

Abstract
Hydrophobic drug candidates require innovative formulation agents. We designed and synthesized lipid‐DNA polymers containing varying numbers of hydrophobic alkyl chains. The hydrophobicity of these amphiphiles is easily tunable by introducing a defined number of alkyl chain‐modified nucleotides during standard solid‐phase synthesis of DNA using an automated DNA synthesizer. We observed that the resulting self‐assembled micelles solubilize the poorly water‐soluble drug, meta‐tetra‐hydroxyphenyl‐chlorin (mTHPC) used in photodynamic therapy (PDT) with high loading concentrations and loading capacities. A cell viability study showed that mTHPC‐loaded micelles exhibit good biocompatibility without irradiation, and high PDT efficacy upon irradiation. Lipid‐DNAs provide a novel class of drug‐delivery vehicle, and hybridization of DNA offers a potentially facile route for further functionalization of the drug‐delivery system with, for instance, targeting or imaging moieties.
Keywords: amphiphiles, drug delivery, lipid-DNA, micelles, photodynamic therapy
With the advent and fast development of high‐throughput (HTS) and ultra‐high‐throughput screening (uHTS) technologies for drug discovery over the past two decades,1 compound libraries have yielded an increasing number of potential candidates that exhibit a high affinity for their targets. A substantial number of these pharmaceutically active compounds, however, suffer from low water solubility, which hinders their development and delays market entry. Even for already marketed drugs, more than 40 % are poorly water‐soluble.2 To enable the use of these active compounds and reduce their side effects, micelles are widely used as drug‐delivery vehicles due to attractive properties such as high solubilizing efficiency, good reproducibility, simple preparation procedures and the possibility to make them stimuli‐responsive.3
Despite various amphiphilic materials being used,4 it is still a challenge to construct a biocompatible, effective and targeted micellar drug‐delivery system. Previous studies showed that amphiphilic DNA‐based copolymers self‐assemble into uniform micelles above their critical micelle concentration (CMC) and are able to accommodate drugs of interest in the hydrophobic core.5 These constructs have several advantages over those formed from synthetic polymers. First, being formed from biomacromolecules, DNA‐based micelles are more biocompatible and biodegradable and have shown no observable toxicity and little immunogenicity.6 Secondly, they can be easily synthesized by automated solid‐phase synthesis.7 Most importantly, DNA‐based micelles can be modified in a straightforward fashion by employing highly specific hybridization, which conveniently endows the system with targeting or imaging moieties.8 All these beneficial properties give them great potential to be used as targeted drug‐delivery vehicles.
meta‐Tetra‐hydroxyphenylchlorin (mTHPC (1), Figure 1 a), also known as Temoporfin and Foscan (as the medicinal product),9 is a poorly water‐soluble second‐generation photosensitizer (PS) that has been widely used in PDT. It has been approved in Europe for the treatment of head and neck carcinoma.10 Conventional formulations, however, are hampered by poor water solubility and tumor‐targeting properties. As a result, novel formulations11 for mTHPC that circumvent these problems and allow for easy functionalization are required.
Figure 1.
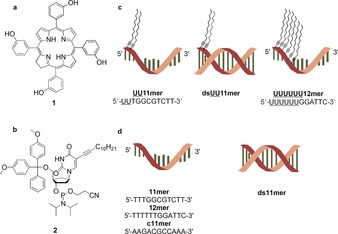
Representation of a) meta‐tetra‐hydroxyphenyl‐chlorin (mTHPC (1)); b) 5‐(dodec‐1‐ynyl)uracil deoxyribophosphoramidite (2) used in solid‐phase synthesis of lipid‐DNAs, this nucleotide building block is abbreviated as U in the corresponding sequences; c) lipid‐DNAs (UU11 mer, double‐stranded UU11 mer (dsUU11 mer) and UUUUUU12 mer) used for the solubilization of 1; d) pristine control DNAs (11 mer, complementary 11 mer (c11 mer), double‐stranded 11 mer (ds11 mer) and 12 mer).
Based on the considerations outlined above, nanocarriers made of lipid‐DNA amphiphiles (Figure 1 c) are excellent candidates to be used as solubilizers for poorly water‐soluble active pharmaceutical ingredients (APIs). Here, we report the successful use of lipid‐DNAs to render mTHPC water‐soluble with high drug loading capacities that (partially) retains the biological activity of the API.
We synthesized lipid‐DNAs with different hydrophobicity by using the alkyl modified 5‐(dodec‐1‐ynyl)uracil phosphoramidite 2 (abbreviated U in the resulting DNA sequence, Figure 1 b) using standard solid‐phase synthesis. It was reported that lipid‐DNAs can form micellar aggregates with comparatively low CMCs and the alkyl chains did not influence the hybridization of the DNA.5b We designed and synthesized two random sequences without any self‐complementarity employing an automated DNA synthesizer. The first sequence, an 11‐mer (UU11 mer, 5′‐UUTGGCGTCTT‐3′), contains two modified uracil bases and the second oligonucleotide, a 12‐mer (UUUUUU12 mer, 5′‐UUUUUUGGATTC‐3′) (Figure 1 c), is comprised of six modified uracil bases. The CMCs are 29 and 24 μm for UU11 mer and UUUUUU12 mer, respectively.
To identify the specific solubilizers for mTHPC, we screened the micelles resulting from three different types of lipid‐DNAs: single‐stranded (ss) UU11 mer, double‐stranded (ds) UU11 mer (dsUU11 mer) and ss UUUUUU12 mer (Figure 1 c). The pristine DNA counterparts with the same nucleic acid sequences, but in which the modified uracils were replaced by thymines, served as controls. This includes the ss 11 mer (5′‐TTTGGCGTCTT‐3′), ss complementary 11 mer (c11 mer, 5′‐AAGACGCCAAA‐3′), ds 11 mer (ds11 mer) and the ss 12 mer (5′‐TTTTTTGGATTC‐3′) (Figure 1 d). Samples for the screening for solubilizers contain a concentration of 50 μm both for lipid‐DNAs and controls. The formation of micellar aggregates is ensured as the concentration was set higher than the CMC of UU11 mer and UUUUUU12 mer. Incubating the aqueous solutions of DNA with the solid mTHPC ensures incorporation of mTHPC into the micelles. Centrifugation allowed for separation of the mTHPC‐loaded samples (supernatant) from the non‐solubilized mTHPC (pellet) for further characterization.
We visualized and characterized the unloaded lipid‐DNA micellar aggregates by cryogenic electron microscopy (cryo‐EM) and dynamic light scattering (DLS) in terms of their size and morphology. As expected, the cryo‐EM images (Figure 2) show in the absence of mTHPC the formation of micellar aggregates with narrow size distributions and rather uniform shapes for UU11 mer, dsUU11 mer and UUUUUU12 mer. No obvious aggregation is visible for UU11 mer and dsUU11 mer micelles (Figure 2 a,b), while bigger aggregates form for UUUUUU12 mer (Figure 2 c), as confirmed by DLS displaying a characteristic slow mode of large amplitude, which might be ascribed to the hydrophobic interactions of the six alkyl chains. Interestingly, the diameter of all aggregates are within the experimental error within the same range as UUUUUU12 mer with six alkyl chains gives 8.2±1.8 nm (Figure 2 c), and both UU11 mer (Figure 2 a) and dsUU11 mer (Figure 2 b) have 9.8±1.0 nm and 9.9±2.0 nm, irrespectively of the two alkyl chains (Table S1). It indicates that hydrophobicity does not seem to play a critical role with respect to the micelle size, which might be due to the small size of the alkyl chains. DLS experiments performed on solutions are in agreement with cryo‐EM and give the hydrodynamic radius (R h) 10.53±1 nm and 9.54±1 nm for UU11 mer and dsUU11 mer, respectively. A slow mode of small amplitude corresponding to larger aggregates is also visible in the long‐time range of the correlation function. However, this minority population can be neglected (≈0.1 % in mass), and UU11 mer and dsUU11 mer solutions can be considered as monodisperse. Apparently, the hydrophilic DNA segments are in all cases sufficient to stabilize the polar/non‐polar interfaces nonetheless of the DNA is hybridized or not. Only for UUUUUU12 mer agglomeration is visible (Figure 2), which might indicate a borderline stabilization of the six alkyl chains by the six polar nucleotides.
Figure 2.

Cryo‐EM images of micellar aggregates of UU11 mer (a), dsUU11 mer (b), UUUUUU12 mer (c) prior loading and mTHPC‐loaded micellar aggregates of UU11 mer (d), dsUU11 mer (e) and UUUUUU12 mer (f) (non‐stained samples and image acquisition was achieved with a 2 μm defocus; scale bar=50 nm).
We screened solubilizers for mTHPC by using UV/Vis spectroscopy. Based on the absorption spectra, all mTHPC‐loaded lipid‐DNA (UU11 mer, dsUU11 mer and UUUUUU12 mer) supernatants show typical absorption of mTHPC at 417 nm, which demonstrates the incorporation of mTHPC into the aqueous solutions (Figure 3). In contrast, the pristine DNAs do not show any mTHPC absorption, indicating no solubilization of the drug. The observed differences in mTHPC absorbance values can be attributed to the varied abilities to solubilize mTHPC. In this regard, the dsUU11 mer micelles are most efficient in incorporating the compound, followed by UU11 mer and finally UUUUUU12 mer.
Figure 3.
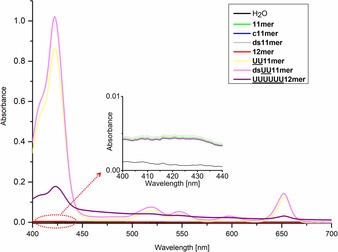
Absorption spectra of mTHPC‐solubilized supernatants for solubilizer‐screening experiments. The inset shows the region where mTHPC exhibits an absorption maximum (417 nm).
To determine the maximum loading capacities of the various lipid‐DNAs, they were incubated with a suspension of mTHPC in a mixture of H2O and EtOH, followed by lyophilization and redissolution in H2O.12 Centrifugation enabled removal of any undissolved mTHPC, affording maximum mTHPC‐loaded lipid‐DNA micelles. During the experiments, H2O and an aqueous solution of MgCl2 (Mg2+ was added to stabilize the double‐helix of the ds DNA) served as controls. We determined the loading concentrations and loading capacities by RP‐HPLC (Figure S4), adopting a method of lyophilization‐redissolution and using a calibration curve (Figure S5). The loading concentrations and loading capacities of lipid‐DNAs are presented in Table S2. As is visible, mTHPC is efficiently loaded into UU11 mer, dsUU11 mer and UUUUUU12 mer micellar aggregates and is found at high concentrations and corresponding loading capacities, while the controls show minimal solubilization of mTHPC. The maximum mTHPC loading concentration of dsUU11 mer (40.0 μm, 1:1.25 mTHPC/carrier ratio) is markedly higher than that of UU11 mer (31.1 μm, 1:1.61 mTHPC/carrier ratio) and UUUUUU12 mer (16.7 μm, 1:2.99 mTHPC/carrier ratio), which is in good agreement with the results from the solubilizer‐screening experiment. In contrast, UU11 mer achieved a higher loading capacity (11.7 %, w/w) than that of dsUU11 mer (7.8 %, w/w) due to the higher molecular weight of the double‐stranded DNA, which is a similar value to that of Pluronic® F68 (11.9 %, w/w) and conventional polymeric delivery systems.12, 13 Moreover, compared to conventional micelles, hybridization of DNA provides a facile approach for further functionalization of lipid‐DNAs with targeting groups or imaging agents. To further confirm the high loading concentrations, we recorded fluorescence‐emission spectra of the maximum mTHPC‐loaded micelles and their dilutions with equivalent volumes of EtOH (Figure S6). Abrupt increments are observed after dilution with EtOH, which illustrates the intermolecular quenching of mTHPC caused by the high concentrations inside the lipid‐DNA micelles. Cryo‐EM images of mTHPC‐loaded micelles (Figure 2 d,e,f) show the formation of micelles with a narrow size distribution and regular shape after mTHPC maximum loading. By analogy to unloaded micelles, no obvious aggregation is visible for UU11 mer and dsUU11 mer micelles, while big aggregates are formed for UUUUUU12 mer, as confirmed by DLS displaying a slow mode associated to these aggregates and masking the signal of the micelles in the short‐time range. As expected, the diameters of all the lipid‐DNA micelles increase after mTHPC loading. Although this trend is also observed with DLS giving R h 11.33±1 nm and 10.84±1 nm for loaded UU11 mer and dsUU11 mer, respectively, changes remain within the error bar. Interestingly, the aggregate sizes are not dramatically changing during mTHPC‐loading as only an increase in diameter of 0.8–1.3 nm could be found. Considering the significant differences in loading capacities of the three different carriers UU11 mer, dsUU11 mer and UUUUUU12 mer only slight hydrophobic swelling of the micellar aggregates by the mTHPC but not dramatic reorganization seems to be evident. While this might indicate the general stability of aggregates of lipid‐DNA, it also indirectly confirms a progressively loose packing of the hydrophobic alkyl chains in the core from UUUUUU12 mer to UU11 mer to dsUU11 mer. Hence, this observation suggests that the lipid‐DNA micelles with a less hydrophobic core or more hydrophilic corona feature bigger core spaces, enabling more efficient solubilization of mTHPC without dramatic increase of the aggregate sizes.
Given that singlet oxygen (1O2) plays a key role in killing tumor cells during PDT, we evaluated the activities of mTHPC‐loaded lipid‐DNA micelles by measuring the 1O2 generation, which we monitored by near‐infrared (NIR) emission spectroscopy of 1O2 generated at 1270 nm. To facilitate the detection of 1O2, we performed all measurements in D2O, which elongates the 1O2 lifetime compared to H2O.14 For this purpose, we characterized mTHPC‐loaded micellar aggregates of UU11 mer and dsUU11 mer in D2O, by UV/Vis spectroscopy and RP‐HPLC before 1O2 generation experiments. The measured absorption spectra (Figure S7) confirm that independent of D2O, the dsUU11 mer solubilizes a higher amount of mTHPC than UU11 mer. By using RP‐HPLC (Figure S4f), we found that 12 % more mTHPC is solubilized in dsUU11 mer (2.8 μm) than in UU11 mer micelles (2.4 μm) (Table S3), which is in line with the results from the absorption spectra (Figure S7). The 1O2 phosphorescence spectra by sensitization of mTHPC‐loaded UU11 mer or dsUU11 mer micelles in D2O demonstrate that mTHPC (partially) retains its activity despite micellar solubilization (Figure 4). The observed quantum yields for singlet oxygen generation are estimated to be 0.05–0.1 for both mTHPC‐loaded UU11 mer or dsUU11 mer micelles in D2O.
Figure 4.
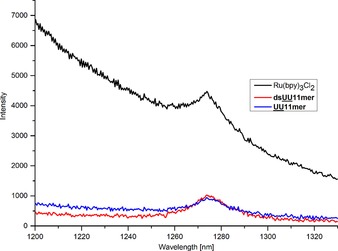
Singlet oxygen luminescence spectra of mTHPC‐loaded UU11 mer and dsUU11 mer micelles compared to reference compound (Ru(bpy)3Cl2) in D2O.
To demonstrate the PDT efficacy in vitro, we determined cell phototoxicity and dark toxicity of mTHPC (2 and 10 μm)‐loaded UU11 mer after 24 h incubation in six different cell lines, including human epidermoid carcinoma A253, human epithelial carcinoma A431, human oral adenosquamous carcinoma CAL27, murine hematopoiesis monocytic macrophages J774A.1, murine fibroblasts L929 and human colorectal adenocarcinoma HT29 cells, and followed by irradiation with laser light (Figure 5). As controls, we investigated cell phototoxicity and dark toxicity of empty UU11 mer, free mTHPC in ethanol and cells without photosensitizer following the same protocol (Figures 5 and S7). Figure S7 shows that empty UU11 mer micelles exhibit good biocompatibility in all cell lines, even at higher concentration (80 μm), while free mTHPC shows obvious dark toxicity and higher phototoxicity, demonstrating the PDT efficacy of mTHPC. For mTHPC‐loaded UU11 mer, two major conclusions can be drawn from Figure 5: (1) without irradiation, mTHPC‐loaded UU11 mer is silent and shows reduced dark toxicity in all cell lines in comparison to free mTHPC (Figure S7), which illustrates its good biocompatibility in vitro; (2) upon irradiation, mTHPC‐loaded UU11 mer becomes activated and exhibits as high phototoxicity as free mTHPC (Figure S7) in all cell lines, which demonstrates its PDT efficacy in vitro. The lack of dark toxicity against the whole panel of cells combined with the unusually high phototoxicity is remarkable.
Figure 5.
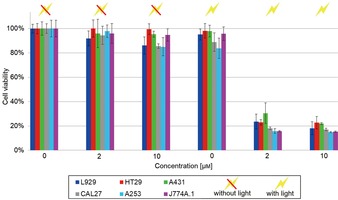
Phototoxicity and dark toxicity of mTHPC (0, 2 and 10 μm)‐loaded UU11 mer in six different cell lines (A431, HT29, L929, J744A.1, CAL27 and A253) after 24 h incubation. The photosensitization was performed at RT with a laser at 652 nm at a dose rate of app. 50 J cm−2. The cell viability was measured with a Tecan Infinite® 200 microplate reader, at a wavelength of 490 nm.
In summary, we have reported herein two types of lipid‐DNA polymers (UU11 mer and UUUUUU12 mer) containing varying numbers of hydrophobic alkyl chains as solubilizers of the poorly water‐soluble drug, mTHPC used in PDT. We designed the sequences and synthesized them through standard solid‐phase synthesis using an automated DNA synthesizer. Having determined their CMC values, we successfully used UU11 mer, dsUU11 mer and UUUUUU12 mer micelles to solubilize mTHPC with high loading concentrations and loading capacities. The dsUU11 mer micelles solubilize the most mTHPC, while UU11 mer has the highest loading capacity due to the lower molecular weight of the non‐hybridized DNA section. We conclude that lipid‐DNA micelles with a less hydrophobic core or more hydrophilic corona result in micellar aggregates and less compact core packings, which leads to enhanced solubilization of mTHPC. In addition, the generated phosphorescence demonstrates that mTHPC (partially) remains active in D2O. Finally, a cell viability study showed that mTHPC‐loaded UU11 mer shows excellent biocompatibility without irradiation, and very high PDT efficacy upon irradiation. Our work illustrates the successful use of lipid‐DNA micelles to solubilize mTHPC with high loading capacities while (partially) retaining the biological activity of the API. Interestingly, the size and morphology of the micelles are related to the hydrophobicity of the corresponding lipid‐DNAs, which can be fine‐tuned by hybridization with the complementary strand or altering the number of incorporated modified uracil nucleotides. Thus, the present results offer a basis for the rational design of a novel class of drug‐delivery vehicle based on lipid‐DNAs. Notably, hybridization offers a facile route for further functionalization of the drug‐delivery system, allowing adding moieties such as targeting groups or imaging reagents by hybridization. Therefore, our lipid‐DNA micellar drug‐delivery system holds great potential for further development and application in the biomedical field.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Y.L. was supported by a PhD fellowship from the Chinese Scholarship Council. Funding was granted by the Netherlands Organisation for Scientific Research (NWO‐CW ECHO‐STIP grant to A. K. H. Hirsch) and by the Dutch Ministry of Education, Culture, Science (gravitation program 024.001.035). HGB would like to acknowledge funding from the European Research Council under the European Union's 7th Framework Program (FP07‐13)/ERC Starting grant “Specifically Interacting Polymers–SIP” (ERC 305064).
Y. Liu, J. W. de Vries, Q. Liu, A. M. Hartman, G. D. Wieland, S. Wieczorek, H. G. Börner, A. Wiehe, E. Buhler, M. C. A. Stuart, W. R. Browne, A. Herrmann, A. K. H. Hirsch, Chem. Eur. J. 2018, 24, 798.
Contributor Information
Prof. Andreas Herrmann, Email: a.herrmann@rug.nl.
Prof. Anna K. H. Hirsch, Email: anna.hirsch@helmholtz-hzi.de.
References
- 1.
- 1a. Mayr L. M., Bojanic D., Curr. Opin. Pharmacol. 2009, 9, 580–588; [DOI] [PubMed] [Google Scholar]
- 1b. White R. E., Annu. Rev. Pharmacol. Toxicol. 2000, 40, 133–157. [DOI] [PubMed] [Google Scholar]
- 2. Loftsson T., Brewster M. E., J. Pharm. Pharmacol. 2010, 62, 1607–1621. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Yu H., Zou Y., Wang Y., Huang X., Huang G., Sumer B. D., Boothman D. A., Gao J., ACS Nano 2011, 5, 9246–9255; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Huang Y., Dong T., Zhu X., Yan D., Soft Matter 2014, 10, 6121–6138; [DOI] [PubMed] [Google Scholar]
- 3c. Lee Y., Kataoka K., Soft Matter 2009, 5, 3810–3817; [Google Scholar]
- 3d. Harada A., Kataoka K., J. Am. Chem. Soc. 1999, 121, 9241–9242; [Google Scholar]
- 3e. Chilkoti A., Dreher M. R., Meyer D. E., Raucher D., Adv. Drug Delivery Rev. 2002, 54, 613–630. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Li Y.-Y., Cheng H., Zhu J.-L., Yuan L., Dai Y., Cheng S.-X., Zhang X.-Z., Zhuo R.-X., Adv. Mater. 2009, 21, 2402–2406; [Google Scholar]
- 4b. Liu Y., Li C., Wang H.-Y., Zhang X.-Z., Zhuo R.-X., Chem. Eur. J. 2012, 18, 2297–2304; [DOI] [PubMed] [Google Scholar]
- 4c. Xu H., Yao Q., Cai C., Gou J., Zhang Y., Zhong H., Tang X., J. Controlled Release 2015, 199, 84–97; [DOI] [PubMed] [Google Scholar]
- 4d. Gaspar V. M., Gonçalves C., De Melo-Diogo D., Costa E. C., Queiroz J. A., Pichon C., Sousa F., Correia I. J., J. Controlled Release 2014, 189, 90–104; [DOI] [PubMed] [Google Scholar]
- 4e. Poelma S. O., Oh S. S., Helmy S., Knight A. S., Burnett G. L., Soh H. T., Hawker C. J., Read de Alaniz J., Chem. Commun. 2016, 52, 10525–10528; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4f. Chung E. J., Cheng Y., Morshed R., Nord K., Han Y., Wegscheid M. L., Auffinger B., Wainwright D. A., Lesniak M. S., Tirrell M. V., Biomaterials 2014, 35, 1249–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Berti D., Barbaro P., Bucci I., Baglioni P., J. Phys. Chem. B 1999, 103, 4916–4922; [Google Scholar]
- 5b. Anaya M., Kwak M., Musser A. J., Müllen K., Herrmann A., Chem. Eur. J. 2010, 16, 12852–12859; [DOI] [PubMed] [Google Scholar]
- 5c. Tang L., Tjong V., Li N., Yingling Y. G., Chilkoti A., Zauscher S., Adv. Mater. 2014, 26, 3050–3054; [DOI] [PubMed] [Google Scholar]
- 5d. Peterson A. M., Heemstra J. M., WIREs Nanomed. Nanobiotechnol. 2015, 7, 282–297; [DOI] [PubMed] [Google Scholar]
- 5e. Pokholenko O., Gissot A., Vialet B., Bathany K., Thiéry A., Barthélémy P., J. Mater. Chem. B 2013, 1, 5329–5334; [DOI] [PubMed] [Google Scholar]
- 5f. Chen T., Wu C. S., Jimenez E., Zhu Z., Dajac J. G., You M., Han D., Zhang X., Tan W., Angew. Chem. Int. Ed. 2013, 52, 2012–2016; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2066–2070. [Google Scholar]
- 6. Zhan P., Jiang Q., Wang Z., Li N., Yu H., Ding B., ChemMedChem 2014, 9, 2013–2020. [DOI] [PubMed] [Google Scholar]
- 7. Bellon L., Wincott F. in Solid-Phase Synthesis. A Practical Guide (Eds.: S. A. Kates, F. Albericio, Marcel Dekker), Marcel Dekker, Inc., New York: 2000, pp. 475–528. [Google Scholar]
- 8. Alemdaroglu F. E., Alemdaroglu N. C., Langguth P., Herrmann A., Adv. Mater. 2008, 20, 899–902. [Google Scholar]
- 9.
- 9a. Senge M. O., Photodiagn. Photodyn. Ther. 2012, 9, 170–179; [DOI] [PubMed] [Google Scholar]
- 9b. Senge M. O., Brandt J. C., Photochem. Photobiol. 2011, 87, 1240–1296. [DOI] [PubMed] [Google Scholar]
- 10. Triesscheijn M., Ruevekamp M., Aalders M., Baas P., Stewart F. A., Photochem. Photobiol. 2005, 81, 1161–1167. [DOI] [PubMed] [Google Scholar]
- 11. Hinger D., Gräfe S., Navarro F., Spingler B., Pandiarajan D., Walt H., Couffin A.-C., Maake C., J. Nanobiotechnol. 2016, 14, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wieczorek S., Krause E., Hackbarth S., Röder B., Hirsch A. K. H., Börner H. G., J. Am. Chem. Soc. 2013, 135, 1711–1714. [DOI] [PubMed] [Google Scholar]
- 13. Syu W.-J., Yu H.-P., Hsu C.-Y., Rajan Y. C., Hsu Y.-H., Chang Y.-C., Hsieh W.-Y., Wang C.-H., Lai P.-S., Small 2012, 8, 2060–2069. [DOI] [PubMed] [Google Scholar]
- 14. Merkel P. B., Nilsson R., Kearns D. R., J. Am. Chem. Soc. 1972, 94, 1030–1031. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary