

Abstract
We previously proposed a role for the two-pore domain potassium (K2P) channel TREK-1 in hyperoxia (HO)-induced lung injury. To determine whether redundancy among the three TREK isoforms (TREK-1, TREK-2, and TRAAK) could protect from HO-induced injury, we now examined the effect of deletion of all three TREK isoforms in a clinically relevant scenario of prolonged HO exposure and mechanical ventilation (MV). We exposed WT and TREK-1/TREK-2/TRAAK-deficient [triple knockout (KO)] mice to either room air, 72-h HO, MV [high and low tidal volume (TV)], or a combination of HO + MV and measured quasistatic lung compliance, bronchoalveolar lavage (BAL) protein concentration, histologic lung injury scores (LIS), cellular apoptosis, and cytokine levels. We determined surfactant gene and protein expression and attempted to prevent HO-induced lung injury by prophylactically administering an exogenous surfactant (Curosurf). HO treatment increased lung injury in triple KO but not WT mice, including an elevated LIS, BAL protein concentration, and markers of apoptosis, decreased lung compliance, and a more proinflammatory cytokine phenotype. MV alone had no effect on lung injury markers. Exposure to HO + MV (low TV) further decreased lung compliance in triple KO but not WT mice, and HO + MV (high TV) was lethal for triple KO mice. In triple KO mice, the HO-induced lung injury was associated with decreased surfactant protein (SP) A and SPC but not SPB and SPD expression. However, these changes could not be explained by alterations in the transcription factors nuclear factor-1 (NF-1), NKX2.1/thyroid transcription factor-1 (TTF-1) or c-jun, or lamellar body levels. Prophylactic Curosurf administration did not improve lung injury scores or compliance in triple KO mice.
Keywords: TREK, TRAAK, lung, lung injury, acute respiratory distress syndrome, ARDS, acute lung injury, ALI, surfactant hyperoxia, mechanical stretch, ventilator-associated injury
INTRODUCTION
We have previously reported the expression of the stretch-activated, two-pore-domain potassium (K2P) channels TREK-1, TREK-2, and TRAAK in cultured alveolar epithelial cells (AECs) (100), and proposed a role for TREK-1 in the development of hyperoxia (HO)-induced lung injury (102). The K2P family consists of six subfamilies (TWIK, TREK, TASK, TALK, THIK, and TRESK) (26). Our group is particularly interested in the role of the TREK subfamily as potential regulators of HO- and mechanical ventilation (MV)-induced lung injury, since these channels are exquisitely sensitive to mechanical stretch (7) and HO (100). In fact, in cultured human AECs and mouse lungs, HO exposure downregulated TREK-1 expression, which was associated with altered inflammatory cytokine secretion both in vitro (99, 101) and in vivo (102). In a previous study, we exposed TREK-1-deficient mice to HO alone for 24 h or to the combination of HO and injurious, high tidal volume MV (MVH; 25 ml/kg). HO exposure by itself significantly increased lung injury in TREK-1-deficient but not in wild-type (WT) control mice, including worsening lung compliance, increased lung injury scores, and increased epithelial cell apoptosis (102). Nevertheless, the contribution of the other members of the TREK subfamily, TREK-2 and TRAAK, to HO- and MV-induced lung injury remains unknown.
This study was designed to determine the role of the entire TREK subfamily of K2P channels (TREK-1, TREK-2, and TRAAK) in the development of HO- and MV-induced lung injury using a clinically relevant model. Most patients with acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are not exposed to short periods of HO and injurious MVH, but rather to prolonged periods of HO and lung protective, low tidal volume MV (MVL). In this study we created a model more consistent with these clinical scenarios by treating WT and TREK-1/TREK-2/TRAAK-deficient (triple KO) mice with either HO alone for 3 days or a combination of HO followed by MVL. In addition to alterations in markers of lung injury, cellular apoptosis, and cytokine profiles in triple KO mice, we were interested in studying changes in surfactant protein production and overall surfactant protein levels in these mice following exposure to HO and MV.
Surfactant molecules are lipoprotein complexes (80% phospholipids, 10% cholesterol, and 10% protein) that are synthesized in alveolar type II (AT II) cells and packaged into lamellar bodies before secretion (131). The four protein subtypes are divided into the hydrophilic surfactant proteins SPA and SPD and hydrophobic SPB and SPC (14). The best known function of surfactant proteins is the reduction of alveolar surface tension at the air-liquid interface, but each surfactant protein exerts several additional important functions that contribute to lung health and disease (126). SPA and SPD are also called collectins and bind pathogens, destroy microbial membranes, and regulate microbial phagocytosis and alveolar macrophage responses (10). SPA is quantitatively the most abundant surfactant protein in the lung and enhances macrophage chemotaxis (24), inhibits TNF-α release in asthmatics (49), promotes T-cell expansion in lipopolysaccharide (28)- and Staphylococcus aureus (104)-induced sepsis, protects against graft vs. host responses (28), and, importantly, counteracts the surfactant-inhibitory effects of plasma proteins released during lung injury and ARDS (117). Furthermore, SPA influences the prevalence, types, and functions of CD4+ T cells in the lung during inflammation (85) and plays an important role in bleomycin-induced lung injury by preventing epithelial cell apoptosis and loss of epithelial integrity, as well as by decreasing proinflammatory cytokine secretion (27). Interestingly, this phenotype can be rescued with intratracheal SPA administration (27). Both SPA and SPD are protective against pulmonary aspergillosis (70), and SPD reduces alveolar macrophage apoptosis (12). SPB and SPC are best known for their ability to regulate lamellar body formation and overall surfactant protein secretion (87). In fact, deficiency of SPB leads to disorganized lamellar bodies and failure to properly package surfactant phospholipids (109). In addition, SPB and SPC may possess immunoregulatory properties similar to SPA and SPD and support the stability of airways (81). However, SPB and SPC are also known to impair the lung’s ability to counteract lipopolysaccharide (22)- and bleomycin (62)-induced inflammation.
Regulation of surfactant protein synthesis and secretion occurs primarily at gene transcription and secretory levels. Several transcription factors are involved in surfactant protein gene expression (130), including NKX2.1/thyroid transcription factor-1 (TTF-1) (25, 32, 74, 120), nuclear factor-1 (NF-1) (3, 120), c-jun (11), and STAT 3 (75, 129). In general, gene transcription is followed by splicing of surfactant protein precursors, packaging into lamellar bodies, translocating these secretory vesicles to the plasma membrane, and secreting surfactant protein into the alveolar space. Importantly, while mechanical stretch has been reported to induce surfactant protein secretion from AT II cells (21, 124), a regulatory role for ion channels, including K2P channels, in this process has never been documented. Here we report that mice lacking the entire TREK subfamily of K2P channels (TREK-1, TREK-2, and TRAAK) suffered increased lung injury during prolonged HO exposure, whereas MVL by itself was tolerated well. The HO-induced lung damage in TREK-1/TREK-2/TRAAK-deficient mice was not prevented by prophylactic administration of exogenous surfactant (Curosurf), which has been previously employed in clinical trials (69).
MATERIALS AND METHODS
Animals.
Mice lacking TREK-1, TREK-2, and TRAAK (triple KO) were generated as described (34). Null mutations were backcrossed against the C57BL/6J inbred strain for 10+ generations before establishment of the breeding cages to generate subjects for this study. Age- and sex-matched C57bl/6J WT mice, aged 9–12 wk, were obtained from Jackson Laboratories (https://www.jax.org/). All animals were housed in same-sex groups of two to five mice after weaning and provided with food and water ad libitum. All experiments were performed in accordance with animal protocols approved by the University of California Los Angeles and the University of Tennessee Health Science Center animal research committees.
Ventilation protocol.
Mice were initially divided into six experimental groups: 1) room air, no MV; 2) exposure to 95% HO for 72 h using a rodent HO chamber (BioSpherix) and a ProOx C21 O2/CO2 controller (BioSpherix); 3) low tidal volume MV (MVL) for 2 h using the Flexivent system (SQIREC) or an Ugo Basile mouse ventilator (Ugo Basile, Comerio, Italy), using a tidal volume of 6 ml/kg, RR of 100 breaths/min, and positive end-expiratory pressure 3 mmH2O); 4) high tidal volume MV (MVH; 25 ml/kg, RR of 100 breaths/min, and positive end-expiratory pressure of 3 mmH2O) for 2 h; 5) 95% HO exposure for 72 h followed by MVL 2-h MVL; and 6) 95% HO exposure for 72 h followed by 2-h MVH. The MVH condition has consistently proven to be injurious and result in a phenotype similar to the one observed in patients with ARDS (71, 72), whereas the MVL condition resembles more closely the clinically employed low tidal volume, lung-protective ventilation strategies (17, 36). Adequate anesthesia was provided with an intraperitoneal injection of a ketamine/xylazine mixture (10 mg/kg ketamine and 20 mg/kg xylazine), and 1–1.5% continuously inhaled isofluorane using a precision gas mixer (PEGAS 400; Columbus Instruments, OH). MVH or MVL was provided using a tracheostomy with an 18-gauge steel needle. Quasistatic lung compliance was measured mice were under general anesthesia. Pressure-volume curves (P-V) were recorded at the beginning and the end of each experiment. Each set of P-V curves was preceded by an inflation maneuver to total lung capacity to ensure equal standard volumes for each experiment. Rectal temperature was maintained within the normal range for mice using a heat lamp. At the end of MV, a second lung compliance measurement was performed and values were calculated by fitting data derived from the P-V curves to the Salazar-Knowles equation as previously described (95). In addition, mice were given intraperitoneal injections of 100 µl of sterile saline every hour on the ventilator to maintain a euvolemic fluid balance and to correct for insensible losses. At the end of each experiment, mice were killed by cardiac puncture and a 4% isofluorane overdose. Compliance data in Fig. 1 are depicted as a box-whisker plot, with median values, first and third quartiles, and maximum and minimum values.
Fig. 1.

Prolonged hyperoxia (HO) exposure caused increased lung injury in TREK-1/TREK-2/TRAAK-deficient mice. A: quaisstatic compliance data of wild-type (WT) control and TREK-1/TREK-2/TRAAK-deficient [triple knockout (TKO)] mice are represented in a box-whisker plot, with median values, 1st and 3rd quartiles, and maximum and minimum values. Exposure to 95% HO exposure for 72 h decreased the quasistatic lung compliance of triple KO but not WT mice, whereas low mechanical ventilation (MV; 6 ml/kg for 2 h) or high mechanical ventilation (MVH; 25 ml/kg for 2 h) alone had no effect in either mouse type. The combination of HO + MVL decreased the quasistatic lung compliance of triple KO but not WT mice. HO + MVH decreased quasistatic lung compliance of WT mice and was lethal for triple KO mice (n = 6–20 animals per group; *comparison of WT and triple KO mice within 1 treatment group; #compared with untreated WT control mice; ^compared with untreated triple KO mice). B: representative hematoxylin and eosin (H&E)-stained lung sections of WT control and triple KO mice after exposure to room air (RA), 95% HO for 72 h, MVL for 2 h, or the combination of HO + MVL, showing increased injury in HO-exposed triple KO mice compared with HO-treated WT mice. WT and triple KO mice exposed to HO + MVL showed a similar increase in lung injury (n = 5–6 for each treatment group; *comparison of WT and triple KO mice within 1 treatment group; #compared with untreated WT control mice; ^compared with untreated triple KO mice). C: summary of the composite lung injury scores (LIS) for each experimental condition shown in B. D: total bronchoalveolar lavage (BAL) fluid protein concentrations (n = 5–7 animals per group; *comparison of WT and triple KO mice within 1 treatment group; #compared with untreated WT control mice; ^compared with untreated triple KO mice).
Lung histology and bronchoalveolar lavage fluid collection.
At the end of the experiment, lung tissue was harvested and processed for histological examination as described previously (72). Briefly, the heart and lungs were removed en bloc and gently retrograde perfused to remove red blood cells from the vasculature using 10 ml PBS in a 10-ml syringe and by applying minimal pressure to avoid interoperator variability. With the use of a 1-ml syringe, bronchoalveolar lavage (BAL) was performed two times with 1ml PBS/0.6 mM EDTA each. The lungs were immediately fixed in 4% formalin, and paraffin-embedded sections were cut into 4-µm thick tissues slices using a microtome. Lung sections were then stained with an hematoxylin and eosin (H&E) stain for histological analysis. Total protein concentrations were measured using the Bradford assay (Bio-Rad, Hercules, CA). Lung injury scores (LIS) were determined by an investigator blinded to the experimental conditions on H&E-stained lung sections using a modified three-criteria version of our previously published four-criteria LIS (72). Our three-criteria score contained the following: 1) interstitial and alveolar edema, 2) cellular infiltrate, and 3) parenchymal and perivascular hemorrhage. Each of the three criteria was assigned a score between 0 and 3, with “0” representing no injury, “1” representing mild injury, “2” representing moderate injury, and “3” representing severe injury. Four random high-power fields per slide were scored under ×40 magnification and averaged for each criterion. The histological composite LIS (see Figs. 1 and 9) represents the mean value of the three parameters on the above scale from 0 to 3 for each mouse. All histological analyses were performed using a Motic AE20/21 inverted microscope.
Fig. 9.
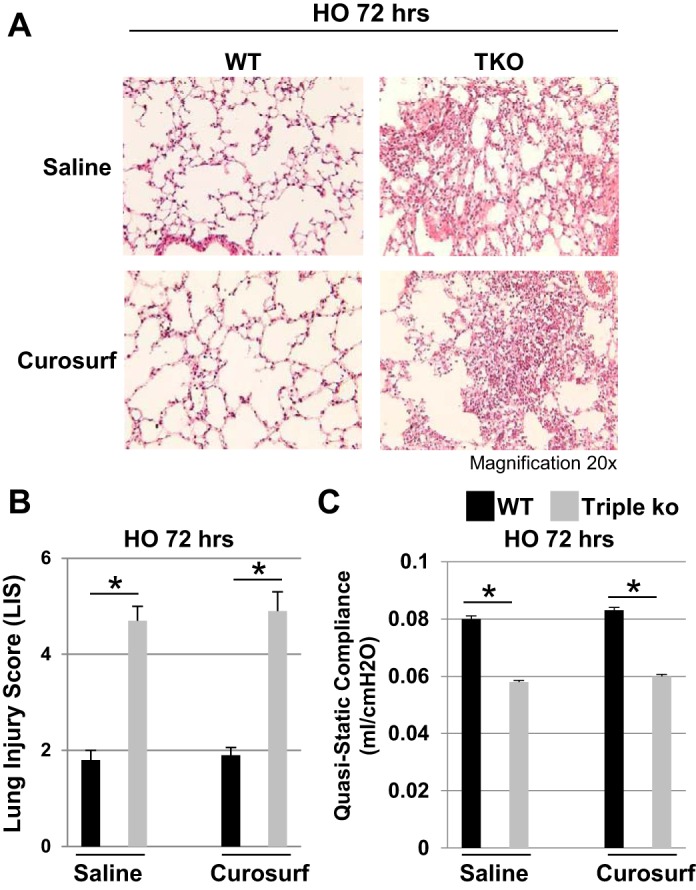
Prophylactic oropharyngeal Curosurf administration did not prevent HO-induced lung injury in TREK-1/TREK-2/TRAAK-deficient mice. A: representative H&E-stained lung sections of HO-treated WT control and triple KO lungs showing that prophylactic Curosurf administration could not prevent the HO-induced lung damage in triple KO mice. B and C: there is no improvement in LIS or quasistatic lung compliance, respectively, in Curosurf-treated triple KO mice (n = 5 mice per group; *comparison between WT control and triple KO mice within the saline-treated or Curosurf-treated groups, respectively).
Confocal immunofluorescence microscopy.
Individual lung lobes were removed from the mice and fixed in 4% paraformaldehyde for 24 h and then transferred into 70% ethanol for storage. Before being sectioned, the lungs were placed into gradually increasing ethanol concentrations ranging from 90 to 95 to 100% for 30 min each time. Lungs sections were then cleared with Citrisolv (Fisher Scientific) in three 30-min washes and embedded in paraffin wax for sectioning. For immunostaining, the sections were incubated with an anti-proSPC antibody (1:300; Millipore, Billerica, MA); an anti-SPA antibody (SFTPA1; 1:200; Sigma, St. Louis); an anti-ATP-binding cassette, subfamily A, member 3 (ABCA3)/lamellar body antibody (1:300; SevenHills Bioreagents, Cincinnati, OH); a T1alpha antibody (1:100; Developmental Studies Hybridoma Bank); and an anti-zonula occludens-1 (ZO1) antibody (1:100; Thermo Fisher), for 24 h at 4°C. Lung sections were washed three times, and the following species-specific secondary antibodies were applied at a 1:500 dilution for 1 h at room temperature: for anti-T1alpha, goat anti-syrian hamster Dylight594 (Jackson Immunoresearch); for anti-SPA, donkey-anti-rabbit FITC (Millipore AP182F); for anti-proSPC, goat anti-rabbit Cy3 (Jackson Immunoresearch); for anti-ABCA3, goat anti-rabbit Cy3 (Jackson Immunoresearch); and for anti-ZO1: goat anti-mouse Cy3 (Abcam 97035). The sections were then mounted in Fluoro Gel II mounting medium with DAPI (EMS, Hatfield, PA).
Terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) staining was performed according to the manufacturer’s instructions using a fluorescence-based In Situ Cell Death Detection kit (Roche, Burlington, NC). Nuclear staining was obtained using Fluoro Gel II mounting medium containing DAPI (Electron Microscopy Sciences, Hatfield, PA). Images were acquired using a Zeiss 710 or Nikon Eclipse E600 imaging system. Emitted fluorescence was collected using a ×20, ×40, or ×63 magnification objective lenses. All images were recorded using Zen 2009 Light Edition software (Zeiss) or Metamorph Software 7.8.6.0.
Western blot analysis.
Tissue samples for Western blot analysis were placed in RIPA buffer (50 mM Tris·HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% Nonidet P-40, and 0.1% SDS) containing a protease inhibitor cocktail (Roche) on ice and stored at −80°C. Frozen lung tissue was then thawed on ice, and whole lung homogenates were prepared using a tissue homogenizer in RIPA buffer containing the protease inhibitor cocktail. Homogenates were centrifuged at 17,000 g for 15 min at 4°C, and total protein concentrations were determined using the Bradford assay (Bio-Rad, Hercules, CA). A total of 50 µg protein of each sample was separated by SDS-PAGE on 4–12% NuPage Bis-Tris gradient gels (Invitrogen) and transferred onto nitrocellulose membranes at 35 mV for 2 h. Membranes were blocked in 5% nonfat dry milk in Tris-buffered saline (Bio-Rad) containing 0.1% Tween-20 (TBST) for 1 h at 37°C. The membranes were then incubated overnight with one of the following commercially available primary antibodies: an anti-proSPC antibody (1:300; Millipore, Billerica, MA); an anti-SPA antibody (SFTPA1; 1:300; Sigma); an anti-total poly (ADP)-ribose polymerase-1 (PARP-1; 1:1,000; Cell Signaling); an anti-cleaved PARP-1 antibody (1:1,00; Cell Signaling); an anti-NF-1 antibody (1:500; Sigma); an anti-NKX2.1/TTF-1 antibody (1:500; Abcam); an anti-total c-jun antibody (1:100; Santa Cruz Biotechnology); an anti-phospho c-jun antibody (1:100; Santa Cruz Biotechnology); an anti-ZO1 antibody (1:250; Thermo Fisher); and ABCA3/lamellar bodies (1:1,000) for 24 h at 4°C. The next day, after three washing steps in TBST, all membranes were incubated for 1 h at room temperature with a species-specific, horseradish peroxidase-conjugated IgG antibody (1:3,000; Cell Signaling). Gels and membranes for a given experiment were processed in parallel to minimize interassay variability. Bands were visualized by enhanced chemiluminescence with ECL SuperSignal West Dura Extended Duration Substrate (Thermo Scientific, Rockford, IL). For each treatment condition, tissue lysates from WT control and triple KO mice were run on the same gel to allow for comparison of protein expression levels between the two groups. Band densitometry measurements to determine relative quantities of protein were performed using ImageJ 1.42 software for Windows.
Surfactant gene expression by real-time PCR.
Tissue samples for real-time PCR were frozen in RNA lysis buffer (Agilent Technologies). Total RNA was isolated from whole lung homogenates using an Absolutely RNA Miniprep kit (Agilent Technologies) according to the manufacturer’s instructions. Contaminating genomic DNA was removed by treatment with a DNA Removal kit (Ambion, Austin, TX) according to the manufacturer’s directions. Single-stranded DNA was synthesized from 1 µg total RNA, and real-time PCR was performed using a Transcriptor First Strand cDNA kit (Roche) according to the manufacturer’s instructions. Real-Time PCR was performed using a TaqMan Gene Expression assay (Roche) using primers specific for each surfactant protein. The following primer sequences were used: SPA forward: ctggagaacatggagacaagg and reverse: aagctcctcatccaggtaagc; SPB forward: ttgtcctccgatgttccact and reverse: tgtcctgtagtggccattctt; SPC forward: ggtcctgatggagagtccac and reverse: gatgagaaggcgtttgaggt; and SPD forward: ggccttaaaaggaaaactacagc and reverse: ccatcagggaacaatgcag. The housekeeping gene HGPRT was used as an internal control. Primers and probes were purchased from Integrated DNA Technologies (Coralville, IA). Based on data from preliminary experiments, genes were amplified for 30 cycles to avoid product saturation. The rate of gene amplification was quantified using the ΔΔCt method as outlined in the manufacturer’s handbook. A greater than fivefold change in gene expression was considered significant.
Cytokine gene expression by conventional PCR.
Total RNA was isolated from whole lung homogenates using an Absolutely RNA Miniprep kit (Agilent Technologies) according to the manufacturer’s instructions. Contaminating genomic DNA was removed by treatment with a DNA Removal kit (Ambion) according to the manufacturer’s directions. Single-stranded DNA was synthesized from 1 µg total RNA, and reverse transcriptase-PCR was performed using a Transcriptor First Strand cDNA kit (Roche) according to the manufacturer’s instructions and then amplified with Taq polymerase for 30 cycles in an Eppendorf Mastercycler EP Thermal Cycler using primers specific for each cytokine. The following primer sequences were used: IL-6 forward: atggatgctaccaaactggat and reverse: ccaggtagttatggtactccaga; monocyte chemotactic protein-1 (MCP-1) forward: agtctctgccgc ccttct and reverse: gtgactggggcattgattg; and IL-18 forward: gcctcaaaccttccaaatca and reverse: tggatccatttcctcttcctcaaagg. The housekeeping gene GAPDH was used as an internal control. After amplification, 16 μl of each reaction mixture were separated by electrophoresis on 1.8% agarose gels and the bands visualized by ethidium bromide staining.
Alveolar type II isolation and culture.
Freshly isolated alveolar type II (AT II) cells from WT control and triple KO mice were obtained according to a previously published method (78). The AT II cell yield per mouse lung was 3–5 million cells with >90% purity assessed by immunostaining for pro-SPC. All experiments were performed in accordance with the Institutional Animal Care and Use Committee at the University of California Los Angeles guidelines and regulations. Freshly isolated AT II cells were seeded to 80–85% confluence at 3.5 × 106 cells/well in six-well tissue culture plates coated with fibronectin in AT II culture medium (DMEM with 10% FBS, 4 mM glutamine, 1% penicillin/streptomycin, and 0.25 µM amphotericin B), and experiments were started on days 2–3 after isolation.
MTT proliferation assay.
A MTT-based proliferation assay was used to determine cell growth rates in AT II cells isolated from untreated WT control and triple KO mice. The MTT assay measures cleavage of the yellow tetrazolium salt MTT to purple formazan crystals by metabolically active cells. The formazan is then solubilized and the concentration is determined by a standard microplate absorbance reader at 540 nm (66). Briefly, 1 × 104 cells/well were seeded into a 96-well plate and grown to 80–90% confluence fibronectin in AT II culture medium (DMEM with 10% FBS, 4 mM glutamine, 1% penicillin/streptomycin, and 0.25 µM amphotericin B). Samples were exposed to either 90–95% HO (with 5% CO2) or room air (with 5% CO2) for 48 h at 37°C. The MTT reagent was purchased from Invitrogen, and the assay was performed according the instructions provided in the Vybrant MTT Cell Proliferation kit (V-13154; Invitrogen). Before the assay was performed, the medium was changed to phenol red-free DMEM (with 10% FBS, 4 mM glutamine, 1% penicillin/streptomycin, and 0.25 µM amphotericin B) to avoid interference of phenol red with the colorimetric analysis, and supplemented with the MTT reagent.
IL-10 measurements by ELISA.
Briefly, BAL was performed as described above by lavaging each lung two times with 1 ml PBS containing 0.6 mM EDTA. The aliquots were combined, and supernatants were collected after ultracentrifugation at 8,000 rpm for 5 min. For IL-10 measurements, 100 μl of BAL supernatant were loaded into a 96-well ELISA plate. IL-10 levels were quantified using BD Bioscience OptEIA species-specific ELISA kits following the manufacturer’s instructions. All samples were run in triplicates. IL-10 amounts were displayed in picograms per milliliters.
Surfactant replacement via oropharyngeal aspiration.
To assure adequate surfactant (Curosurf/poractant alpha; Chiesi Farmaceutici) delivery to the lungs, in preliminary experiments we administered Evans blue dye by oropharyngeal aspiration as previously described in the literature (16, 60) to visualize and confirm the amount and distribution of dye within the lungs. Briefly, mice were anesthetized with inhaled isofuorane until they lost consciousness and then suspended by their cranial incisors on a 3.0-silk suture on a slanted stand. The nares were occluded with a mouse nose clip, and the tongue was gently extracted from the mouth using blunt forceps to visualize the base of the tongue and the retropharynx. Evans blue dye, Curosurf solution, or saline (40 µl each) was placed in the posterior pharynx with a micropipettor. Respiration was monitored to ensure the suspension was fully aspirated before the tongue and the nose clip were released. Mice were allowed to recover under a warming lamp until fully awake. The mice were alert and ambulatory within 4–5 min after the induction of anesthesia. There were no perianesthetic deaths associated with this procedure.
Of note, each 1 ml of Curosurf contains 80 mg of poractant alpha (surfactant extract) that includes 76 mg of phospholipids and 1 mg of protein, which consists of 0.45 mg SPB and 0.55 mg SPC. The amount of phospholipids is calculated from the content of phosphorus and contains 55 mg of phosphatidylcholine, of which 30 mg is dipalmitoylphosphatidylcholine. It is suspended in 0.9% NaCl solution and adjusted with sodium bicarbonate to a pH of 6.2. Once adequate delivery to the lungs was confirmed, we administered 40 μl Curosurf per day to each mouse under sedation for a total of four doses: 1) 24 h before HO exposure, 2) on day 1 of HO exposure, 3) on day 2 of HO exposure, and 4) on day 3 of HO exposure. At the end of the experiment, quasistatic lung compliance was measured using the Flexivent system and lung tissue was harvested for H&E histology and SPC determination by confocal microscopy (as described above).
Statistical analysis.
All values were expressed as means ± SE, except in the box-whisker plot in Fig. 1 where data are expressed as median values, first and third quartiles, and maximum and minimum values. Data were analyzed using the Student’s t-test, multivariate ANOVA, and pair-wise comparison of the means using the Tukey-Kramer method to adjust for multiple comparisons. All statistical analyses were performed using StatPlus software and SAS/STAT software, and P < 0.05 was considered significant. In the quantitative PCR experiments, a change in gene expression of greater than fivefold from baseline gene expression was considered significant.
RESULTS
Prolonged hyperoxia exposure increases lung damage in TREK-1/TREK-2/TRAAK-deficient lungs.
We determined the effects of prolonged HO exposure, alone or in combination with MVL or MVH, on the quasistatic lung compliance of TREK-1/TREK-2/TRAAK-deficient (triple KO) mice. Quasistatic lung compliance is a measure of the elastic recoil pressure of the lungs at a given lung volume, and a decrease in quasistatic lung compliance correlates with worsening lung injury in both patients and in animal models of lung injury (102, 114, 132). At baseline, WT control and triple KO mice had similar quasistatic lung compliance (Fig. 1A). Prolonged HO exposure (95% HO for 72 h) did not affect the quasistatic lung compliance of WT mice but significantly decreased the quasistatic lung compliance of triple KO mice (P < 0.01). Neither MVL (6 ml/kg for 2 h) nor MVH (25 ml/kg for 2 h) alone affected the quasistatic lung compliance of WT or triple KO mice. Preexposure to 72 h of HO followed by 2 h of MVL decreased the quasistatic lung compliance of triple KO mice compared with untreated triple KO mice but had no effect on WT mice. In contrast, preexposure to HO followed by MVH also decreased the quasistatic lung compliance of WT mice compared with untreated WT mice and was lethal for triple KO mice (Fig. 1A). Therefore, for the rest of this study, we eliminated the MVH condition and focused on HO and MVL.
To further define the lung injury observed in triple KO mice, we used histological analysis of H&E-stained lung sections (Fig. 1B) to determine LIS (Fig. 1C) and measured BAL fluid protein concentrations (Fig. 1D), as an indicator of alveolar-capillary barrier dysfunction. Similar to the changes in quasistatic lung compliance, we found an increase in LIS in triple KO mice after HO exposure but not in WT control mice. MVL alone had no effect on LIS in either WT (P = 0.06) or triple KO mice (P = 0.08) (Fig. 1C). The combination of HO + MVL increased the LIS to a similar degree in both WT control and triple KO mice. Furthermore, the BAL fluid protein levels were increased in triple KO but not WT control mice after HO exposure (Fig. 1D). MVL alone did not affect BAL fluid protein concentrations in either mouse type. Exposure to the combination of HO + MVL increased BAL fluid protein levels in both WT control and triple KO mice, but the increase was more pronounced in the triple KO mice.
Prolonged HO exposure decreases surfactant protein A and C levels in TREK-1/TREK-2/TRAAK-deficient lungs.
A reduction in surfactant proteins is well documented in animal models and patients with ALI/ARDS (30, 46). We investigated whether the increased lung injury observed in triple KO mice after prolonged HO exposure was associated with decreased surfactant protein levels. Using confocal immunofluorescence microscopy of lung sections and Western blotting of whole lung homogenates, we measured surfactant protein (SP) A (Fig. 2, A–C) and proSPC levels (Fig. 3, A–C). At baseline and after MVL exposure, lungs from WT and triple KO mice contained similar levels of SPA and proSPC. While HO exposure had no effect on SPA or proSPC levels in WT control mice, in triple KO mice it decreased SPA levels by 36% (Fig. 2, A–C) and proSPC levels by 82% (Fig. 3, A–C), respectively, when compared with untreated control mice for each group. Exposure to the combination of HO + MVL decreased SPA and proSPC levels in WT control and triple KO mice to similar degrees when compared with untreated control mice for each group.
Fig. 2.

HO exposure, but not MVL, decreased surfactant protein A (SPA) levels in TREK-1/TREK-2/TRAAK-deficient lungs. A: representative confocal immunofluorescence microscopy images showing that 95% HO exposure for 72 h decreased SPA protein levels in triple KO but not WT control lungs. MVL alone had no effect on either mouse type. HO + MVL decreased SPA protein levels in both WT control and triple KO lungs. SPA protein is shown in green and nuclei were counterstained in blue with DAPI (n = 5 mice per treatment group). B: representative Western blots using whole lung homogenates showing the effects of the above conditions on SPA protein expression. For a specific treatment condition, tissue lysates from WT control and triple KO lungs were run on the same gel to allow for comparison of protein expression levels between the 2 groups. C: normalized densitometry analysis of Western blot experiments (n = 4–5 mice per group; *comparison of WT control and triple KO mice within 1 treatment group; #compared with untreated WT control mice; ^compared with untreated triple KO mice).
Fig. 3.

HO exposure, but not MVL, decreased proSPC protein levels in TREK-1/TREK-2/TRAAK-deficient lungs. A: representative confocal immunofluorescence microscopy images showing that 95% HO exposure for 72 h decreased proSPC protein levels in triple KO but not WT control lungs. MVL alone had no effect on either mouse type. HO + MVL decreased proSPC protein levels in both WT control and triple KO mice. ProSPC protein is shown in red, and nuclei were counterstained in blue with DAPI (n = 5 mice per treatment group). B: representative Western blots using whole lung homogenates showing the effects of the above conditions on proSPC protein expression. For a specific treatment condition, tissue lysates from WT control and triple KO lungs were run on the same gel to allow for comparison of protein expression levels between the 2 groups. C: normalized densitometry analysis of Western blot experiments (n = 4–5 mice per group; *comparison of WT control and triple KO mice within 1 treatment group; #compared with untreated WT control mice; ^compared with untreated triple KO mice).
Prolonged HO exposure decreases SPA and SPC, but not SPB and SPD, gene expression levels in TREK-1/TREK-2/TRAAK-deficient lungs.
To determine whether the decrease in surfactant proteins following exposure of triple KO mice to prolonged HO was regulated at transcriptional levels and caused by a decrease in surfactant protein gene expression, we measured SPA, SPB, SPC, and SPD mRNA levels using semiquantitative, real-time PCR (Fig. 4, A–D). We found that HO exposure decreased SPA (Fig. 4A) and SPC (Fig. 4B) mRNA levels in triple KO but not WT control lungs, whereas SPB (Fig. 4C) and SPD (Fig. 4D) mRNA levels were not affected by HO exposure in either mouse type. MVL alone decreased SPC mRNA levels in both WT control and triple KO mice but to a similar degree (Fig. 4B). In contrast, MVL alone had no effect on SPA, SPB, or SPD mRNA levels in either mouse type. The combination of HO + MVL decreased SPA and SPC mRNA levels in both WT control and triple KO mice to a similar degree but had no effect on SPB and SPD mRNA levels in either mouse type.
Fig. 4.

HO exposure decreased SPA and SPC, but not SPB and SPD, mRNA levels in TREK-1/TREK-2/TRAAK-deficient lungs. A and B: decreased SPA and SPC mRNA expression, respectively, in triple KO lungs after 72-h HO exposure compared with WT control lungs. MVL alone did not affect SPA or SPC mRNA levels in either mouse type, whereas the combination of HO + MVL decreased SPA and SPC mRNA levels in both WT control and triple KO lungs. In contrast, SPB (C) and SPD (D) mRNA levels were not affected by the above conditions. Data were normalized to the mRNA expression level of each surfactant type in untreated WT control lungs (n = 4–5 mice per group). E, F, and G: representative Western Blot experiments of nuclear factor-1 (NF-1), NKX2.1, and c-jun protein expression in mouse lung homogenates of RA- or HO-treated mice. For a specific treatment condition, tissue lysates from WT control and triple KO lungs were run on the same gel to allow for comparison of protein expression levels between the 2 groups. H, I, and J: semiquantitative densitometry analysis of normalized NF-1, NKX2.1, and c-jun protein expression levels, respectively (n = 4–5 mice per group; *comparison of WT control and triple KO mice within 1 treatment group); #compared with untreated mice; ^compared with untreated triple KO mice.
To determine whether changes in the transcription factors NF-1, NKX2.1/TTF-1, and c-jun were responsible for the observed decrease in SPA and SPC gene expression in triple KO mice, we determined NF-1 (Fig. 4, E and H), NKX2.1 (Fig. 4, F and I), and c-jun (Fig. 4, G and J) gene expression levels by Western blotting in WT control and triple KO lungs after 72-h HO exposure. NF-1 levels were similar between WT control and triple KO mice at baseline and did not change with HO exposure (Figs. 4, E and H). Interestingly, at baseline NKX2.1 levels were higher in triple KO than WT control mice and did not change with HO exposure (Fig. 4, F and I). Phospho/total c-jun ratios were similar in WT control and triple KO mice at baseline but decreased in both mouse types after HO exposure (Fig. 4, G and J). These data suggest that neither NF-1, NKX2.1, nor c-jun mediated the decrease in SPA and SPC mRNA levels in HO-exposed triple KO lungs.
TREK-1/TREK-2/TRAAK-deficient lungs contain lower ABCA3/lamellar body levels than WT control lungs at baseline but not after HO exposure.
To determine whether lamellar body formation was compromised in triple KO mice, potentially explaining the decreased SPA and SPC levels in these mice, we measured ABCA3 protein levels (a lung-specific phospholipid transporter and membrane marker of lamellar bodies; Ref. 86) in lung homogenates from WT control and triple KO mice at baseline and after HO exposure. Using Western blotting (Fig. 5A), semiquantitative optical densitometry measurements (Fig. 5B), and confocal immunofluorescence microscopy (Fig. 5C), we found that at baseline triple KO mice contained lower ABCA3 protein levels than WT controls. However, HO exposure increased ABCA3 protein levels in both mouse types resulting in no further differences between WT control and triple KO mice. Therefore, it is unlikely that the decreased SPA and SPC levels in HO-exposed triple KO mice were due to alterations in lamellar body levels.
Fig. 5.

TREK-1/TREK-2/TRAAK-deficient lungs contained lower ATP-binding cassette, subfamily A, member 3 (ABCA3)/lamellar body levels than WT controls at baseline but not after HO exposure. At baseline, triple KO lungs contain lower ABCA3/lamellar body levels than WT controls, but HO exposure increased ABCA3/lamellar body levels in both WT control and triple KO lungs as shown by representative Western blot (A), semiquantitative optical densitometry measurements (B), and confocal immunofluorescence microscopy (C). ABCA3 protein is shown in red, and nuclei were counterstained in blue with DAPI (n = 5 mice per each group; *comparison of WT control and triple KO mice within 1 treatment group; #compared with untreated WT control mice; ^compared with untreated triple KO mice).
HO exposure increased cell apoptosis in TREK-1/TREK-2/TRAAK-deficient lungs.
To investigate whether cell apoptosis contributed to the decreased SPA and SPC levels observed in triple KO mice after HO exposure, we determined total and cleaved PARP-1 levels using Western blotting (Fig. 6A) and quantified PARP-1 activation by calculating cleaved/total PARP-1 ratios (Fig. 6B). HO exposure increased cleaved/total PARP-1 levels in triple KO lungs compared with untreated control mice by 60%, whereas MVL alone had no effect on PARP-1 activation in WT control or triple KO mice. Exposure to the combination of HO + MVL increased cleaved/total PARP-1 levels in both WT control and triple KO lungs to a similar degree when compared with their respective untreated control mice (Fig. 6, A and B). Similar to PARP-1 activation, TUNEL staining revealed an increased number of apoptotic cells following HO exposure in both WT control and triple KO lungs when compared with their respective untreated control mice, but the effect was more pronounced in triple KO lungs (Fig. 6C).
Fig. 6.

HO exposure increased cellular apoptosis in TREK-1/TREK-2/TRAAK-deficient lungs. A: representative Western blot showing an increase in cleaved poly (ADP)-ribose polymerase-1 (PARP-1) levels in HO-exposed triple KO lungs compared with WT control lungs. For a specific treatment condition, tissue lysates from WT control and triple KO lungs were run on the same gel to allow for comparison of protein expression levels between the 2 groups. B: densitometry analysis of cleaved/total PARP-1 ratios. C: quantification of terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL)-positive (apoptotic) cells in RA- and HO-exposed WT control and triple KO lungs (n = 4–5 mice per group). D: MTT proliferation assay of alveolar type II cells isolated from untreated WT control and untreated triple KO lungs and cultured for 48 h under either RA or 95% HO conditions (AT II cells were isolated from n = 5 mice for each treatment group; *comparison between HO-exposed AT II cells from WT control and triple KO mice; #comparison of HO-exposed WT control AT II cells to RA-exposed WT control cells). E and F: representative confocal immunofluorescence images of TUNEL-positive AT I cells (T1alpha-positive) and AT II cells (proSPC-positive) in lung sections of WT control and triple KO lung sections (n = 4–5; *comparison of WT control and triple KO mice within 1 treatment group; #compared with untreated WT control mice; ^compared with untreated triple KO mice).
To assess whether both AT I and AT II cells undergo apoptosis following HO exposure, we used confocal immunofluorescence microscopy and triple-stained WT control and triple KO lung sections for TUNEL + T1alpha + DAPI (Fig. 6E) or TUNEL + proSPC + DAPI (Fig. 6F) to detect AT I and AT II cells, respectively. We found that 72-h HO exposure caused apoptosis in both cell types.
To determine whether alterations in epithelial cell proliferation contribute to the increased injury seen in triple KO lungs, we compared cell proliferation rates of AT II cells isolated from WT control and triple KO mice that were cultured either at room air or under HO conditions (48 h; Fig. 6D). AT II cells from WT control and triple KO mice showed similar proliferation rates when cultured at room air, whereas HO exposure induced AT II cell proliferation in WT-derived but not triple KO-derived AT II cells.
We also measured Gadd45a levels (a marker induced by DNA damage and other stress signals associated with growth arrest and apoptosis; Ref. 96) by Western blot since in preliminary experiments we found a fivefold upregulation of Gadd45a mRNA in triple KO lungs after 72-h HO exposure (unpublished data). However, in contrast to Gadd45a gene expression, we found no changes in Gadd45a protein expression levels in lung homogenates of WT control or triple KO mice following HO, MVL, or HO + MVL exposure (data not shown). Therefore, in contrast to PARP-1, changes in Gadd45a expression are unlikely contributing to the increased cell death found in triple KO lungs following HO exposure.
Cytokine levels and tight junction protein expression in HO-exposed TREK-1/TREK-2/TRAAK-deficient lungs.
To determine whether an exacerbated inflammatory cytokine response contributed to the increased injury seen in HO-exposed triple KO lungs, we measured IL-6, MCP-1, IL-18, and IL-22 mRNA levels (Fig. 7, A and B) in whole lung homogenates, as well as IL-10 protein levels in the BAL fluid of WT control and triple KO lungs (Fig. 7C). Interestingly, IL-6 mRNA levels were increased in the lung homogenates of triple KO mice both at baseline and after HO exposure, potentially making these mice more vulnerable to HO-induced injury. MCP-1 mRNA levels were undetectable at baseline in both WT control and triple KO lungs but increased in triple KO lungs after HO exposure. In contrast, IL-18 mRNA levels were similar both at baseline and after HO exposure in both mouse types. Baseline IL-22 mRNA levels were similar in lung homogenates from WT control and triple KO lungs (Fig. 7B). However, HO exposure decreased IL-22 mRNA levels in triple KO but not WT control lung homogenates. Interestingly, in contrast to lung homogenate IL-22 mRNA levels, IL-22 protein levels in the BAL fluid were either very low or undetectable by ELISA (data not shown), suggesting that IL-22 exerts its function predominantly in extra-alveolar regions. Baseline IL-10 protein levels were similar in the BAL fluid of untreated control WT control and triple KO lungs (Fig. 7C). After HO exposure, BAL IL-10 levels decreased in triple KO but not WT control lungs. Taken together, these cytokine changes point toward an exaggerated proinflammatory response in triple KO mice following HO exposure.
Fig. 7.

Cytokine and zonula occludens-1 (ZO1) expression in TREK-1/TREK-2/TRAAK-deficient lungs. A: Representative images of IL-6, monocyte chemotactic protein-1 (MCP-1), and IL-18 mRNA levels detected in whole lung homogenates of WT control and triple KO lungs by conventional PCR. B: IL-22 levels measured in whole lung homogenates of WT control and triple KO mice by semiquantitative, real-time PCR (data are depicted as fold-change in gene expression normalized to WT RA). C: IL-10 levels in BAL fluid of WT control and triple KO lungs measured by ELISA. D: similar levels of ZO1 expression by confocal immunofluorescent microscopy in WT control and triple KO lung slices at baseline and after 72-h HO exposure (n = 5–6 mice per group; *comparison of WT control and triple KO mice within 2 treatment groups; ^compared with untreated triple KO mice).
Since triple KO lungs contained higher BAL fluid protein levels suggesting an impairment in alveolar barrier function, we studied ZO1 expression in WT control and triple KO lungs before and after HO exposure using confocal immunofluorescence microscopy (Fig. 7D). We found similar levels of ZO1 expression at baseline in both mouse types and no significant changes with HO treatment.
Surfactant repletion did not prevent HO-induced lung injury in TREK-1/TREK-2/TRAAK-deficient lungs.
To counteract the decrease in SPA and SPC levels seen in HO-exposed triple KO lungs and to prevent the increased lung injury in these mice, we administered a total of four doses of Curosurf (40 μl/dose) to WT control and triple KO mice. Details of this protocol are depicted in the schematic timeline in Fig. 8A. The first dose was given 24 h before HO exposure. The following three Curosurf doses (each 40 μl/dose) were given in 24-h intervals on each day of HO exposure (for 3 days). Curosurf is a surfactant mixture commonly used in clinical practice containing SPB and SPC proteins. To confirm adequate delivery to the lungs by oropharyngeal aspiration, in a preliminary experiment we used Evans blue dye to visualize effective and bilateral distribution of the aspirated fluid into the lungs (Fig. 8B). Following Curosurf administration, proSPC levels were determined by confocal immunofluorescence microscopy and compared with saline-injected WT control mice (Fig. 8C). As expected, after HO exposure proSPC levels were decreased in saline-treated triple KO but not WT control mice. In contrast, Curosurf treatment replenished the depleted proSPC stores in HO-exposed triple KO lungs, whereas proSPC levels in Curosurf-treated WT control mice remained similar to the levels in saline-treated WT mice. However, despite repletion of proSPC levels, Curosurf administration did neither prevent nor ameliorate the HO-induced lung damage in triple KO mice, as evidenced by a lack of improvement in lung histology (Fig. 9A), LIS (Fig. 9B), or in quasistatic lung compliance measurements (Fig. 9C).
Fig. 8.
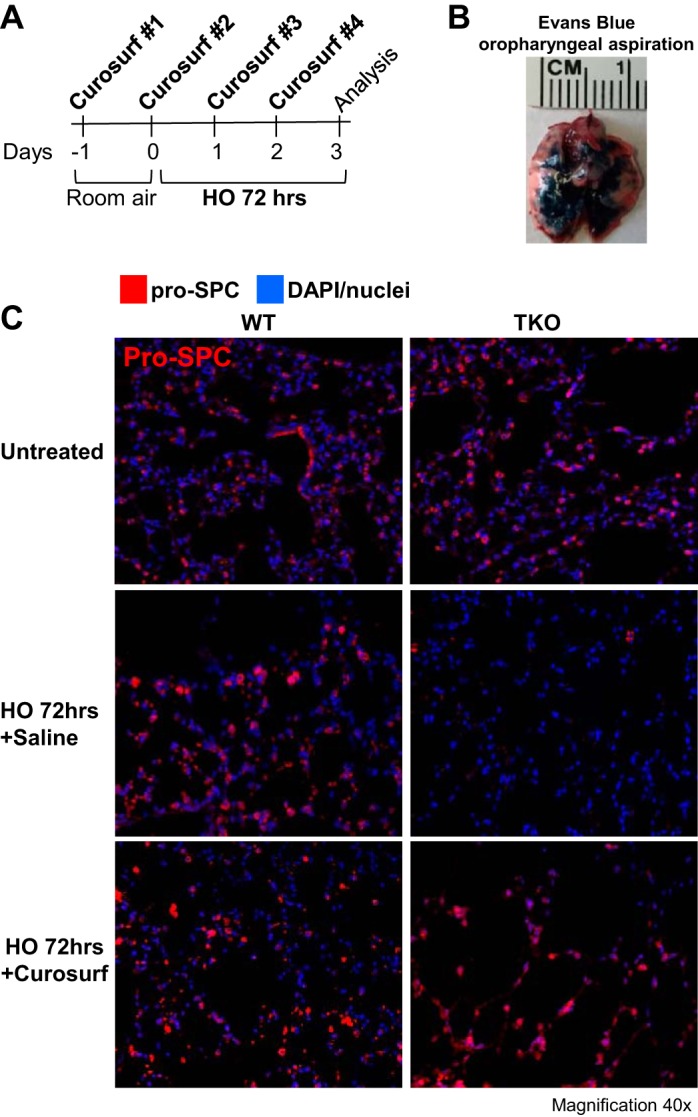
Oropharyngeal Curosurf administration restored proSPC levels in HO-treated lungs of TREK-1/TREK-2/TRAAK-deficient mice. A: experimental design of the Curosurf repletion experiments. B: oropharyngeal aspiration constitutes an adequate delivery system of a test solution (Evans blue dye) to the lungs (representative image of 6–8 separate experiments). C: representative confocal immunofluorescence microscopy images of proSPC protein levels in HO-treated WT control and triple KO lungs after oropharyngeal administration of either saline (vehicle control) or Curosurf (n = 5 mice per group). ProSPC protein is shown in red, and nuclei were counterstained in blue with DAPI.
DISCUSSION
We previously provided evidence supporting the expression of TREK K+ channels (TREK-1, TREK-2, and TRAAK) in lung alveolar and bronchial epithelial cells (100) and proposed a role for TREK-1 channels in inflammatory cytokine secretion and HO-induced lung injury using in vitro and in vivo models of ALI/ARDS (94, 99–102). In addition to TREK-1, the TREK subfamily of K2P channels includes two other closely related members, namely TREK-2 and TRAAK. In this study, we investigated whether potential redundancy between these channels could provide protection against HO-induced lung injury. We used the triple KO model (TREK-1/TREK-2/TRAAK-deficient) to explore the effects of HO alone and in combination with low (MVL) and high tidal volume (MVH) mechanical ventilation on triple KO lungs. Interestingly, neither MVH nor MVL alone (without HO) altered quasistatic lung compliance in WT control or triple KO mice (Fig. 1), but it is possible that a longer duration of MV would have led to lung injury. We chose 2 h of MV because in the triple KO group the combination of HO + 2 h of MVL already caused substantial lung injury and HO + MVH was lethal for triple KO mice. Interestingly, others have observed a moderate increase in lung injury markers (although a 100% survival) in C57/bl WT mice after 3 days of HO exposure (79), while we only detected mild weight loss (<10%; data not shown) but no obvious changes in lung histology or compliance. The reason for this difference remains speculative.
Although HO exposure caused lung injury in both TREK-1 and triple KO mice (102), one key difference between the two models is that after HO exposure TREK-1-deficient mice maintained their alveolar-capillary barrier function (no increase in BAL protein), whereas BAL protein was increased in HO-exposed triple KO mice, suggesting an impairment in alveolar-capillary barrier integrity (Fig. 1). This could be explained by the shorter HO exposure in the TREK-1 study. Another possibility is that deficiency of all three TREK channel isoforms (TREK-1/TREK-2/TRAAK) is required to disrupt the alveolar-capillary barrier. We speculate that TREK-driven alterations in cytoplasmic, or even local, Ca2+ concentrations (97), or K+-mediated TRPV channel activation (53, 128) could lead to epithelial (and possibly endothelial) tight junction dysfunction, thus facilitating BAL protein leakage. Increased cell death, as seen predominantly in the AT I cell population, could also lead to barrier dysfunction in triple KO lungs (Fig. 5), although TREK-1-deficient mice also showed increased cellular apoptosis (AT I and AT II cells) after HO exposure without any concomitant changes in BAL protein levels (102). Furthermore, we found that HO induced AT II cell proliferation in WT-derived but not triple KO-derived AT II cells (Fig. 6D). This may be a protective response to HO exposure in WT mice that is defective in triple KO mice, potentially contributing to the increased lung damage after HO exposure.
Another reason for the increased lung injury seen in HO-exposed triple KO mice could be related to a shift toward a more proinflammatory cytokine profile in the lungs of triple KO mice following HO exposure as supported by our findings of increased IL-6 and MCP-1 but decreased IL-10 and IL-22 expression levels (Fig. 7). Interestingly, IL-6 mRNA levels were elevated even at baseline, which could potentially lower the threshold for a pronounced proinflammatory response under HO conditions. Both IL-10 and IL-22 belong to the IL-10 superfamily and share a common receptor (IL-10R2) (88, 125). The decreased expression levels of IL-10 superfamily members could contribute to a decreased counterregulatory response to IL-6- and MCP-1-driven pathways in HO-exposed triple KO mice. IL-18 is a cytokine of the IL-1 superfamily and an important regulator of innate immune responses (29). In general it promotes T-cell and macrophage/inflammasome activation (23, 127, 133), but in the lung in particular both pro (105)- and anti-inflammatory (42) effects have been assigned to IL-18. Since we found no changes in its expression levels between WT or triple KO mice with or without HO exposure, lL-18 is unlikely to contribute to the observed effects. Although the increase in BAL fluid protein suggests a loss of alveolar barrier function in triple KO lungs after HO exposure, we found similar levels of ZO1 in both mouse types at baseline and after HO exposure (Fig. 7D). Nevertheless, despite similar total ZO1 levels, assembly and disassembly of tight junctions may still be impaired in triple KO mice and expression of other tight junctions proteins, including claudin, occluding, and JAM-A, needs to be studied in future experiments.
One of the most interesting findings of our study is the remarkable deficiency in SPA and SPC proteins found in triple KO lungs after HO exposure (Figs. 2 and 3). Since the 1990s, evidence has accumulated from human and animal studies linking surfactant dysfunction to the pathogenesis of ALI/ARDS (30, 33, 43, 44, 47, 63). In the field of neonatology, the protective properties of surfactant on premature lung development and surfactant repletion as a treatment for acute hypoxemic respiratory failure and persistent pulmonary hypertension are well recognized (2, 61, 82). Similarly, in children with acute, hypoxic respiratory failure due to ALI/ARDS, pneumonia, or bronchiolitis studies have consistently shown improved oxygenation, shortened duration of ventilation, and better survival after surfactant administration (39, 40, 67–69, 80, 123), although one recent trial using calfactant showed no oxygenation benefit despite more hospital-free days (121). In adults, the recent Adult Calfactant in Acute Respiratory Distress Syndrome Trial found no improvement in survival, length of stay, or oxygenation with surfactant therapy (122), and five other randomized trials were also unable to show any measurable benefit of exogenous surfactant administration (1, 31, 54, 107, 108). Given our data, it would have been nice to compare Curosurf, which contains SPB and SPC, to a surfactant mix that also contains SPA. Nevertheless, Curosurf (Poractant) is widely employed clinically, especially in neonates and children. Unfortunately, another commercially available product, Survanta (Beractant), also contains only SPB and SPC but no SPA. It is possible that the lack of benefit of surfactant therapy in clinical ARDS trials and in our study is in part due to a lack of SPA in the mixture (in addition to dosing, timing, and frequency of administration.
Although in our ALI/ARDS model prophylactic administration of Curosurf had no beneficial effect, together with other studies it shows that functional surfactant can be safely and repeatedly instilled into injured lungs (91). Interestingly, analysis of crude surfactant pellets from ARDS and pneumonia patients showed decreased SPA and SPB levels and fewer large (surface-active) surfactant aggregates (30, 33), consistent with our findings in triple KO mice. Others found a decrease in SPA, but not SPB, levels in ARDS patients (115). In a fatty acid-induced rabbit lung injury model and in trauma-induced ARDS patients, the overall composition and phospholipid content of BAL fluid did not change as the injury progressed, suggesting surfactant dysfunction rather than decreased levels (35, 89). These and other studies indicate that in different ALI/ARDS models surfactant could contribute to lung injury via at least two mechanisms: 1) a decrease in surfactant protein production and secretion, and 2) a loss of surfactant protein function despite normal levels. Interestingly, in certain rodent and rabbit models of HO-induced lung injury, an initial increase in surfactant protein levels was often observed after HO exposure (15, 19, 84, 113), whereas we found a decrease in tissue mRNA and protein expression levels of SPA and SPC but no changes in SPB and SPD levels. Besides species-specific differences (mouse vs. rat vs. rabbit), these differences could be due to the fact that the above models often used lower HO concentrations (80–85%) compared with our 95% HO and different timing of measurements (the above reports range from 1 to 7 days HO exposure) and the fact that we studied whole lung homogenate mRNA and protein levels whereas the above studies often measured BAL fluid surfactant protein concentrations.
In general, surfactant production is primarily regulated at the transcriptional and secretory level (lamellar bodies) (4, 13), although it can also be modified by developmental, physiological, humoral, and inflammatory stimuli (120), all of which are relevant in ALI/ARDS. A single dose of intratracheal TNF-α was found to decrease SPA and SPC mRNA levels in mice (90), similar to our findings in HO-exposed triple KO mice. Several transcription factors regulate surfactant gene transcription, in particular NF-1, NKX2.1/TTF-1, and c-jun (3, 6, 52). Although we found differences in the baseline expression levels of NKX2.1 between WT control and triple KO mice and a decrease in c-jun levels after HO exposure in both mouse types, none of these three transcription factors (NF-1, NKX2.1, or c-jun) appear to mediate the decreased SPA and SPC mRNA levels observed in triple KO mice after HO exposure. Other transcription factors such as FoxA/HNF-3 and AP-1 have also been implicated in surfactant regulation and should be examined in future studies (5, 6, 11, 116). In contrast to our findings in mice, exposure of rats to HO stimulated SPA protein secretion after 3 days (83) and induced de novo SPA, SPB, and SPC synthesis after 5 days (84), despite a concomitant decrease in lung compliance. These differences between mice and rats could be species related, but it is also possible that longer exposure of our mice to HO could eventually upregulate surfactant gene transcription. Such differences in surfactant sensitivity to HO among species are supported by a study in rabbits showing an increase in SPA and SPB levels in Clara cells but not AT II cells, while SPC remained unaffected (48). Unfortunately, given the degree of lung injury observed in our triple KO mice after 3 days of HO, it is unlikely that these mice would survive a longer HO exposure period. Furthermore, to determine whether the decrease in SPA and SPC levels in triple KO lungs could be related to impaired surfactant packaging, we measured ABCA3 protein/lamellar body levels in WT and triple KO lungs. ABCA3 is a lung-specific phospholipid transporter critical for intracellular surfactant synthesis and storage of lamellar bodies (86). Mutations in the ABCA3 gene cause fatal surfactant deficiency in newborns (106), and ABCA3 insufficiency is a risk factor for HO- and MV-induced lung injury in mice (38). Interestingly, at baseline triple KO lungs contain lower ABCA3/lamellar body levels than WT controls (Fig. 5). However, HO exposure increases ABCA3/lamellar body levels in both WT control and triple KO lungs and this difference is lost. It is possible, however, that secretory mechanisms are altered in triple KO mice impeding fusion of lamellar bodies with the plasma membrane despite equal intracellular ABCA3/lamellar body levels after HO exposure. The fact that SPA and SPC protein levels are equal at baseline between WT control and triple KO mice despite the lower ABCA3/lamellar body levels in triple KO mice may suggest that 1) not all SPA and SPC proteins are packed in lamellar bodies, or 2) individual surfactant protein concentrations per lamellar body are higher in triple KO lungs. Despite its semiquantitative limitations, our confocal immunofluorescent staining revealed no significant differences in SPA or SPC concentrations in individual AT II cells from WT and triple KO lungs, making this second option less likely.
Other potential mechanisms leading to decreased SPC and SPA levels in triple KO lungs include alterations in surfactant metabolism, degradation, or inactivation. Plasma proteins such as albumin and hemoglobin, which are increased in the BAL fluid of patients and animal models of ALI/ARDS (76, 77), can impair surfactant activity (44, 45). Interestingly, this inhibitory effect can be overcome in vitro by increasing the phospholipid concentrations (45) or the administration of exogenous surfactant (44). These findings suggest that surfactant repletion strategies may be a potential therapeutic option against ALI/ARDS. Importantly, the inhibitory effect of albumin on surfactant activity is regulated by Ca2+ ions (55), and Ca2+ gradients, in turn, can be regulated by TREK-mediated changes in the membrane potential (8, 56). Another mechanism of surfactant inactivation was increased conversion from heavy (surface-active) to light (less surface active) forms, as reported in a rabbit model of ARDS, and was counteracted by the addition of exogenous SPA (41). Interestingly, pretreatment of rabbits with exogenous surfactant showed impaired functional properties of subsequently recovered surfactant from injured lungs (64), which could, at least in part, explain the lack of protective effects of surfactant administration in our study.
Despite the interesting findings in HO- and MV-exposed triple KO lungs, it remains difficult to conclusively determine whether the deficiency in surfactant protein in triple KO lungs was one of the causes leading to increased lung injury or whether it occurred secondary to the activation of proinflammatory pathways by HO. The fact that HO exposure of triple KO mice 1) downregulated SPA and SPC mRNA expression, and 2) prophylactic administration of functional surfactant did not prevent HO-induced lung injury, would support the first hypothesis that TREK-1/TREK-2/TRAAK deficiency decreased SPA and SPC expression, but we cannot exclude that it also accelerated surfactant degradation. If increased AT II cell death was a major contributor to the decreased surfactant levels found in HO-exposed triple KO lungs, one would expect not only a decrease in SPA and SPC but also SPB and SPD levels. At least at the mRNA level, we found no differences in SPB and SPD between HO-exposed WT and triple KO mice. SPA could be particularly important in this context since it is the most abundant soluble pulmonary collectin and can act as a cytokine through interactions with an AT II cell surface receptor (SPAR) (57). Binding of SPA to SPAR activates the phosphatidylinositol 3-kinase-PKB-Akt pathway and prevents AT II cell apoptosis (118, 119). Interestingly, in our model, decreased SPA levels in HO-exposed triple KO lungs were associated with increase cell apoptosis (TUNEL staining and cleaved PARP-1). Furthermore, given the fact that SPA has antioxidant properties, decreased SPA levels in triple KO mice could result in impaired protection from HO-induced oxygen free radicals resulting in increased lung injury (37). Although SPA gene-deficient mice appear to have normal pulmonary function at baseline (50, 58), these properties of SPA could become quite important in the context of HO-induced lung injury and MV. In fact, surfactant obtained from the SPA-deficient mice is more sensitive to inhibition by plasma proteins than surfactant from WT mice (51), and decreased SPA levels can be found in both infants with RDS (18) and adults with ARDS (59).
Although still poorly defined, extrapulmonary functions of surfactant could also be of interest to our study since the triple KO mice are whole body deficient in TREK-1/TREK-2/TRAAK channels (20, 55, 65, 92). In particular, abnormalities in bone marrow-derived immune and inflammatory cells or defective programming of T cells in the thymus and spleen could affect the lung’s response to HO and mechanical stretch. This hypothesis is supported by our previous finding of altered macrophage activation in TREK-1-deficient mice (102).
An important point to address in future studies is whether the effects of TREK deficiency on lung injury and surfactant biology are related to alterations in cellular K+ handling or whether TREK channels exert functions that are unrelated to their role as K+ conductances, as, for example, described for cystic fibrosis transmembrane conductance regulator (73, 112). We previously showed that TREK-1 regulates the mechanobiological properties of AT II cells by altering F-actin metabolism and overall cellular stiffness (94). Unfortunately, the overall the molecular mechanisms that regulate surfactant biology are still poorly understood. This is in part due to the large variety of surfactant secretagogues, their receptors and different signaling mechanisms, in addition to nonreceptor-mediated surfactant secretion (9). If TREK proteins regulate surfactant secretion via changes in K+ currents, then these effects are most likely mediated via K+-induced changes in Ca2+ gradients. In fact, surfactant secretion induced by most secretagogues, including purines, β-adrenergic agents, or Ca2+ ionophores, is induced by rapid changes in intracellular Ca2+ (98, 103). During surfactant secretion from AT II cells, an initial release of Ca2+ from intracellular stores is followed by activation of plasma membrane Ca2+ channels (110), and K+ channels are powerful regulators of Ca2+ gradients (93). TREK channels in particular are known for their resting membrane potential stabilizing properties and by maintaining the membrane potential at negative values can create a driving force for Ca2+ entry (56). Interestingly, once SPA binds to its receptor (SPAR) on AT II cells, it blocks Ca2+ release from intracellular stores but not the influx of extracellular Ca2+ (111). Of note, however, phorbol esters (PMA) can induce surfactant secretion from AT II cells independently of Ca2+ signaling (110).
In conclusion, this study shows a heightened sensitivity of TREK-1/TREK-2/TRAAK-deficient mice to HO exposure, which is associated with a decrease in SPA and SPC levels. Further details of the molecular mechanisms underlying these observations need to be explored in upcoming studies.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grants K08-HL-118118-3 (to A. Schwingshackl) and HL-123540 (to C. M. Waters) and Agence Nationale de la Recherche, Laboratory of Excellence “Ion Channel Science and Therapeutics“ Grant ANR-11-LABX-0015-01 (to F. Lesage).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.S., B.L., B.T., and C.M.W. conceived and designed research; A.S., B.L., B.T., and C.L.L. performed experiments; A.S., B.L., and B.T. analyzed data; A.S., F.L., J.B., R.O., and C.M.W. interpreted results of experiments; A.S. prepared figures; A.S. drafted manuscript; A.S., F.L., J.B., R.O., and C.M.W. edited and revised manuscript; A.S., B.L., B.T., C.L.L., F.L., J.B., R.O., and C.M.W. approved final version of manuscript.
REFERENCES
- 1.Anzueto A, Baughman RP, Guntupalli KK, Weg JG, Wiedemann HP, Raventós AA, Lemaire F, Long W, Zaccardelli DS, Pattishall EN; Exosurf Acute Respiratory Distress Syndrome Sepsis Study Group . Aerosolized surfactant in adults with sepsis-induced acute respiratory distress syndrome. N Engl J Med 334: 1417–1421, 1996. doi: 10.1056/NEJM199605303342201. [DOI] [PubMed] [Google Scholar]
- 2.Auten RL, Notter RH, Kendig JW, Davis JM, Shapiro DL. Surfactant treatment of full-term newborns with respiratory failure. Pediatrics 87: 101–107, 1991. [PubMed] [Google Scholar]
- 3.Bachurski CJ, Kelly SE, Glasser SW, Currier TA. Nuclear factor I family members regulate the transcription of surfactant protein-C. J Biol Chem 272: 32759–32766, 1997. doi: 10.1074/jbc.272.52.32759. [DOI] [PubMed] [Google Scholar]
- 4.Boggaram V. Regulation of lung surfactant protein gene expression. Front Biosci 8: d751–d764, 2003. doi: 10.2741/1062. [DOI] [PubMed] [Google Scholar]
- 5.Boggaram V. Thyroid transcription factor-1 (TTF-1/Nkx2.1/TITF1) gene regulation in the lung. Clin Sci (Lond) 116: 27–35, 2009. doi: 10.1042/CS20080068. [DOI] [PubMed] [Google Scholar]
- 6.Bohinski RJ, Di Lauro R, Whitsett JA. The lung-specific surfactant protein B gene promoter is a target for thyroid transcription factor 1 and hepatocyte nuclear factor 3, indicating common factors for organ-specific gene expression along the foregut axis. Mol Cell Biol 14: 5671–5681, 1994. doi: 10.1128/MCB.14.9.5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brohawn SG, Su Z, MacKinnon R. Mechanosensitivity is mediated directly by the lipid membrane in TRAAK and TREK1 K+ channels. Proc Natl Acad Sci USA 111: 3614–3619, 2014. doi: 10.1073/pnas.1320768111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Budde T, Daut J, Kurtz A, Pape HC. Two-pore-domain potassium channels: regulators of many cellular functions. Pflugers Arch 467: 865–866, 2015. doi: 10.1007/s00424-015-1699-z. [DOI] [PubMed] [Google Scholar]
- 9.Chander A, Fisher AB. Regulation of lung surfactant secretion. Am J Physiol Lung Cell Mol Physiol 258: L241–L253, 1990. [DOI] [PubMed] [Google Scholar]
- 10.Chroneos ZC, Sever-Chroneos Z, Shepherd VL. Pulmonary surfactant: an immunological perspective. Cell Physiol Biochem 25: 13–26, 2010. doi: 10.1159/000272047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chuang CY, Chen TL, Chen RM. Molecular mechanisms of lipopolysaccharide-caused induction of surfactant protein-A gene expression in human alveolar epithelial A549 cells. Toxicol Lett 191: 132–139, 2009. doi: 10.1016/j.toxlet.2009.08.015. [DOI] [PubMed] [Google Scholar]
- 12.Clark H, Palaniyar N, Strong P, Edmondson J, Hawgood S, Reid KB. Surfactant protein D reduces alveolar macrophage apoptosis in vivo. J Immunol 169: 2892–2899, 2002. doi: 10.4049/jimmunol.169.6.2892. [DOI] [PubMed] [Google Scholar]
- 13.Clements JA. Lung surfactant: a personal perspective. Annu Rev Physiol 59: 1–21, 1997. doi: 10.1146/annurev.physiol.59.1.1. [DOI] [PubMed] [Google Scholar]
- 14.Creuwels LA, van Golde LM, Haagsman HP. The pulmonary surfactant system: biochemical and clinical aspects. Lung 175: 1–39, 1997. doi: 10.1007/PL00007554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D’Angio CT, Finkelstein JN, Lomonaco MB, Paxhia A, Wright SA, Baggs RB, Notter RH, Ryan RM. Changes in surfactant protein gene expression in a neonatal rabbit model of hyperoxia-induced fibrosis. Am J Physiol Lung Cell Mol Physiol 272: L720–L730, 1997. [DOI] [PubMed] [Google Scholar]
- 16.De Vooght V, Vanoirbeek JA, Haenen S, Verbeken E, Nemery B, Hoet PH. Oropharyngeal aspiration: an alternative route for challenging in a mouse model of chemical-induced asthma. Toxicology 259: 84–89, 2009. doi: 10.1016/j.tox.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 17.Delong P, Murray JA, Cook CK. Mechanical ventilation in the management of acute respiratory distress syndrome. Semin Dial 19: 517–524, 2006. doi: 10.1111/j.1525-139X.2006.00215.x. [DOI] [PubMed] [Google Scholar]
- 18.deMello DE, Phelps DS, Patel G, Floros J, Lagunoff D. Expression of the 35kDa and low molecular weight surfactant-associated proteins in the lungs of infants dying with respiratory distress syndrome. Am J Pathol 134: 1285–1293, 1989. [PMC free article] [PubMed] [Google Scholar]
- 19.Dombrowsky H, Tschernig T, Vieten G, Rau GA, Ohler F, Acevedo C, Behrens C, Poets CF, von der Hardt H, Bernhard W. Molecular and functional changes of pulmonary surfactant in response to hyperoxia. Pediatr Pulmonol 41: 1025–1039, 2006. doi: 10.1002/ppul.20443. [DOI] [PubMed] [Google Scholar]
- 20.Dutton JM, Goss K, Khubchandani KR, Shah CD, Smith RJ, Snyder JM. Surfactant protein A in rabbit sinus and middle ear mucosa. Ann Otol Rhinol Laryngol 108: 915–924, 1999. doi: 10.1177/000348949910801001. [DOI] [PubMed] [Google Scholar]
- 21.Edwards YS, Sutherland LM, Power JH, Nicholas TE, Murray AW. Cyclic stretch induces both apoptosis and secretion in rat alveolar type II cells. FEBS Lett 448: 127–130, 1999. doi: 10.1016/S0014-5793(99)00357-9. [DOI] [PubMed] [Google Scholar]
- 22.Epaud R, Ikegami M, Whitsett JA, Jobe AH, Weaver TE, Akinbi HT. Surfactant protein B inhibits endotoxin-induced lung inflammation. Am J Respir Cell Mol Biol 28: 373–378, 2003. doi: 10.1165/rcmb.2002-0071OC. [DOI] [PubMed] [Google Scholar]
- 23.Esquerdo KF, Sharma NK, Brunialti MKC, Baggio-Zappia GL, Assunção M, Azevedo LCP, Bafi AT, Salomao R. Inflammasome gene profile is modulated in septic patients, with a greater magnitude in non-survivors. Clin Exp Immunol 189: 232–240, 2017. doi: 10.1111/cei.12971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foley JP, Lam D, Jiang H, Liao J, Cheong N, McDevitt TM, Zaman A, Wright JR, Savani RC. Toll-like receptor 2 (TLR2), transforming growth factor-β, hyaluronan (HA), and receptor for HA-mediated motility (RHAMM) are required for surfactant protein A-stimulated macrophage chemotaxis. J Biol Chem 287: 37406–37419, 2012. doi: 10.1074/jbc.M112.360982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galambos C, Levy H, Cannon CL, Vargas SO, Reid LM, Cleveland R, Lindeman R, deMello DE, Wert SE, Whitsett JA, Perez-Atayde AR, Kozakewich H. Pulmonary pathology in thyroid transcription factor-1 deficiency syndrome. Am J Respir Crit Care Med 182: 549–554, 2010. doi: 10.1164/rccm.201002-0167CR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.González C, Baez-Nieto D, Valencia I, Oyarzún I, Rojas P, Naranjo D, Latorre R. K(+) channels: function-structural overview. Compr Physiol 2: 2087–2149, 2012. doi: 10.1002/cphy.c110047. [DOI] [PubMed] [Google Scholar]
- 27.Goto H, Ledford JG, Mukherjee S, Noble PW, Williams KL, Wright JR. The role of surfactant protein A in bleomycin-induced acute lung injury. Am J Respir Crit Care Med 181: 1336–1344, 2010. doi: 10.1164/rccm.200907-1002OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gowdy KM, Cardona DM, Nugent JL, Giamberardino C, Thomas JM, Mukherjee S, Martinu T, Foster WM, Plevy SE, Pastva AM, Wright JR, Palmer SM. Novel role for surfactant protein A in gastrointestinal graft-versus-host disease. J Immunol 188: 4897–4905, 2012. doi: 10.4049/jimmunol.1103558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gracie JA, Robertson SE, McInnes IB. Interleukin-18. J Leukoc Biol 73: 213–224, 2003. doi: 10.1189/jlb.0602313. [DOI] [PubMed] [Google Scholar]
- 30.Gregory TJ, Longmore WJ, Moxley MA, Whitsett JA, Reed CR, Fowler AA 3rd, Hudson LD, Maunder RJ, Crim C, Hyers TM. Surfactant chemical composition and biophysical activity in acute respiratory distress syndrome. J Clin Invest 88: 1976–1981, 1991. doi: 10.1172/JCI115523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gregory TJ, Steinberg KP, Spragg R, Gadek JE, Hyers TM, Longmore WJ, Moxley MA, Cai GZ, Hite RD, Smith RM, Hudson LD, Crim C, Newton P, Mitchell BR, Gold AJ. Bovine surfactant therapy for patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 155: 1309–1315, 1997. doi: 10.1164/ajrccm.155.4.9105072. [DOI] [PubMed] [Google Scholar]
- 32.Guillot L, Carré A, Szinnai G, Castanet M, Tron E, Jaubert F, Broutin I, Counil F, Feldmann D, Clement A, Polak M, Epaud R. NKX2-1 mutations leading to surfactant protein promoter dysregulation cause interstitial lung disease in “Brain-Lung-Thyroid Syndrome”. Hum Mutat 31: E1146–E1162, 2010. doi: 10.1002/humu.21183. [DOI] [PubMed] [Google Scholar]
- 33.Günther A, Siebert C, Schmidt R, Ziegler S, Grimminger F, Yabut M, Temmesfeld B, Walmrath D, Morr H, Seeger W. Surfactant alterations in severe pneumonia, acute respiratory distress syndrome, and cardiogenic lung edema. Am J Respir Crit Care Med 153: 176–184, 1996. doi: 10.1164/ajrccm.153.1.8542113. [DOI] [PubMed] [Google Scholar]
- 34.Guyon A, Tardy MP, Rovère C, Nahon JL, Barhanin J, Lesage F. Glucose inhibition persists in hypothalamic neurons lacking tandem-pore K+ channels. J Neurosci 29: 2528–2533, 2009. doi: 10.1523/JNEUROSCI.5764-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hall SB, Notter RH, Smith RJ, Hyde RW. Altered function of pulmonary surfactant in fatty acid lung injury. J Appl Physiol (1985) 69: 1143–1149, 1990. 2246164 [DOI] [PubMed]
- 36.Hanson JH, Flori H. Application of the acute respiratory distress syndrome network low-tidal volume strategy to pediatric acute lung injury. Respir Care Clin N Am 12: 349–357, 2006. doi: 10.1016/j.rcc.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 37.Hayakawa H, Myrvik QN, St Clair RW. Pulmonary surfactant inhibits priming of rabbit alveolar macrophage. Evidence that surfactant suppresses the oxidative burst of alveolar macrophage in infant rabbits. Am Rev Respir Dis 140: 1390–1397, 1989. doi: 10.1164/ajrccm/140.5.1390. [DOI] [PubMed] [Google Scholar]
- 38.Herber-Jonat S, Mittal R, Huppmann M, Hammel M, Liebisch G, Yildirim AO, Eickelberg O, Schmitz G, Hrabé de Angelis M, Flemmer AW, Holzinger A. Abca3 haploinsufficiency is a risk factor for lung injury induced by hyperoxia or mechanical ventilation in a murine model. Pediatr Res 74: 384–392, 2013. doi: 10.1038/pr.2013.127. [DOI] [PubMed] [Google Scholar]
- 39.Hermon MM, Golej J, Burda G, Boigner H, Stoll E, Vergesslich K, Strohmaier W, Pollak A, Trittenwein G. Surfactant therapy in infants and children: three years experience in a pediatric intensive care unit. Shock 17: 247–251, 2002. doi: 10.1097/00024382-200204000-00001. [DOI] [PubMed] [Google Scholar]
- 40.Herting E, Moller O, Schiffmann JH, Robertson B. Surfactant improves oxygenation in infants and children with pneumonia and acute respiratory distress syndrome. Acta Paediatr 91: 1174–1178, 2002. [DOI] [PubMed] [Google Scholar]
- 41.Higuchi R, Lewis J, Ikegami M. In vitro conversion of surfactant subtypes is altered in alveolar surfactant isolated from injured lungs. Am Rev Respir Dis 145: 1416–1420, 1992. doi: 10.1164/ajrccm/145.6.1416. [DOI] [PubMed] [Google Scholar]
- 42.Ho LP, Davis M, Denison A, Wood FT, Greening AP. Reduced interleukin-18 levels in BAL specimens from patients with asthma compared to patients with sarcoidosis and healthy control subjects. Chest 121: 1421–1426, 2002. doi: 10.1378/chest.121.5.1421. [DOI] [PubMed] [Google Scholar]
- 43.Holm BA, Enhorning G, Notter RH. A biophysical mechanism by which plasma proteins inhibit lung surfactant activity. Chem Phys Lipids 49: 49–55, 1988. doi: 10.1016/0009-3084(88)90063-1. [DOI] [PubMed] [Google Scholar]
- 44.Holm BA, Notter RH. Effects of hemoglobin and cell membrane lipids on pulmonary surfactant activity. J Appl Physiol (1985) 63: 1434–1442, 1987. [DOI] [PubMed] [Google Scholar]
- 45.Holm BA, Notter RH, Finkelstein JN. Surface property changes from interactions of albumin with natural lung surfactant and extracted lung lipids. Chem Phys Lipids 38: 287–298, 1985. doi: 10.1016/0009-3084(85)90022-2. [DOI] [PubMed] [Google Scholar]
- 46.Holm BA, Notter RH, Siegle J, Matalon S. Pulmonary physiological and surfactant changes during injury and recovery from hyperoxia. J Appl Physiol (1985) 59: 1402–1409, 1985. [DOI] [PubMed] [Google Scholar]
- 47.Holm BA, Wang Z, Notter RH. Multiple mechanisms of lung surfactant inhibition. Pediatr Res 46: 85–93, 1999. doi: 10.1203/00006450-199907000-00015. [DOI] [PubMed] [Google Scholar]
- 48.Horowitz S, Watkins RH, Auten RL Jr, Mercier CE, Cheng ER. Differential accumulation of surfactant protein A, B, and C mRNAs in two epithelial cell types of hyperoxic lung. Am J Respir Cell Mol Biol 5: 511–515, 1991. doi: 10.1165/ajrcmb/5.6.511. [DOI] [PubMed] [Google Scholar]
- 49.Hsia BJ, Ledford JG, Potts-Kant EN, Nikam VS, Lugogo NL, Foster WM, Kraft M, Abraham SN, Wright JR. Mast cell TNF receptors regulate responses to Mycoplasma pneumoniae in surfactant protein A (SP-A)−/− mice. J Allergy Clin Immunol 130: 205–14.e2, 2012. doi: 10.1016/j.jaci.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 50.Ikegami M, Jobe AH, Whitsett J, Korfhagen T. Tolerance of SP-A-deficient mice to hyperoxia or exercise. J Appl Physiol (1985) 89: 644–648, 2000. [DOI] [PubMed] [Google Scholar]
- 51.Ikegami M, Korfhagen TR, Whitsett JA, Bruno MD, Wert SE, Wada K, Jobe AH. Characteristics of surfactant from SP-A-deficient mice. Am J Physiol Lung Cell Mol Physiol 275: L247–L254, 1998. [DOI] [PubMed] [Google Scholar]
- 52.Kamio K, Usuki J, Azuma A, Matsuda K, Ishii T, Inomata M, Hayashi H, Kokuho N, Fujita K, Saito Y, Miya T, Gemma A. Nintedanib modulates surfactant protein-D expression in A549 human lung epithelial cells via the c-Jun N-terminal kinase-activator protein-1 pathway. Pulm Pharmacol Ther 32: 29–36, 2015. doi: 10.1016/j.pupt.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 53.Ke SK, Chen L, Duan HB, Tu YR. Opposing actions of TRPV4 channel activation in the lung vasculature. Respir Physiol Neurobiol 219: 43–50, 2015. doi: 10.1016/j.resp.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 54.Kesecioglu J, Beale R, Stewart TE, Findlay GP, Rouby JJ, Holzapfel L, Bruins P, Steenken EJ, Jeppesen OK, Lachmann B. Exogenous natural surfactant for treatment of acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med 180: 989–994, 2009. doi: 10.1164/rccm.200812-1955OC. [DOI] [PubMed] [Google Scholar]
- 55.Khubchandani KR, Snyder JM. Surfactant protein A (SP-A): the alveolus and beyond. FASEB J 15: 59–69, 2001. doi: 10.1096/fj.00-0318rev. [DOI] [PubMed] [Google Scholar]
- 56.Kim D, Kang D. Role of K2p channels in stimulus-secretion coupling. Pflugers Arch 467: 1001–1011, 2015. doi: 10.1007/s00424-014-1663-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kishore U, Greenhough TJ, Waters P, Shrive AK, Ghai R, Kamran MF, Bernal AL, Reid KB, Madan T, Chakraborty T. Surfactant proteins SP-A and SP-D: structure, function and receptors. Mol Immunol 43: 1293–1315, 2006. doi: 10.1016/j.molimm.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 58.Korfhagen TR, Bruno MD, Ross GF, Huelsman KM, Ikegami M, Jobe AH, Wert SE, Stripp BR, Morris RE, Glasser SW, Bachurski CJ, Iwamoto HS, Whitsett JA. Altered surfactant function and structure in SP-A gene targeted mice. Proc Natl Acad Sci USA 93: 9594–9599, 1996. doi: 10.1073/pnas.93.18.9594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuroki Y, Takahashi H, Chiba H, Akino T. Surfactant proteins A and D: disease markers. Biochim Biophys Acta 1408: 334–345, 1998. doi: 10.1016/S0925-4439(98)00079-9. [DOI] [PubMed] [Google Scholar]
- 60.Lakatos HF, Burgess HA, Thatcher TH, Redonnet MR, Hernady E, Williams JP, Sime PJ. Oropharyngeal aspiration of a silica suspension produces a superior model of silicosis in the mouse when compared to intratracheal instillation. Exp Lung Res 32: 181–199, 2006. doi: 10.1080/01902140600817465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lakshminrusimha S, Konduri GG, Steinhorn RH. Considerations in the management of hypoxemic respiratory failure and persistent pulmonary hypertension in term and late preterm neonates. J Perinatol 36, Suppl 2: S12–19, 2016. doi: 10.1038/jp.2016.44. [DOI] [PubMed] [Google Scholar]
- 62.Lawson WE, Polosukhin VV, Stathopoulos GT, Zoia O, Han W, Lane KB, Li B, Donnelly EF, Holburn GE, Lewis KG, Collins RD, Hull WM, Glasser SW, Whitsett JA, Blackwell TS. Increased and prolonged pulmonary fibrosis in surfactant protein C-deficient mice following intratracheal bleomycin. Am J Pathol 167: 1267–1277, 2005. doi: 10.1016/S0002-9440(10)61214-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lewis JF, Ikegami M, and Jobe AH. Altered surfactant function and metabolism in rabbits with acute lung injury. J Appl Physiol (1985) 69: 2303–2310, 1990. [DOI] [PubMed] [Google Scholar]
- 64.Lewis JF, Ikegami M, Jobe AH. Metabolism of exogenously administered surfactant in the acutely injured lungs of adult rabbits. Am Rev Respir Dis 145: 19–23, 1992. doi: 10.1164/ajrccm/145.1.19. [DOI] [PubMed] [Google Scholar]
- 65.Lichtenberger LM, Graziani LA, Dial EJ, Butler BD, Hills BA. Role of surface-active phospholipids in gastric cytoprotection. Science 219: 1327–1329, 1983. doi: 10.1126/science.6828859. [DOI] [PubMed] [Google Scholar]
- 66.Liu Y, Peterson DA, Kimura H, Schubert D. Mechanism of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction. J Neurochem 69: 581–593, 1997. doi: 10.1046/j.1471-4159.1997.69020581.x. [DOI] [PubMed] [Google Scholar]
- 67.López-Herce J, de Lucas N, Carrillo A, Bustinza A, Moral R. Surfactant treatment for acute respiratory distress syndrome. Arch Dis Child 80: 248–252, 1999. doi: 10.1136/adc.80.3.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Luchetti M, Casiraghi G, Valsecchi R, Galassini E, Marraro G. Porcine-derived surfactant treatment of severe bronchiolitis. Acta Anaesthesiol Scand 42: 805–810, 1998. doi: 10.1111/j.1399-6576.1998.tb05326.x. [DOI] [PubMed] [Google Scholar]
- 69.Luchetti M, Ferrero F, Gallini C, Natale A, Pigna A, Tortorolo L, Marraro G. Multicenter, randomized, controlled study of porcine surfactant in severe respiratory syncytial virus-induced respiratory failure. Pediatr Crit Care Med 3: 261–268, 2002. [DOI] [PubMed] [Google Scholar]
- 70.Madan T, Kishore U, Singh M, Strong P, Clark H, Hussain EM, Reid KB, Sarma PU. Surfactant proteins A and D protect mice against pulmonary hypersensitivity induced by Aspergillus fumigatus antigens and allergens. J Clin Invest 107: 467–475, 2001. doi: 10.1172/JCI10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Makena PS, Gorantla VK, Ghosh MC, Bezawada L, Balazs L, Luellen C, Parthasarathi K, Waters CM, Sinclair SE. Lung injury caused by high tidal volume mechanical ventilation and hyperoxia is dependent on oxidant-mediated c-Jun NH2-terminal kinase activation. J Appl Physiol (1985) 111: 1467–1476, 2011. doi: 10.1152/japplphysiol.00539.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Makena PS, Luellen CL, Balazs L, Ghosh MC, Parthasarathi K, Waters CM, Sinclair SE. Preexposure to hyperoxia causes increased lung injury and epithelial apoptosis in mice ventilated with high tidal volumes. Am J Physiol Lung Cell Mol Physiol 299: L711–L719, 2010. doi: 10.1152/ajplung.00072.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marino GI, Kotsias BA. Cystic fibrosis transmembrane regulator (CFTR) in human trophoblast BeWo cells and its relation to cell migration. Placenta 35: 92–98, 2013. doi: 10.1016/j.placenta.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 74.Marten E, Nielsen HC, Dammann CE. Interdependent TTF1-ErbB4 interactions are critical for surfactant protein-B homeostasis in primary mouse lung alveolar type II cells. J Cell Commun Signal 9: 207–215, 2015. doi: 10.1007/s12079-015-0299-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Matsuzaki Y, Besnard V, Clark JC, Xu Y, Wert SE, Ikegami M, Whitsett JA. STAT3 regulates ABCA3 expression and influences lamellar body formation in alveolar type II cells. Am J Respir Cell Mol Biol 38: 551–558, 2008. doi: 10.1165/rcmb.2007-0311OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meduri GU, Headley S, Tolley E, Shelby M, Stentz F, Postlethwaite A. Plasma and BAL cytokine response to corticosteroid rescue treatment in late ARDS. Chest 108: 1315–1325, 1995. doi: 10.1378/chest.108.5.1315. [DOI] [PubMed] [Google Scholar]
- 77.Mei SH, McCarter SD, Deng Y, Parker CH, Liles WC, Stewart DJ. Prevention of LPS-induced acute lung injury in mice by mesenchymal stem cells overexpressing angiopoietin 1. PLoS Med 4: e269, 2007. doi: 10.1371/journal.pmed.0040269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Messier EM, Mason RJ, Kosmider B. Efficient and rapid isolation and purification of mouse alveolar type II epithelial cells. Exp Lung Res 38: 363–373, 2012. doi: 10.3109/01902148.2012.713077. [DOI] [PubMed] [Google Scholar]
- 79.Mizushina Y, Shirasuna K, Usui F, Karasawa T, Kawashima A, Kimura H, Kobayashi M, Komada T, Inoue Y, Mato N, Yamasawa H, Latz E, Iwakura Y, Kasahara T, Bando M, Sugiyama Y, Takahashi M. NLRP3 protein deficiency exacerbates hyperoxia-induced lethality through Stat3 protein signaling independent of interleukin-1β. J Biol Chem 290: 5065–5077, 2015. doi: 10.1074/jbc.M114.603217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Möller JC, Schaible T, Roll C, Schiffmann JH, Bindl L, Schrod L, Reiss I, Kohl M, Demirakca S, Hentschel R, Paul T, Vierzig A, Groneck P, von Seefeld H, Schumacher H, Gortner L; Surfactant ARDS Study Group . Treatment with bovine surfactant in severe acute respiratory distress syndrome in children: a randomized multicenter study. Intensive Care Med 29: 437–446, 2003. doi: 10.1007/s00134-003-1650-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mulugeta S, Beers MF. Surfactant protein C: its unique properties and emerging immunomodulatory role in the lung. Microbes Infect 8: 2317–2323, 2006. doi: 10.1016/j.micinf.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 82.Natarajan CK, Sankar MJ, Jain K, Agarwal R, Paul VK. Surfactant therapy and antibiotics in neonates with meconium aspiration syndrome: a systematic review and meta-analysis. J Perinatol 36, Suppl 1: S49–54, 2016. doi: 10.1038/jp.2016.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nogee LM, Wispe JR. Effects of pulmonary oxygen injury on airway content of surfactant-associated protein A. Pediatr Res 24: 568–573, 1988. doi: 10.1203/00006450-198811000-00006. [DOI] [PubMed] [Google Scholar]
- 84.Nogee LM, Wispé JR, Clark JC, Weaver TE, Whitsett JA. Increased expression of pulmonary surfactant proteins in oxygen-exposed rats. Am J Respir Cell Mol Biol 4: 102–107, 1991. doi: 10.1165/ajrcmb/4.2.102. [DOI] [PubMed] [Google Scholar]
- 85.Pastva AM, Mukherjee S, Giamberardino C, Hsia B, Lo B, Sempowski GD, Wright JR. Lung effector memory and activated CD4+ T cells display enhanced proliferation in surfactant protein A-deficient mice during allergen-mediated inflammation. J Immunol 186: 2842–2849, 2011. doi: 10.4049/jimmunol.0904190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Peca D, Cutrera R, Masotti A, Boldrini R, Danhaive O. ABCA3, a key player in neonatal respiratory transition and genetic disorders of the surfactant system. Biochem Soc Trans 43: 913–919, 2015. doi: 10.1042/BST20150100. [DOI] [PubMed] [Google Scholar]
- 87.Pérez-Gil J. Structure of pulmonary surfactant membranes and films: the role of proteins and lipid-protein interactions. Biochim Biophys Acta 1778: 1676–1695, 2008. doi: 10.1016/j.bbamem.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 88.Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB. Interleukin-10 and related cytokines and receptors. Annu Rev Immunol 22: 929–979, 2004. doi: 10.1146/annurev.immunol.22.012703.104622. [DOI] [PubMed] [Google Scholar]
- 89.Pison U, Seeger W, Buchhorn R, Joka T, Brand M, Obertacke U, Neuhof H, Schmit-Neuerburg KP. Surfactant abnormalities in patients with respiratory failure after multiple trauma. Am Rev Respir Dis 140: 1033–1039, 1989. doi: 10.1164/ajrccm/140.4.1033. [DOI] [PubMed] [Google Scholar]
- 90.Pryhuber GS, Bachurski C, Hirsch R, Bacon A, Whitsett JA. Tumor necrosis factor-alpha decreases surfactant protein B mRNA in murine lung. Am J Physiol Lung Cell Mol Physiol 270: L714–L721, 1996. [DOI] [PubMed] [Google Scholar]
- 91.Puntorieri V, Hiansen JQ, McCaig LA, Yao LJ, Veldhuizen RA, Lewis JF. The effects of exogenous surfactant administration on ventilation-induced inflammation in mouse models of lung injury. BMC Pulm Med 13: 67, 2013. doi: 10.1186/1471-2466-13-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rapport PN, Lim DJ, Weiss HS. Surface-active agent in Eustachian tube function. Arch Otolaryngol 101: 305–311, 1975. [DOI] [PubMed] [Google Scholar]
- 93.Ridge FP, Duszyk M, French AS. A large conductance, Ca2+-activated K+ channel in a human lung epithelial cell line (A549). Biochim Biophys Acta 1327: 249–258, 1997. doi: 10.1016/S0005-2736(97)00073-4. [DOI] [PubMed] [Google Scholar]
- 94.Roan E, Waters CM, Teng B, Ghosh M, Schwingshackl A. The 2-pore domain potassium channel TREK-1 regulates stretch-induced detachment of alveolar epithelial cells. PLoS One 9: e89429, 2014. doi: 10.1371/journal.pone.0089429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Salazar E, Knowles JH. An alaysis of pressure-volume characteristics of the lungs. J Appl Physiol 19: 97–104, 1964. [DOI] [PubMed] [Google Scholar]
- 96.Salvador JM, Brown-Clay JD, Fornace AJ Jr. Gadd45 in stress signaling, cell cycle control, and apoptosis. Adv Exp Med Biol 793: 1–19, 2013. doi: 10.1007/978-1-4614-8289-5_1. [DOI] [PubMed] [Google Scholar]
- 97.Samak G, Chaudhry KK, Gangwar R, Narayanan D, Jaggar JH, Rao R. Calcium/Ask1/MKK7/JNK2/c-Src signalling cascade mediates disruption of intestinal epithelial tight junctions by dextran sulfate sodium. Biochem J 465: 503–515, 2015. doi: 10.1042/BJ20140450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sano K, Voelker DR, Mason RJ. Effect of secretagogues on cytoplasmic free calcium in alveolar type II epithelial cells. Am J Physiol Cell Physiol 253: C679–C686, 1987. [DOI] [PubMed] [Google Scholar]
- 99.Schwingshackl A, Teng B, Ghosh M, Waters CM. Regulation of monocyte chemotactic protein-1 secretion by the two-pore-domain potassium (K2P) channel TREK-1 in human alveolar epithelial cells. Am J Transl Res 5: 530–542, 2013. [PMC free article] [PubMed] [Google Scholar]
- 100.Schwingshackl A, Teng B, Ghosh M, West AN, Makena P, Gorantla V, Sinclair SE, Waters CM. Regulation and function of the two-pore-domain (K2P) potassium channel Trek-1 in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 302: L93–L102, 2012. doi: 10.1152/ajplung.00078.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schwingshackl A, Teng B, Ghosh MC, Lim KG, Tigyi GJ, Narayanan D, Jaggar JH, Waters CM. Regulation of interleukin-6 secretion by the two-pore-domain potassium (K2P) channel Trek-1 in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 304: L276–L86, 2013. doi: 10.1152/ajplung.00299.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schwingshackl A, Teng B, Makena P, Ghosh M, Sinclair SE, Luellen C, Balasz L, Rovnaghi C, Bryan RM, Lloyd EE, Fitzpatrick E, Saravia JS, Cormier SA, Waters CM. Deficiency of the two-pore-domain potassium channel TREK-1 promotes hyperoxia-induced lung injury. Crit Care Med 42: e692–e701, 2014. doi: 10.1097/CCM.0000000000000603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sen N, Chander A. Alkalosis- and ATP-induced increases in the diacyglycerol pool in alveolar type II cells are derived from phosphatidylcholine and phosphatidylinositol. Biochem J 298: 681–687, 1994. doi: 10.1042/bj2980681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sever-Chroneos Z, Krupa A, Davis J, Hasan M, Yang CH, Szeliga J, Herrmann M, Hussain M, Geisbrecht BV, Kobzik L, Chroneos ZC. Surfactant protein A (SP-A)-mediated clearance of Staphylococcus aureus involves binding of SP-A to the staphylococcal adhesin eap and the macrophage receptors SP-A receptor 210 and scavenger receptor class A. J Biol Chem 286: 4854–4870, 2011. doi: 10.1074/jbc.M110.125567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shigehara K, Shijubo N, Ohmichi M, Yamada G, Takahashi R, Okamura H, Kurimoto M, Hiraga Y, Tatsuno T, Abe S, Sato N. Increased levels of interleukin-18 in patients with pulmonary sarcoidosis. Am J Respir Crit Care Med 162: 1979–1982, 2000. doi: 10.1164/ajrccm.162.5.9911113. [DOI] [PubMed] [Google Scholar]
- 106.Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, Dean M. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med 350: 1296–1303, 2004. doi: 10.1056/NEJMoa032178. [DOI] [PubMed] [Google Scholar]
- 107.Spragg RG, Lewis JF, Walmrath HD, Johannigman J, Bellingan G, Laterre PF, Witte MC, Richards GA, Rippin G, Rathgeb F, Häfner D, Taut FJ, Seeger W. Effect of recombinant surfactant protein C-based surfactant on the acute respiratory distress syndrome. N Engl J Med 351: 884–892, 2004. doi: 10.1056/NEJMoa033181. [DOI] [PubMed] [Google Scholar]
- 108.Spragg RG, Taut FJ, Lewis JF, Schenk P, Ruppert C, Dean N, Krell K, Karabinis A, Günther A. Recombinant surfactant protein C-based surfactant for patients with severe direct lung injury. Am J Respir Crit Care Med 183: 1055–1061, 2011. doi: 10.1164/rccm.201009-1424OC. [DOI] [PubMed] [Google Scholar]
- 109.Stahlman MT, Gray MP, Falconieri MW, Whitsett JA, Weaver TE. Lamellar body formation in normal and surfactant protein B-deficient fetal mice. Lab Invest 80: 395–403, 2000. doi: 10.1038/labinvest.3780044. [DOI] [PubMed] [Google Scholar]
- 110.Strayer DS, Hoek JB, Thomas AP, White MK. Cellular activation by Ca2+ release from stores in the endoplasmic reticulum but not by increased free Ca2+ in the cytosol. Biochem J 344: 39–46, 1999. [PMC free article] [PubMed] [Google Scholar]
- 111.Strayer DS, Pinder R, Chander A. Receptor-mediated regulation of pulmonary surfactant secretion. Exp Cell Res 226: 90–97, 1996. doi: 10.1006/excr.1996.0206. [DOI] [PubMed] [Google Scholar]
- 112.Suaud L, Yan W, Carattino MD, Robay A, Kleyman TR, Rubenstein RC. Regulatory interactions of N1303K-CFTR and ENaC in Xenopus oocytes: evidence that chloride transport is not necessary for inhibition of ENaC. Am J Physiol Cell Physiol 292: C1553–C1561, 2007. doi: 10.1152/ajpcell.00064.2006. [DOI] [PubMed] [Google Scholar]
- 113.Sugahara K, Iyama K, Sano K, Kuroki Y, Akino T, Matsumoto M. Overexpression of surfactant protein SP-A, SP-B, and SP-C mRNA in rat lungs with lipopolysaccharide-induced injury. Lab Invest 74: 209–220, 1996. [PubMed] [Google Scholar]
- 114.Vanoirbeek JA, Rinaldi M, De Vooght V, Haenen S, Bobic S, Gayan-Ramirez G, Hoet PH, Verbeken E, Decramer M, Nemery B, Janssens W. Noninvasive and invasive pulmonary function in mouse models of obstructive and restrictive respiratory diseases. Am J Respir Cell Mol Biol 42: 96–104, 2010. doi: 10.1165/rcmb.2008-0487OC. [DOI] [PubMed] [Google Scholar]
- 115.Veldhuizen RA, McCaig LA, Akino T, Lewis JF. Pulmonary surfactant subfractions in patients with the acute respiratory distress syndrome. Am J Respir Crit Care Med 152: 1867–1871, 1995. doi: 10.1164/ajrccm.152.6.8520748. [DOI] [PubMed] [Google Scholar]
- 116.Wan H, Kaestner KH, Ang SL, Ikegami M, Finkelman FD, Stahlman MT, Fulkerson PC, Rothenberg ME, Whitsett JA. Foxa2 regulates alveolarization and goblet cell hyperplasia. Development 131: 953–964, 2004. doi: 10.1242/dev.00966. [DOI] [PubMed] [Google Scholar]
- 117.Weaver TE, Whitsett JA. Function and regulation of expression of pulmonary surfactant-associated proteins. Biochem J 273: 249–264, 1991. doi: 10.1042/bj2730249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.White MK, Baireddy V, Strayer DS. Natural protection from apoptosis by surfactant protein A in type II pneumocytes. Exp Cell Res 263: 183–192, 2001. doi: 10.1006/excr.2000.5120. [DOI] [PubMed] [Google Scholar]
- 119.White MK, Strayer DS. Survival signaling in type II pneumocytes activated by surfactant protein-A. Exp Cell Res 280: 270–279, 2002. doi: 10.1006/excr.2002.5646. [DOI] [PubMed] [Google Scholar]
- 120.Whitsett JA, Glasser SW. Regulation of surfactant protein gene transcription. Biochim Biophys Acta 1408: 303–311, 1998. doi: 10.1016/S0925-4439(98)00076-3. [DOI] [PubMed] [Google Scholar]
- 121.Willson DF, Thomas NJ, Tamburro R, Truemper E, Truwit J, Conaway M, Traul C, Egan EE. Pediatric calfactant in acute respiratory distress syndrome trial. Pediatr Crit Care Med 14: 657–665, 2013. doi: 10.1097/PCC.0b013e3182917cb5. [DOI] [PubMed] [Google Scholar]
- 122.Willson DF, Truwit JD, Conaway MR, Traul CS, Egan EE. The Adult Calfactant in Acute Respiratory Distress Syndrome Trial. Chest 148: 356–364, 2015. doi: 10.1378/chest.14-1139. [DOI] [PubMed] [Google Scholar]
- 123.Willson DF, Zaritsky A, Bauman LA, Dockery K, James RL, Conrad D, Craft H, Novotny WE, Egan EA, Dalton H; Members of the Mid-Atlantic Pediatric Critical Care Network . Instillation of calf lung surfactant extract (calfactant) is beneficial in pediatric acute hypoxemic respiratory failure. Crit Care Med 27: 188–195, 1999. doi: 10.1097/00003246-199901000-00050. [DOI] [PubMed] [Google Scholar]
- 124.Wirtz HR, Dobbs LG. Calcium mobilization and exocytosis after one mechanical stretch of lung epithelial cells. Science 250: 1266–1269, 1990. doi: 10.1126/science.2173861. [DOI] [PubMed] [Google Scholar]
- 125.Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity 21: 241–254, 2004. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 126.Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol 5: 58–68, 2005. doi: 10.1038/nri1528. [DOI] [PubMed] [Google Scholar]
- 127.Xu D, Trajkovic V, Hunter D, Leung BP, Schulz K, Gracie JA, McInnes IB, Liew FY. IL-18 induces the differentiation of Th1 or Th2 cells depending upon cytokine milieu and genetic background. Eur J Immunol 30: 3147–3156, 2000. doi:. [DOI] [PubMed] [Google Scholar]
- 128.Xu R, Li Q, Zhou J, Zhou XD, Perelman JM, Kolosov VP. The degradation of airway tight junction protein under acidic conditions is probably mediated by transient receptor potential vanilloid 1 receptor. Biosci Rep 33: 33, 2013. doi: 10.1042/BSR20130087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Xu Y, Ikegami M, Wang Y, Matsuzaki Y, Whitsett JA. Gene expression and biological processes influenced by deletion of Stat3 in pulmonary type II epithelial cells. BMC Genomics 8: 455, 2007. doi: 10.1186/1471-2164-8-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Xu Y, Zhang M, Wang Y, Kadambi P, Dave V, Lu LJ, Whitsett JA. A systems approach to mapping transcriptional networks controlling surfactant homeostasis. BMC Genomics 11: 451, 2010. doi: 10.1186/1471-2164-11-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yamano G, Funahashi H, Kawanami O, Zhao LX, Ban N, Uchida Y, Morohoshi T, Ogawa J, Shioda S, Inagaki N. ABCA3 is a lamellar body membrane protein in human lung alveolar type II cells. FEBS Lett 508: 221–225, 2001. doi: 10.1016/S0014-5793(01)03056-3. [DOI] [PubMed] [Google Scholar]
- 132.Zhao Z, Guttmann J, Moller K. Assessment of a volume-dependent dynamic respiratory system compliance in ALI/ARDS by pooling breathing cycles. Physiol Meas 33: N61-67, 2012. doi: 10.1088/0967-3334/33/8/N61. [DOI] [PubMed] [Google Scholar]
- 133.Zhou F, Zhang Y, Chen J, Hu X, Xu Y. Liraglutide attenuates lipopolysaccharide-induced acute lung injury in mice. Eur J Pharmacol 791: 735–740, 2016. doi: 10.1016/j.ejphar.2016.10.016. [DOI] [PubMed] [Google Scholar]