect of chronic A61603 stimulation. Specifically, we reported that the α1-AR inotropic response was increased in the failing RV (33) and that of the two major α1-AR subtypes on cardiac myocytes, only the α1A-subtype is responsible for this effect (8). Thus, chronic A61603 treatment might have a direct inotropic effect to augment RV pump function. In the bleomycin lung injury model, systemic hypoxia may also contribute to RV oxidative stress (5). A direct inotropic action of A61603 could augment RV function and lead to decreased hypoxia and reduced oxidative stress and thereby also contribute to reducing the level of oxidative stress-mediated RV injury.
Fig. 10.
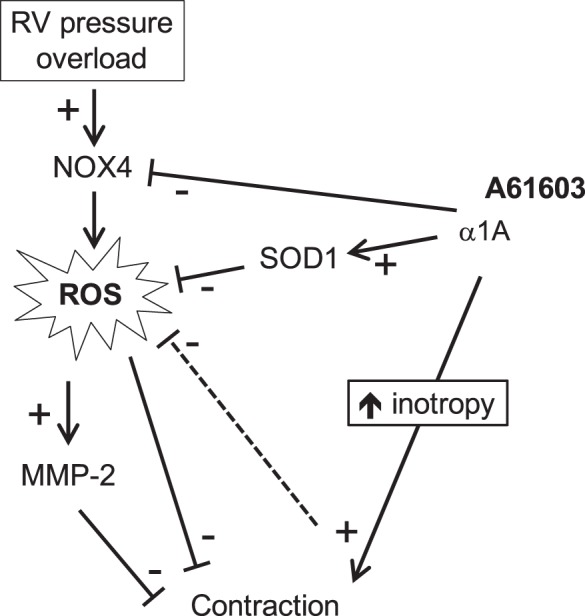
Hypothetical scheme linking chronic A61603 treatment to a beneficial effect on the failing right ventricle (RV) via lowering of oxidative stress. NOX4, NAD(P)H oxidase 4; ROS, reactive oxygen species; SOD1, superoxide dismutase 1; MMP-2, matrix metalloproteinase-2.
The findings in this study are consistent with growing experimental and clinical evidence suggesting that α1-ARs mediate cardioprotective effects (3, 14, 23). Moreover, these findings are consistent with recent studies suggesting that chronic α1A-subtype stimulation is beneficial for improving LV function in the doxorubicin-induced cardiotoxicity model (4, 20).
Limitations.
Confounding factors related to the bleomycin model used in this study include pulmonary fibrosis, impaired gas exchange, hypoxia, and altered lung mechanics.
This study found beneficial effects of A61603 treatment in the mouse bleomycin model of RV failure. However, it is unclear if these findings are relevant to other models of RV failure and other species. In previous studies, other models of RV failure have been used, including pulmonary artery constriction and sugen-hypoxia, and it will be important to evaluate the efficacy of A61603 treatment in other RV failure models.
The hypothesis for the mechanisms for RV injury and the beneficial effect of A61603 treatment are based on associations between cell injury and factors related to redox stress and MMP-2 isoforms. However, it remains to be demonstrated which of these associations are causally related.
The mechanisms by which A61603 could mediate beneficial effects on oxidative stress are not resolved. For example, how does α1A-subtype stimulation leads to higher levels of SOD1 and lower levels of NOX4?
The mechanisms causing effects on myofilament force (decreased Fmax in the failing RV and higher Fmax with A61603 treatment) are unknown.
This project shows that chronic A61603 treatment initiated concurrently with bleomycin is beneficial for preventing RV damage. However, it is not known if A61603 treatment that is initiated after RV dysfunction has already developed will be able to reverse RV damage. Therefore, it will be important to use a more clinically relevant rescue/reversal study design to determine if A61603 treatment can rescue RV function in a chronic model of already established RV failure.
We found that the beneficial effect of A61603 treatment is associated with decreased oxidative stress in the RV. Lung weight was increased by bleomycin, and A61603 treatment did not normalize lung weight. Nevertheless, it is possible that A61603 treatment reduced some aspect of lung injury, which contributed to the beneficial effect on the RV. Use of an additional RV-specific failure model, such as pulmonary artery banding, could confirm if the therapeutic effect of A61603 is due solely to effects on the RV.
Conclusions.
Chronic α1A-subtype stimulation had a beneficial effect on RV function and multiple markers of RV injury in the mouse bleomycin model of RV failure. In the context of pulmonary fibrosis, the α1A-subtype is a potential therapeutic target to treat the failing RV.
GRANTS
This work was supported by Department of Veterans Affairs Merit Review Awards I01BX000740 (to A. Baker) and I01BX001970 (to P. Simpson), National Heart, Lung, and Blood Institute Grant HL-31113 (to P. Simpson), and American Heart Association Postdoctoral Fellowship 16POST30970031 (to P. Cowley), Grant-In-Aid 14GRNT20380813 (to D. Lovett), and Grant-In-Aid 15GRNT25550041 (to A. Baker).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
P.M.C., G.-Y.W., D.H.L., P.C.S., and A.J.B. conceived and designed research; P.M.C., G.-Y.W., S.K.J., and P.M.S. performed experiments; P.M.C., G.-Y.W., S.K.J., P.M.S., and A.J.B. analyzed data; P.M.C., G.-Y.W., S.K.J., D.H.L., P.C.S., and A.J.B. interpreted results of experiments; P.M.C., S.K.J., P.M.S., and A.J.B. prepared figures; P.M.C., D.H.L., P.C.S., and A.J.B. edited and revised manuscript; P.M.C., G.-Y.W., S.K.J., P.M.S., D.H.L., P.C.S., and A.J.B. approved final version of manuscript; A.J.B. drafted manuscript.
REFERENCES
- 1.Aberle NS II, Picklo MJ Sr, Amarnath V, Ren J. Inhibition of cardiac myocyte contraction by 4-hydroxy-trans-2-nonenal. Cardiovasc Toxicol 4: 21–28, 2004. doi: 10.1385/CT:4:1:21. [DOI] [PubMed] [Google Scholar]
- 2.Ali MA, Fan X, Schulz R. Cardiac sarcomeric proteins: novel intracellular targets of matrix metalloproteinase-2 in heart disease. Trends Cardiovasc Med 21: 112–118, 2011. doi: 10.1016/j.tcm.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 3.Baker AJ. Adrenergic signaling in heart failure: a balance of toxic and protective effects. Pflugers Arch 466: 1139–1150, 2014. doi: 10.1007/s00424-014-1491-5. [DOI] [PubMed] [Google Scholar]
- 4.Beak J, Huang W, Parker JS, Hicks ST, Patterson C, Simpson PC, Ma A, Jin J, Jensen BC. An oral selective alpha-1A adrenergic receptor agonist prevents doxorubicin cardiotoxicity. JACC Basic Transl Sci 2: 39–53, 2017. doi: 10.1016/j.jacbts.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, Ockaili R, McCord JM, Voelkel NF. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 120: 1951–1960, 2009. doi: 10.1161/CIRCULATIONAHA.109.883843. [DOI] [PubMed] [Google Scholar]
- 6.Cheung PY, Sawicki G, Wozniak M, Wang W, Radomski MW, Schulz R. Matrix metalloproteinase-2 contributes to ischemia-reperfusion injury in the heart. Circulation 101: 1833–1839, 2000. doi: 10.1161/01.CIR.101.15.1833. [DOI] [PubMed] [Google Scholar]
- 7.Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol 147: 325–338, 1995. [PMC free article] [PubMed] [Google Scholar]
- 8.Cowley PM, Wang G, Chang AN, Makwana O, Swigart PM, Lovett DH, Stull JT, Simpson PC, Baker AJ. The α1A-adrenergic receptor subtype mediates increased contraction of failing right ventricular myocardium. Am J Physiol Heart Circ Physiol 309: H888–H896, 2015. doi: 10.1152/ajpheart.00042.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeCoux A, Lindsey ML, Villarreal F, Garcia RA, Schulz R. Myocardial matrix metalloproteinase-2: inside out and upside down. J Mol Cell Cardiol 77: 64–72, 2014. doi: 10.1016/j.yjmcc.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghio S, Gavazzi A, Campana C, Inserra C, Klersy C, Sebastiani R, Arbustini E, Recusani F, Tavazzi L. Independent and additive prognostic value of right ventricular systolic function and pulmonary artery pressure in patients with chronic heart failure. J Am Coll Cardiol 37: 183–188, 2001. doi: 10.1016/S0735-1097(00)01102-5. [DOI] [PubMed] [Google Scholar]
- 11.Greyson CR. Pathophysiology of right ventricular failure. Crit Care Med 36, Suppl: S57–S65, 2008. doi: 10.1097/01.CCM.0000296265.52518.70. [DOI] [PubMed] [Google Scholar]
- 12.Hemnes AR, Zaiman A, Champion HC. PDE5A inhibition attenuates bleomycin-induced pulmonary fibrosis and pulmonary hypertension through inhibition of ROS generation and RhoA/Rho kinase activation. Am J Physiol Lung Cell Mol Physiol 294: L24–L33, 2008. doi: 10.1152/ajplung.00245.2007. [DOI] [PubMed] [Google Scholar]
- 13.Huang Y, Wright CD, Merkwan CL, Baye NL, Liang Q, Simpson PC, O’Connell TD. An α1A-adrenergic-extracellular signal-regulated kinase survival signaling pathway in cardiac myocytes. Circulation 115: 763–772, 2007. doi: 10.1161/CIRCULATIONAHA.106.664862. [DOI] [PubMed] [Google Scholar]
- 14.Jensen BC, OʼConnell TD, Simpson PC. Alpha-1-adrenergic receptors in heart failure: the adaptive arm of the cardiac response to chronic catecholamine stimulation. J Cardiovasc Pharmacol 63: 291–301, 2014. doi: 10.1097/FJC.0000000000000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci USA 107: 15565–15570, 2010. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lovett DH, Chu C, Wang G, Ratcliffe MB, Baker AJ. A N-terminal truncated intracellular isoform of matrix metalloproteinase-2 impairs contractility of mouse myocardium. Front Physiol 5: 363, 2014. doi: 10.3389/fphys.2014.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lovett DH, Mahimkar R, Raffai RL, Cape L, Maklashina E, Cecchini G, Karliner JS. A novel intracellular isoform of matrix metalloproteinase-2 induced by oxidative stress activates innate immunity. PLoS One 7: e34177, 2012. doi: 10.1371/journal.pone.0034177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lovett DH, Mahimkar R, Raffai RL, Cape L, Zhu BQ, Jin ZQ, Baker AJ, Karliner JS. N-terminal truncated intracellular matrix metalloproteinase-2 induces cardiomyocyte hypertrophy, inflammation and systolic heart failure. PLoS One 8: e68154, 2013. doi: 10.1371/journal.pone.0068154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacFarlane NG, Miller DJ. Depression of peak force without altering calcium sensitivity by the superoxide anion in chemically skinned cardiac muscle of rat. Circ Res 70: 1217–1224, 1992. doi: 10.1161/01.RES.70.6.1217. [DOI] [PubMed] [Google Scholar]
- 20.Montgomery MD, Chan T, Swigart PM, Myagmar BE, Dash R, Simpson PC. An alpha-1A adrenergic receptor agonist prevents acute doxorubicin cardiomyopathy in male mice. PLoS One 12: e0168409, 2017. doi: 10.1371/journal.pone.0168409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nadrous HF, Pellikka PA, Krowka MJ, Swanson KL, Chaowalit N, Decker PA, Ryu JH. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Chest 128: 2393–2399, 2005. doi: 10.1378/chest.128.4.2393. [DOI] [PubMed] [Google Scholar]
- 22.Nazarewicz RR, Bikineyeva A, Dikalov SI. Rapid and specific measurements of superoxide using fluorescence spectroscopy. J Biomol Screen 18: 498–503, 2013. doi: 10.1177/1087057112468765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Connell TD, Jensen BC, Baker AJ, Simpson PC. Cardiac alpha1-adrenergic receptors: novel aspects of expression, signaling mechanisms, physiologic function, and clinical importance. Pharmacol Rev 66: 308–333, 2013. doi: 10.1124/pr.112.007203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reddy S, Bernstein D. Molecular mechanisms of right ventricular failure. Circulation 132: 1734–1742, 2015. doi: 10.1161/CIRCULATIONAHA.114.012975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reddy S, Bernstein D. The vulnerable right ventricle. Curr Opin Pediatr 27: 563–568, 2015. doi: 10.1097/MOP.0000000000000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sasse J, Gallagher SR. Detection of proteins on blot transfer membranes. Curr Protoc Mol Biol chapt. 10: Unit 10.7, 2008. doi: 10.1002/0471142727.mb1007s84. [DOI] [PubMed] [Google Scholar]
- 27.Sawicki G, Leon H, Sawicka J, Sariahmetoglu M, Schulze CJ, Scott PG, Szczesna-Cordary D, Schulz R. Degradation of myosin light chain in isolated rat hearts subjected to ischemia-reperfusion injury: a new intracellular target for matrix metalloproteinase-2. Circulation 112: 544–552, 2005. doi: 10.1161/CIRCULATIONAHA.104.531616. [DOI] [PubMed] [Google Scholar]
- 28.Schulz R. Intracellular targets of matrix metalloproteinase-2 in cardiac disease: rationale and therapeutic approaches. Annu Rev Pharmacol Toxicol 47: 211–242, 2007. doi: 10.1146/annurev.pharmtox.47.120505.105230. [DOI] [PubMed] [Google Scholar]
- 29.Shimkunas R, Makwana O, Spaulding K, Bazargan M, Khazalpour M, Takaba K, Soleimani M, Myagmar BE, Lovett DH, Simpson PC, Ratcliffe MB, Baker AJ. Myofilament dysfunction contributes to impaired myocardial contraction in the infarct border zone. Am J Physiol Heart Circ Physiol 307: H1150–H1158, 2014. doi: 10.1152/ajpheart.00463.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor SC, Posch A. The design of a quantitative Western blot experiment. BioMed Res Int 2014: 361590, 2014. doi: 10.1155/2014/361590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Viappiani S, Nicolescu AC, Holt A, Sawicki G, Crawford BD, León H, van Mulligen T, Schulz R. Activation and modulation of 72 kDa matrix metalloproteinase-2 by peroxynitrite and glutathione. Biochem Pharmacol 77: 826–834, 2009. doi: 10.1016/j.bcp.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 32.Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR, Dupuis J, Long CS, Rubin LJ, Smart FW, Suzuki YJ, Gladwin M, Denholm EM, Gail DB. Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute Working Group on Cellular and Molecular Mechanisms of Right Heart Failure. Circulation 114: 1883–1891, 2006. doi: 10.1161/CIRCULATIONAHA.106.632208. [DOI] [PubMed] [Google Scholar]
- 33.Wang GY, Yeh CC, Jensen BC, Mann MJ, Simpson PC, Baker AJ. Heart failure switches the RV α1-adrenergic inotropic response from negative to positive. Am J Physiol Heart Circ Physiol 298: H913–H920, 2010. doi: 10.1152/ajpheart.00259.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang GY, Bergman MR, Nguyen AP, Turcato S, Swigart PM, Rodrigo MC, Simpson PC, Karliner JS, Lovett DH, Baker AJ. Cardiac transgenic matrix metalloproteinase-2 expression directly induces impaired contractility. Cardiovasc Res 69: 688–696, 2006. doi: 10.1016/j.cardiores.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 35.Wang GY, McCloskey DT, Turcato S, Swigart PM, Simpson PC, Baker AJ. Contrasting inotropic responses to α1-adrenergic receptor stimulation in left versus right ventricular myocardium. Am J Physiol Heart Circ Physiol 291: H2013–H2017, 2006. doi: 10.1152/ajpheart.00167.2006. [DOI] [PubMed] [Google Scholar]
- 36.Wang W, Schulze CJ, Suarez-Pinzon WL, Dyck JR, Sawicki G, Schulz R. Intracellular action of matrix metalloproteinase-2 accounts for acute myocardial ischemia and reperfusion injury. Circulation 106: 1543–1549, 2002. doi: 10.1161/01.CIR.0000028818.33488.7B. [DOI] [PubMed] [Google Scholar]