


Abstract
The alkylation of the specific higher-order nucleic acid structures is of great significance in order to control its function and gene expression. In this report, we have described the T–T mismatch selective alkylation with a vinyldiaminotriazine (VDAT)–acridine conjugate. The alkylation selectively proceeded at the N3 position of thymidine on the T–T mismatch. Interestingly, the alkylated thymidine induced base flipping of the complementary base in the duplex. In a model experiment for the alkylation of the CTG repeats DNA which causes myotonic dystrophy type 1 (DM1), the observed reaction rate for one alkylation increased in proportion to the number of T–T mismatches. In addition, we showed that primer extension reactions with DNA polymerase and transcription with RNA polymerase were stopped by the alkylation. The alkylation of the repeat DNA will efficiently work for the inhibition of replication and transcription reactions. These functions of the VDAT–acridine conjugate would be useful as a new biochemical tool for the study of CTG repeats and may provide a new strategy for the molecular therapy of DM1.
INTRODUCTION
Higher-order structures of nucleic acids, such as the mismatched, bulge and G-quadruplex structures, are significant research targets in nucleic acid chemistry because the structures are involved in the genetic control and diseases (1,2). Reactive molecules, which recognize the specific structure and form a covalent bond, have been developed as a strong inhibitor and biochemical research tool (3,4). While several excellent molecules, which can selectively form the covalent bond with the higher-order structures of nucleic acids were reported (5–13), the reactive moiety with a high selectivity and efficiency to the target is quite limited.
In our previous study, we developed 2-amino-6-vinylpurine (AVP) as a reactive moiety for the selective alkylation of the target thymine base (14). The AVP moiety was conjugated with Hoechst 33258 (Figure 1A), and the selective alkylation of the thymine base opposite an amino purine (AP) site was achieved by a proximity effect. In order to expand this alkylation ability, we investigated the suitable DNA or RNA binding molecule which is applicable for the vinyl alkylation chemistry. The triaminotriazine (TAT)–acridine conjugate 1 developed by Zimmerman is a T–T or U–U mismatch binding molecule which stabilizes the complex by the intercalation of the acridine and hydrogen bonds (15). In our study, we designed the vinyldiaminotriazine (VDAT)–acridine conjugate 2 for the selective alkylation of the T–T mismatched DNA or U–U mismatched RNA. Based on the paper reported by Zimmerman (16), we expected that the VDAT–acridine conjugate would recognize the T–T or U–U by the stable complex formation with hydrogen bonds as shown in Figure 1 and selectively react with the T or U base by the proximity effect.
Figure 1.

Molecular design of the small molecule for the selective alkylation of the T–T mismatched DNA. (A) The structure of Hoechst–AVP conjugate. (B) The structure of TAT–acridine conjugate 1 reported by Zimmerman. (C) The structure of VDAT–acridine conjugate 2 designed in this study. (D) The conceptual scheme of the T–T mismatched DNA alkylation. (E) The predicted complex between the ligand and T–T mismatched DNA. (F) The predicted complex between the ligand and U–U mismatched RNA.
Generally, the T–T mismatch in DNA and U–U mismatch in RNA can be formed in the abnormal expanded trinucleotide repeat sequence (1). The myotonic dystrophy type 1 (DM1) is one of the trinucleotide repeat disorders caused by an abnormal expansion of the CTG trinucleotide repeats in the 3′-UTR of the DMPK gene. While the normal expansion of the CTG is 5–37 repeats, the abnormal one reaches 50–2000 repeats. This repeat DNA produces the toxic CUG repeat RNA which forms a complex with the alternative-splicing regulator muscleblind-like protein (MBNL), leading to splicing defects and disease symptoms. Thus, abnormal extended CTG or CUG repeats are considered to be one of the important targets for DM1 and several promising compounds have been developed (17,18). For example, TAT derivatives, which can bind the CTG or CUG repeats, strongly inhibit the transcription or formation of the complex with MBNL (19). Disney’s group developed bis-Hoechst derivatives conjugated with chlorambucil for the covalent bond formation, bleomycin for the RNA cleavage or azide-alkyne moieties for in situ probe synthesis (7). Berglund’s group found that actinomycin D (20) and the diamidine derivatives (21) can reduce the abnormal extended CUG RNA levels. Nakatani’s group developed new small molecules targeting the CUG repeats by the rational molecular design (22). In this study, we aimed to develop a reactive small molecule with the ability of the T–T or U–U mismatch selective alkylation for DM1 research.
MATERIALS AND METHODS
The general chemicals were purchased from Wako Pure Chemical, Aldrich or the Tokyo Chemical Institute. The target oligo DNAs and RNAs were purchased from JBioS (Japan). The oligo sequences used in this study are shown in Table 1. The 1H NMR spectra (400 MHz) were recorded by a Bruker 400 spectrometer. The 1H NMR spectra (600 MHz) and 13C NMR spectra (150 MHz) were recorded by a Bruker AVANCE III 600 spectrometer. The high resolution electrospray mass analysis was performed by a Bruker MicrOTOFQ II. The high performance liquid chromatography (HPLC) purification was performed by a JASCO HPLC System (PU-2089Plus, UV-2075Plus, FP-2015Plus and CO-2065Plus) using a reverse-phase C18 column (COSMOSIL 5C18-AR-II, Nacalai tesque, 4.6 × 250 or 10 × 250 mm for ligand purification and CAPCELL PAK C18 MGII, Shiseido, 4.6 × 250 or 10 × 250 mm for oligo DNA purification). MALDI-TOF MS measurements were performed by a Bruker Autoflex speed instrument using a 3-hydroxypicolinic acid/diammonium hydrogen citrate matrix. The gel imaging and quantification were performed by a FLA-5100 (Fujifilm Co.).
Table 1. ODN and ORN sequences used in this study.
| Entry | Sequences (5′-3′) |
|---|---|
| ODN1 | Alexa647-CTGGCXGCGC (X = dA, dG, dC, dT or dU) |
| ODN2 | FAM-GCGCYGCCAG (Y = dA, dG, dC, dT or dU) |
| ODN3 | FAM-CTGGCTGCGC |
| ODN4 | GCGCYGCCAG (Y = dA, dG, dC or dT) |
| ODN5 | FAM-CTGGCT*GCGC |
| ODN6 | GCGCT*GCCAG |
| ODN7 | CTGGCXGCGC (X = dA, dG, dC, dT or AP) |
| ODN8 | FAM-GC(CTG)12GC |
| ODN9 | FAM-TAATACGACTCACTATAGGG |
| ODN10 | (CTG)20CCCTATAGTGAGTCGTATTA |
| ODN11 | (CAG)20CCCTATAGTGAGTCGTATTA |
| ODN12 | TAATACGACTCACTATAGGG(CAG)20 |
| ODN13 | TAATACGACTCACTATAGGG(CTG)20 |
| ORN1 | Alexa647-CUGGCXGCGC (X = rA, rG, rC, rU or rT) |
| ORN2 | FAM-GCGCYGCCAG (Y = rA, rG, rC, rU or rT) |
| ORN3 | CUGGCXGCGC (X = rA, rG, rC or rU) |
Synthesis of tert-butyl (4-aminobutyl)carbamate (4)
To a vigorously stirred solution of butan-1,4-diamine (5.00 g, 56.7 mmol) in CHCl3 (40 ml) was slowly added a solution of Boc2O (2.48 g, 11.3 mmol) in CHCl3 (30 ml). The white suspension was stirred at room temperature overnight. The solvent was removed under reduced pressure, and 10% aqueous Na2CO3 (100 ml) was added. The mixture was then extracted with CH2Cl2 (80 ml × 3). The organic layer was dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by column chromatography (CHCl3: MeOH: NH4OH = 89: 10: 1) to give 4 as a pale yellow oil (2.00 g, 94%). 1H NMR (600 MHz, CDCl3) δ (ppm) 1.19 (brs, 2H), 1.44 (s, 9H), 1.46–1.55 (m, 4H), 2.71 (t, J = 7.2 Hz, 2H), 3.12–3.14 (m, 2H), 4.74 (brs, 1H). 13C NMR (150 MHz, CDCl3) δ (ppm) 27.5, 28.4, 30.9, 40.4, 41.9, 79.0, 156.0. ESI–HRMS (m/z) calcd for C9H20N2O2 [M+H]+ 189.1598, found 189.1597.
Synthesis of tert-butyl (4-((4-amino-6-(2-(methylthio)ethyl)-1,3,5-triazin-2-yl)amino)butyl)carbamate (5)
To a solution of 4, 6-dichloro-1, 3, 5-triazin-2-amine 3 (0.500 g, 3.03 mmol) and K2CO3 (0.628 g, 4.55 mmol) in 1,4-dioxane (5.0 ml) was added a solution of 4 (0.616 g, 3.27 mmol) in 1,4-dioxane (12.0 ml). The mixture was stirred overnight at 50°C. To the mixture were added H2O (5.0 ml), 1,4-dioxane (3.0 ml), Pd(PPh3)4 (0.350 g, 0.303 mmol), K2CO3 (0.209 g, 1.52 mmol) and the vinylboronic anhydride pyridine complex (1.09 g, 4.55 mmol). The mixture was refluxed for 140 min, then cooled to room temperature. To the mixture, NaSCH3 (0.425 g, 6.06 mmol) was added and the mixture was stirred overnight at room temperature. The solvent was removed under reduced pressure, and the residue was dissolved in EtOAc (100 ml). The solution was washed with water (50 ml × 2) and brine (50 ml × 3). The organic phase was dried over Na2SO4, and concentrated under reduced pressure. The crude was purified by column chromatography (CH2Cl2: ethyl acetate = 3: 2→ ethyl acetate only) to give 5 as a yellow foam (0.612 g, 57%). 1H NMR (400 MHz, CDCl3) δ (ppm) 1.45 (s, 9H), 1.53–1.62 (m, 4H), 2.14 (s, 3H), 2.76 (t, J = 7.6 Hz, 2H), 2.87 (t, J = 7.6 Hz, 2H), 3.20 (m, 2H), 3.39 (m, 2H), 4.87–5.17 (m, 4H). 13C NMR (150 MHz, CDCl3) δ (ppm) 15.6, 26.5, 27.1, 28.5, 31.5, 38.4, 39.9, 40.2, 79.4, 156.0, 166.1, 167.0, 176.8. ESI-HRMS (m/z) calcd for C15H28N6O2S [M+H]+ 357.2067, found 357.2084.
Synthesis of N2-(4-((6-chloro-2-methoxyacridin-9-yl)amino)butyl)-6-(2-(methylthio)ethyl)-1,3,5-triazine-2,4-diamine (6)
To a solution of 5 (0.050 g, 0.140 mmol) and triethylsilane (0.14 ml) in CH2Cl2 (0.20 ml) was dropwise added TFA (0.60 ml) and the mixture was stirred at room temperature for 1 h. The reaction mixture was diluted with toluene (3.0 ml) and concentrated under reduced pressure. The residue was co-evaporated with toluene (3.0 ml) twice. This crude was used without further purification. The mixture of the deprotected triazine unit (0.140 mmol), phenol (1.0 g) and 6-chloro-2-methoxy-9-phenoxyacridine (0.071 g, 0.21 mmol) was stirred at 90°C for 5 h. The reaction mixture was diluted with Et2O (100 ml) and washed with 3 M aqueous NaOH (100 ml × 3) and brine (100 ml × 2). The organic layer was concentrated under reduced pressure. The crude product was purified by column chromatography (CH2Cl2: CH3OH: NH4OH = 97: 2: 1) to give 6 as a yellow solid (0.045 g, 58%). Compound 6 was co-evaporated with MeCN containing 0.1% TFA. The TFA salt was dissolved in H2O containing 0.1% TFA and purified by RP-HPLC. The concentration of the pure compound 6 TFA salt was quantified by nuclear magnetic resonance (NMR) using maleic acid as an internal standard. 1H NMR (600 MHz, CDCl3) δ (ppm) 1.71 (quint, J = 7.2 Hz, 2H), 1.81 (quint, J = 7.2 Hz, 2H), 2.13 (s, 3H), 2.76–2.77 (m, 2H), 2.86 (t, J = 7.2 Hz, 2H), 3.39–3.47 (m, 2H), 3.73–3.75 (m, 2H), 3.95 (s, 3H), 4.70–5.17 (m, 4H), 7.20 (d, J = 2.4 Hz, 1H), 7.31 (dd, J = 9.0, 2.4 Hz, 1H), 7.43 (dd, J = 9.0, 2.4 Hz, 1H), 7.99 (d, J = 9.0 Hz, 1H), 8.00 (d, 9.0 Hz, 1H), 8.08 (s, 1H). 13C NMR (150 MHz, CDCl3) δ (ppm) 15.6, 27.0, 29.0, 31.4, 38.4, 40.1, 50.4, 55.6, 99.1, 116.2, 118.3, 123.9, 124.5, 124.8, 128.5, 131.7, 134.8, 146.9, 148.4, 149.5, 156.2, 166.2, 167.0, 177.0. ESI–HRMS (m/z) calcd for C24H28ClN7OS [M+H]+ 498.1837, found 498.1844.
Synthesis of VDAT–acridine conjugate (2)
To a solution of 6 (25 nmol) in dimethyl sulfoxide (DMSO) (1.0 μl) was added a solution of magnesium monoperphtalate (MMPP) (125 nmol) in water (8.0 μl) and the mixture was stored at room temperature for 45 min. 1.0 M aqueous NaOH (5.0 μl) and DMSO (4.0 μl) were then added and stored at room temperature for 45 min. 1.0 M acetic acid was added to neutralize the solution, and water was added to prepare a 1.0 mM solution of 2. The solution was used for the alkylations without further purification. For 1H-NMR and ESI–HRMS measurements, 2 was purified by RP-HPLC. 1H NMR (600 MHz, CD3OD) δ (ppm) 1.80 (quint, J = 7.2 Hz, 2H), 2.06 (quint, J = 7.2 Hz, 2H), 3.46 (t, J = 7.2 Hz, 2H), 4.00 (s, 3H), 4.21 (t, J = 7.2 Hz, 2H), 5.74 (d, J = 8.4 Hz, 0.25H), 5.90 (d, J = 8.4 Hz, 0.75H), 6.33 (dd, J = 16.8, 8.4 Hz, 0.25H), 6.40 (dd, J = 16.8, 8.4 Hz, 0.75H), 6.52 (d, J = 16.8 Hz, 0.25H), 6.58 (d, J = 16.8 Hz, 0.75H), 7.49 (d, J = 9.0 Hz, 1H), 7.67 (d, J = 9.0 Hz, 1H), 7.75 (d, J = 9.0 Hz, 1H), 7.78 (s, 1H), 7.81 (s, 1H), 8.46 (d, J = 9.0 Hz, 1H). ESI–HRMS (m/z) calcd for C23H28ClN7O [M+H]+ 450.1804, found 450.1813.
Alkylation to DNA
A solution (50 μl) of ODN1 (10 μM) and ODN2 (10 μM) in MES buffer (100 mM) containing NaCl (200 mM) was heated at 90°C for 5 min, then gradually cooled to room temperature. To the ODN solution (5.0 μl) were added water (4.0 μl) and a 1.0 mM solution of 2 (1.0 μl), and the mixture was incubated at 37°C. The aliquots (1.0 μl each) were removed from the reaction mixture at various time points, quenched by a loading buffer (80% formamide, 10 mM ethylenediaminetetraacetic acid (EDTA), 4 μl), then cooled to 0°C. Electrophoresis was performed on a 16% denaturing polyacrylamide gel containing 20% formamide with 1 × TBE and 6.0 M urea at 450 V for 90 min.
Alkylation to RNA
The reaction was performed with ORN1 (5 μM) and ORN2 (5 μM) in the same manner as the alkylation to DNA.
Enzymatic hydrolysis of the alkylated ODN5 and ODN6
The alkylated ODN5 and ODN6 were synthesized with ODN3 (600 nmol), ODN4 (Y = dT) (600 nmol) and VDAT–acridine conjugate 2 (3.0 μmol) in the same manner as the alkylation conditions with ODN1 (X = dT) and ODN2 (Y = dT) and purified by RP-HPLC (CAPCELL PAL MG-II, Shiseido, 4.6 × 250 mm, solvent A: 50 mM TEAA, solvent B: MeCN, linear gradient: B 10–30%/ 20 min, flow rate: 1.0 ml/min, temperature: 40°C). A solution (100 μl) of the alkylated ODN5 or ODN6 (100 μM), alkaline phosphatase (0.03 U/μl, Takara Bio Inc.) and phosphodiesterase I (0.03 U/μl, Worthington Biochemical Corp.) in alkaline phosphatase buffer was incubated at 37°C for 1 h. The reaction mixture was heated at 90°C for 3 min, cooled to 0°C for 3 min and analyzed by RP-HPLC (CAPCELL PAL MG-II, Shiseido, 4.6 × 250 mm, solvent A: 50 mM TEAA for analysis or HCOONH4 for purification, solvent B: MeCN, linear gradient: B 5–40%/ 30 min, flow rate: 1.0 ml/min).
Determination of the alkylation position of dT* by NMR
The alkylated thymidine dT* obtained by enzymatic hydrolysis of the alkylated ODN5 and ODN6 was dissolved in DMSO-d6 containing 10 μM 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS), the final concentration being 400 μM. DSS was used as an internal chemical shift reference. The COSY, edited-HSQC and HMBC spectra were recorded at 37°C using Bruker BioSpin DRX 600 and AVANCE III HD 600 spectrometers equipped with a cryogenic probe and a Z-gradient.
Melting temperature (Tm) measurement
A mixture of the DNA–DNA or DNA–RNA duplex (2.0 μM) in MES buffer (50 mM, pH 7.0) containing NaCl (100 mM) was transferred to a microquartz cell with a 1-cm path length. The melting temperature was then measured under UV absorption at 260 nm from 25 to 85°C at the rate of 1°C/min. The measurements were carried out three times per each sample and averaged for obtaining the final value. The melting temperature measurement was performed by a DU-800 (Beckman-coulter) equipped with a temperature controller.
Fluorescence measurement with 2-aminopurine-containing ODN
A mixture of the duplex (5.0 μM) in MES buffer (50 mM, pH 7.0) containing NaCl (100 mM) was transferred to a quartz cell with a 3-mm path length. The emission spectra were obtained with an excitation wavelength at 310 nm. The fluorescence measurement was performed by a FP-6500 (JASCO Corporation).
Alkylation to CTG repeats DNA
A solution (150 μl) of (CTG)12 ODN8 (5.0 μM) and VDAT–acridine conjugate 2 (100 μM) in MES buffer (50 mM, pH 7.0) containing NaCl (100 mM) and 2% DMSO was incubated at 37°C. The aliquots (20 μl each) were removed from the reaction mixture at various time points and purified by RP-HPLC to remove the ligand 2. The conversion was analyzed by a MALDI-TOF/MS measurement (linear negative mode) because the non-alkylated and alkylated ODN8 were not separated in the HPLC profile. The percentage of the residual (CTG)12 was quantified from the ratio of the peak area. The percentages were calculated from three separate experiments. The observed first-order reaction rate constant (kobs) of the alkylation reaction to (CTG)12 ODN8 was graphically obtained from the first-order kinetic plot (Equation 1).
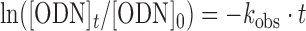 |
(1) |
Primer extension reaction using CTG repeats DNA
A solution (20 μl) of (CTG)20 ODN10 (5.0 μM) and VDAT–acridine conjugate 2 (100 μM) in MES buffer (50 mM, pH 7.0) containing NaCl (100 mM) and 2% DMSO was incubated at 37°C for 6 or 24 h. The mixture was used as the alkylated template ODN10. A solution (30 μl) of the primer ODN9 (0.17 μM), the alkylated template ODN10 (0.5 μM) and internal standard ODN2 (Y = dT) (0.07 μM) in NE buffer 2 (50 mM NaCl, 10 mM Tris–HCl, 10 mM MgCl2, 1 mM DTT, pH 7.9, New England Biolabs) was heated at 93°C and gradually cooled to room temperature for the annealing. To the solution were added dNTP (2.4 μl, final 0.2 mM) and the Klenow Fragment (exo−) (0.6 μl, final 0.1 U/μl, New England Biolabs), then the mixture was incubated at 37°C for 10 min. The reaction mixture was quenched by a loading buffer (80% formamide, 10 mM EDTA, 30 μl), then cooled to 0°C. Electrophoresis was performed on a 14% denaturing polyacrylamide gel containing 30% formamide with 1 × TBE and 5.3 M urea at 300 V and 40°C for 25 min.
Transcription reaction using CTG repeats DNA
A solution (7.8 μl) of ODN12 and the alkylated template ODN10 in buffer (40 mM Tris–HCl, 8 mM MgCl2, 2 mM spermidine, 5 mM DTT, pH 8.0, Takara Bio Inc.) was heated at 93°C and gradually cooled to room temperature for the annealing. A solution (10 μl) of the annealed duplex (0.05 μM), T7 RNA polymerase (0.01 U/μl, Takara Bio, Inc.) and NTP (2 mM) in buffer (40 mM Tris–HCl, 8 mM MgCl2, 2 mM spermidine, 5 mM DTT, pH 8.0, Takara Bio Inc.) was incubated at 37°C for 1 h, then to the mixture was added DNase I (0.2 μl, final 0.1 U/μl, Takara Bio Inc.). The mixture was incubated at 37°C for 20 min, quenched by a loading buffer (80% formamide, 10 mM EDTA, 10 μl), then cooled to 0°C. Electrophoresis was performed on a 20% denaturing polyacrylamide gel with 1 × TBE and 7.5 M urea at 300 V and 15°C for 3.5 h. The gel was stained with SYBR Green II for the RNA product detection.
RESULTS AND DISCUSSION
Alkylation with VDAT–acridine conjugate
The designed VDAT–acridine conjugate 2 was synthesized via the SMe precursor 6. First, the triazine unit 5 was synthesized from 4, 6-dichloro-1, 3, 5-triazin-2-amine 3 by the substitution reaction with the amine linker 4, vinylation and the addition of methanethiol (Scheme 1). After deprotection of the Boc group, the triazine unit was conjugated with 6-chloro-2-methoxyacridine to give the SMe precursor 6. Compound 6 was oxidized with MMPP and the resulting sulfoxide 7 was converted to the vinyl compound 2 under alkaline conditions (Scheme 2). The conversion from 6 to 2 was confirmed by HPLC and ESI–HRMS (Supplementary Figure S1), and the product was used for the alkylation.
Scheme 1.

Synthesis of VDAT–acridine conjugate precursor.
Scheme 2.

Synthesis of VDAT–acridine conjugate.
Alkylation with the VDAT–acridine conjugate was performed using the ODN1 and ODN2 with the different fluorescent labeling by Alexa647 and 6-FAM, respectively (Figure 2A). The reactions were evaluated by detecting the different fluorescent labeling on a denaturing polyacrylamide gel (Figure 2B and Supplementary Figure S2). A clear band shift was observed only when the T–T mismatched DNA was used. The alkylation yields were calculated by combining the yield of each strand. The reaction yield to the T–T mismatch reached 93% in 120 h and a slight reaction was observed to the C–C mismatch (Figure 2C and D). The reaction yields to other mismatches, full matches and single strands were quite low, indicating that the selectivity of the VDAT–acridine conjugate was significantly high. Next, the reactions to the RNA target were performed in the same manner as the DNA target. A slight alkylation was observed to the U–U mismatched RNA and the yield reached 20% in 120 h (Figure 3). The reaction efficiency to the U–U mismatched RNA was much lower than that to the T–T mismatched DNA. Clear band shifts were not observed for the other base pairs (Supplementary Figure S3). To check the difference in the nucleophilicity between T and U, the alkylation was carried out using ODN1(X = dU)-ODN2(Y = dU) for the DNA duplex or ORN1(X = rT)-ORN2(Y = rT) for the RNA duplex (Supplementary Figure S4). While the reaction with the dU–dU mismatched DNA proceeded, no reaction occurred with the rT–rT mismatched RNA. Thus, the large difference in the reaction efficiency will depend on the structure difference between the DNA and RNA duplex rather than the difference in the nucleophilicity. According to the results using the TAT–acridine conjugate 1, the dissociation constant (Kd) to T–T mismatch (0.39 μM) was lower than that to the U–U mismatch (2.1 μM) (15). In addition, the dimethylation of the amino group on the triazine significantly decreased the U–U mismatched RNA binding affinity (16). These results suggest that the VDAT–acridine conjugate 2 would bind to the T–T mismatched DNA more tightly than the U–U mismatched RNA probably due to the difference in the duplex conformation. Thus, the reaction efficiency of the U–U mismatch would be much lower than that to the T–T mismatch.
Figure 2.

Alkylation to DNA using VDAT–acridine conjugate 2. The reaction was carried out with the duplex DNA (5 μM) and VDAT–acridine conjugate 2 (100 μM) in MES buffer (50 mM, pH 7.0) containing NaCl (100 mM) and 2% DMSO at 37°C. (A) The sequence of the target duplex DNA. (B) Gel image of the alkylation. The electrophoresis was performed on a 16% denaturing polyacrylamide gel containing 20% formamide. The red or green bases indicate X in ODN1 or Y in ODN2, respectively. (C) Time course of the reaction yields. The alkylation yields were calculated by combining the yield of each strand. (D) The reaction yields after 48 h to the mismatched duplex DNA.
Figure 3.

Gel image of the alkylation to RNA using VDAT–acridine conjugate 2. The reaction was carried out with the duplex RNA (ORN1–ORN2) (5 μM) and VDAT–acridine conjugate 2 (100 μM) in MES buffer (50 mM, pH 7.0) containing NaCl (100 mM) and 2% DMSO at 37°C. The red or green bases indicate X in ORN1 or Y in ORN2, respectively.
Determination of the alkylated thymidine structure
To determine the alkylated nucleoside structure, the alkylated DNA was synthesized on a large scale. The reaction was carried out using the FAM-labeled ODN3 and non-labeled ODN4(Y = dT). FAM was labeled to easily discriminate another strand by HPLC. After 5 days of reaction, two new peaks, ODN5 and ODN6, were observed in the HPLC profile (Supplementary Figure S5). MALDI-TOF/MS measurement of the purified products showed that the ODNs were the alkylated ODN with the VDAT–acridine conjugate.
The alkylated ODNs were next enzymatically hydrolyzed with alkaline phosphatase and phosphodiesterase I (Figure 4A). In addition to the native nucleosides and FAM, a new peak dT* was observed in the HPLC profiles. The peaks of dT*1 and dT*2 were collected and the products were analyzed by ESI-MS. Both values of dT*1 (692.7204) and dT*2 (692.7201) corresponded to the calculated value of the thymidine alkylated with the VDAT–acridine conjugate (692.7206). Additionally, the co-injection of dT*1 and dT*2 showed only one peak (Figure 4B). These results indicated that the alkylation proceeded at the thymidine base and the structure of dT*1 and dT*2 would be the same.
Figure 4.
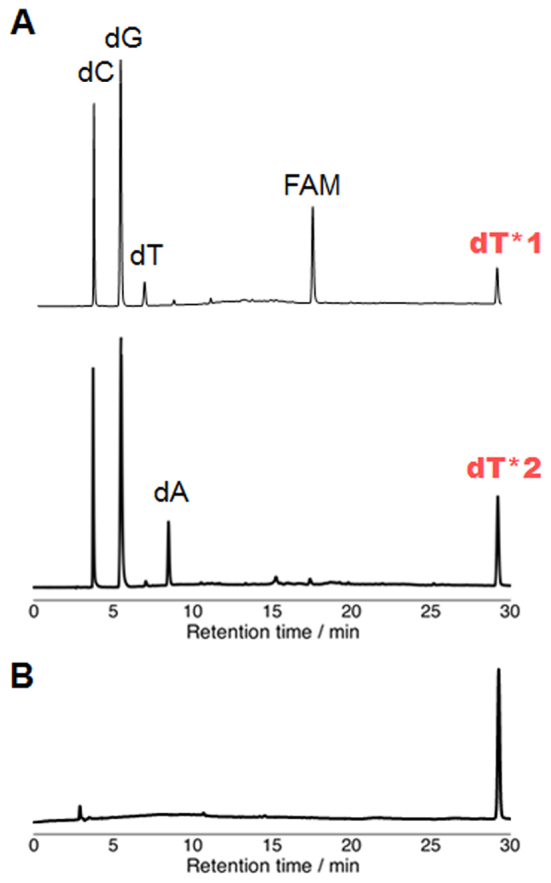
HPLC profiles of the enzymatic hydrolysis products. (A) HPLC analysis after enzymatic hydrolysis of ODN5 (top) and ODN6 (bottom). The enzymatic hydrolysis was performed with ODN5 or ODN6 (100 μM), alkaline phosphatase (0.03 U/μl) and phosphodiesterase I (0.03 U/μl) in alkaline phosphatase buffer at 37°C for 1 h. (B) Co-injection of dT*1 and dT*2.
According to the papers by Singer, the O2 and O4-alkylated dTs are hydrolyzed under acidic conditions (23), in contrast, the N3-alkylated dT is stable under the same conditions (24). The alkylated thymidine dT*1 and 2 were combined and the mixture was treated with 0.1 M HCl at 50°C for 5 min and at 70°C for 60 min. An HPLC analysis showed that dT* was stable (Supplementary Figure S6), suggesting that dT* would be the N3 alkylated product.
To clearly determine the alkylation position, the COSY, edited-HSQC and HMBC NMR spectra of dT* were recorded (Figure 5 and Supplementary Figure S7). The proton resonances of dT*, including Ha, were assigned on the basis of the COSY spectrum (Supplementary Figure S7). The assignment of Ha, which is one of ethylene protons between the thymine base and triazine, was further confirmed by the edited-HSQC spectrum, the sign of the corresponding correlation peak being opposite to the sign of the correlation peaks of CHs. Ha gave the HMBC correlation peaks to two carbon resonances around 160 ppm, i.e. C2 and C4 (Figure 5). If the position of the alkylation of dT* is either O2 or O4, these HMBC correlation peaks cannot be observed. Thus, the position of the alkylation of dT* was concluded to be the N3 position.
Figure 5.

Elucidation of the alkylated position of dT* by NMR. (A) Chemical structure of dT* alkylated at the N3 position. (B) The edited-HSQC spectrum of dT* is overlaid on its HMBC spectrum for two portions. Positive and negative peaks of the edited-HSQC spectrum are indicated by blue and green, respectively, while the peaks of the HMBC spectrum are indicated by red.
Thermal stability of the alkylated duplex and base flipping structure
To investigate the property of the alkylated ODN6, the Tm values of the dT*-containing DNA–DNA and DNA–RNA duplex were investigated (Figure 6). For the native duplex, ODN7 (X = dA, dG, dC or dT)–ODN4(Y = dT) or ORN3(X = rA, rG, rC or rU)–ODN4(Y = dT), only the A-T base pair showed a high Tm value. Interestingly, for the alkylated duplex, ODN7(X = dA, dG, dC or dT)–ODN6 or ORN3(X = rA, rG, rC or rU)–ODN6, all the Tm values were comparable to the native full match duplex. We hypothesized that the alkylated ODN6 would stabilize the duplex by the intercalation of the acridine part and flipping out the complementary base. The base flipping structure model of the alkylated duplex DNA is shown in Figure 7. To confirm the base flipping, the fluorescence emission assay using 2-AP) was performed (Figure 8) (25,26). AP is a purine base with a strong fluorescence and used as a fluorescent probe. Since the AP fluorescence is highly quenched in a duplex due to the stacking interactions, we can confirm whether the AP base is flipped out of the DNA helix by checking the fluorescence increase. The fluorescence of ODN6(dT*)–ODN7(X = AP) was significantly high, in contrast, ODN4(Y = dT)-ODN7(X = AP) or ODN6(dT*)–ODN7(X = dA) did not show any fluorescence, clearly indicating that dT* can induce the flipping of the AP base.
Figure 6.

T m values of dT*-containing DNA–DNA or DNA–RNA duplex. (A) ODN7–ODN4 (Y = dT) (left blue bar) or −ODN6 (right red bar) duplex. (B) ORN3–ODN4 (Y = dT) (left blue bar) or −ODN6 (right red bar) duplex. The Tm values were measured using duplex (2.0 μM) in MES buffer (20 mM, pH 7.0) containing NaCl (100 mM).
Figure 7.
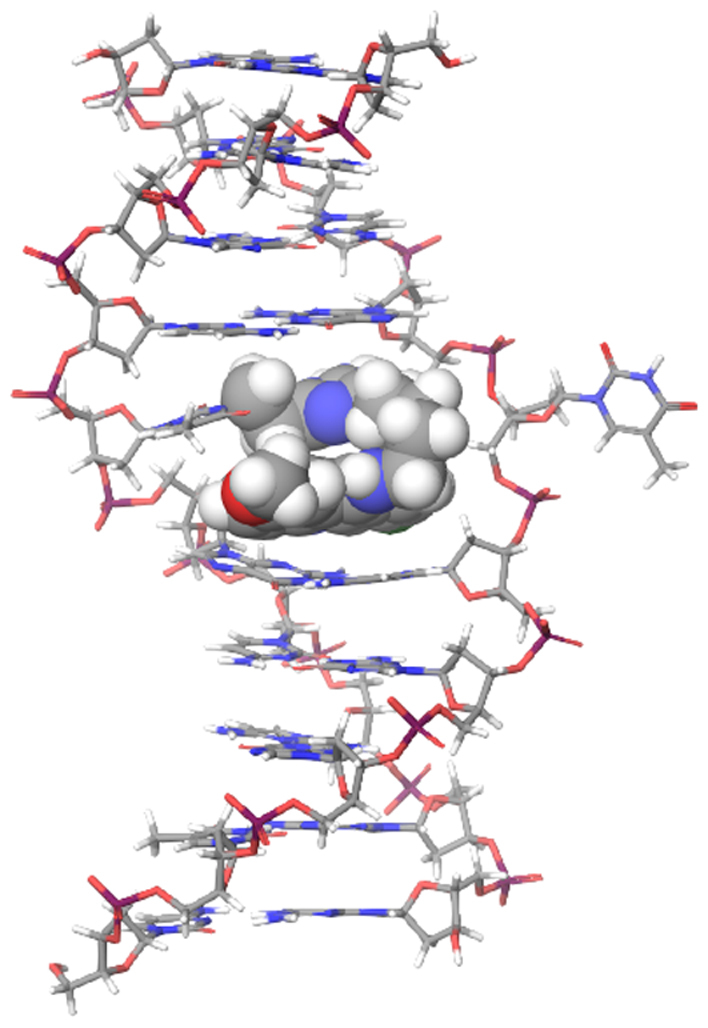
The base flipping structure of the alkylated duplex DNA. The model was calculated using the MacroModel software with the OPLS3 force field. The alkylating ligand is shown as the CPK model.
Figure 8.

Fluorescence emission spectra excited at 310 nm to confirm the base flipping. All measurements were carried out using the duplex (5.0 μM) in MES buffer (50 mM, pH 7.0) containing 100 mM NaCl at room temperature. (A) ODN6 (dT*)–ODN7 (X = AP) duplex, (B) ODN4 (Y = dT)–ODN7 (X = AP) duplex, (C) ODN6 (dT*)–ODN7 (X = dA) duplex.
Based on the results for the structural determination with the adduct and the property of the alkylated ODN, we propose the mechanism for the alkylation to the T–T mismatched duplex (Figure 9). Generally, the T–T mismatched DNA forms a wobble base pair (27). The VDAT–acridine conjugate will bind to the mismatched base pair and form the initial complex with four hydrogen bonds (16). In the equilibrium, one thymidine base might gradually flip out and enol form of the thymidine base would slowly react with VDAT to give the N3-adduct. The O2-adduct was not observed probably due to the poor nucleophilicity.
Figure 9.

The proposed alkylation mechanism to T–T mismatched duplex.
Alkylation to CTG repeats model DNA
It is considered that the expanded (CTG)·(CAG) repeats sequence forms T–T mismatches by forming slipped DNA structures (1,28,29). We used (CTG)12 ODN8 as the model for the alkylation to the CTG repeats DNA (Figure 10A). The reaction was observed by MALDI-TOF/MS measurements because ODN8 was too large to discriminate the alkylated products from the non-reacted ODN8 on a denaturing polyacrylamide gel electrophoresis and by an HPLC analysis. In the MS spectrum after 6 h, a clear 1-adduct peak was observed (Figure 10B). After 24 h, the 2-adduct and 3-adduct peaks were observed in addition to the 1-adduct peak. The percentage of the residual (CTG)12 ODN8 was calculated from the peak area of each peak, and the time course is shown in Figure 10C. The observed reaction rate (kobs) of (CTG)12 ODN8 was calculated by treating the reaction as a pseudo first-order reaction (Figure 10D). The kobs of (CTG)12 (26.4 × 10−6 s−1) was six times higher than that of (CTG)2 (ODN1(X = T)-ODN2(Y = T) duplex) (4.2 × 10−6 s−1), suggesting that the efficiency of one alkylation would increase in proportion to the number of the T–T mismatches. This means that the alkylation efficiency to the abnormal expansion repeats target can be expected to increase in proportion to the number of CTGs. For DM1, the abnormal expansion of the CTG is 50–2000 repeats. Given that T–T mismatches are formed in the expanded (CTG)·(CAG) repeats sequence as reported (1,28,29), the efficient and selective alkylation to the long CTG repeats sequence might be possible using the VDAT–acridine conjugate 2.
Figure 10.

Alkylation to (CTG)12 ODN8 using VDAT–acridine conjugate 2. The reaction was carried out with ODN8 (5 μM) and VDAT–acridine conjugate 2 (100 μM) in MES buffer (50 mM, pH 7.0) containing NaCl (100 mM) and 2% DMSO at 37°C. (A) The sequence of (CTG)12 ODN8. (B) MALDI-TOF/MS spectra after running the reaction for 1, 6 and 24 h. (C) Time course of the residual (CTG)12. (D) Calculation of the observed first-order reaction rate constant (kobs) of the alkylation reaction to (CTG)12 ODN8 or (CTG)2 ODN1(X = T)–ODN2(Y = T) duplex.
The alkylation of DNA should strongly inhibit the replication and transcription. We initially investigated the alkylation selectivity and the effect of the alkylation on the DNA polymerase reaction (Figure 11). The alkylation was carried out with (CTG)20 ODN10 and the VDAT–acridine conjugate 2 at 37°C for 6 or 24 h. (CAG)20 ODN11 was used as the control to check the alkylation selectivity. The alkylated ODN was annealed with the primer ODN9 and the primer extension reactions were performed with the Klenow Fragment (exo−) as a DNA polymerase at 37°C for 10 min. When the alkylated (CTG)20 ODN10 was used as the template, the ratio of the full length production of the primer extension reaction drastically decreased with the alkylation time. The short length products were observed like a ladder instead of the full length product. The ratio did not change when (CAG)20 ODN11 was used, indicating that the alkylation selectivity was significantly high. In addition, the non-reactive TAT–acridine conjugate 1 did not inhibit the primer extension reaction. These results suggest that the VDAT–acridine conjugate 2 would be able to selectively and strongly inhibit the replication reaction.
Figure 11.

The effect of the alkylation on the primer extension reaction. (A) Gel image of primer extension reaction. The alkylation was initially carried out with (CTG)20 ODN10 or (CAG)20 ODN11 (5 μM) and VDAT–acridine conjugate 2 (100 μM) in MES buffer (50 mM, pH 7.0) containing NaCl (100 mM) and 2% DMSO at 37°C for 6 h (lane 2 or 6) or 24 h (lane 3 or 7). For TAT–acridine conjugate 1, the mixture was incubated for 24 h (lane 4 or 8). The ODNs were then annealed with the primer ODN9. The primer extension reactions were performed with the template ODN10 or ODN11 (0.5 μM) and primer ODN9 (0.17 μM), Klenow Fragment (exo−) (0.1 U/μl) and 0.2 mM dNTP in buffer (pH 7.9) at 37°C for 10 min. (B) The ratio of full length production.
Next, we investigated the effect of the alkylation on the transcription reaction (Figure 12). The alkylated ODN was annealed with the sense ODN and the transcription reactions were performed with the T7 RNA polymerase at 37°C for 1 h. When the alkylated (CTG)20 ODN10 was used as the template, the ratio of the full length production of the transcription reaction drastically decreased, the same as the DNA polymerase reaction. The ratio did not significantly change when (CAG)20 ODN11 was used. The TAT–acridine conjugate 1 did not inhibit the transcription reaction. Although this alkylation was performed with single strand (CTG)20 ODN10 not (CTG)·(CAG) repeats duplex, these results suggest that the alkylated thymidine would strongly inhibit the transcription reaction.
Figure 12.

The effect of the alkylation on the transcription reaction. (A) Gel image of full length product in transcription reaction. The alkylation was initially carried out with (CTG)20 ODN10 or (CAG)20 ODN11 (5 μM) and VDAT–acridine conjugate 2 (100 μM) in MES buffer (50 mM, pH 7.0) containing NaCl (100 mM) and 2% DMSO at 37°C for 6 h (lane 2 or 6) or 24 h (lane 3 or 7). For TAT–acridine conjugate 1, the mixture was incubated for 24 h (lane 4 or 8). The ODNs were annealed with the sense ODN12 or ODN13. The transcription reactions were performed with the template ODN10 or ODN11 (0.05 μM), sense ODN12 or ODN13 (0.05 μM), T7 RNA polymerase (0.01 U/μl) and 2 mM NTP in buffer (pH 8.0) at 37°C for 1 h. (B) The ratio of the full length production.
CONCLUSION
We have described the T–T mismatch selective alkylation with the VDAT–acridine conjugate 2. The selectivity was significantly high while the yield to other mismatches was quite low even to the U–U mismatched RNA. From the NMR study of the alkylated thymidine, the alkylation selectively proceeded at the N3 position of dT. Interestingly, the alkylated thymidine induced the base flipping of the complimentary base by the stabilization of the duplex with the acridine part and the steric hindrance of the triazine part alkylated at the N3 position of dT. This information would be useful to develop the base flipping induced ODN, such as the phenylurea derivative of deoxyadenosine developed by Sugimoto's group (26,30). Importantly, the observed reaction rate for one alkylation increased in proportion to the number of T–T mismatches. This indicates that the efficiency for one alkylation would increase to the abnormal extended CTG repeats DNA. In addition, the alkylation of the CTG repeats DNA led to strongly inhibiting the primer extension reaction and transcription which the TAT–acridine conjugate 1 could not inhibit. The VDAT–acridine conjugate 2 would be a new biochemical tool for a CTG repeats study and may give a new strategy for the molecular therapy of DM1.
From the results of the T–T mismatch alkylation, we consider that a reactive vinyl group modified on a DNA or RNA binding molecule is a promising and broad-applicable alkylation moiety for nucleic acid chemistry. The alkylation and modification of the various higher-order structures of nucleic acid using vinyl-modified small molecules are ongoing in our laboratory.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Japan Society for the Promotion of Science (JSPS), Grant-in-Aid for Scientific Research on Innovative Areas ‘‘Middle Molecular Strategy’ [JP15H05838]; Research Program of ‘Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials’ (in part). Funding for open access charge: JSPS, Grant-in-Aid for Scientific Research on Innovative Areas ‘Middle Molecular Strategy’ [JP15H05838].
Conflict of interest statement. None declared.
REFERENCES
- 1. Mirkin S.M. Expandable DNA repeats and human disease. Nature. 2007; 447:932–940. [DOI] [PubMed] [Google Scholar]
- 2. Cech T.R., Steitz J.A.. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014; 157:77–94. [DOI] [PubMed] [Google Scholar]
- 3. Singh J., Petter R.C., Baillie T.A., Whitty A.. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011; 10:307–317. [DOI] [PubMed] [Google Scholar]
- 4. Bauer R.A. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today. 2015; 20:1061–1073. [DOI] [PubMed] [Google Scholar]
- 5. Yang W.Y., Wilson H.D., Velagapudi S.P., Disney M.D.. Inhibition of non-ATG translational events in cells via covalent small molecules targeting RNA. J. Am. Chem. Soc. 2015; 137:5336–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Velagapudi S.P., Cameron M.D., Haga C.L., Rosenberg L.H., Lafitte M., Duckett D.R., Phinney D.G., Disney M.D.. Design of a small molecule against an oncogenic noncoding RNA. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:5898–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rzuczek S.G., Colgan L.A., Nakai Y., Cameron M.D., Furling D., Yasuda R., Disney M.D.. Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol. 2017; 13:188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Di Antonio M., McLuckie K.I., Balasubramanian S.. Reprogramming the mechanism of action of chlorambucil by coupling to a G-quadruplex ligand. J. Am. Chem. Soc. 2014; 136:5860–5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen C.H., Hu T.H., Huang T.C., Chen Y.L., Chen Y.R., Cheng C.C., Chen C.T.. Delineation of G-Quadruplex Alkylation Sites Mediated by 3,6-Bis(1-methyl-4-vinylpyridinium iodide)carbazole-Aniline Mustard Conjugates. Chem. Eur. J. 2015; 21:17379–17390. [DOI] [PubMed] [Google Scholar]
- 10. Tishinov K., Schmidt K., Haussinger D., Gillingham D.G.. Structure-selective catalytic alkylation of DNA and RNA. Angew. Chem. Int. Ed. 2012; 51:12000–12004. [DOI] [PubMed] [Google Scholar]
- 11. Spitale R.C., Crisalli P., Flynn R.A., Torre E.A., Kool E.T., Chang H.Y.. RNA SHAPE analysis in living cells. Nat. Chem. Biol. 2013; 9:18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Flynn R.A., Zhang Q.C., Spitale R.C., Lee B., Mumbach M.R., Chang H.Y.. Transcriptome-wide interrogation of RNA secondary structure in living cells with icSHAPE. Nat. Protoc. 2016; 11:273–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lonnberg T., Hutchinson M., Rokita S.. Selective alkylation of C-rich bulge motifs in nucleic acids by quinone methide derivatives. Chem. Eur. J. 2015; 21:13127–13136. [DOI] [PubMed] [Google Scholar]
- 14. Sato N., Tsuji G., Sasaki Y., Usami A., Moki T., Onizuka K., Yamada K., Nagatsugi F.. A new strategy for site-specific alkylation of DNA using oligonucleotides containing an abasic site and alkylating probes. Chem. Commun. 2015; 51:14885–14888. [DOI] [PubMed] [Google Scholar]
- 15. Arambula J.F., Ramisetty S.R., Baranger A.M., Zimmerman S.C.. A simple ligand that selectively targets CUG trinucleotide repeats and inhibits MBNL protein binding. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:16068–16073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wong C.H., Richardson S.L., Ho Y.J., Lucas A.M., Tuccinardi T., Baranger A.M., Zimmerman S.C.. Investigating the binding mode of an inhibitor of the MBNL1.RNA complex in myotonic dystrophy type 1 (DM1) leads to the unexpected discovery of a DNA-selective binder. Chembiochem. 2012; 13:2505–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Konieczny P., Selma-Soriano E., Rapisarda A.S., Fernandez-Costa J.M., Perez-Alonso M., Artero R.. Myotonic dystrophy: candidate small molecule therapeutics. Drug Discov. Today. 2017; 22:1740–1748. [DOI] [PubMed] [Google Scholar]
- 18. Thornton C.A., Wang E., Carrell E.M.. Myotonic dystrophy: approach to therapy. Curr. Opin. Genet. Dev. 2017; 44:135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nguyen L., Luu L.M., Peng S., Serrano J.F., Chan H.Y., Zimmerman S.C.. Rationally designed small molecules that target both the DNA and RNA causing myotonic dystrophy type 1. J. Am. Chem. Soc. 2015; 137:14180–14189. [DOI] [PubMed] [Google Scholar]
- 20. Siboni R.B., Nakamori M., Wagner S.D., Struck A.J., Coonrod L.A., Harriott S.A., Cass D.M., Tanner M.K., Berglund J.A.. Actinomycin D specifically reduces expanded CUG repeat RNA in myotonic dystrophy models. Cell Rep. 2015; 13:2386–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Siboni R.B., Bodner M.J., Khalifa M.M., Docter A.G., Choi J.Y., Nakamori M., Haley M.M., Berglund J.A.. Biological efficacy and toxicity of diamidines in myotonic dystrophy type 1 models. J. Med. Chem. 2015; 58:5770–5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li J., Matsumoto J., Bai L.P., Murata A., Dohno C., Nakatani K.. A ligand that targets CUG trinucleotide repeats. Chem. Eur. J. 2016; 22:14881–14889. [DOI] [PubMed] [Google Scholar]
- 23. Singer B., Kroger M., Carrano M.. O2- and O4-alkyl pyrimidine nucleosides: stability of the glycosyl bond and of the alkyl group as a function of pH. Biochemistry. 1978; 17:1246–1250. [DOI] [PubMed] [Google Scholar]
- 24. Kusmierek J.T., Singer B.. Reaction of diazoalkanes with 1-substituted 2, 4-dioxopyrimidines. Formation of O2, N-3 and O4-alkyl products. Nucleic Acids Res. 1976; 3:989–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Holz B., Klimasauskas S., Serva S., Weinhold E.. 2-Aminopurine as a fluorescent probe for DNA base flipping by methyltransferases. Nucleic Acids Res. 1998; 26:1076–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nakano S., Uotani Y., Uenishi K., Fujii M., Sugimoto N.. DNA base flipping by a base pair-mimic nucleoside. Nucleic Acids Res. 2005; 33:7111–7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. He G., Kwok C.K., Lam S.L.. Preferential base pairing modes of T.T mismatches. FEBS Lett. 2011; 585:3953–3958. [DOI] [PubMed] [Google Scholar]
- 28. Lopez Castel A., Cleary J.D., Pearson C.E.. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat. Rev. Mol. Cell Biol. 2010; 11:165–170. [DOI] [PubMed] [Google Scholar]
- 29. Pearson C.E., Tam M., Wang Y., Montgomery S.E., Dar A.C., Cleary J.D., Nichol K.. Slipped-strand DNAs formed by long (CAG)·(CTG) repeats: slipped-out repeats and slip-out junctions. Nucleic Acids Res. 2002; 30:989–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nakano S., Uotani Y., Uenishi K., Fujii M., Sugimoto N.. Site-selective RNA cleavage by DNA bearing a base pair-mimic nucleoside. J. Am. Chem. Soc. 2005; 127:518–519. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.