

Abstract
Dysregulation of sodium (Na+) balance is a major cause of hypertensive cardiovascular disease. The current dogma is that interstitial Na+ readily equilibrates with plasma and that renal excretion and reabsorption is sufficient to regulate extracellular fluid volume and control blood pressure. These ideas have been recently challenged by the discovery that Na+ accumulates in tissues without commensurate volume retention and activates immune cells, leading to hypertension and autoimmune disease. However, objections have been raised to this new paradigm, with some investigators concerned about where and how salt is stored in tissues. Further concerns also include how Na+ is mobilized from tissue stores and how it interacts with various organ systems to cause hypertension and end-organ damage. This review assesses these two paradigms of Na+ regulation in the context of inflammation-mediated hypertension and cardiovascular disease pathogenesis. Also highlighted are future perspectives and important gaps in our understanding of how Na+ is linked to inflammation and hypertension. Understanding mechanisms of salt and body fluid regulation is the sine qua non of research efforts to identify therapeutic targets for hypertension and cardiovascular disease.
Keywords: sodium regulation, inflammation, hypertension
regulation of salt and body fluids is critical for blood pressure control. High blood pressure is the major risk factor for cardiovascular disease and the leading cause of morbidity and mortality due to myocardial infarction, stroke, heart failure, and chronic kidney disease. Cardiovascular disease causes one in six American deaths annually. The prevalence of hypertension among United States children and adolescents 8 to 17 yr old is 11% (2). Thirty one percent of adults over 18 yr of age in the United States are hypertensive and this prevalence increases to ~70% by age 65 (2). In clinical trials, antihypertensive treatment results in a substantial reduction in the incidence of myocardial infarction (20–25%), stroke (35–40%), and heart failure (>50%) (52). However, despite intense efforts over the past several decades to understand underlying mechanisms, the pathogenesis of essential hypertension remains elusive and blood pressure control remains suboptimal in the general population (4). Here, I review mechanisms of salt homeostasis and its linkage to blood pressure, inflammation, and end-organ damage. Understanding these mechanisms could have a global impact in developing targeted approaches to prevent and treat cardiovascular disease.
Sodium and Hypertension
Na+ is the major cation that maintains extracellular fluid volume and promotes adequate tissue perfusion and normal cell metabolism. According to the American Heart association, the ideal amount of daily Na+ intake is 1.5 g/day, with a maximum recommended intake of 2.3 g/day. Until recently, Na+ consumption was below this level and the incidence of hypertension very low (27). However, the Center of Disease Control (CDC) recently reported that in 2013–2014, Americans consumed ~3.4 g of Na+ daily, more than double the ideal recommendations. In fact, less than 10% of the US population observes the recommended guidelines for Na+ intake (7, 27).
Numerous epidemiological, clinical, and experimental studies have demonstrated that excess dietary salt intake contributes to hypertension and cardiovascular disease. A recent meta-analysis showed that in the United States, modest reductions in salt intake would lower blood pressure and reduce the annual number of new cases of coronary heart disease and stroke by 20% (15). However, accomplishing this goal is difficult as over 70% of dietary Na+ is added to food outside of the home (14). In addition, there is heterogeneity in susceptibility to salt-induced hypertension and thus controversy regarding recommendations to reduce salt intake in the general population. In a recent publication in the FASEB Journal (34a), analysis of 2,632 normotensive adults in the Framingham Offspring Study found that low Na+ intake is not associated with lower blood pressure. These paradoxical results are likely due to the fact that only a minority of individuals exhibit salt-sensitive hypertension and that potential beneficial effects of low Na+ intake are masked by the salt resistant majority (27).
Salt-sensitive hypertension, defined as a change in blood pressure greater than 10% in response to either increased or reduced salt intake, has important prognostic and therapeutic implications. Unlike trends observed in the entire population, the prevalence of salt-sensitive hypertension is higher in African Americans and the elderly (29). It is associated with increased endothelial dysfunction, insulin resistance, congestive heart failure, and renal damage. In addition, salt-sensitive hypertension increases the risk of mortality in both normotensive and hypertensive subjects, suggesting that it is an independent cardiovascular risk factor (27, 47). While several animal models, including the Dahl salt-sensitive rat developed in the 1960s by Lewis Dahl, have provided great insight into the consequences of salt-sensitive hypertension, much research still needs to be done to understand the underlying mechanisms.
A Brief Historical Perspective of Salt Regulation in Hypertension
The discovery of the renal pressure-natriuresis relationship is credited to Dr. Carl Ludwig who first described the concept of glomerular filtration and the effect of blood pressure to increase urine outflow in his 1842 Habilitation thesis (28). Ernest Starling advanced this relationship into a mechanism for controlling blood pressure when he described the Starling’s principle of transvascular fluid exchange in his 1909 Herter Lecture “The Fluids of the Body” (13). In 1963, Drs. Borst and Borst-de Geus extrapolated Starling's work and hypothesized that the kidneys are the setpoint for blood pressure control (3). Drs. Guyton and Coleman in 1969 developed a computer model that integrated this setpoint into a global blood pressure control system (9). This model predicted that there has to be a shift in the pressure natriuresis setpoint to sustain elevated blood pressure. Thus, under healthy conditions, increased Na+ and water intake is accompanied by increased excretion by the kidney, and normal blood pressure is maintained under a wide range of dietary Na+ intake concentrations (10). Dr. Guyton and colleagues proposed a systems analysis approach whereby hypertension is caused by two main scenarios: first, retention of volume and Na+ due to perturbed renal function, and second, excess water and Na+ intake (11). The authors also proposed that a multitude of other systems including the renin-angiotensin-aldosterone system (RAAS), atrial natriuretic peptide (ANP), and sympathetic outflow control Na+ balance and blood pressure. These systems respond to changes in Na+ intake to achieve balance without changing blood pressure. Hypertension only occurs if there is a perturbation in any of these systems and a decline in renal excretory capability leading to Na+ retention and plasma volume expansion. In keeping with this concept, subsequent studies found that in human subjects, increasing Na+ intake increased systemic blood pressure (35). However, these studies were short term and did not address consequences of long-term intake of excessive amounts of salt. In addition, changes in plasma Na+ due to diet are small and plasma Na+ concentration is maintained relatively constant. For example, only a 3 mmol/l decrease in Na+ concentration is achieved in humans following a large reduction in Na+ intake from 350 mmol/day to 10 mmol/day) for 5 days (16). It is still not clear whether such small changes in plasma Na+ concentrations directly affect blood pressure.
A New Paradigm for Sodium Regulation
Previously, the assumption was that during high Na+ intake, the increases in the Na+ concentration in the interstitial tissues are the same as those observed in plasma. New findings suggest that Na+ accumulates in the interstitium in concentrations exceeding those of plasma and suggest existence of extra-renal regulatory mechanisms. Below, I discuss these findings and how they are changing our thinking of how Na+ is regulated.
Long-term Na+ balance studies in humans suggest that high dietary Na+ consumption does not necessarily promote expansion of the extracellular volume (17). In 1968, Garnett et al. found that in human subjects, levels of exchangeable Na+ decrease during the first week of starvation and then increase progressively to values similar to those before starvation, despite the negative Na+ balance, suggesting mobilization of osmotically inactive Na+ (8). Subsequent studies by Titze et al. found that the skin serves as a reservoir for osmotically inactive Na+ and that such Na+ reserves are mobilized during salt restriction, aging, and in ovariectomized rats (39, 42–44). Gender differences have been identified in Na+ storage between muscle and skin: men exhibit higher Na+ content in skin than in muscle, whereas women accumulate Na+ to greater levels in muscle (46). The mechanisms underlying these gender differences and their consequences are not known. While these studies do not contradict proposed mechanisms of pressure natriuresis and renal Na+ regulation, they show that Na+ can be stored in tissues independent of water retention and suggest existence of extrarenal mechanisms for Na+ homeostasis.
Mobilization and Storage of Sodium in Tissues
In 2004, Titze et al. found that osmotically inactive Na+ associates with glycosaminoglycans (GAGs) in the skin (45). GAGs consist of alternating copolymers of uronic acids and amino sugars, and their polyanionic property attracts cations, particularly Na+ (26). A high-salt diet results in increased GAG polymerization with a resulting increase in the negative interstitial charge density and a parallel increase in Na+ storage in skin (45). In contrast, a low-salt diet reduces GAG polymerization, decreasing negative charge density with subsequent release of Na+ (45). In a recent study by Fischereder et al., Na+ storage was found to be regulated by GAG expression in humans (6). Interactions between GAGs and Na+ are presumed to be electrostatic and therefore water free and osmotically inactive (45).
In 2009, Machnik et al. found that feeding rodents excess dietary salt increases interstitial concentrations of NaCl in the skin to 190 mM without changing plasma concentrations (31). Subsequent studies using 23Na MRI found that comparable concentrations are reached in the skin and skeletal muscle interstitium of humans with hypertension and during aging (25). These studies suggest that Na+ can be stored in tissues without commensurate water retention and that interventions to modulate GAG negative charge density could control release and storage of Na+ in the skin.
Regulation of Tissue Sodium by the Immune System
In the kidney, regulation of Na+ balance, primarily by the renin angiotensin aldosterone system, is well studied. However, molecular mechanisms governing regulation of tissue Na+ and how they contribute to salt-sensitive hypertension remain unclear. Machnik et al. reported that high salt intake and subsequent accumulation of Na+ in skin are associated with increased macrophage infiltration in the interstitium (31). The macrophages express and activate the transcription factor tonicity-responsive enhancer binding protein (TonEBP), which binds to and enhances expression of the vascular endothelial growth factor (VEGF) C gene. VEGFC stimulates lymphatic capillary network formation and increases expression of endothelial nitric oxide synthase (eNOS) in interstitial cells, resulting in increased Na+ clearance from the skin (Fig. 1) (31). Macrophage depletion and blockade of interstitial TonEBP/VEGF-C signaling induces salt-sensitive hypertension in rats (30). Thus, the immune system, in particular macrophages, acts as an extrarenal regulator of interstitial Na+ and water homeostasis. The mechanisms by which macrophages sense salt are unclear, and it is not known whether other immune cell types participate in regulation of tissue Na+.
Fig. 1.
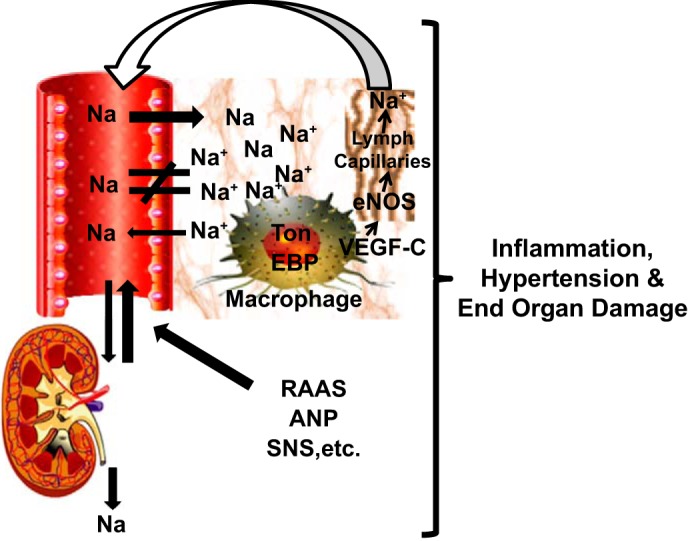
A new paradigm of sodium regulation. Sodium accumulates in tissues in concentrations exceeding those in plasma. In addition to kidney and other regulatory systems including the renin-angiotensin-aldosterone system (RAAS), atrial natriuretic peptide (ANP), and the sympathetic nervous system (SNS), macrophages function in an extrarenal sodium regulatory mechanism in part by expressing and activating the transcription factor tonicity-responsive enhancer binding protein (TonEBP), which binds to and upregulates expression of the vascular endothelial growth factor (VEGF) C gene. VEGFC stimulates lymphatic capillary network formation and increases expression of endothelial nitric oxide synthase (eNOS) in interstitial cells, increasing Na+ clearance from the skin.
Inflammation and Salt-Sensitive Hypertension
Inflammatory cells, including macrophages and T cells, infiltrate the kidney and vasculature and contribute to hypertension. In the 1960s, Grollman and colleagues found that immunosuppression attenuates hypertension in rats with partial renal infarction and that transfer of lymph node cells from rats with renal infarction into naïve recipient rats induced hypertension (37, 49). Subsequent studies in the 1970s and 1980s found that the thymus is important for development of hypertension in rodents (1, 40, 41). In 2007, Guzik et al. found that T lymphocytes are required for full development of hypertension (12). Subsequently, Crowley et al. reported that mice with severe combined immunodeficiency exhibit reduced blood pressure and associated renal injury, confirming a critical role for adaptive immunity in hypertension (5). Moreover, immune cells are known to play a role in salt-sensitive hypertension (33). In 2013, Mattson et al. found that Dahl-salt sensitive rats in which the RAG1 gene has been deleted using the zinc-finger DNAse technology develop blunted hypertension in response to salt feeding (34). Cytokines, particularly interleukin 17A (IL17A), released from T cells promote vascular and kidney injury leading to hypertension (32, 36).
In addition to the adaptive immune system, cells of the innate immune system, including monocytes, macrophages, and dendritic cells (DCs), contribute to hypertension. Wenzel et al. found that deletion of monocytes attenuates development of hypertension and vascular dysfunction (48). Recently, we demonstrated that antigen-presenting DCs play a critical role in development of both angiotensin II and deoxycorticosterone acetate (DOCA)-salt hypertension (22). In this study, we found that DCs accumulate isolevuglandin (IsoLG)-protein adducts, which are immunogenic and activate T cells, leading to hypertension.
Increasing evidence indicates that excess dietary salt promotes tissue inflammation and autoimmune disease (38, 51). In a 205-day salt balance study, Yi et al. found that healthy human subjects fed a high-salt diet exhibited a marked increase in the number monocytes and levels of proinflammatory cytokines including IL-6, TNF-α, and IL-23, when compared with low-salt diet controls (50). Recently, Jantsch et al. found that microbial infection is associated with high Na+ concentration in the tissue microenvironment, which enhances macrophage function and microbicidal activity via increased nitric oxide production (18). Moreover, exposure to high salt polarizes immune cells toward an inflammatory phenotype, enhancing IL-17 production (19, 24, 53). These studies indicate that salt activates immune cells, which lead to hypertension.
In summary, Na+ can accumulate in the tissue interstitium in concentrations exceeding those of plasma due to its association with negatively charged GAGs. This interaction is water free and therefore osmotically inactive. Such high concentrations of Na+ activate T cells via signaling molecules such as NFAT5 and the salt-sensing kinase 1 or SGK1 (24). Once activated, immune cells infiltrate the perivascular space and the kidneys, and release cytokines that promote vascular dysfunction and Na+ and water retention leading to hypertension (20).
Perspectives and Significance
Current antihypertensive therapies clearly do not adequately meet the needs of the general population. Alongside intense efforts to develop therapies effective to treat resistant hypertension by focusing on the kidney as the master regulator of Na+, current research efforts are increasingly considering tissue Na+ accumulation and its effects on the immune system as an important target. This approach is particularly critical given that over 70% of extracellular fluid is interstitial and thereby not directly controlled by renal salt and water excretion. Thus, it is important to understand extrarenal mechanisms of Na+ regulation and how their disruption may contribute to the pathogenesis of inflammation, hypertension, and cardiovascular disease.
Immunotherapies, including checkpoint inhibitors, have been widely exploited to treat cancer and autoimmune disease. For example, enhancement of macrophage function via inhibitors of the receptor CD47 has an anticancer effect (21). However, immunotherapies have not been applied to treatment of hypertension. Given that macrophages play a critical role in clearing Na+ from the interstitium, modulation of macrophage function may provide an important novel target to treat hypertension and the associated end-organ damage. Several currently existing medications, including diuretics, appear to exhibit therapeutic benefit beyond what can be explained by lowering blood pressure, possibly due to antioxidant or anti-inflammatory activations directly on immune cells. Our discovery that oxidative stress in DCs leads to posttranslational modification of proteins by IsoLGs, which are immunogenic (22), further supports this hypothesis.
Understanding mechanisms by which immune cells sense Na+ and how they are activated in hypertension will advance our understanding of the pathophysiology of this disease and lead to development of novel therapeutics. To precisely define how extrarenal Na+ contributes to inflammation and the pathophysiology of hypertension, novel, direct, and validated assays to measure tissue Na+ such as energy dispersive X-ray microanalysis are required. Moreover, efforts to assess the importance of tissue Na+ and water homeostasis in blood pressure regulation have predominantly focused on skin. Additional research focusing on other tissues is required to confirm these findings and determine specific contributions to blood pressure control. In addition, it is not known how the GAG-associated osmotically inactive Na+ activates immune cells.
GRANTS
This work was supported by the National Institutes of Health Grant K01HL130497.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author.
AUTHOR CONTRIBUTIONS
A.K. prepared figures; A.K. drafted manuscript; A.K. edited and revised manuscript; A.K. approved final version of manuscript.
ACKNOWLEDGMENTS
Graphics were produced using the online ePath3D toll (Protein Lounge, Inc.).
REFERENCES
- 1.Bataillard A, Freiche JC, Vincent M, Sassard J, Touraine JL. Antihypertensive effect of neonatal thymectomy in the genetically hypertensive LH rat. Thymus 8: 321–330, 1986. [PubMed] [Google Scholar]
- 2.Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P; American Heart Association Statistics Committee and Stroke Statistics Subcommittee . Heart Disease and Stroke Statistics-2017 Update: A report from the American Heart Association. Circulation 135: e146–e603, 2017. [Erratum: Correction to: Heart Disease and Stroke Statistics-2017 Update: A report From the American Heart Association. Circulation 135: e646, 2017.] 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borst JG, Borst-De Geus A. Hypertension explained by Starling’s theory of circulatory homoeostasis. Lancet 1: 677–682, 1963. doi: 10.1016/S0140-6736(63)91443-0. [DOI] [PubMed] [Google Scholar]
- 4.Chobanian AV. Shattuck Lecture. The hypertension paradox–more uncontrolled disease despite improved therapy. N Engl J Med 361: 878–887, 2009. doi: 10.1056/NEJMsa0903829. [DOI] [PubMed] [Google Scholar]
- 5.Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol 298: R1089–R1097, 2010. doi: 10.1152/ajpregu.00373.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fischereder M, Michalke B, Schmoeckel E, Habicht A, Kunisch R, Pavelic I, Szabados B, Schonermarck U, Nelson P, Stangl M. Sodium storage in human tissues is mediated by glycosaminoglycan expression. Am J Physiol Renal Physiol 313: F319–F325, 2017. doi: 10.1152/ajprenal.00703.2016. [DOI] [PubMed] [Google Scholar]
- 7.Frisoli TM, Schmieder RE, Grodzicki T, Messerli FH. Salt and hypertension: is salt dietary reduction worth the effort? Am J Med 125: 433–439, 2012. doi: 10.1016/j.amjmed.2011.10.023. [DOI] [PubMed] [Google Scholar]
- 8.Garnett ES, Ford J, Golding PL, Mardell RJ, Whyman AE. The mobilizaton of osmotically inactive sodium during total starvation in man. Clin Sci 35: 93–103, 1968. [PubMed] [Google Scholar]
- 9.Guyton AC, Coleman TG. Quantitative analysis of the pathophysiology of hypertension. Circ Res 24, Suppl: 1–19, 1969. [PubMed] [Google Scholar]
- 10.Guyton AC, Coleman TG, Cowley AV Jr, Scheel KW, Manning RD Jr, Norman RA Jr. Arterial pressure regulation. Overriding dominance of the kidneys in long-term regulation and in hypertension. Am J Med 52: 584–594, 1972. doi: 10.1016/0002-9343(72)90050-2. [DOI] [PubMed] [Google Scholar]
- 11.Guyton AC, Coleman TG, Cowley AW Jr, Liard JF, Norman RA Jr, Manning RD Jr. Systems analysis of arterial pressure regulation and hypertension. Ann Biomed Eng 1: 254–281, 1972. doi: 10.1007/BF02584211. [DOI] [PubMed] [Google Scholar]
- 12.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hall JE. Historical perspective of the renin-angiotensin system. Methods Mol Med 51: 3–21, 2001. [DOI] [PubMed] [Google Scholar]
- 14.Harnack LJ, Cogswell ME, Shikany JM, Gardner CD, Gillespie C, Loria CM, Zhou X, Yuan K, Steffen LM. Sources of sodium in US adults from 3 geographic regions. Circulation 135: 1775–1783, 2017. doi: 10.1161/CIRCULATIONAHA.116.024446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He FJ, Li J, Macgregor GA. Effect of longer-term modest salt reduction on blood pressure. Cochrane Database Syst Rev 4: CD004937, 2013. [DOI] [PubMed] [Google Scholar]
- 16.He FJ, Markandu ND, Sagnella GA, de Wardener HE, MacGregor GA. Plasma sodium: ignored and underestimated. Hypertension 45: 98–102, 2005. doi: 10.1161/01.HYP.0000149431.79450.a2. [DOI] [PubMed] [Google Scholar]
- 17.Heer M, Baisch F, Kropp J, Gerzer R, Drummer C. High dietary sodium chloride consumption may not induce body fluid retention in humans. Am J Physiol Renal Physiol 278: F585–F595, 2000. [DOI] [PubMed] [Google Scholar]
- 18.Jantsch J, Schatz V, Friedrich D, Schröder A, Kopp C, Siegert I, Maronna A, Wendelborn D, Linz P, Binger KJ, Gebhardt M, Heinig M, Neubert P, Fischer F, Teufel S, David JP, Neufert C, Cavallaro A, Rakova N, Küper C, Beck FX, Neuhofer W, Muller DN, Schuler G, Uder M, Bogdan C, Luft FC, Titze J. Cutaneous Na+ storage strengthens the antimicrobial barrier function of the skin and boosts macrophage-driven host defense. Cell Metab 21: 493–501, 2015. doi: 10.1016/j.cmet.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jörg S, Kissel J, Manzel A, Kleinewietfeld M, Haghikia A, Gold R, Müller DN, Linker RA. High salt drives Th17 responses in experimental autoimmune encephalomyelitis without impacting myeloid dendritic cells. Exp Neurol 279: 212–222, 2016. doi: 10.1016/j.expneurol.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 20.Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, Delpire E, Harrison DG, McDonough AA. Renal transporter activation during angiotensin-II hypertension is blunted in interferon-γ−/− and interleukin-17A−/− mice. Hypertension 65: 569–576, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim D, Wang J, Willingham SB, Martin R, Wernig G, Weissman IL. Anti-CD47 antibodies promote phagocytosis and inhibit the growth of human myeloma cells. Leukemia 26: 2538–2545, 2012. doi: 10.1038/leu.2012.141. [DOI] [PubMed] [Google Scholar]
- 22.Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, Trott DW, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen SC, Mernaugh RL, Laffer CL, Elijovich F, Davies SS, Moreno H, Madhur MS, Roberts J II, Harrison DG. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest 124: 4642–4656, 2014. doi: 10.1172/JCI74084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, Hafler DA. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496: 518–522, 2013. doi: 10.1038/nature11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kopp C, Linz P, Dahlmann A, Hammon M, Jantsch J, Müller DN, Schmieder RE, Cavallaro A, Eckardt KU, Uder M, Luft FC, Titze J. 23Na magnetic resonance imaging-determined tissue sodium in healthy subjects and hypertensive patients. Hypertension 61: 635–640, 2013. doi: 10.1161/HYPERTENSIONAHA.111.00566. [DOI] [PubMed] [Google Scholar]
- 26.Lesperance LM, Gray ML, Burstein D. Determination of fixed charge density in cartilage using nuclear magnetic resonance. J Orthop Res 10: 1–13, 1992. doi: 10.1002/jor.1100100102. [DOI] [PubMed] [Google Scholar]
- 27.Lev-Ran A, Porta M. Salt and hypertension: a phylogenetic perspective. Diabetes Metab Res Rev 21: 118–131, 2005. doi: 10.1002/dmrr.539. [DOI] [PubMed] [Google Scholar]
- 28.Ludwig CF. 1842–a landmark in nephrology: Carl Ludwig’s revolutionary concept of renal function. Kidney Int Suppl 46: 1–23, 1994. [PubMed] [Google Scholar]
- 29.Luft FC, Miller JZ, Grim CE, Fineberg NS, Christian JC, Daugherty SA, Weinberger MH. Salt sensitivity and resistance of blood pressure. Age and race as factors in physiological responses. Hypertension 17, Suppl: I102–I108, 1991. doi: 10.1161/01.HYP.17.1_Suppl.I102. [DOI] [PubMed] [Google Scholar]
- 30.Machnik A, Dahlmann A, Kopp C, Goss J, Wagner H, van Rooijen N, Eckardt KU, Müller DN, Park JK, Luft FC, Kerjaschki D, Titze J. Mononuclear phagocyte system depletion blocks interstitial tonicity-responsive enhancer binding protein/vascular endothelial growth factor C expression and induces salt-sensitive hypertension in rats. Hypertension 55: 755–761, 2010. doi: 10.1161/HYPERTENSIONAHA.109.143339. [DOI] [PubMed] [Google Scholar]
- 31.Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, Park JK, Beck FX, Müller DN, Derer W, Goss J, Ziomber A, Dietsch P, Wagner H, van Rooijen N, Kurtz A, Hilgers KF, Alitalo K, Eckardt KU, Luft FC, Kerjaschki D, Titze J. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med 15: 545–552, 2009. doi: 10.1038/nm.1960. [DOI] [PubMed] [Google Scholar]
- 32.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 55: 500–507, 2010. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mattson DL. Infiltrating immune cells in the kidney in salt-sensitive hypertension and renal injury. Am J Physiol Renal Physiol 307: F499–F508, 2014. doi: 10.1152/ajprenal.00258.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H. Genetic mutation of recombination activating gene 1 in Dahl salt-sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol 304: R407–R414, 2013. doi: 10.1152/ajpregu.00304.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34a.Moore LL, Singer RM, Bradlee ML. Low sodium intakes are not associated with lower blood pressure levels among Framingham Offspring Study adults. FASEB J 31, Suppl: 446.6, 2017. [Google Scholar]
- 35.Murray RH, Luft FC, Bloch R, Weyman AE. Blood pressure responses to extremes of sodium intake in normal man. Proc Soc Exp Biol Med 159: 432–436, 1978. doi: 10.3181/00379727-159-40364. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res 97: 696–704, 2013. doi: 10.1093/cvr/cvs422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okuda T, Grollman A. Passive transfer of autoimmune induced hypertension in the rat by lymph node cells. Tex Rep Biol Med 25: 257–264, 1967. [PubMed] [Google Scholar]
- 38.Salgado E, Bes-Rastrollo M, de Irala J, Carmona L, Gómez-Reino JJ. High sodium intake is associated with self-reported rheumatoid arthritis: a cross sectional and case control analysis within the SUN cohort. Medicine (Baltimore) 94: e0924, 2015. doi: 10.1097/MD.0000000000000924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schafflhuber M, Volpi N, Dahlmann A, Hilgers KF, Maccari F, Dietsch P, Wagner H, Luft FC, Eckardt KU, Titze J. Mobilization of osmotically inactive Na+ by growth and by dietary salt restriction in rats. Am J Physiol Renal Physiol 292: F1490–F1500, 2007. doi: 10.1152/ajprenal.00300.2006. [DOI] [PubMed] [Google Scholar]
- 40.Svendsen UG. Evidence for an initial, thymus independent and a chronic, thymus dependent phase of DOCA and salt hypertension in mice. Acta Pathol Microbiol Scand A 84: 523–528, 1976. [DOI] [PubMed] [Google Scholar]
- 41.Svendsen UG. The importance of thymus in the pathogenesis of the chronic phase of hypertension in mice following partial infarction of the kidney. Acta Pathol Microbiol Scand A 85: 539–547, 1977. [DOI] [PubMed] [Google Scholar]
- 42.Titze J, Krause H, Hecht H, Dietsch P, Rittweger J, Lang R, Kirsch KA, Hilgers KF. Reduced osmotically inactive Na storage capacity and hypertension in the Dahl model. Am J Physiol Renal Physiol 283: F134–F141, 2002. doi: 10.1152/ajprenal.00323.2001. [DOI] [PubMed] [Google Scholar]
- 43.Titze J, Lang R, Ilies C, Schwind KH, Kirsch KA, Dietsch P, Luft FC, Hilgers KF. Osmotically inactive skin Na+ storage in rats. Am J Physiol Renal Physiol 285: F1108–F1117, 2003. doi: 10.1152/ajprenal.00200.2003. [DOI] [PubMed] [Google Scholar]
- 44.Titze J, Luft FC, Bauer K, Dietsch P, Lang R, Veelken R, Wagner H, Eckardt KU, Hilgers KF. Extrarenal Na+ balance, volume, and blood pressure homeostasis in intact and ovariectomized deoxycorticosterone-acetate salt rats. Hypertension 47: 1101–1107, 2006. doi: 10.1161/01.HYP.0000221039.17735.1a. [DOI] [PubMed] [Google Scholar]
- 45.Titze J, Shakibaei M, Schafflhuber M, Schulze-Tanzil G, Porst M, Schwind KH, Dietsch P, Hilgers KF. Glycosaminoglycan polymerization may enable osmotically inactive Na+ storage in the skin. Am J Physiol Heart Circ Physiol 287: H203–H208, 2004. doi: 10.1152/ajpheart.01237.2003. [DOI] [PubMed] [Google Scholar]
- 46.Wang P, Deger MS, Kang H, Ikizler TA, Titze J, Gore JC. Sex differences in sodium deposition in human muscle and skin. Magn Reson Imaging 36: 93–97, 2017. doi: 10.1016/j.mri.2016.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weinberger MH, Fineberg NS, Fineberg SE, Weinberger M. Salt sensitivity, pulse pressure, and death in normal and hypertensive humans. Hypertension 37: 429–432, 2001. doi: 10.1161/01.HYP.37.2.429. [DOI] [PubMed] [Google Scholar]
- 48.Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, Karbach SH, Schwenk M, Yogev N, Schulz E, Oelze M, Grabbe S, Jonuleit H, Becker C, Daiber A, Waisman A, Münzel T. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation 124: 1370–1381, 2011. doi: 10.1161/CIRCULATIONAHA.111.034470. [DOI] [PubMed] [Google Scholar]
- 49.White FN, Grollman A. Autoimmune Factors Associated with Infarction of the Kidney. Nephron 1: 93–102, 1964. doi: 10.1159/000179322. [DOI] [PubMed] [Google Scholar]
- 50.Yi B, Titze J, Rykova M, Feuerecker M, Vassilieva G, Nichiporuk I, Schelling G, Morukov B, Choukèr A. Effects of dietary salt levels on monocytic cells and immune responses in healthy human subjects: a longitudinal study. Transl Res 166: 103–110, 2015. doi: 10.1016/j.trsl.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yilmaz R, Akoglu H, Altun B, Yildirim T, Arici M, Erdem Y. Dietary salt intake is related to inflammation and albuminuria in primary hypertensive patients. Eur J Clin Nutr 66: 1214–1218, 2012. doi: 10.1038/ejcn.2012.110. [DOI] [PubMed] [Google Scholar]
- 52.Yoon PW, Gillespie CD, George MG, Wall HK; Centers for Disease Control and Prevention (CDC) . Control of hypertension among adults–National Health and Nutrition Examination Survey, United States, 2005-2008. MMWR Suppl 61: 19–25, 2012. [PubMed] [Google Scholar]
- 53.Zhang WC, Zheng XJ, Du LJ, Sun JY, Shen ZX, Shi C, Sun S, Zhang Z, Chen XQ, Qin M, Liu X, Tao J, Jia L, Fan HY, Zhou B, Yu Y, Ying H, Hui L, Liu X, Yi X, Liu X, Zhang L, Duan SZ. High salt primes a specific activation state of macrophages, M(Na). Cell Res 25: 893–910, 2015. doi: 10.1038/cr.2015.87. [DOI] [PMC free article] [PubMed] [Google Scholar]