

Abstract
To characterize somatic alterations in colorectal cancer (CRC), we conducted a genome‐scale analysis of 106 CRC specimens. We assessed comprehensive somatic copy number alterations (SCNAs) in these CRC specimens. In addition, we examined microsatellite instability (MSI; low and high), genetic mutations (KRAS, BRAF, TP53, and PIK3CA), and DNA methylation status (classified into low, intermediate, and high type). We stratified molecular alterations in the CRCs using a hierarchical cluster analysis. The examined CRCs could be categorized into three subgroups using hierarchical cluster analysis. Tumors in subgroup 1 were characterized by a low frequency of SCNAs and a high frequency of MSI‐high status, whereas tumors in subgroups 2 and 3 were closely associated with a high frequency of SCNAs. Tumors in subgroup 1 were preferentially present in the right‐sided colon and showed frequent MSI‐high status. Subgroup 3 was distinguished by specific alterations, including gains at 1q23‐44, 1p11‐36, 10q11‐26, 10p11‐13, 12q24‐24, and 13q33‐33. In contrast, tumors in subgroup 2 were characterized by copy‐neutral LOH at 12p12‐13, 1q24‐25, and 10q22. In addition, KRAS mutations were more frequently found in subgroup 3 than in subgroup 1. TP53 mutations and intermediate levels of DNA methylation were common alterations in the three subgroups. SCNAs contributed to sporadic CRC, and there were three subgroups based on SCNAs that played a different role in driving the development of this disease.
Keywords: colorectal cancer, comprehensive genomic analysis, copy number alteration, microsatellite instability, mutation
1. INTRODUCTION
Colorectal cancer (CRC) is the third most common cancer worldwide and the fourth most common cause of cancer‐related death.1 Dramatic advancements in medical science have been made in the field of CRC. For example, previous studies have shown that there are three distinct molecular pathways contributing to CRC, including chromosomal instability (CIN), microsatellite instability (MIN; MSI), and CpG island methylator phenotype (CIMP).2, 3, 4 Moreover, most cases of CRC can be explained by a combination of these three pathways occurring in the colorectum.3, 4, 5, 6 Many advances in molecular analyses have also been made in CRC.7 Recent studies have described novel molecular classifications based on gene expression, including consensus molecular subtypes and colorectal subtyping associated with cellular phenotypes and responses to therapy.8, 9, 10 These hypotheses provide novel insights into the molecular features of colorectal carcinogenesis.7, 8, 9
Accumulation of genomic alterations is closely associated with the progression of CRC.11 For example, somatic copy number alterations (SCNAs) that characterize the CIN pathway are involved in the progression of CRC.11, 12 Specific mutations, including mutations in APC, KRAS, PIK3CA, and TP53, are frequently detected in CRC with the CIN‐phenotype and may be associated with accumulation of SCNAs, playing only a minor role in the progression of CRC with the MIN type13 because accumulation of numerous mutations within the whole genome, rather than SCNAs, is important for the progression of the MIN type of CRC.6, 7 Specific mutations in BRAF and KRAS have also been shown to be closely related to CRC with the MIN‐phenotype.6, 7 CIMP‐low or ‐intermediate status is primarily found in CIN‐type CRC,13 whereas acquisition of the CIMP‐high status is closely associated with MIN‐type CRC.13 CIMP markers have been developed for prediction of genome‐wide methylation levels in tumor cells,14, 15, 16 and a two‐step panel for classification of the CIMP status of tumor cells has been shown to be a good predictor of CIMP status.15, 16 Elucidation of the relationships among SCNAs and the progression of CRC with regard to specific mutations, microsatellite status (MSI or microsatellite stable [MSS]), and methylation status is essential for identification of the causes of colorectal carcinogenesis.
Although the molecular features of colorectal carcinogenesis have been extensively studied, here, we conducted genome‐wide SCNA pattern analysis in CRC cells to identify the molecular characteristics of colorectal carcinogenesis and the role of SCNA patterns in the progression of CRC. In addition, we attempted to analyze the associations of SCNA patterns with well‐known molecular alterations, including specific mutations, MS status, and methylation status in CRCs.
2. METHODS
2.1. Patients
A total of 106 samples (excluding intramucosal and submucosal cancers) were obtained from patients with newly diagnosed primary CRC at Iwate Medical University between 2011 and 2016. These samples were from resected specimens, and the patients had not received prior chemotherapy.
The pathological diagnosis and staging were determined according to a combination of a Japanese classification and the modified Dukes' classification.17 The detailed clinicopathological findings are listed in Table 1.
Table 1.
Clinicopathological features of 106 colorectal cancers examined
| Characteristics | Colorectal cancers (%) |
|---|---|
| Total | 106 |
| Gender | |
| Male | 52 (49.1) |
| Female | 54 (50.9) |
| Age | |
| Median (range) | 71 (39‐90) |
| Location | |
| Right‐sided | 39 (36.8) |
| Left‐sided | 67 (63.2) |
| Differentiation | |
| WDA | 6 (5.7) |
| MDA | 92 (86.8) |
| Other | 8 (7.5) |
| Lymphatic invasion | |
| Negative or low | 98 (92.5) |
| High | 8 (7.5) |
| Venous invasion | |
| Negative or low | 93 (87.7) |
| High | 13 (12.3) |
| TNM stage | |
| I | 18 (17.0) |
| II | 31 (29.2) |
| III | 42 (39.6) |
| IV | 15 (14.2) |
This study was approved by the Ethical Research Committee, Iwate Medical University.
2.2. Crypt isolation
Fresh tumor and normal tissues were obtained from resected CRC tissues. Normal colonic mucosa was collected from the most distal portion of the colon.
Crypt isolation from the tumor and normal mucosa was performed as previously reported.18 Briefly, fresh mucosa and tumor tissues were minced with a razor into small pieces and incubated at 37°C for 30 min in calcium‐ and magnesium‐free Hanks' balanced salt solution (CMF) containing 30 mM ethylenediaminetetraacetic acid (EDTA). The isolated crypts were immediately fixed in 70% ethanol and stored at 4°C until used for DNA extraction.
The fixed isolated crypts were observed under a dissecting microscope (SZ60; Olympus, Tokyo, Japan). The tumor samples were collected primarily from the central area of tumor ulceration because the invasive front, which was thought to determine the invasive ability of the tumor cells, was observed in the central area.19 In addition, considerable cancerous glands were present in this area.
Some isolated crypts were routinely processed by histopathological analysis to confirm the histological nature of the isolated glands. Contamination, such as interstitial cells, was not evident in any of our 106 samples.
2.3. DNA extraction
DNA from normal and tumor glands was extracted by standard SDS proteinase K treatment. DNA extracted from the samples was resuspended in TE buffer (10 mM Tris‐HCl, 1 mM EDTA [pH 8.0]).
2.4. Analysis of MSI
The DNA was amplified by polymerase chain reaction (PCR) with fluorescent dye‐labeled primers targeting five microsatellites, including BAT25, BAT26, D5S346, D2S123, and D17S250. The DNA was analyzed using a DNA sequencer (ABI PRISM‐310 Genetic Analyzer; ABI, Foster City CA), and microsatellite fragment analyses were performed using a GeneScan software (ABI). MSI status was determined by the presence of additional bands in the PCR product from tumor DNA. MSI‐high (MSI‐H) status was defined as instability in at least two of the five microsatellite loci, whereas MSI‐low (MSI‐L) was defined as instability in only one locus. In addition, MSI‐negative was defined as having no shifted loci according to NCI criteria.20 However, in the present study, tumors categorized as MSI‐L and MSI‐negative were considered MSS in this analysis.
2.5. Analysis of KRAS and BRAF mutations
Mutations in KRAS (codons 12 and 13) and BRAF (V600E) genes were analyzed using a CE‐IVD marked PyroMark (Qiagen, Hilden, Germany) according to the manufacturer's protocols (Therascreen KRAS Pyro Kit Handbook, version 1, July 2011). The primers used in the present study were described previously.21 The cut‐off value for the mutation assay was 15% mutant alleles. Each PCR product was examined by pyrosequencing using PyroMark Gold Q96 reagents, Streptavidin Sepharose High Performance (GE Healthcare Bio‐Science AB, Uppsala, Sweden) and a PyroMark Q24 instrument (Qiagen) with PyroMark Q24 1.0.6.3 software.
2.6. Analysis of TP53 and PIK3CA mutations
Mutations in the TP53 gene in exons 5‐8 and the PIK3CA gene in exons 9 and 20 were examined by PCR single‐stranded conformation polymorphism (PCR‐SSCP), with some modifications.12 Mutations obtained by SSCP in the TP53 and PIK3CA genes were then analyzed by sequence analysis. Briefly, PCR was used to amplify exons 5‐8 of the TP53 gene and exons 9 and 20 of the PIK3CA gene. The conditions for PCR were described previously.22 Briefly, the PCR products (2 µL) were mixed with 10 µL of gel loading solution (9.5% deionized formamide, 20 mM EDTA‐Na, 0.05% xylene cyanol, and bromphenol blue), denatured at 95°C for 5 min, and then kept on ice until loading. Nondenaturing 7.5% polyacrylamide gels were used for electrophoresis, which was performed at 260‐300 V and 22°C for 3‐12 h using a temperature controller (Resolmax, ATTO Co., Tokyo). The gels were visualized by silver staining and photographed. The migrated band was removed from the gel, and the DNA was extracted. The sequence was determined by direct sequencing, as previously described.
2.7. Pyrosequencing for evaluation of methylation
The DNA methylation status was determined by PCR analysis of bisulfite‐modified genomic DNA (EpiTect Bisulfite Kit; Qiagen) using pyrosequencing for quantitative methylation analysis (Pyromark Q24; Qiagen NV). The primers used in this study were designed previously.13, 21
DNA methylation was quantified using six specific promoters originally described by Yagi et al and Kaneda and Yagi.15, 16 Briefly, after methylation analysis of the first panel of three markers (RUNX3, MINT31, and LOX), tumors with hypermethylated epigenomes (HMEs) were identified based on methylation with at least two methylated markers. The remaining tumors were examined using a second panel of three markers (NEUROG1, ELMO1, and THBD). Tumors with intermediate methylated epigenomes (IMEs) were defined as those with at least two methylated markers, whereas tumors not classified as having HMEs or IMEs were designated as showing hypomethylated epigenomes (LMEs); that for the methylation assay was 30% of tumor cells, as previously reported.15, 16
2.8. SCNA analysis
Extracted DNA was adjusted to a concentration of 50 ng/µL. All 106 paired samples were assayed using an Infinium HumanCytoSNP‐12v2.1 BeadChip (Illumina, San Diego, CA), which contains 299 140 single nucleotide polymorphism (SNP) loci, according to the Illumina Infinium HD assay protocol.12 BeadChips were scanned using iScan (Illumina) and analyzed using GenomeStudio software (v.2011.1; Illumina). The log R ratio (LRR) and B allele frequency (BAF) for each sample were exported from normalized Illumina data using GenomeStudio. Data analysis was performed using KaryoStudio 1.4.3 (CNV Plugin v3.0.7.0; Illumina) with default parameters. CNVs were classified as described below. In the classification of chromosome copy number variations by CNV partition algorithms, LRR 0 indicated a normal diploid region, LRR greater than 0 indicated a copy number gain, and LRR less than 0 indicated a copy number loss‐of‐heterozygosity (LOH). BAF values ranged from 0 to 1; homozygous SNPs had BAFs near 0 (A‐allele) or 1 (B‐allele), and heterozygous diploid region SNPs had BAFs near 0.5 (AB genotype). Additionally, LRR and BAF data were used to identify regions of hemizygous and copy‐neutral LOH.
In the present study, we classified copy number alterations into three types, including copy number gains (CN‐gains), copy number‐loss of heterozygosity (LOH), and copy neutral‐LOH.12
2.9. Statistical analysis
Hierarchical analysis was performed for clustering the samples according to the SCNA pattern in order to achieve maximal homogeneity for each group and the highest difference between groups. The clustering algorithm was set to centroid linkage clustering, the standard hierarchical clustering method used in biological analyses. The method was described elsewhere.
Data obtained for histological features, mutations, methylation, and SCNA status based on each subgroup were analyzed using chi‐square tests with Yates' corrections with the aid of Stat Mate‐III software (Atom, Tokyo, Japan). Differences in age distributions between the two groups were analyzed using Mann‐Whitney U tests (PRISM6; GraphPad software, La Jolla, CA). Differences with P values of less than 0.05 were considered significant.
3. RESULTS
In the present study, hierarchical clustering analysis based on the SCNA pattern, including gains, LOHs, and copy‐neutral LOHs, was carried out to examine differences in genetic alterations in samples from patients with CRC.
Three distinct subgroups were categorized, as shown in Figure 1. The vertical line shows SCNAs, and the horizontal lines denote “relatedness” between samples and SCNAs at the chromosomal loci. The CRCs examined in this study were categorized into three distinct patterns in the cluster analysis.
Figure 1.
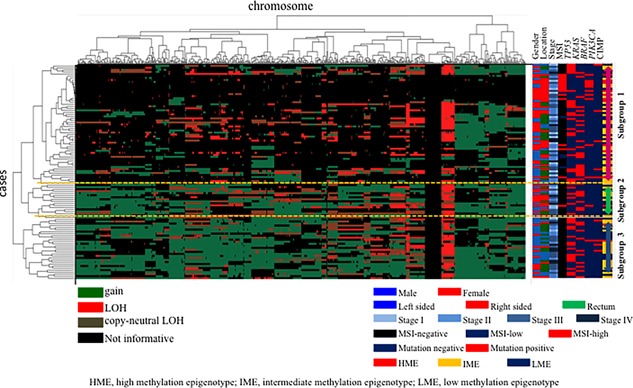
Hierarchical cluster analysis based on copy number alterations in 106 colorectal cancer specimens
The clinical findings in each subgroup categorized based on SCNAs are listed in Table 2. The frequency of right‐sided CRC was significantly higher in subgroup 1 than in subgroup 3.
Table 2.
Clinicopathological features in each subgroup categorized based on copy number alterations
| Characteristics | Subgroup 1 | Subgroup 2 | Subgroup 3 | P‐value |
|---|---|---|---|---|
| Total | 59 (%) | 17 (%) | 30 (%) | |
| Gender | ||||
| Male | 25 (42.4) | 7 (41.2) | 20 (66.7) | NS |
| Female | 34 (57.6) | 10 (58.8) | 10 (33.3) | |
| Age | ||||
| Median (range) | 73 (39‐90) | 70 (40‐88) | 72 (47‐87) | NS |
| Location | ||||
| Right sided | 26 (44.1) | 9 (52.9) | 4 (13.3) | <0.01 |
| Left sided | 33 (55.9) | 8 (47.1) | 26 (86.7) | |
| Differentiation | ||||
| WDA | 5 (8.5) | 1 (5.9) | 0 (0) | NS |
| MDA | 48 (81.4) | 15 (88.2) | 29 (96.7) | |
| Other | 3 (5.1) | 1 (5.9) | 1 (3.3) | |
| Lymphatic invasion | ||||
| Negative or low | 56 (94.9) | 15 (88.2) | 27 (90.0) | NS |
| High | 3 (5.1) | 2 (11.8) | 3 (10.0) | |
| Venous invasion | ||||
| Negative or low | 54 (91.5) | 12 (70.6) | 27 (90.0) | NS |
| High | 5 (8.5) | 5 (29.4) | 3 (10.0) | |
| TNM stage | ||||
| I | 12 (20.3) | 4 (23.5) | 2 (6.7) | NS |
| II | 19 (32.2) | 3 (17.7) | 9 (30.0) | |
| III | 23 (39.0) | 6 (35.3) | 13 (43.3) | |
| IV | 5 (8.5) | 4 (23.5) | 6 (20.0) | |
3.1. SCNAs in CRCs we examined
The mean total number of chromosomal aberrations per patient was 548, with an average of 419 gains (range: 0‐583), 48 LOHs (range: 0‐157), and 81.7 copy‐neutral LOHs (range: 0‐328).
Regions of gain detected in more than 50% of cases were located at 7p22.1, 7q11‐12, 7q21.12, 7q33, 8q22.1, 8q23.2, 13q11‐13, 13q21‐22, 13q31.2‐32.3. 13q34, 20p11.21, 20q11.23, and 20q21‐23 in the CRCs examined in this study. Additionally, regions of LOH (more than 50% of cases) were at 18p11.32, 18p21‐23, and 18q in the CRCs. No copy‐neutral LOHs showing more than 50% of cases were found in the examined CRCs. The average frequencies of SCNAs across the entire genome are shown in Figure 2.
Figure 2.
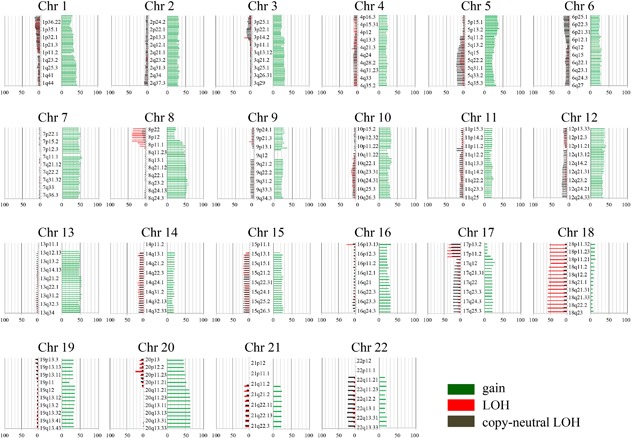
Ideogram of copy number alterations in 106 colorectal cancers specimens. Chromosomes are ordered from 1 to 22. The colored horizontal lines represent the frequencies of gains, LOHs, and CNLOHs. Lines on the left indicate losses (red, copy neutral LOH; gray, LOH), and those on the right (green) indicate gains
3.2. SCNAs in subgroups 1, 2, and 3
The SCNAs of all chromosomes according to each subgroup are shown in Figure 3A‐C. The mean total number of chromosomal aberrations per patient was 198, with an average of 104 gains (range: 0‐291), 50 LOHs (range: 0‐184), and 44 copy‐neutral LOHs (range: 0‐292) in subgroup 1. In subgroup 3, the mean total number of chromosomal aberrations per patient was 529, with an average of 415 gains (range: 271‐583), 47 LOHs (range: 1‐127), and 67 copy‐neutral LOHs (range: 0‐182). Finally, in subgroup 2, the mean total number of chromosomal aberrations per patient was 530, with an average of 350 gains (range: 186‐482), 46 LOHs (rang: 2‐157), and 134 copy‐neutral LOHs (range: 9‐328). There were significant differences in the total numbers of CNAs between subgroups 1 and 2 or 3 (P < 0.01). Moreover, significant differences were observed in the average numbers of CN gains between subgroups 1 and 2 or 3 (P < 0.01). Although LOH was common between the three subgroups, there were significant differences in the average numbers of CN LOHs between subgroups 2 and 1 or 3 (P < 0.01).
Figure 3.

Ideogram of copy number alterations in three subgrouops categorized based on copy number alteation pattern. Chromosomes are ordered from 1 to 22. The colored horizontal lines represent the frequencies of gains, LOHs, and CNLOHs. Lines on the left indicate losses (red, copy neutral LOH; gray, LOH), and those on the right (green) indicate gains. A, Ideogram of copy number alterations in subgroup 1. B, Ideogram of copy number alterations in subgroup 2. C, Ideogram of copy number alterations in subgroup 3
Regions of gains detected in more than 50% of cases were located at 20q11.21‐13.33 in subgroup 1; 13q, 2q, 2p, 3q, 3p, 5p, 20q, 20p, 8q, 19q, 19p, 21q, 7q, 4p, and 4q (in decreasing order of frequency) in subgroup 2; and 1q, 1p, 10q, 10p, 21q, 2q0p, 8q, 8p, 12q, 12p, 5p, 3q, 7p, 7q, 9p21‐11‐22.32, 15q21.2‐26.3, 19q, 19p, and 16q12.1‐.21 in subgroup 3. LOHs detected in more than 50% of cases were found at 18q21.33‐22.3 in subgroup 1, 18q, and 18p in subgroups 2 and 3, and 8p12.22 in subgroup 3. Copy‐neutral LOH was not detected in any subgroup. These results are summarized in Table 3.
Table 3.
Comparison of regions frequent CNAs in each subgroup
| Chromosomal regions | Subgroup 1, n = 59 (%) | Chromosomal regions | Subgroup 2, n = 17 (%) | Chromosomal regions | Subgroup 3, n = 30 (%) | |
|---|---|---|---|---|---|---|
| Gain | 20q11.21‐13.33 | 30‐32 (50.8‐54.2) | 13q | 15‐16 (88.2‐94.1) | 1q | 23‐28 (76.7‐93.3) |
| 2q | 12‐14 (70.6‐82.4) | 1p | 20‐28 (66.7‐93.3) | |||
| 2p | 11‐13 (64.7‐76.5) | 10q | 19‐24 (63.3‐80.0) | |||
| 3q | 11‐12 (64.7‐70.6) | 10p | 22‐24 (73.3‐80.0) | |||
| 3p | 9‐12 (52.9‐70.6) | 20q | 23‐24 (76.7‐80.0) | |||
| 5p | 11‐12 (64.7‐70.6) | 20p | 19‐23 (63.3‐76.7) | |||
| 20q | 11 (64.7) | 8q | 19‐23 (63.3‐76.7) | |||
| 20p | 10‐11 (58.8‐64.7) | 8p | 16‐21 (53.3‐70.0) | |||
| 8q | 9‐13 (52.9‐76.5) | 12q | 20‐22 (66.7‐73.3) | |||
| 19q | 9‐11 (52.9‐64.7) | 12p | 21‐22 (70.0‐73.3) | |||
| 19p | 9‐10 (52.9‐58.8) | 13q | 16‐21 (53.3‐70.0) | |||
| 21q | 9‐10 (52.9‐58.8) | 5p | 20 (66.7) | |||
| 7q | 9‐10 (52.9‐58.8) | 3q | 16‐19 (53.3‐63.3) | |||
| 4q | 9 (52.9) | 7q | 17‐18 (56.7‐63.3) | |||
| 4p | 9 (52.9) | 7p | 17‐19 (56.7‐63.3) | |||
| 9q21.11‐22.32 | 17 (56.7) | |||||
| 15q21.2‐26.3 | 16‐18 (53.3‐60.0) | |||||
| 19q | 16‐18 (53.3‐60.0) | |||||
| 19p | 17 (56.7) | |||||
| 16q12.1‐21 | 16 (53.3) | |||||
| LOH | 18q21.33‐22.3 | 30‐31 (50.8‐52.5) | 18q | 12 (70.6) | 18q | 19 (63.3) |
| 18p | 10 (58.8) | 18p | 16‐17 (53.3‐56.7) | |||
| 8p12‐22 | 16 (53.3) | |||||
| Copy‐neutral LOH | None | None | None |
3.3. Differences in SCNAs between subgroups
Next, we examined differences in SCNAs between the three subgroups. Regions of gain detected in more than 50% of cases were selected for comparison of each group. We also examined regions of loss found in more than around 30% of cases in the three subgroups.
Significant differences in gains between subgroups 2 and 3 were found at 1q23.1‐44, 1p11.2‐36.33, 10q11.23‐26.3, 10p11.21‐13, 12q24.21‐24.32, and 13q33.1‐33.3 (subgroup 2 < subgroup 3; Table 4). Significant differences in the frequencies of copy‐neutral LOH between subgroups 2 and 3 were found at 12p12.2–13.1, 1q24.3–25.1, and 10q22.1 (subgroup 3 < subgroup 2; Table 4). However, no differences in the frequency of LOH were found between the three subgroups.
Table 4.
Significant differences in the frequencies of CNAs between subgroup 2 and subgroup 3
| Chromosomal regions | Subgroup 2, n = 17 (%) | Subgroup 3, n = 30 (%) | P value | |
|---|---|---|---|---|
| Gain | 1q23.1‐44 | 7‐8 (41.2‐47.1) | 26‐28 (86.7‐93.3) | <0.01 |
| 1p11.2‐36.33 | 3‐4 (17.6‐23.5) | 20‐28 (66.7‐93.3) | <0.01 | |
| 10q11.23‐26.3 | 4‐5 (23.5‐29.4) | 22‐25 (73.3‐83.3) | <0.01 | |
| 10p11.21‐13 | 6 (35.3) | 24 (80.0) | <0.01 | |
| 12q24.21‐24.32 | 5 (29.4) | 22 (73.3) | <0.01 | |
| 13q33.1‐33.3 | 16 (94.1) | 16 (53.3) | <0.01 | |
| LOH | None | |||
| Copy‐neutral LOH | 12p12.2‐13.1 | 6 (35.3) | 0 (0) | <0.01 |
| 1q24.3‐25.1 | 5 (29.4) | 0 (0) | 0.01 | |
| 10q22.1 | 5 (29.4) | 0 (0) | 0.01 |
Significant differences in the frequencies of CN gains were observed between subgroups 1 and 2 at 2q, 2p, 3q, 3p, 5p, 4q, 4p, 21q, 19q, and 19p (Supplementary Table S1). Although no significant differences in LOH were observed between subgroups 1 and 2, significant differences in copy‐neutral LOH at 5q31‐33, 5q21‐22, 22q11‐12, 17q21‐25, 17p11‐13, and 12p11‐12 were found between subgroups 1 and 2.
Finally, we examined significant differences in the frequencies of SCNAs between subgroups 1 and 3. Significant differences in CN gains between subgroups 1 and 3 were observed at 1p, 1q, 2p, 2q, 3q, 3q, 4q, 5p, 5q, 6p, 6q, 7p, 7q, 8p, 8q, 9p, 9q, 10p, 11p, 11q, 12p, 12q, 13q, 14q, 16p, 16q, 19p, 19q, 20q, 21q, and 22q (subgroup 1 < subgroup 3) (Supplementary Table S2). There were significant differences in the frequencies of CN‐LOH at 17q24‐25 and 17p11‐13 between subgroups 1 and 3. However, no significant differences in LOH were found between subgroups 1 and 3.
When regions of gains detected in more than 40% of cases were selected for comparisons of the groups, common SCNAs between the three subgroups were gains at 7p15.2, 7q11‐12, 8q21‐24, 20q11‐13, and LOHs of 17p11, and 18p/q.
3.4. Differences in MSI, mutations in cancer‐related genes, and methylation statuses between subgroups 1, 2, and 3
The frequency of MSI‐high status was significantly higher in subgroup 1 than in subgroup 3. However, there were no significant differences in the frequencies of MSI‐high status between subgroups 2 and 3. Next, we examined mutations in TP53, KRAS, BRAF, and PIK3CA in subgroups 1, 2, and 3. The frequency of KRAS mutation was significantly higher in subgroup 3 (20/30, 66.7%) than in subgroup 1 (21/59, 35.6%; P = 0.02). In addition, TP53, BRAF, and PIK3CA mutations were not correlated with subgroups 1, 2, or 3. Finally, we analyzed methylation statuses in subgroups 1, 2, and 3 and found that there were no significant differences in the frequencies of methylation statuses among the subgroups. These results are summarized in Table 5.
Table 5.
Frequencies of microsatellite phenotype, mutations, and DNA methylation status in each subgroup
| Subgroup 1 | Subgroup 2 | Subgroup 3 | P‐value | |
|---|---|---|---|---|
| 5‐a | ||||
| Microsatellite phenotype | ||||
| Total | 59 (%) | 17 (%) | 30 (%) | |
| High | 14 (23.7) | 1 (5.8) | 0 (0) | <0.001 |
| Low | 5 (8.5) | 0 (0) | 0 (0) | |
| Negative | 40 (67.8) | 16 (94.2) | 30 (100) | |
| 5‐b | ||||
| Mutated gene | ||||
| Total | 59 (%) | 17 (%) | 30 (%) | |
| TP53 positive | 22 (37.3) | 6 (35.3) | 14 (46.7) | NS |
| KRAS positive | 21 (35.6) | 8 (47.1) | 20 (66.7) | 0.02 |
| BRAF positive | 10 (16.9) | 1 (5.8) | 1 (3.3) | NS |
| PIK3CA positive | 5 (8.5) | 0 (0) | 4 (13.3) | NS |
| 5‐c | ||||
| Methylation status | ||||
| Total | 59 (%) | 17 (%) | 30 (%) | |
| HME | 10 (17.0) | 1 (5.8) | 2 (6.6) | NS |
| IME | 30 (50.8) | 8 (47.1) | 17 (56.7) | |
| LME | 19 (32.2) | 8 (47.1) | 11 (36.7) | |
HME, high methylation phenotype; IME, intermediate methylation phenotype; LME, low methylation phenotype.
4. DISCUSSION
CRC results from the accumulation of somatic CNAs.23, 24, 25 Genome‐wide analysis of SCNAs provides opportunities for identifying cancer driver genes in an unbiased manner.11 Recent studies have shown that many molecular mechanisms affect the development of CRC,7, 8, 10, 12, 13 contributing to advancements in approaches for the clinical diagnosis and treatment of CRC.8, 10 Our current data indicated that there were three molecular patterns based on SCNAs, as identified by high‐throughput genomic analysis. Thus, analysis of the molecular patterns of CRC according to SCNAs may help us to understand the characteristics of colorectal carcinogenesis.11 We showed that the three patterns established based on SCNA patterns were closely associated with clinicopathological and molecular findings in CRCs. Moreover, our study confirmed that molecular alterations based on SCNAs converged into three different patterns during colorectal progression.
A previous study showed that there are two molecular types of colorectal carcinogenesis, including MSS and MIN.2, 3 The majority of CRC cases are classified into the MSS phenotype (approximately 80‐90%), and the remaining CRC cases (approximately 10‐20%) are assigned into the MIN phenotype.2, 3, 4 However, CRC with the MSS phenotype is thought to be a heterogeneous disease. We showed that CRC with the MSS phenotype could be largely classified into two subtypes in terms of SCNAs (low frequency SCNAs and high frequency SCNAs). The latter subgroup can be further sub‐classified into two subgroups based on differences in SCNAs in the tumor cells. In the present study, subgroup 3 was distinguished from subgroup 2 by gains at 1q23.1‐44, 1p11.2‐36.33, 10q11.23‐26.3, 10p11.21‐13, 12q24.21‐24.32 (subgroup 2 < subgroup 3), and 13q33.1‐33.3 and copy‐neutral LOH at 12p12.2‐13.1, 1q24.3‐25.1, and 10q22.1 (subgroup 2 > subgroup 3). In addition, there were significant differences in the average numbers of CN‐LOHs between subgroups 2 and 1 or 3. These findings suggest that CN‐LOH or CN‐LOH on the specific chromosomal loci (12p12.2‐13.1, 1q24.3‐25.1, and 10q22.1) played certain roles in the development of CRC with the MSS phenotype. Thus, these findings revealed that although CRC was a complex disease with regard to pathological and molecular findings,3, 4, 5 CRC with the MSS phenotype, which is the major phenotype of CRC, could result in a more simple disease in terms of SCNAs (CRC could be represented by only three subgroups). The molecular profiles established by the SCNA patterns described in this study provide important insights into the molecular processes that are influenced by a wide range of complex alterations and that can be used as a basis for the development of new strategies to predict and prevent CRCs.7
Vogelstein et al26 first proposed a multistep genetic model of colorectal carcinogenesis defined by multiple LOHs and genetic mutations (APC, KRAS, DPC, and TP53 genes). Although there continue to be refinements to the original model, several key principles have been established, including that multiple SCNAs are required for the progression of CRC.27, 28 The detection of aberrant SCNAs may provide novel markers for the early diagnosis and personalized treatment of CRC.11 A major challenge in array‐based profiling of SCNAs is to distinguish the alterations that play causative roles from the random alterations that accumulate during colorectal carcinogenesis.11 In the present study, we described frequent SCNAs that may characterize tumor progression in CRC, including regions of gains at 7p22.1, 7q11‐12, 7q21.12, 7q33, 8q22.1, 8q23.2, 13q11‐13, 13q21‐22, 13q31.2‐32.3, 13q34, 20p11.21, 20q11.23, and 20q21‐23 and regions of LOH at 18p11.32, 18p21‐23, and 18q in the CRCs we examined (more than 50% of cases). Although these findings were mostly consistent with previous studies, the molecular profiles we proposed in this study were different from those of other previous studies.7, 23, 24 Our hypothesis was characterized by multiple CN gains and few CN losses (8p [around 40%], 17p [approximately 40%], and 18p/q). This current hypothesis suggested that CN gains may contribute to the progression of CRC rather than CN losses, as supported by a previous study showing the presence of many CN gains in genome‐wide analysis in CRCs.17 Thus, we suggest that the main alteration governing the molecular mechanisms of CRC is oncogenic potential.
Most CRCs examined in this study were subgroup 1 tumors characterized by a low frequency of SCNAs. In addition, subgroup 1 was closely associated with a high frequency of MSI‐high, BRAF mutations, and high methylation status compared with subgroups 2 and 3, although differences in the frequencies of BRAF mutations and HME were not significant. CN gains at 7p15.2, 7q11‐12, 8q21‐24, and 20q11‐13 and LOHs at 17p11 and 18p/q were frequently found in subgroup 1. However, both CRC with the MSI‐high phenotype and CRC with the MSS phenotype were involved in subgroup 1. This finding suggested that CRC with the MSS phenotype in subgroup 1, which accounted for about half of all CRC cases examined in this study, showed a pattern similar to the MSI‐high phenotype found in subgroup 1 in terms of SCNA pattern and that these molecular alterations may characterize right‐sided CRC. Thus, clustering of SCNA patterns could improve our understanding of the molecular carcinogenesis of CRC.
Previous studies have shown that multiple SCNAs are frequently found in CRCs.23, 24, 25 In the present study, we found significant differences in gains and LOHs between subgroups 2 and 3, which were characterized by multiple SCNAs. This finding suggested that different patterns existed in terms of progressive accumulation of multiple SCNAs during CRC development. In the present study, gains at 1q23.1‐44, 1p11.2‐36.33, 10q11.23‐26.3, 10p11.21‐13, 12q24.21‐24.32, and 13q33.1‐33.3 may be key genetic events characterizing tumors in subgroup 3. Of the chromosomal alleles showing CN gains, MAP3K8 (Mitogen‐Activated Protein Kinase Kinase Kinase8; 10q11),28 5‐lipoxygenases (Loxs; ALOX5, Arachidonate 15‐Lipoxygenase at 10q11),29 LIG4 (DNA ligase 4; 12q23),30 and MSI1 (Musashi‐1; 12q24)31 may be suitable as candidate genes that are responsible for the gains. Notably, a prior report showed that overexpression of MAP3K8 is correlated with poor prognosis in patients with early‐onset CRC.28 Additionally, a separate study showed that 5‐Lox is upregulated in CRC and that inhibition of 5‐Lox expression may be valuable for the prevention and treatment of CRC.29 Another previous study revealed that LIG4, a DNA ligase in DNA double‐strand break repair, is a direct target of β‐catenin and that blocking LIG4 sensitizes CRC cells to radiation.30 Moreover, LIG4 is highly upregulated in human CRC cells through β‐catenin hyperactivation, which is closely associated with Wnt signal activation.30 This finding suggests that LIG4 plays a specific role in Wnt signaling‐induced radioresistance in CRC.30 Finally, a recent study showed that the stem cell marker MSI1 is overexpressed in many cancer types, including CRC.31, 32 MSI1 is highly expressed in primary colon tumors and metastatic lesions in the lymph nodes as compared with that in paired adjacent normal colon mucosal tissue.31, 32 We suggest that these candidate genes located at 10q11 and 12q23‐24 may contribute to the development of CRC.
Copy‐neutral LOHs at 12p12.2‐13.1, 1q24.3‐25.1, and 10q22 contributed to clustering of tumors in subgroup 2. Copy‐neutral LOH, referred to as uniparental disomy, leads to LOH by duplication of a maternal or paternal chromosome or chromosomal region33, 34 this process should be studied in greater detail given that mutations occurring in duplicated alleles by CN LOH enhance oncogenic potential.33, 34 Among frequent copy‐neutral LOHs at 12p12.2‐13.1, 1q24.3‐25.1, and 10q22 occurring in tumors in subgroup 3, that at 12p12‐13 was interesting because the oncogene KRAS is mapped to 12p12.1 and is closely associated with colorectal carcinogenesis. KRAS mutations could be activated by amplification, resulting in duplication of the KRAS gene through CN LOH. If this hypothesis is correct, further studies are needed to assess KRAS mutations in colorectal carcinogenesis. In addition, KRAS mutations were more frequent in tumors in subgroup 3 than in tumors in subgroup 1. The mutation status of the KRAS gene in tumors may affect the response to cetuximab and could have treatment‐independent prognostic value.35 Thus, our results suggested that the therapeutic effects of cetuximab may be inhibited by double restriction of KRAS mutations (tumors in subgroup 3) and frequent CN LOH at 12p12.1 containing KRAS (tumors in subgroup 2) in tumors in subgroups 2 and 3.
According to the multistep process of carcinogenesis established by Vogelstein et al26 TP53 mutations play a central role in colorectal carcinogenesis. In the present study, TP53 mutations were commonly observed in tumors in all three subgroups. Mutations in TP53 are closely associated with genomic instability related to multiple SCNAs.3, 5 However, the present data showed that common alterations found in tumors in subgroups 2 and 3 were characterized by multiple SCNAs and that tumors in subgroup 1 showed a low frequency of SCNAs. This finding suggested that TP53 mutations occurred prior to progression of CRC to an advanced stage, emerging during the early stage of colorectal carcinogenesis.
Epigenetic alterations, particularly DNA methylation in selected gene promoters, are important molecular alterations in CRCs.14, 15 In the present study, each subgroup shared an intermediate level of DNA methylation in the whole genome of CRC cells. This finding was consistent with a previous study reported by Kaneda and Yagi16 suggested that considerable DNA methylation may be required for the development of CRC. In addition, our data showed that CRC arose as a consequence of the accumulation of genetic and epigenetic alterations during neoplastic transformation.
There are some limitations to our study. First, The Cancer Genome Atlas (TCGA) provides representative data for CRCs.7 Although TCGA platform is different from that of the present study,7 it is important to compare molecular alterations from TCGA database with those of the present study. The molecular patterns we proposed were different from those of TCGA, as shown in our current findings. In TCGA, molecular alterations were also subclassified into hypermutated and non‐hypermutated type.7 Our data indicated that although molecular pathways could be divided into three subgroups based on SCNAs, hypermutated, and non‐hypermutated types corresponded to the MSI type in subgroup 1 and the remaining tumors in subgroup 1 plus tumors in subgroups 2 and 3, respectively. In addition, our classification was characterized by distinct clinicopathological and molecular findings. We believe that the present findings are useful to evaluate the molecular basis of colorectal carcinogenesis. Second, association of the present molecular classification with patient prognosis could not be examined, given that most cases of CRC examined in this study had been diagnosed within the last 5 years. Future studies are needed to identify the relationships between the current classifications and patient prognosis in CRC.
In conclusion, we used a high‐density professional whole‐genome SCNA array that covered more than approximately 300 000 SNPs to define a comprehensive allelotype for a heterogeneous group of sporadic CRCs based on SCNAs. In the present study, we found that CRC could be classified into three phenotypes (subgroups 1, 2, and 3) based on SCNAs. Taken together, our findings showed that subgroup 1 was characterized by a low frequency of SCNAs and a high frequency of MSI‐high status, and subgroups 2 and 3 were closely associated with a high frequency of SCNAs. Subgroup 3 was distinguished by specific alterations, including gains at 1q23.1‐44, 1p11.2‐36.33, 10q11.23‐26.3, 10p11.21‐13, 12q24.21‐24.32, and 13q33.1‐33.3, and copy‐neutral LOH at 12p12.2‐13.1, 1q24.3‐25.1, and 10q22.1. In addition, KRAS mutations were more frequent in subgroup 3 than in subgroup 1. TP53 mutations and intermediate levels of DNA methylation were common alterations in the three subgroups. Thus, our findings suggested that SCNAs played an important role in the progression of CRC. However, further studies are necessary to determine the impact of these SCNAs on the outcomes of the disease.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was approved by the Ethical Research Committee, Iwate Medical University.
AVAILABILITY OF DATA AND MATERIAL
All data generated or analyzed during this study are included in this published article (and its supplementary information files).
COMPETING INTERESTS
There were no financial or non‐financial competing interests.
AUTHORS’ CONTRIBUTIONS AND DISCLOSURE STATEMENT
ST, who is the first and corresponding author, contributed to the preparation of the manuscript, including all aspects of data collection and analysis. TY, EM, FY, and SR constructed the figures and tables and performed statistical analysis. HW, MT, and SH provided input during the preparation of the manuscript. YE provided support for experiments involving molecular technologies. OK and SA assisted with clinical data and experiments. We guarantee that (a) the work is original; (b) the work has not been, and will not be, published in whole or in part in any other journal; and (c) all of the authors have agreed to the contents of the manuscript in its submitted form.
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article.
Table S1. Significant Differences of gain CN between subgroup 1 and 2.
Table S2. Significant Differences of gain CN between subgroup 1 and 3.
ACKNOWLEDGMENTS
We gratefully acknowledge the technical assistance of Ms. E. Sugawara. We also thank members of the Department of Molecular Diagnostic Pathology, Iwate Medical University for their support.
Sugai T, Takahashi Y, Eizuka M, et al. Molecular profiling and genome‐wide analysis based on somatic copy number alterations in advanced colorectal cancers. Molecular Carcinogenesis. 2018;57: 451–461. https://doi.org/10.1002/mc.22769
REFERENCES
- 1. Haggar FA, Boushey RP. Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. Clin Colon Rectal Surg. 2009; 22:191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997; 386:623–627. [DOI] [PubMed] [Google Scholar]
- 3. Jass JR, Whitehall VL, Young J, Leggett BA. Emerging concepts in colorectal neoplasia. Gastroenterology. 2002; 123:862–876. [DOI] [PubMed] [Google Scholar]
- 4. Ogino S, Goel A. Molecular classification and correlates in colorectal cancer. J Mol Diagn. 2008; 10:13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. 2010; 138:2059–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology. 2010; 138:2088–2100. [DOI] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015; 21:1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Müller MF, Ibrahim AE, Arends MJ. Molecular pathological classification of colorectal cancer. Virchows Arch. 2016; 469:125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sadanandam A, Lyssiotis CA, Homicsko K, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013; 19:619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang H, Liang L, Fang JY, Xu J. Somatic gene copy number alterations in colorectal cancer: new quest for cancer drivers and biomarkers. Oncogene. 2016; 35:2011–2019. [DOI] [PubMed] [Google Scholar]
- 12. Takahashi Y, Sugai T, Habano W, et al. Molecular differences in the microsatellite stable phenotype between left‐sided and right‐sided colorectal cancer. Int J Cancer. 2016; 139:2493–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sugai T, Eizuka M, Takahashi Y, et al. Molecular subtypes of colorectal cancers determined by PCR‐based analysis. Cancer Sci. 2017; 108:427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Toyota M, Ahuja N, Ohe‐Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA. 1999; 96:8681–8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yagi K, Takahashi H, Akagi K, et al. Intermediate methylation epigenotype and its correlation to KRAS mutation in conventional colorectal adenoma. Am J Pathol. 2012; 180:616–625. [DOI] [PubMed] [Google Scholar]
- 16. Kaneda A, Yagi K. Two groups of DNA methylation markers to classify colorectal cancer into three epigenotypes. Cancer Sci. 2011; 102:18–24. [DOI] [PubMed] [Google Scholar]
- 17. Japanese society for cancer of the colon and rectum Japanese Classification of Colorectal Carcinoma. 2nd English ed Tokyo: Kanehara Co; 2009;30–63. [Google Scholar]
- 18. Nakamura S, Goto J, Kitayama M, Kino I. Application of the crypt‐isolation technique to flow‐cytometric analysis of DNA content in colorectal neoplasms. Gastroenterology. 1994; 106:100–107. [DOI] [PubMed] [Google Scholar]
- 19. Sugai T, Yamada N, Eizuka M, et al. Vascular invasion and stromal S100A4 expression at the invasive front of colorectal cancer are novel determinants and tumor prognostic markers. J Cancer. 2017; 8:1552–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998; 58:5248–5257. [PubMed] [Google Scholar]
- 21. Yamamoto E, Suzuki H, Yamano HO, et al. Molecular dissection of premalignant colorectal lesions reveals early onset of the CpG island methylator phenotype. Am J Pathol. 2012; 181:1847–1861. [DOI] [PubMed] [Google Scholar]
- 22. Sugai T, Habano W, Nakamura S, Uesugi N, Sasou S, Itoh C. A unique method for mutation analysis of tumor suppressor genes in colorectal carcinomas using a crypt isolation technique. Arch Pathol Lab Med. 2000; 124:382–386. [DOI] [PubMed] [Google Scholar]
- 23. Goossens‐Beumer IJ, Oosting J, Corver WE, et al. Copy number alterations and allelic ratio in relation to recurrence of rectal cancer. BMC Genomics. 2015; 16:438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin CH, Lin JK, Chang SC,C, et al. Molecular profile and copy number analysis of sporadic colorectal cancer in Taiwan. J Biomed Sci. 2011; 18:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zack TI, Schumacher SE, Carter SL, et al. Pan‐cancer patterns of somatic copy number alteration. Nat Genet. 2013; 45:1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal tumor development. N Eng J Med. 1988; 319:525–532. [DOI] [PubMed] [Google Scholar]
- 27. Eizuka M, Sugai T, Habano W, et al. Molecular alterations in colorectal adenomas and intramucosal adenocarcinomas defined by high‐density single‐nucleotide polymorphism arrays. J Gastroenterol. 2017; 52:1158–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tunca B, Tezcan G, Cecener G, et al. Overexpression of CK20, MAP3K8 and EIF5A correlates with poor prognosis in early‐onset colorectal cancer patients. J Cancer Res Clin Oncol. 2013; 139:691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Soumaoro LT, Iida S, Uetake H, et al. Expression of 5‐lipoxygenase in human colorectal cancer. World J Gastroenterol. 2006; 12:6355–6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jun S, Jung YS, Suh HN, et al. LIG4 mediates Wnt signalling‐induced radioresistance. Nat Commun. 2016; 7:10994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smith AR, Marquez RT, Tsao WC, et al. Tumor suppressive microRNA‐137 negatively regulates Musashi‐1 and colorectal cancer progression. Oncotarget. 2015; 6:12558–12573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li D, Peng X, Yan D, et al. Msi‐1 is a predictor of survival and a novel therapeutic target in colon cancer. Ann Surg Oncol. 2011; 18:2074–2083. [DOI] [PubMed] [Google Scholar]
- 33. O'Keefe C, McDevitt MA, Maciejewski JP. Copy neutral loss of heterozygosity: a novel chromosomal lesion in myeloid malignancies. Blood. 2010; 115:2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Melcher R, Hartmann E, Zopf W, et al. LOH and copy neutral LOH (cnLOH) act as alternative mechanism in sporadic colorectal cancers with chromosomal and microsatellite instability. Carcinogenesis. 2011; 32:636–642. [DOI] [PubMed] [Google Scholar]
- 35. Yokota T. Are KRAS/BRAF mutations potent prognostic and/or predictive biomarkers in colorectal cancers? Anticancer Agents Med Chem. 2012; 12:163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found online in the supporting information tab for this article.
Table S1. Significant Differences of gain CN between subgroup 1 and 2.
Table S2. Significant Differences of gain CN between subgroup 1 and 3.