

Summary
Acute RyR2 activation by exchange protein directly activated by cAMP (Epac) reversibly perturbs myocyte Ca2+ homeostasis, slows myocardial action potential conduction, and exerts pro‐arrhythmic effects. Loose patch‐clamp studies, preserving in vivo extracellular and intracellular conditions, investigated Na+ current in intact cardiomyocytes in murine atrial and ventricular preparations following Epac activation. Depolarising steps to varying test voltages activated typical voltage‐dependent Na+ currents. Plots of peak current against depolarisation from resting potential gave pretreatment maximum atrial and ventricular currents of −20.23 ± 1.48 (17) and −29.8 ± 2.4 (10) pA/μm2 (mean ± SEM [n]). Challenge by 8‐CPT (1 μmol/L) reduced these currents to −11.21 ± 0.91 (12) (P < .004) and −19.3 ± 1.6 (11) pA/μm2 (P < .04) respectively. Currents following further addition of the RyR2 inhibitor dantrolene (10 μmol/L) (−19.91 ± 2.84 (13) and −26.6 ± 1.7 (17)), and dantrolene whether alone (−19.53 ± 1.97 (8) and −27.6 ± 1.9 (14)) or combined with 8‐CPT (−19.93 ± 2.59 (12) and −29.9 ± 2.5(11)), were indistinguishable from pretreatment values (all P >> .05). Assessment of the inactivation that followed by applying subsequent steps to a fixed voltage 100 mV positive to resting potential gave concordant results. Half‐maximal inactivation voltages and steepness factors, and time constants for Na+ current recovery from inactivation in double‐pulse experiments, were similar through all the pharmacological conditions. Intracellular sharp microelectrode membrane potential recordings in intact Langendorff‐perfused preparations demonstrated concordant variations in maximum rates of atrial and ventricular action potential upstroke, (dV/dt)max. We thus demonstrate an acute, reversible, Na+ channel inhibition offering a possible mechanism for previously reported pro‐arrhythmic slowing of AP propagation following modifications of Ca2+ homeostasis, complementing earlier findings from chronic alterations in Ca2+ homeostasis in genetically‐modified RyR2‐P2328S hearts.
Keywords: 8‐(4‐chlorophenylthio)‐2′‐O‐methyladenosine‐3′,5′‐cyclic monophosphate; arrhythmic substrate; Ca2+ homeostasis; conduction velocity; dantrolene; Epac; Na+ current; ryanodine receptor
1. INTRODUCTION
Cardiac arrhythmias result from disruptions in the normal excitable activity propagating through successive structures in the heart. They arise from altered function in surface membrane ionic channels whose successive activation and inactivation underlies the production and propagation of cardiac action potentials (AP). Sustained arrhythmia may require both triggering events and arrhythmic substrate to maintain the resulting abnormal electrical activity.1, 2, 3 Arrhythmic substrate arises either from slowed myocardial AP conduction or activation, exemplified by Brugada Syndrome,4, 5 or altered AP recovery, reflected in altered AP duration and/or refractoriness, exemplified by long QT syndrome.6 Arrhythmic risk is also associated with dysregulated intracellular Ca2+ homeostasis. This may arise from abnormal ryanodine receptor‐2 (RyR2)‐mediated sarcoplasmic reticular Ca2+ release. The resulting elevations in diastolic [Ca2+] and spontaneous propagated intracellular Ca2+ waves increase electrogenic Na+‐Ca2+ exchanger activity thereby driving delayed after‐depolarisations that may trigger premature ventricular beats.7
Murine hearts carrying genetically altered RyR2‐Ca2+ release channel and SR Ca2+ storage protein calsequestrin‐2 have successfully modelled such triggering events. They recapitulate mutations associated with the chronic pro‐arrhythmic condition of human catecholaminergic polymorphic ventricular tachycardia (CPVT).8, 9 Triggering events have also been reported following acute adrenergic activation produced by modifications of RyR2‐mediated SR Ca2+ release10 and surface Ca2+ channel properties in wild‐type hearts expressing normal RyR2 and calsequestrin‐2.11, 12 More recent studies selectively activated RyR2‐Ca2+ release channels using the phosphokinase A (PKA)‐independent exchange protein directly activated by cAMP (Epac) pathway.13, 14, 15 This increased Ca2+ spark frequencies in adult rat cardiac myocytes16 and amplitudes of Ca2+‐dependent Ca2+ release after isoproterenol treatment17 in murine ventricular cardiomyocytes. They also increased the amplitudes and frequencies of spontaneous Ca2+ release.18 These changes correlated with increases in triggered activity and ventricular tachycardia (VT) in murine hearts.18
Fewer studies have explored arrhythmic substrate under conditions of altered Ca2+ homeostasis. Neither chronic modifications in Ca2+ homeostasis in RyR2‐P2328S models nor acute manipulations of Ca2+ homeostasis in WT hearts altered AP recovery characteristics as reflected in AP durations (APD), refractory periods (ERP), or the relationships between these.8, 12, 19 However, murine RyR2‐P2328S CPVT cardiac models showed reduced atrial20 and ventricular conduction velocities in common with Nav1.5‐haploinsufficient Scn5a+/− hearts modelling the Brugada Syndrome.21 Pharmacological inhibition of RyR2‐mediated Ca2+ release with flecainide partly rescued these effects.22, 23, 24, 25 Furthermore, selective, acute RyR2 activation through the Epac pathway produced parallel pro‐arrhythmic effects.18 It correspondingly produced decreases in AP conduction velocities that were partially reversed by the RyR2 antagonist dantrolene, with an absence of alterations in AP recovery characteristics.19
The mechanism for the conduction velocity changes in RyR2‐P2328S hearts was identified as the direct action of intracellular Ca2+ on Nav1.5 function20, 21, 26 and/or Nav1.5 membrane expression.27 However, the mechanisms by which acute manipulations of intracellular Ca2+ homeostasis, particularly Epac activation, alter AP conduction have not been investigated. The present experiments assessed Nav1.5 activation, inactivation, and recovery from inactivation following acute rather than chronic manipulations of Ca2+ homeostasis, and in WT rather than genetically‐modified hearts. They employed the loose patch technique for voltage‐clamping of Na+ current. This apposes an electrode containing extracellular solution against an intact cell surface membrane without accessing intracellular space. Studies were thus performed in cardiomyocytes in intact murine atrial and ventricular preparations without perturbing extracellular [Na+] and intracellular Ca2+ homeostasis21, 28, 29 as opposed to following cardiomyocyte isolation necessitated by conventional whole‐cell patch clamp techniques.30, 31
Recent cardiomyocyte studies involving reversible manipulations of loose patch pipette [Na+] had identified early inward currents in response to step depolarisations with Na+ currents responsible for AP conduction and the maximum upstroke rate, (dV/dt)max, of the cardiac action potential.21 The corresponding changes in such (dV/dt)max were accordingly determined by independent experiments performing intracellular sharp electrode recordings of membrane potential in intact atrial and ventricular preparations. These explored the extent to which changes in observed (dV/dt)max, paralleled corresponding changes in Na+ currents in the loose‐patch experiments with similar pharmacological manipulations. Such correlations would be consistent with previously reported relationships between (dV/dt)max and peak Na+ currents (I Na).32
We thus studied electrophysiological effects of pharmacological manipulations of Ca2+ homeostasis through Epac activation on Na+ currents in a near physiological environment for the first time. The Epac activator (8‐pCPT‐2′‐O‐Me‐cAMP: 8‐(4‐chlorophenylthio)‐2′‐O‐methyladenosine‐3′,5′‐cyclic monophosphate)13 at approximately 1 μmol/L offered 300‐fold preferential selectivity for Epac over PKA action.13, 33, 34, 35 The RyR2 antagonist dantrolene inhibits RyR2‐mediated diastolic Ca2+ release, decreases frequencies and durations of aberrant Ca2+ sparks in myocyte cultures modelling CPVT36 and cardiac failure37, 38 and increases thresholds for Ca2+‐induced sarcoplasmic reticular Ca2+‐release in myocytes from failing but not normal rabbit hearts.38 Thus, both 8‐CPT and dantrolene act on intracellular targets following access across cardiomyocyte surface membrane as a whole whether during loose‐patch or sharp electrode recordings. Finally, the results obtained with the approach adopted here could be directly compared to corresponding explorations in intact Langendorff perfused murine hearts. In addition to establishing the expected alterations in cellular Ca2+ homeostasis, they used identical Epac activators and RyR2 antagonists in identical pharmacological protocols exploring the effects of Epac activation and their reversibility applying these to examine changes in AP conduction velocity and arrhythmic substrate.18, 19
2. RESULTS
2.1. Currents obtained in the combined pulse procedure
Figures 1 and 2 illustrate results obtained from the isolated atrial (Figure 1) and ventricular (Figure 2) preparations subject to a combined pulse procedure (Panel Aa) designed to explore both activation and inactivation properties of Na+ currents. Cells were first held at the resting membrane potential (RMP) for 5 ms from the beginning of the recording period. A prepulse of duration 5 ms to V 0 = (RMP−40) mV was then applied to remove any residual Na+ current inactivation and standardise the initial activation state of Na+ channels within the patch. Na+ current activation properties were then investigated using the initial depolarising test voltage steps made to a voltage varied with the 13 successive sweeps between V 1 = RMP to (RMP + 120) mV in +10 mV increments. These currents, following correction for residual leakage by a P/4 protocol, provided a family of records reflecting the voltage dependence of Na+ channel activation (Panels Ab and Ba). Inward currents are represented as downward, negative deflections. Records typically began with a consistent small upward deflection in response to the −40 mV prepulse. The subsequent voltage steps to level V 1 yielded a family of inward currents initially increasing with time to a peak value that increased as V 1 was made more positive. This was followed by a decay reflecting channel inactivation whose extent and kinetics was similarly determined by the voltage V 1.
Figure 1.
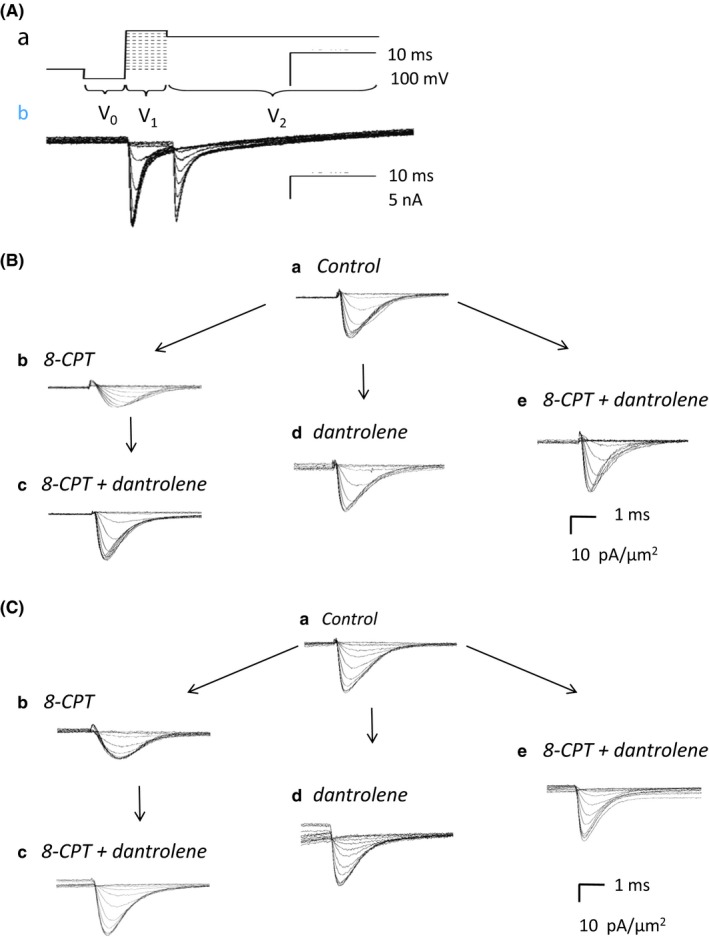
Ionic currents from an isolated atrial preparation studied under loose‐patch clamp. (Aa) Pulse protocol designed to investigate Na+ channel activation and inactivation. (Ab) Typical recordings obtained using with a pipette of 28 μm tip diameter. Inward currents in nA are represented as negative. All currents shown in subsequent panels converted to current density in units of (pA/μm2) to account for pipette diameters, typically 28‐32 μm. (B, C) Na+ currents observed upon (B) activation by depolarisation to a level V 1, before and following pharmacological challenge and (C) in response to the voltage step from levels V 1 to the final level V 2. The latter permitted assessment of Na+ channel inactivation produced by the voltage step to level V 1. Recordings made (a) under pretreatment conditions, when no drug was present, (b) in the presence of 8‐CPT (1 μmol/L), (c) in the presence of a combination of 8‐CPT (1 μmol/L) and dantrolene (10 μmol/L) after the addition of 8‐CPT, (d) after the addition of dantrolene (10 μmol/L), and (e) in the presence of a combination of 8‐CPT (1 μmol/L) and dantrolene (10 μmol/L). (d) and (e) were obtained following direct introduction of pharmacological agents following pretreatment conditions in the absence of any preceding application of 8‐CPT
Figure 2.
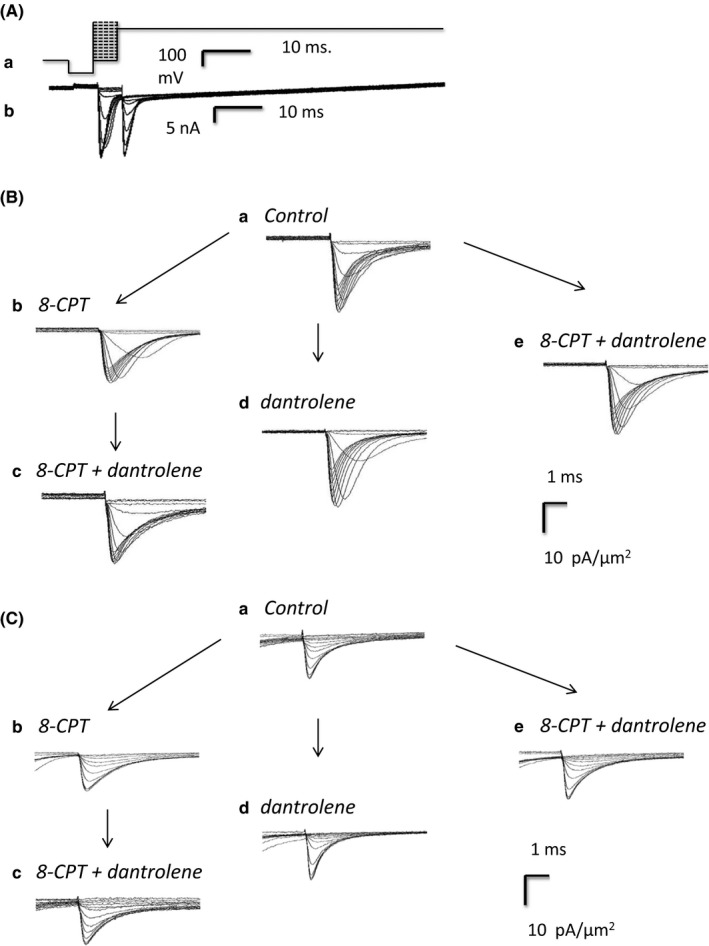
Ionic currents from an isolated ventricular preparation studied under loose‐patch clamp. (Aa) Pulse protocol designed to investigate Na+ channel activation and inactivation. (Ab) Typical recordings obtained using with a pipette of tip diameter 30 μm. Inward currents in nA represented as negative. All currents shown in subsequent figures have been converted to current density in units of (pA/μm2) to account for pipette diameters, typically 28‐32 μm. (B, C) Na+ currents observed upon (B) activation by depolarisation to a level V 1, before and following pharmacological challenge and (C) in response to the voltage step from levels V 1 to the final level V 2. The latter permitted assessment of Na+ channel inactivation produced by the voltage step to level V 1. Recordings made (a) under pretreatment conditions, when no drug was present, (b) in the presence of 8‐CPT (1 μmol/L), (c) in the presence of a combination of 8‐CPT (1 μmol/L) and dantrolene (10 μmol/L) after the addition of 8‐CPT, (d) after the addition of dantrolene (10 μmol/L), and (e) in the presence of a combination of 8‐CPT (1 μmol/L) and dantrolene (10 μmol/L). (d) and (e) were obtained following direct introduction of pharmacological agents following pretreatment conditions in the absence of any preceding application of 8‐CPT
The subsequent voltage steps in the pulse protocol were used to assess properties of the Na+ current inactivation resulting from the voltage step to V 1. Thus, 5 ms following imposition of the latter step, an additional step was applied to a fixed voltage of V 2 = (RMP + 100) mV that would result in a peak Na+ current reflecting the extent of the preceding channel inactivation (Panels Ab and Ca). The step to voltage V 2 thus elicited a second family of currents which decreased in amplitude with increasing V 1. Both sets of responses showed nonlinear increments in the magnitude of currents obtained at successive voltages reflecting the nonlinear dependence of either activation or inactivation about the voltage V 1.
2.2. Currents reflecting Na+ channel activation
Figures 1B and 2B show typical families of current records illustrating Na+ current activation obtained in response to the initial steps between voltages V 0 and V 1 in atrial (Figure 1) and ventricular (Figure 2) preparations. In both panels B and C currents are normalised to the area subtended by the pipette tip lumen (in μm2) to give current densities. Recordings were made before (pretreatment: Panel Ba), and following challenge by 8‐CPT (1 μmol/L) (Bb), and then 8‐CPT (1 μmol/L) combined with dantrolene (10 μmol/L) (Bc). These were compared with records obtained following challenge by dantrolene (10 μmol/L) alone (Bd) and in combination with 8‐CPT (1 μmol/L) (Be).
The pretreatment Na+ currents showed typical activation and inactivation time courses with their amplitude nonlinearly graded with the size of the depolarising step (Ba). 8‐CPT markedly decreased the Na+ current amplitudes at all test voltages in both atrial and ventricular preparations (Bb). However, further inclusion of dantrolene in the test solutions rescued the Na+ current, reversing the inhibitory action of 8‐CPT alone (Bc). These findings were corroborated by another control procedure. Thus, families of currents before treatment (Ba) were recorded and the pretreatment solution then replaced by one including dantrolene either alone (Bd) or combined with 8‐CPT (Be). All these conditions (Ba, Bd, Be) gave similar Na+ currents. Thus, 8‐CPT reduces Na+ current in both atrial and ventricular preparations, but this action was reversed or absent in the presence of dantrolene whether by itself or in combination with 8‐CPT.
2.3. Currents reflecting Na+ channel inactivation
Records demonstrating typical families of Na+ current inactivation from atrial and ventricular preparations are shown in Figures 1C and 2C. Each family of records was obtained from an individual patch in response to the steps between the voltages V 1 and V 2 (panel Aa). These illustrate the extent of Na+ channel inactivation resulting from the step to voltage V 1. Only the fraction of channels spared such inactivation would then be activated by the step to the fixed voltage V 2. The Na+ current amplitudes would therefore permit quantification of the voltage dependence of Na+ channel inactivation at the voltage V 1. Records were obtained before 8‐CPT challenge (pretreatment) (Ca), following 8‐CPT challenge (Cb), and following challenge by 8‐CPT combined with dantrolene (Cc). They were compared with results of transferring pretreatment preparations directly to solutions containing dantrolene alone (Cd) or dantrolene combined with 8‐CPT (Ce). The resulting Na+ currents showed typical activation and inactivation time courses, but these decreased in amplitude the more depolarised the voltage V 1 (Ca). Again, 8‐CPT markedly decreased Na+ current amplitudes at all voltages tested (Cb), and further inclusion of dantrolene restored the Na+ currents (Cc). Na+ currents following challenge by dantrolene alone (Cd) or dantrolene in combination with 8‐CPT (Ce) were similar in amplitude and waveform as pretreatment records. Thus, the pulse procedures assessing current inactivation similarly demonstrated that 8‐CPT reduced Na+ current amplitude and this action was rescued by further addition of dantrolene.
2.4. Voltage dependences of atrial Na+ current activation and inactivation
Figures 3 and 4 illustrate voltage‐dependences of atrial (Figure 3) and ventricular (Figure 4) Na+ current activation (A) and inactivation (B) under each of the experimental conditions described above (a–c). They plot the peak Na+ current (mean ± standard error of the mean [SEM]) against the voltage V 1. Results are shown for pretreatment conditions (open symbols) and following addition of 8‐CPT (filled triangles) and 8‐CPT combined with dantrolene (filled circles) (a). These results were compared with findings obtained before and following addition of dantrolene alone (pretreatment: open symbols; dantrolene: filled diamonds) (b) and before and following addition of dantrolene in combination with 8‐CPT (pretreatment: open symbols; 8 CPT with dantrolene, filled circles) (c). In both atrial (Figure 3) and the ventricular preparations (Figure 4) peak current increased with the amplitudes of depolarising steps exceeding +10 mV in size to a maximum value at a voltage excursion around +80 mV. They then decreased with further depolarisation as expected with approach of V1 towards the Na+ current reversal potential. Table 1 summarizes the results of applying voltage steps of a fixed +80 mV amplitude, employing 12 signal‐averaged records, to explore the effect of the different pharmacological agents on such maximum peak current under the different pharmacological conditions. The presence of significant differences in both atrial (F = 3.462, P = .013) and ventricular findings between experimental groups (F = 4.071, P = .006) prompted pairwise testing demonstrating differences between results of exposure to 8‐CPT compared to findings from untreated patches in both atrial (P < .04) and ventricular cardiomyocytes (P < .004; α = .05). In contrast, there were no detectable differences (P > .2) of findings obtained with challenge by 8‐CPT followed by 8‐CPT combined with dantrolene, and dantrolene whether alone, or combined with 8‐CPT, when all were compared from data from untreated patches.
Figure 3.
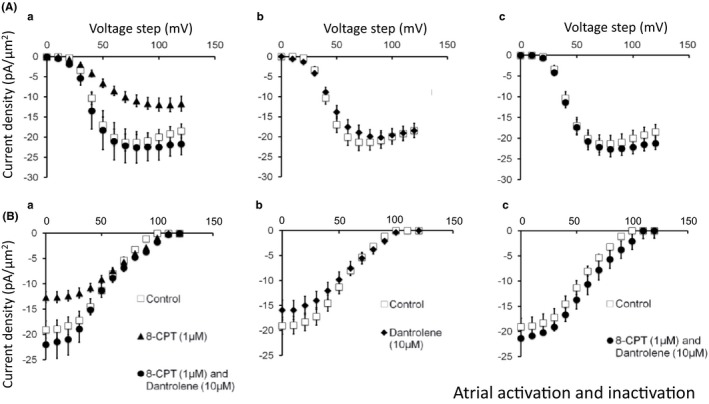
Voltage dependence of atrial Na+ current activation and inactivation under the different pharmacological conditions employing 8‐CPT and dantrolene. Dependences of (A) Na+ current activation (mean ± SEM) upon voltage step V 1, before (pretreatment) and following pharmacological challenge and (B) Na+ current inactivation (mean ± SEM) as a function of voltage step V 1, assessed using the further voltage step V 2, before and after pharmacological challenge. Results obtained (a) before (open squares) and following introduction of 8‐CPT (filled triangles) and a combination of 8‐CPT and dantrolene (filled circles), (b) before (open squares) and following introduction of dantrolene (filled diamonds), (c) before (open squares) and following introduction of a combination of 8‐CPT and dantrolene (filled circles)
Figure 4.
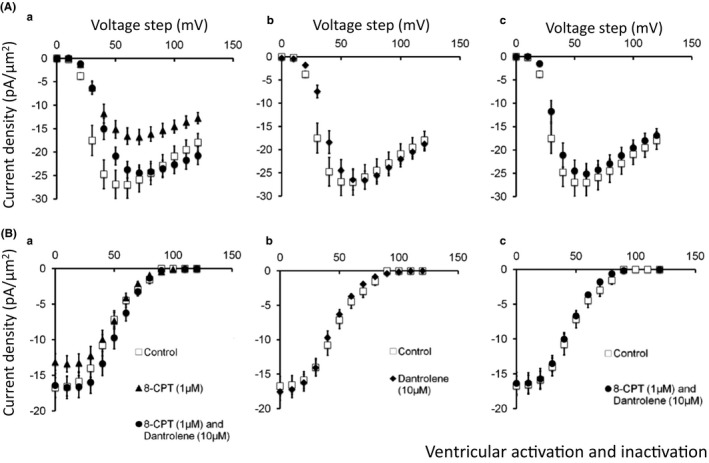
Voltage dependence of ventricular Na+ current activation and inactivation under the different pharmacological conditions employing 8‐CPT and dantrolene. Dependences of (A) Na+ current activation (mean ± SEM) upon voltage step V1, before (pretreatment) and following pharmacological challenge and (B) Na+ current inactivation (mean ± SEM) as a function of voltage step V 1 assessed using the further voltage step V 2, before and after pharmacological challenge. Results obtained (a) before (open squares) and following introduction of 8‐CPT (filled triangles) and a combination of 8‐CPT and dantrolene (filled circles), (b) before (open squares) and following introduction of dantrolene (filled diamonds), (c) before (open squares) and following introduction of a combination of 8‐CPT and dantrolene (filled circles)
Table 1.
Maximum inward currents in atrial and ventricular preparations under the tested pharmacological conditions
| Pharmacological condition | Maximum currents, (Mean ± SEM (n–value), pA/μm2 | |
|---|---|---|
| Atrial preparation | Ventricular preparation | |
| (a) Untreated | −20.23 ± 1.48 (n = 17)*** | −29.8 ± 2.4 (n = 10)** |
| (b) 8–CPT | −11.21 ± 0.91 (n = 12)*** | −19.3 ± 1.6 (n = 11)** |
| (c) 8–CPT followed by 8–CPT + dantrolene | −19.91 ± 2.84 (n = 13) | −26.6 ± 1.7 (n = 17) |
| (d) Dantrolene alone | −19.53 ± 1.97 (n = 8) | −27.6 ± 1.9 (n = 14) |
| (e) 8–CPT + dantrolene | −19.93 ± 2.59 (n = 12) | −29.9 ± 2.5 (n = 11) |
8–(4–chlorophenylthio) adenosine 3′,5′–cyclic monophosphate (8–CPT, 1 μmol/L); dantrolene (10 μmol/L). One‐way ANOVA: atrial preparations: F = 3.462, P = .013; ventricular preparations: F = 4.071, P = .006. Posthoc comparisons (after Bonferroni correction for four pairwise tests): ** P < .04; *** P < .004, relative to untreated group; no significant changes in all remaining groups (c)–(e) compared to control: P > .2.
These findings from Table 1 were corroborated by the corresponding atrial and ventricular Na+ current‐voltage curves. Peak atrial currents increased with depolarisation to approximately −18.5 ± 1.8 pA/μm2 (n = 13) (Figure 3Aa) and −13.5 ± 1.3 pA/μm2 (n = 14) before and following 8‐CPT challenge. In contrast, currents following further addition of dantrolene (−22.5 ± 3.9 pA/μm2 [n = 6]) (Aa), or exposure to dantrolene either alone (−20.2 ± 1.9 pA/μm2 [n = 11]) (Ab), or combined with 8‐CPT (−22.7 ± 1.9 pA/μm2 [n = 7]) (Ac) were similar to pretreatment values. Similar results emerged from atrial Na+ current inactivation studies of peak Na+ current in response to voltage steps from V 1 to the fixed level V 2, plotted against inactivating voltage V 1 (Figure 3B). Maximum currents at the most negative V 1 before treatment (−19.1 ± 1.7 pA/μm2 [n = 13]) fell (to −13.6 ± 0.9 pA/μm2 [n = 14] (P < .01)) with 8‐CPT challenge, whereas currents observed following further addition of dantrolene (−22.0 ± 3.3 pA/μm2 [n = 5]) (Ba), or exposure to dantrolene whether alone (Bb), or combined with 8‐CPT (Bc) (−19.4 ± 1.8 pA/μm2 [n = 11] and −21.4 ± 1.8 pA/μm2 [n = 8] respectively) approximated pretreatment values. These changes occurred in an absence of alterations in fits of a Boltzmann function to the inactivation curves. Neither half‐maximal voltages, V* (54.9 ± 2.1 mV [pretreatment], 55.8 ± 2.1 mV (8‐CPT); 55.7 ± 6.8 mV [addition of dantrolene to 8‐CPT]; 55.7 ± 3.2 mV [dantrolene] and 58.6 ± 2.2 mV [dantrolene with 8‐CPT]), nor steepness factors, k, (13.2 ± 0.6 mV, 15.3 ± 0.7 mV, 14.4 ± 0.83 mV, 14.1 ± 0.6 mV, and 15.1 ± 0.5 mV respectively) showed significant differences between groups.
2.5. Voltage dependences of ventricular Na+ current activation and inactivation
Current‐voltage curves describing voltage dependences of ventricular Na+ current activation (Figure 4A) similarly corroborate results in Table 1. Peak Na+ currents increased with depolarisation to −27.0 ± 2.8 pA/μm2 (n = 11) (Figure 4Aa) under pretreatment conditions but only to −16.8 ± 1.6 pA/μm2 (n = 10) in the presence of 8‐CPT. However, currents following further addition of dantrolene (−24.4 ± 1.7 pA/μm2 [n = 10]) (Aa), or exposure to dantrolene whether alone (−26.6 ± 2.0 pA/μm2) (n = 12) (Ab), or combined with 8‐CPT (−25.1 ± 2.2 pA/μm2) (n = 11) (Ac) approximated pretreatment values. Protocols exploring Na+ current inactivation with voltage V 1, reflected in currents observed at voltage V 2 demonstrated currents at the most negative V 1 before treatment (−16.7 ± 1.4 pA/μm2 [n = 7]) falling (to −13.1 ± 1.1 pA/μm2 [n = 9]) with 8‐CPT challenge. However, currents following further addition of dantrolene (−16.4 ± 1.7 pA/μm2 [n = 7]) (Ba), or exposure to dantrolene whether alone (Bb), or combined with 8‐CPT (Bc) (−17.6 ± 1.2 pA/μm2 [n = 12] and −16.3 ± 1.6 pA/μm2 [n = 8] respectively) approximated pretreatment values (all P > .05). These changes occurred in an absence of alterations in fits of a Boltzmann function to the inactivation curves. Neither V* nor k showed significant differences between experimental groups (46.0 ± 3.5, 11.2 ± 1.4 [pretreatment]; 50.3 ± 2.3, 10.0 ± 0.4 [8‐CPT]; 53.4 ± 2.1, 9.8 ± 0.5 [addition of dantrolene to 8‐CPT]; 42.5 ± 1.4, 11.7 ± 0.6 [dantrolene alone] and 45.6 ± 1.9, 10.8 ± 0.5 mV [dantrolene with 8‐CPT]).
2.6. Time course of Na+ channel recovery from inactivation
The subsequent protocols investigated timecourse of Na+ channel recovery from inactivation following atrial or ventricular repolarisation (Figures 5A and 6A) under the same set of pharmacological conditions as described above. The voltage was held at the RMP for 1 ms from the beginning of the recording period before imposition of a hyperpolarising prepulse to voltage V 0 = (RMP−40) mV for 4 ms. This established a consistent baseline level of Na+ current inactivation as in the previous protocol (Figures 5 and 6; panel Aa). A P1 conditioning step between V 0 and V 1 = (RMP + 80) mV of 5 ms duration then elicited activation of an initial Na+ current followed by its inactivation decay (Figures 5 and 6, panel Ab, B). Depolarising P2 steps of 5 ms duration to voltage V 3 = (RMP + 80) mV were then imposed following different time intervals, T, which varied between 5 to 65 ms in 5 ms increments through the 12 successive sweeps making up the protocol. These P2 steps could be used to assess the recovery with time of the peak Na+ current from inactivation (Ab). The peak currents are normalized to their values obtained in the P1 step (Figures 5C and 6C). Fits of time constants to this recovery gave statistically indistinguishable results (P > .05) in each case (a)–(e) (atria: 4.58 ± 0.31 ms; 3.99 ± 0.38 ms; 3.90 ± 0.22 ms; 3.92 ± 0.38 ms and 3.91 ± 0.25 ms; ventricles: 4.6 ± 0.4 ms; 4.5 ± 0.3 ms; 4.7 ± 0.2 ms; 4.7 ± 0.3 ms and 4.8 ± 0.3 ms, respectively).
Figure 5.
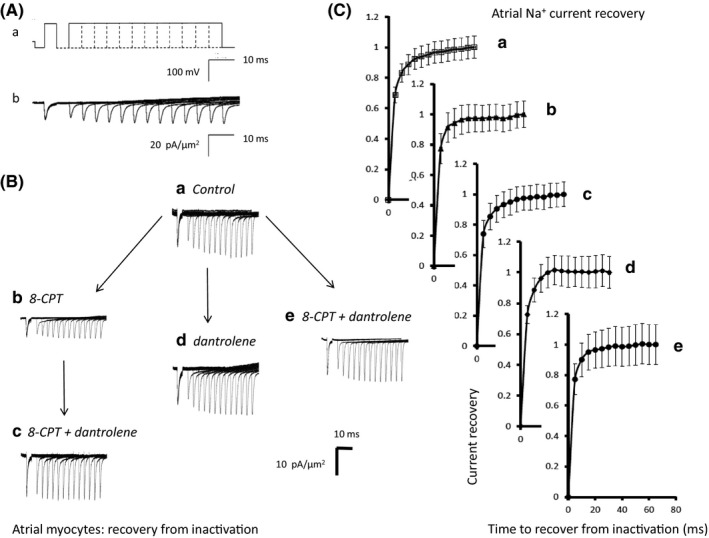
Time course of atrial Na+ current recovery from inactivation. (Aa) Pulse protocols used to investigate the time course of Na+ current recovery from inactivation (Ab) Recovery of Na+ currents with time after inactivation. A pipette of tip diameter 28 μm was used to obtain the currents shown in (b). (B) Families of recovery currents recorded (a) when no drug was present (pretreatment), (b) in the presence of 8‐CPT (1 μmol/L), (c) in the presence of a combination of 8‐CPT (1 μmol/L) and dantrolene (10 μmol/L) after the addition of 8‐CPT, (d) after the addition of dantrolene (10 μmol/L), and (e) in the presence of a combination of 8‐CPT (1 μmol/L) and dantrolene (10 μmol/L). (d) and (e) were obtained following direct introduction of pharmacological agents following pretreatment conditions in the absence of any preceding application of 8‐CPT. (C) Dependences of Na+ current recovery (mean ± SEM) with recovery time before and following pharmacological challenge. The graphs show results (a) before and (b) following introduction of 8‐CPT followed by (c) a combination of 8‐CPT and dantrolene, (d) dantrolene alone and (e) a combination of 8‐CPT and dantrolene
Figure 6.
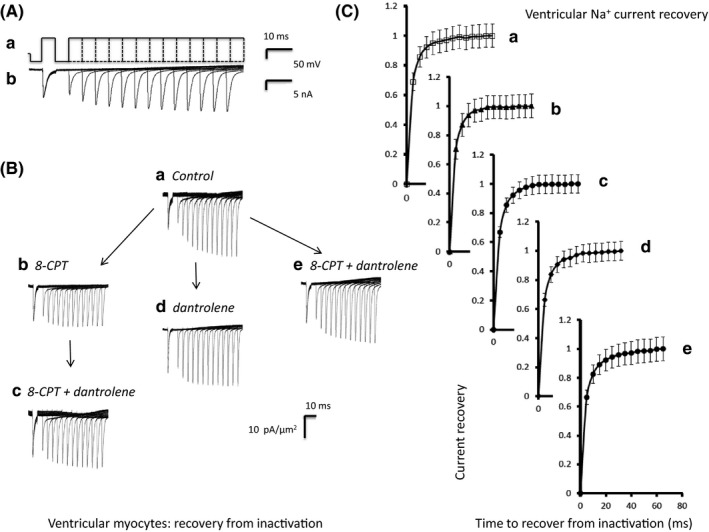
Time course of ventricular Na+ current recovery from inactivation. (Aa) Pulse protocols used to investigate the time course of Na+ current recovery from inactivation (Ab) Recovery of Na+ currents with time after inactivation. A pipette of tip diameter 28 μm was used to obtain the currents shown in (b). (B) Families of recovery currents recorded (a) when no drug was present (pretreatment), (b) in the presence of 8‐CPT (1 μmol/L), (c) in the presence of a combination of 8‐CPT (1 μmol/L) and dantrolene (10 μmol/L) after the addition of 8‐CPT, (d) after the addition of dantrolene (10 μmol/L), and (e) in the presence of a combination of 8‐CPT (1 μmol/L) and dantrolene (10 μmol/L). (d) and (e) were obtained following direct introduction of pharmacological agents following pretreatment conditions in the absence of any preceding application of 8‐CPT. (C) Dependences of Na+ current recovery (mean ± SEM) with recovery time before and following pharmacological challenge. The graphs show results (a) before and (b) following introduction of 8‐CPT followed by (c) a combination of 8‐CPT and dantrolene, (d) dantrolene alone and (e) a combination of 8‐CPT and dantrolene
2.7. Microelectrode recordings of AP characteristics
Reductions of Na+ current of the kind described above have been associated with corresponding reductions in maximum action potential upstroke rate, (dV/dt)max in turn resulting in the pro‐arrhythmic slowing of AP conduction32 reported in previous explorations of effects of Epac modulation.19 Figures 7 and 8 illustrate typical traces of atrial and ventricular action potential waveforms in response to regular 6 Hz pacing (A) and their first derivatives whose peaks reflect (dV/dt)max (B). In comparison with (i) untreated atrial (Figure 7) and ventricular cardiomyocytes (Figure 8), (ii) 8‐CPT challenge reduced (dV/dt)max. This was reversed by further addition of (iii) dantrolene, which when applied by itself (iv) similarly gave (dV/dt)max resembling those of untreated hearts.
Figure 7.
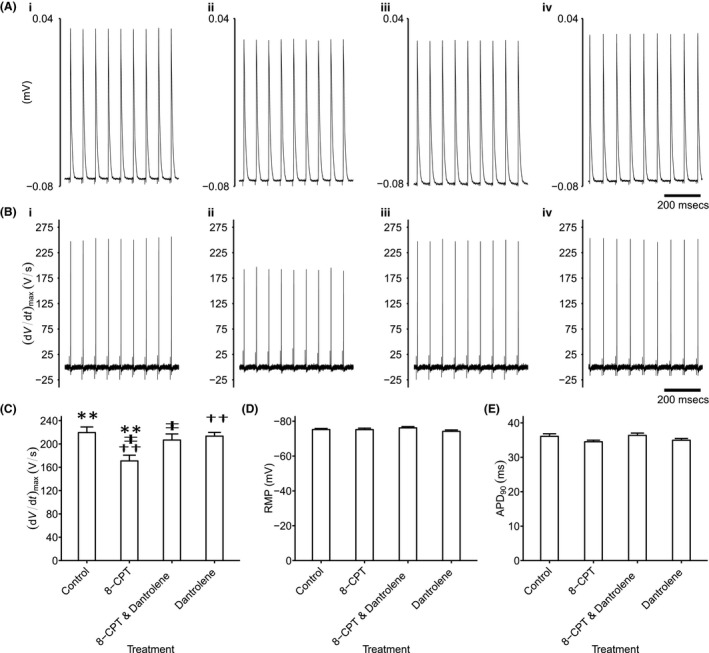
Analysis of atrial action potential waveforms. Intracellular recording of atrial action potential waveforms during regular 6 Hz pacing (A) and their first derivatives reflecting (dV/dt) (B) in (i) untreated cardiomyocytes, (ii) with 8‐CPT challenge followed by (iii) further addition of dantrolene, as well as (iv) dantrolene alone. These recordings yielded values of (dV/dt)max (C) that could be compared with remaining features of the resting potential (RMP) (D) and APD 90 (E). Single, double and triple symbols denote significantly different pairs of values, to a significance value of P < .05, .01 and .001 respectively
Figure 8.
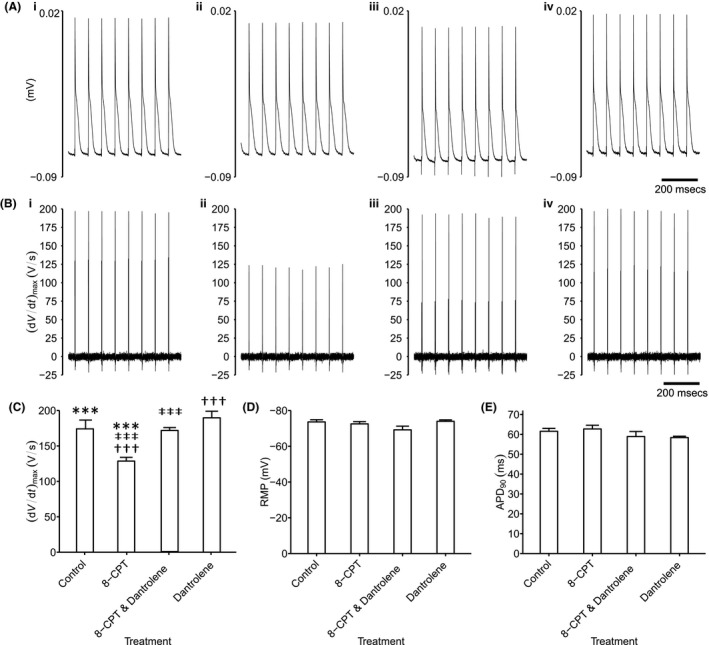
Analysis of ventricular action potential waveforms. Intracellular recording of ventricular action potential waveforms during regular 6 Hz pacing (A) and their first derivatives reflecting (dV/dt) (B) in (i) untreated cardiomyocytes, (ii) with 8‐CPT challenge followed by (iii) further addition of dantrolene, as well as (iv) dantrolene alone. These recordings yielded values of (dV/dt)max (C) that could be compared with remaining features of the resting potential (RMP) (D) and APD 90 (E). Single, double and triple symbols denote significantly different pairs of values, to a significance value of P < .05, .01 and .001 respectively
Significant differences in both atrial and ventricular (dV/dt)max values were thus detected amongst findings obtained in the absence (atria: 219.56 ± 9.64 V/s [n = 23]; ventricles: 174.11 ± 6.21 V/s [n = 17]), and following challenge with 8‐CPT before (atria: 170.97 ± 9.78 V/s [n = 20]; ventricles: 128.64 ± 5.26 V/s [n = 14]), and following further addition of dantrolene (atria: 206.74 ± 10.47 V/s [n = 22]; ventricles: 170.90 ± 4.16 V/s [n = 20]) and of dantrolene applied alone (atria: 213.48 ± 6.39 V/s [n = 29]; ventricles: 189.70 ± 9.28 V/s [n = 20]) (atria: F = 5.41; P = .0018; ventricles: F = 12.98; P = 8.8 × 10−7). There were significant pairwise (dV/dt)max differences in cardiomyocytes studied before, and following 8‐CPT challenge (atria: P = .002; ventricles: P = .0002) but not following further addition of dantrolene (atria: P = .75; ventricles: P = .99) or addition of dantrolene alone (atria: P = .96; ventricles: P = .36). Thus, further addition of dantrolene restored Na+ current previously reduced by 8‐CPT (atria: P = .042; ventricles: P = .0004; Figures 7C and 8C). The similar findings in both atrial and ventricular preparations thus paralleled corresponding observations in loose patch clamp Na+ currents. In contrast, resting membrane potentials showed no significant differences between groups (atria: F = 1.24, P = .30; ventricles: F = 1.18, P = .32), falling close to −75 mV in both atrial and ventricular cardiomyocytes (Figures 7D and 8D). Similarly, action potential durations at 90% recovery showed no significant differences between experimental groups (atria: F = 2.10, P = .11; ventricles: F = 1.24; P = .296), falling close to ~35 and ~60 ms in atrial and ventricular cardiomyocytes respectively (Figures 7E and 8E).
3. DISCUSSION
The experiments here investigated for acute effects of Epac activation, known to acutely perturb cellular Ca2+ homeostasis,18 upon Na+ currents in wild‐type murine cardiomyocytes. The loose patch clamp method detected current flowing into an electrode apposed to the extracellular surface of a fixed area of intact cell membrane, yielding membrane currents normalised to the area of voltage‐clamped cell surface (pA/μm2). In contrast, conventional patch clamp methods typically normalise measured currents to background membrane capacitance (pA/pF). Atrial and ventricular cells showed differing Na+ current magnitudes (e.g. ~−42.6 ± 2.9 vs −27.5 ± 1.8 pA/pF;39 ~−89.59 ± 41.05 vs −50.20 ± 3.34 pA/pF respectively40), but total membrane capacitances differing in the opposite direction (~43 vs ~90 pF).39 Their total capacitances also differ in their relative contributions of surface to tubular membrane41, 42 that in turn express differing channel densities.43, 44 Nevertheless, the loose‐patch clamp permitted cardiomyocyte recordings to be made in intact superfused atrial and ventricular preparations, avoiding disruptions of intracellular Ca2+ homeostasis necessitated by the isolation and intracellular Ca2+ chelation otherwise required in conventional whole‐cell patch‐clamp recordings.30, 31 Loose‐patch clamp recordings also allowed employment of in vivo rather than reduced extracellular [Na+] levels sparing Na+‐Ca2+ exchange. Previous reports had identified early inward currents obtained with this technique with Na+ currents mediating action potential (AP) conduction and upstroke.21
The experiments followed directly from findings that acute Epac (exchange protein directly activated by cAMP) activation: (i) produced phosphokinase A (PKA)‐independent activation of RyR2‐mediated Ca2+ release,13, 14, 15 increasing Ca2+ spark frequencies in adult rat cardiomyocytes,16 Ca2+‐dependent Ca2+ release amplitudes after isoproterenol treatment,17 and amplitudes and frequencies of spontaneous Ca2+ release in mouse ventricular cardiomyocytes.18 It correspondingly, (ii) increased incidences of both triggered activity and ventricular tachycardia (VT).18 However, it also (iii) associated such pro‐arrhythmic effects with decreased conduction velocities.19 Furthermore, (iv) these electrophysiological characteristics were rescued by the RyR2‐Ca2+ release channel blocker dantrolene, despite (v) all these changes being associated with unaltered recovery characteristics as reflected in the cardiac action potential (AP) durations and ventricular effective refractory periods.18, 19
Such findings suggest that acutely altered Ca2+ homeostasis might result in arrhythmic substrate arising from delayed conduction, as previously suggested for the pro‐arrhythmic Brugada syndrome1, 2, 3, 4, 5 in contrast to the recovery abnormalities associated with the LQT syndromes.6, 45 The pro‐arrhythmic effects of acute abnormalities in cardiomyocyte Ca2+ homeostasis provoked by adrenergic stimulation, caffeine‐mediated RyR2 stimulation10 or modified extracellular Ca2+ entry,11, 12 have hitherto been primarily associated with arrhythmic triggering by delayed afterdepolarisation effects arising from the consequently altered Na+–Ca2+ exchanger activity.
Early Na+ current is a primary determinant of cardiac AP conduction velocity. The present experiments are the first time Na+ current measurements have been made during Epac activation using an experimental design that permits comparisons with those previous studies.20, 21, 26 Thus, in addition to employing similar methods of Na+ current measurement: (i) They also (cf.18) investigated effects of Epac activation using the agonist 8‐CPT13 at ~1 μmol/L, a concentration range known to provide a 300‐fold preferential selectivity for Epac over PKA pathways;13, 33, 34 8‐CPT only inhibits phosphodiesterase isoforms at considerably higher concentrations.35 (ii) The reversibility of the pharmacological effects of 8‐CPT were tested for by the RyR2‐inhibiting agent dantrolene (cf.19) thought to stabilize RyR2 closed states by enhancing interactions between its N‐terminal and the central domains particularly under conditions of increased open channel probability.36, 37, 46 Dantrolene thus inhibits RyR2‐mediated diastolic Ca2+ release and decreases frequencies and durations of aberrant Ca2+ sparks in cardiomyocyte models for CPVT36 and cardiac failure.37, 38 (iii) Effects of dantrolene were further controlled for in experiments involving direct exposure to dantrolene, both by itself and in combination with 8‐CPT (cf.19), and (iv) The experiments similarly measured the time courses and steady‐state voltage‐dependences of Na+ current activation and inactivation (cf.21), in both atrial and ventricular preparations from WT murine hearts.
The variation of the test step to voltage V 1 allowed measurement of the peak amplitudes of Na+ current and yielded current‐voltage activation relationships. These peaks were followed by decays to a steady‐state inactivation level whose dependence upon the voltage V 1 was assessed by further voltage steps to a standard depolarised voltage V 2. 8‐CPT markedly reduced the Na+ currents in both procedures. The inhibitory effect of 8‐CPT was reversed by further addition of dantrolene. In contrast, transfer of cardiomyocytes from pretreatment conditions to solutions containing dantrolene alone or dantrolene combined with 8‐CPT left the Na+ currents intact. Furthermore, detailed parameters describing the voltage dependence of current inactivation were unchanged through these pharmacological conditions, as were the time courses of recovery from such inactivation.
These findings thus demonstrate that acutely altered Ca2+ homeostasis following Epac‐mediated RyR2‐activation reversibly reduces Na+ channel function. Previous reversible manipulations of loose patch pipette [Na+] had identified early inward currents following step depolarisations with cardiomyocyte Na+ currents driving the maximum upstroke rate, (dV/dt)max, and in turn conduction of the cardiac action potential21. The present experiments accordingly went on to investigate such corresponding changes in action potential activation reflected in (dV/dt)max measurements from intracellular sharp electrode recordings of membrane potential in intact atrial and ventricular preparations under comparable pharmacological conditions. The observed (dV/dt)max were reduced with 8‐CPT challenge; this effect was absent with dantrolene whether applied alone or in combination with 8‐CPT. This was in direct agreement with the observed changes in Na+ currents under corresponding pharmacological conditions. Together these findings implicate such Na+ current alterations with the pro‐arrhythmic reductions in AP conduction velocity observed in previous explorations of the effects of Epac modulation in murine hearts.19
The present findings thus implicate conduction velocity secondary to compromised Na+ current as a source for arrhythmic substrate under conditions of acutely perturbed cytosolic Ca2+ homeostasis, reconstructing the altered conduction previously reported in Nav1.5 haplo‐insufficient, Scn5a +/− murine models for Brugada Syndrome.1, 2, 3, 4, 5 They also complement previous findings in murine hearts chronically modelling catecholaminergic polymorphic ventricular tachycardia (CPVT). Cardiomyocytes in the latter systems similarly showed diastolic episodes, or propagating waves, of RyR2‐mediated Ca2+ release, as well as afterdepolarisation and triggering phenomena.8, 9 RyR2‐P2328S hearts additionally showed parallel reductions in atrial21 and ventricular action potential conduction velocities, the latter particularly following catcholaminergic challenge.20 These changes accompanied chronically downregulated Nav1.5 expression.21, 27 Furthermore, WT rat cardiomyocytes increased their expression of functionally active surface membrane Nav1.5, Nav1.5 mRNA and total Nav1.5 protein following verapamil challenge and decreased their surface membrane Nav1.5 expression following calcimycin challenge.47, 48 In parallel with the present findings, RyR2‐P2328S cardiomyocytes also demonstrated acutely reduced Nav1.5 function.20, 21, 26 This was partially rescued by pharmacological interventions reducing RyR2‐mediated Ca2+ release.22, 23, 24, 25 These findings together suggest direct effects of altered cytosolic [Ca2+] upon Nav1.5 function.49, 50, 51 Certainly, patch‐clamped WT myocytes show respective reductions, or increases in Na+ current and (dV/dt)max, with increases in, or sequestration of, the pipette [Ca2+].52 Structural evidence suggests direct and/or indirect Ca2+ binding sites on Nav1.5 whose occupancy might modify Nav1.5 channel function. Close to the Nav1.5 carboxy‐terminal, direct Ca2+ binding occurring at an EF hand motif may increase Na+ channel activity.53 In contrast, an additional indirect ‘IQ’ domain binding site permits Ca2+/calmodulin (CaM) binding. Finally, multiple phosphorylatable sites in the DI‐II linker region, including serines 516 and 571, and threonine 594, are targeted by calmodulin kinase II (CaMKII)54, 55, 56. The latter two binding mechanisms require prior Ca2+ binding to EF hand motifs in Ca2+/CaM or CaMKII and compromise Na+ channel activity.50, 51
4. MATERIALS AND METHODS
4.1. Solutions
Krebs–Henseleit (KH) solution was prepared (mmol/L: NaCl, 119; NaHCO3, 25; KCl, 4.0; KH2PO4, 1.2; MgCl2, 1.0; CaCl2, 1.8; glucose, 10; and Na‐pyruvate, 2.0; pH adjusted to 7.4 and bubbled with 95% O2/5% CO2 (British Oxygen Company, Manchester, UK)) for all tissue preparations. Chemical agents were purchased from Sigma‐Aldrich (Poole, UK) unless otherwise stated. A modified KH solution containing 10 mmol/L 2,3‐butanedione monoxime (BDM) and 10 μmol/L blebbistatin (Cayman Chemical Company, Ann Arbor, MI, USA) was also prepared for Langendorff perfusion of isolated hearts to electromechanically uncouple the heart prior to isolation of right ventricular preparations. KH solution containing 8‐(4‐chlorophenylthio)adenosine 3′,5′‐cyclic monophosphate sodium salt (8‐CPT, 1 μmol/L) and/or dantrolene (10 μmol/L) (LKT Laboratories Inc, St Paul, MN, USA) was prepared and filtered to remove particles greater than 10 μm in diameter with standard filtration paper (Millipore, Bedford, MA, USA).
4.2. Tissue preparation
This research has been regulated under the Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012 following ethical review by the University of Cambridge Animal Welfare and Ethical Review Body (AWERB). It also conformed to the Guide for the Care and Use of Laboratory Animals, U.S. National Institutes of Health (NIH Publication No. 85‐23, revised 1996). C57BL6 WT mice aged 3‐6 months were used in all experiments. Mice were housed at room temperature in a licensed facility, allowed free access to sterile rodent chow and water, and subjected to 12 hours light/dark cycles. Mice were killed by cervical dislocation (Schedule 1 of the UK Animals (Scientific Procedures) Act 1986), following administration of 200 IU intraperitoneal heparin (ACROS Organics. ThermoFisher, Loughborough, Leicestershire, UK) 10 minutes prior to death. The heart was immediately excised and transferred into ice‐cold KH solution. The aorta was cannulated with a trimmed and filed 21G hypodermic needle, and secured onto the cannula with an aneurysm clip and 5‐0 braided silk suture. The heart was then perfused retrogradely in a Langendorff system under constant flow (2 mL/min) by a Watson‐Marlow (Falmouth, UK) peristaltic pump with 75 mL KH‐BDM/blebbistatin solution to electromechanically uncouple the heart. Following cessation of contractions, the heart was immediately transferred into ice‐cold KH‐BDM/blebbistatin solution. For the loose patch clamp experiments, the atria and right ventricle were dissected from the rest of the heart, mounted onto Sylgard (Dow Chemical Company, Staines, UK) then placed in the bath containing filtered KH buffer solution maintained at 27°C. Experiments performing intracellular electrophysiological measurements transferred the hearts to a Langendorff perfusion system for the electrophysiological studies described below.
4.3. Loose patch clamp recording
Pipettes were pulled from borosilicate glass capillaries (GC150‐10 specification Harvard Apparatus, Cambourne, Cambridge, UK) using a micropipette puller (Flaming/Brown micropipette puller; model P‐97, Sutter Instrument Co. Novato, CA, USA). Pipettes were then mounted under a microscope at 250× magnification with a calibrated eyepiece graticule, and fractured with a diamond knife under visual control. Transverse force applied to the distal tip of the pipette resulted in fracture perpendicular to the axis of the micropipette. Selected pipettes were fire‐polished using an electrically heated nichrome filament under visual guidance at 400× magnification. The pipette tips were then bent to ~45° from the shaft of the pipette to allow the membrane to be approached vertically when mounted on the recording amplifier headstage. Maximum internal tip diameters were measured at 1000× magnification. All pipettes were of 28‐32 μm in diameter following polishing.
The distal half of the micropipettes were filled with KH buffer and mounted onto a pipette holder connected to a headstage. Connection of an air filled line to the pipette holder allowed for application of suction through a syringe to form membrane seals. Ag/AgCl electrodes maintained electrical connections to the organ bath and pipette. Loose patch‐clamp experiments were performed as previously described.21, 24, 27 The pipette was lowered onto the membrane surface and gentle suction applied to allow seal formation around the patch of membrane. Voltage‐clamp steps were delivered under computer control relative to the resting membrane potential. The loose patch clamp method applies voltage steps to the extracellular surface of membrane within the seal. Positive and negative voltage steps applied through the pipette respectively hyperpolarise and depolarise the membrane potential relative to the initial cell resting membrane potential (RMP). In the text membrane potentials are therefore stated in terms of their displacement from the RMP in the same convention adopted by earlier studies using this technique.29, 57 The loose patch clamp configuration results in larger leakage currents than the conventional patch clamp due to the comparatively low seal resistance. A custom‐built amplifier was used to compensate for the majority of the leakage current, series resistance errors and the displacement current through the pipette capacitance.28 Residual leakage and capacitative currents were corrected for using reference records from subsequent P/4 control protocols applying steps whose amplitudes were scaled down by a factor of four and of the opposite sign relative to the test steps, as fully described previously.29, 57
Experiments were first performed under pretreatment conditions using preparations superfused with KH solution. Measurements were then made with KH solution containing 8‐CPT (1 μmol/L), then with KH solution containing both 8‐CPT (1 μmol/L) and dantrolene (10 μmol/L). To control for effects of dantrolene, further experiments superfused preparations with KH solution containing dantrolene (10 μmol/L) both alone and in combination with 8‐CPT (1 μmol/L). Patch‐clamp studies took place under standardised electrophysiological conditions following similar intervals after each solution change. Each patch was subject only to a single application of the pulse protocols. These made differences between results attributable to prolonged changes in the patch such as bleb formation unlikely.58
4.4. Whole heart intracellular microelectrode recordings
Microelectrode studies were performed using a modified horizontal Langendorff perfusion system incorporating a light microscope (objective ×5, eyepiece ×5, W. Watson and Sons Limited, London, UK), and warmed tissue chamber housed within a Faraday cage. Stimulating and recording electrodes were positioned on the heart using micromanipulators (Prior Scientific Instruments, Cambridge, UK). Sharp microelectrodes drawn from borosilicate glass pipettes (OD 1.2 mm, ID 0.69 mm, Harvard Apparatus, Cambridge, UK) using a homebuilt microelectrode puller, filled with 3 M KCl (tip resistance 15‐25 MΩ), and mounted onto a right‐angled microelectrode holder containing a Ag/AgCl half‐cell were connected to a high‐input impedance direct‐current microelectrode amplifier system (University of Cambridge, Cambridge, UK). Following band‐pass filtering (between 0 and 2 kHz), signals were subject to analogue‐to‐digital conversion for recording at a sampling frequency of 10 kHz (1401; Spike software: Cambridge Electronic Design, Cambridge, UK). Impalements were consistently made in the proximal left ventricle to minimise confounds from regional variations in AP characteristics. Intracellular voltages were measured relative to that of a bath Ag/AgCl reference electrode. Impalements were used for detailed recordings where they resulted in abrupt appearance of a resting membrane potential (RMP) between −65 and −90 mV and APs of normal amplitude >75 mV. Hearts were paced by a bipolar platinum‐coated stimulating electrode (NuMed, New York, USA) applied to the right ventricle lateral surface using a DS2A‐Mk.II stimulator (Digitimer, Welwyn Garden City, Herts., UK) applying a stimulus voltage twice diastolic excitation threshold plus 0.5 mV, at a regular 6 Hz pacing frequency. Maximum rates of action potential depolarisation (dV/dt)max were obtained from the first time differential of the intracellular AP waveform. These were measured along with cardiomyocyte resting potentials, RMP, with AP amplitude measured from the RMP to the peak AP voltage. Action potential duration was measured as the time from the AP peak to 90% repolarisation to baseline.
4.5. Analysis of results
The currents obtained from the loose patch procedure are dimensioned in units of current (nA). Subsequent analysis converted the units of current (nA) to current densities (pA/μm2) using the formula:
Current‐voltage curves describing Na+ current activation and inactivation and current‐time relationships plotting the recovery timecourse of Na+ current from inactivation were obtained from the corresponding measurements of peak Na+ currents (means ± standard error of the mean [SEM]) in the relevant protocol (see Figure 1Aa). Peak Na+ currents, I, reflecting inactivation properties were empirically related to the inactivating voltage V = V 1 by a Boltzmann function: I = I max/{1 + exp(V−V*/k)}. Peak currents obtained during determinations of the timecourse, T, of recovery from inactivation were described by the exponential function I = I max(1−exp(−T/τ)). The curve‐fitting procedures employed the open source fitting algorithms QtiPlot (Version 0.9.9‐rc9).
4.6. Statistical analysis of results
Owing to the limited recording time permitted following establishment of each loose patch,58 each pharmacological condition was tested using different patches giving unpaired comparisons of results obtained (i) before, and following (ii) 8‐CPT treatment, (iii) 8‐CPT followed by addition of dantrolene, (iv) 8‐CPT combined with dantrolene, and (v) dantrolene alone initially using one‐way analysis of variance (ANOVA) to an α < .05 significance level. Subsequent posthoc comparisons suggested by a presence of significant differences employed a Bonferroni method in view of its application to (i) a subset of four rather than all possible pairwise comparisons with (ii) the number of contrasts to be estimated close to the number of factors (NIST/SEMATECH e‐Handbook of Statistical Methods, http://www.itl.nist.gov/div898/handbook/).
DISCLOSURE
None declared.
ACKNOWLEDGEMENTS
We are grateful for financial support from the Medical Research Council (MR/M001288/1), the Wellcome Trust (105727/Z/14/Z), British Heart Foundation (PG/14/79/31102 and PG/15/12/31280), the Fundamental Research Grant Scheme Ministry of Education, Malaysia (FRGS/2/2014/SKK01/PERDANA/02/1), and the McVeigh Benefaction and Sudden Arrhythmic Death Syndrome SADS UK. We are also grateful to Paul Frost and Vicky Johnson for their technical support.
Valli H, Ahmad S, Sriharan S, et al. Epac‐induced ryanodine receptor type 2 activation inhibits sodium currents in atrial and ventricular murine cardiomyocytes. Clin Exp Pharmacol Physiol. 2018;45:278‐292. https://doi.org/10.1111/1440-1681.12870
REFERENCES
- 1. Sabir IN, Killeen MJ, Grace AA, Huang CL‐H. Ventricular arrhythmogenesis: Insights from murine models. Prog Biophys Mol Biol. 2008;98:208‐218. [DOI] [PubMed] [Google Scholar]
- 2. Kalin A, Usher‐Smith J, Jones VJ, Huang CL‐H, Sabir IN. Cardiac arrhythmia: A simple conceptual framework. Trends Cardiovasc Med. 2010;20:103‐107. [DOI] [PubMed] [Google Scholar]
- 3. Huang CL‐H. Murine models of cardiac arrhythmogenesis. Physiol Rev. 2017;97:283‐409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Martin CA, Guzadhur L, Grace AA, Lei M, Huang CL‐H. Mapping of reentrant spontaneous polymorphic ventricular tachycardia in a Scn5a+/‐ mouse model. Am J Physiol Heart Circ Physiol. 2011;300:H1853‐H1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Martin CA, Grace AA, Huang CL‐H. Refractory dispersion promotes conduction disturbance and arrhythmias in a Scn5a (+/‐) mouse model. Pflugers Arch. 2011;462:495‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sabir IN, Fraser JA, Cass TR, Grace AA, Huang CL‐H. A quantitative analysis of the effect of cycle length on arrhythmogenicity in hypokalaemic Langendorff‐perfused murine hearts. Pflugers Arch. 2007;454:925‐936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure roles of sodium‐calcium exchange, inward rectifier potassium current, and residual β‐adrenergic responsiveness. Circ Res. 2001;88:1159‐1167. [DOI] [PubMed] [Google Scholar]
- 8. Goddard CA, Ghais NS, Zhang Y, et al. Physiological consequences of the P2328S mutation in the ryanodine receptor (RyR2) gene in genetically modified murine hearts. Acta Physiol. 2008;194:123‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jiang D, Wang R, Xiao B, et al. Enhanced store overload‐induced Ca(2+) release and channel sensitivity to luminal Ca(2+) activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005;97:1173‐1181. [DOI] [PubMed] [Google Scholar]
- 10. Balasubramaniam R, Chawla S, Grace AA, Huang CL‐H. Caffeine‐induced arrhythmias in murine hearts parallel changes in cellular Ca(2+) homeostasis. Am J Physiol Heart Circ Physiol. 2005;289:H1584‐H1593. [DOI] [PubMed] [Google Scholar]
- 11. Balasubramaniam R, Chawla S, Mackenzie L, Schwiening CJ, Grace AA, Huang CL‐H. Nifedipine and diltiazem suppress ventricular arrhythmogenesis and calcium release in mouse hearts. Pflugers Arch. 2004;449:150‐158. [DOI] [PubMed] [Google Scholar]
- 12. Ghais NS, Zhang Y, Grace AA, Huang CL‐H. Arrhythmogenic actions of the Ca(2+) channel agonist FPL‐64176 in Langendorff‐perfused murine hearts. Exp Physiol. 2009;94:240‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Holz GG, Kang G, Harbeck M, Roe MW, Chepurny OG. Cell physiology of cAMP sensor Epac. J Physiol. 2006;577:5‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kawasaki H, Springett GM, Mochizuki N, et al. A family of cAMP‐binding proteins that directly activate Rap1. Science. 1998;282:2275‐2279. [DOI] [PubMed] [Google Scholar]
- 15. De Rooij J, Zwartkruis FJ, Verheijen MH, et al. Epac is a Rap1 guanine‐nucleotide‐exchange factor directly activated by cyclic AMP. Nature. 1998;396:474‐477. [DOI] [PubMed] [Google Scholar]
- 16. Pereira L, Metrich M, Fernandez‐Velasco M, et al. The cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J Physiol. 2007;583:685‐694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oestreich EA, Wang H, Malik S, et al. Epac‐mediated activation of phospholipase Cϵ plays a critical role in β‐adrenergic receptor‐dependent enhancement of Ca2+ mobilization in cardiac myocytes. J Biol Chem. 2007;282:5488‐5495. [DOI] [PubMed] [Google Scholar]
- 18. Hothi SS, Gurung IS, Heathcote JC, et al. Epac activation, altered calcium homeostasis and ventricular arrhythmogenesis in the murine heart. Pflugers Arch. 2008;457:253‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li M, Hothi S, Salvage S, Jeevaratnam K, Grace A, Huang C. Arrhythmic effects of Epac‐mediated ryanodine receptor activation in Langendorff‐perfused murine hearts are associated with reduced conduction velocity. Clin Exp Pharmacol Physiol. 2017;44:686‐692. 2017 Mar 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Y, Wu J, Jeevaratnam K, et al. Conduction slowing contributes to spontaneous ventricular arrhythmias in intrinsically active murine RyR2‐P2328S hearts. J Cardiovasc Electrophysiol. 2013;24:210‐218. [DOI] [PubMed] [Google Scholar]
- 21. King J, Wickramarachchi C, Kua K, et al. Loss of Nav1.5 expression and function in murine atria containing the RyR2‐P2328S gain‐of‐function mutation. Cardiovasc Res. 2013;99:751‐759. [DOI] [PubMed] [Google Scholar]
- 22. Watanabe H, Van Der Werf C, Roses‐Noguer F, et al. Effects of flecainide on exercise‐induced ventricular arrhythmias and recurrences in genotype‐negative patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2013;10:542‐547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Salvage S, Chandrasekharan KH, Jeevaratnam K, et al. Multiple targets for flecainide action: Implications for cardiac arrhythmogenesis. Br J Pharmacol. 2017; doi: 10.1111/bph.13807 (PMID: 28369767). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Salvage SC, King JH, Chandrasekharan KH, et al. Flecainide exerts paradoxical effects on sodium currents and atrial arrhythmia in murine RyR2‐P2328S hearts. Acta Physiol. 2015;214:361‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Knollmann BC, Blatt SA, Horton K, et al. Inotropic stimulation induces cardiac dysfunction in transgenic mice expressing a troponin T (I79N) mutation linked to familial hypertrophic cardiomyopathy. J Biol Chem. 2001;276:10039‐10048. [DOI] [PubMed] [Google Scholar]
- 26. King J, Zhang Y, Lei M, Grace A, Huang C‐H, Fraser J. Atrial arrhythmia, triggering events and conduction abnormalities in isolated murine RyR2‐P2328S hearts. Acta Physiol. 2013;207:308‐323. [DOI] [PubMed] [Google Scholar]
- 27. Ning F, Luo L, Ahmad S, et al. The RyR2‐P2328S mutation downregulates Nav1.5 producing arrhythmic substrate in murine ventricles. Pflügers Arch Eur J Physiol. 2016;468:655‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stühmer W, Roberts WM, Almers W. The loose patch clamp In: Sakmann B, Neher E, eds. Single‐Channel Recording. Dordrecht: Springer; 1983:123‐132. [Google Scholar]
- 29. Almers W, Stanfield PR, Stuhmer W. Lateral distribution of sodium and potassium channels in frog skeletal muscle: measurements with a patch‐clamp technique. J Physiol. 1983;336:261‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martin CA, Siedlecka U, Kemmerich K, et al. Reduced Na(+) and higher K(+) channel expression and function contribute to right ventricular origin of arrhythmias in Scn5a+/‐ mice. Open Biol. 2012;2:120072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lei M, Goddard C, Liu J, et al. Sinus node dysfunction following targeted disruption of the murine cardiac sodium channel gene Scn5a. J Physiol. 2005;567:387‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. King J, Huang CL‐H, Fraser JA. Determinants of myocardial conduction velocity: Implications for arrhythmogenesis. Front Physiol 2013;4:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Christensen AE, Selheim F, de Rooij J, et al. cAMP analog mapping of Epac1 and cAMP kinase discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC‐12 cell neurite extension. J Biol Chem. 2003;278:35394‐35402. [DOI] [PubMed] [Google Scholar]
- 34. Fujita T, Umemura M, Yokoyama U, Okumura S, Ishikawa Y. The role of Epac in the heart. Cell Mol Life Sci. 2017;74:591‐606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Poppe H, Rybalkin SD, Rehmann H, et al. Cyclic nucleotide analogs as probes of signaling pathways. Nat Methods. 2008;5:277‐278. [DOI] [PubMed] [Google Scholar]
- 36. Jung CB, Moretti A, Mederos y Schnitzler, M , et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient‐specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med. 2012;4:180‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kobayashi S, Yano M, Suetomi T, et al. Dantrolene, a therapeutic agent for malignant hyperthermia, markedly improves the function of failing cardiomyocytes by stabilizing inter‐domain interactions within the ryanodine receptor. J Am Coll Cardiol. 2009;53:1993‐2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maxwell JT, Domeier TL, Blatter LA. Dantrolene prevents arrhythmogenic Ca2+ release in heart failure. Am J Physiol Heart Circ Physiol. 2012;302:H953‐H963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li GR, Lau CP, Shrier A. Heterogeneity of sodium current in atrial vs epicardial ventricular myocytes of adult guinea pig hearts. J Mol Cell Cardiol. 2002;34:1185‐1194. [DOI] [PubMed] [Google Scholar]
- 40. Burashnikov A, Di Diego JM, Zygmunt AC, Belardinelli L, Antzelevitch C. Atrium‐selective sodium channel block as a strategy for suppression of atrial fibrillation: Differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation. 2007;116:1449‐1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Orchard C, Brette F. t‐Tubules and sarcoplasmic reticulum function in cardiac ventricular myocytes. Cardiovasc Res. 2008;77:237‐244. [DOI] [PubMed] [Google Scholar]
- 42. Moore L, Schmid A, Isenberg G. Linear electrical properties of isolated cardiac cells. J Membr Biol. 1984;81:29‐40. [DOI] [PubMed] [Google Scholar]
- 43. Gadeberg HC, Kong CHT, Bryant SM, James AF, Orchard CH. Sarcolemmal distribution of ICa and INCX and and Ca2+ autoregulation in mouse ventricular myocytes. Am J Physiol ‐ Heart Circ Physiol. 2017;313:H190‐H199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chase A, Orchard CH. Ca efflux via the sarcolemmal Ca ATPase occurs only in the t‐tubules of rat ventricular myocytes. J Mol Cell Cardiol. 2011;50:187‐193. [DOI] [PubMed] [Google Scholar]
- 45. Sabir IN, Li LM, Jones VJ, Goddard CA, Grace AA, Huang CL‐H. Criteria for arrhythmogenicity in genetically‐modified Langendorff‐perfused murine hearts modelling the congenital long QT syndrome type 3 and the Brugada syndrome. Pflugers Arch Eur J Physiol. 2008;455:637‐651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Paul‐Pletzer K, Yamamoto T, Ikemoto N, et al. Probing a putative dantrolene‐binding site on the cardiac ryanodine receptor. Biochem J. 2005;387:905‐909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Offord J, Catterall WA. Electrical activity, cAMP, and cytosolic calcium regulate mRNA encoding sodium channel alpha subunits in rat muscle cells. Neuron. 1989;2:1447‐1452. [DOI] [PubMed] [Google Scholar]
- 48. Duff HJ, Offord J, West J, Catterall WA. Class I and IV antiarrhythmic drugs and cytosolic calcium regulate mRNA encoding the sodium channel alpha subunit in rat cardiac muscle. Mol Pharmacol. 1992;42:570‐574. [PubMed] [Google Scholar]
- 49. Aiba T, Hesketh GG, Liu T, et al. Na+ channel regulation by Ca(2+)/calmodulin and Ca(2+)/calmodulin‐dependent protein kinase II in guinea‐pig ventricular myocytes. Cardiovasc Res. 2010;85:454‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ashpole NM, Herren AW, Ginsburg KS, et al. Ca2+/calmodulin‐dependent protein kinase II (CaMKII) regulates cardiac sodium channel Nav1.5 gating by multiple phosphorylation sites. J Biol Chem. 2012;287:19856‐19869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tan HL, Kupershmidt S, Zhang R, et al. A calcium sensor in the sodium channel modulates cardiac excitability. Nature. 2002;415:442‐447. [DOI] [PubMed] [Google Scholar]
- 52. Casini S, Verkerk AO, van Borren MMGJ, et al. Intracellular calcium modulation of voltage‐gated sodium channels in ventricular myocytes. Cardiovasc Res. 2009;81:72‐81. [DOI] [PubMed] [Google Scholar]
- 53. Wingo TL, Shah VN, Anderson ME, Lybrand TP, Chazin WJ, Balser JR. An EF‐hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat Struct Mol Biol. 2004;11:219‐225. [DOI] [PubMed] [Google Scholar]
- 54. Grandi E, Herren AW. CaMKII‐dependent regulation of cardiac Na(+) homeostasis. Front Pharmacol. 2014;5:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mori M, Konno T, Ozawa T, Murata M, Imoto K, Nagayama K. Novel interaction of the voltage‐dependent sodium channel (VDSC) with calmodulin: Does VDSC acquire calmodulin‐mediated Ca(2+)‐sensitivity? Biochemistry. 2000;39:1316‐1323. [DOI] [PubMed] [Google Scholar]
- 56. Wagner S, Ruff HM, Weber SL, et al. Reactive oxygen species‐activated Ca/calmodulin kinase IIδ is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ Res. 2011;108:555‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Almers W, Stanfield PR, Stühmer W. Slow changes in currents through sodium channels in frog muscle membrane. J Physiol. 1983;339:253‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Milton RL, Caldwell JH. Na current in membrane blebs: Implications for channel mobility and patch clamp recording. J Neurosci. 1990;10:885‐893. [DOI] [PMC free article] [PubMed] [Google Scholar]