


Abstract
DNA self-assembly has proved to be a useful bottom-up strategy for the construction of user-defined nanoscale objects, lattices and devices. The design of these structures has largely relied on exploiting simple base pairing rules and the formation of double-helical domains as secondary structural elements. However, other helical forms involving specific non-canonical base-base interactions have introduced a novel paradigm into the process of engineering with DNA. The most notable of these is a three-stranded complex generated by the binding of a third strand within the duplex major groove, generating a triple-helical (‘triplex’) structure. The sequence, structural and assembly requirements that differentiate triplexes from their duplex counterparts has allowed the design of nanostructures for both dynamic and/or structural purposes, as well as a means to target non-nucleic acid components to precise locations within a nanostructure scaffold. Here, we review the properties of triplexes that have proved useful in the engineering of DNA nanostructures, with an emphasis on applications that hitherto have not been possible by duplex formation alone.
INTRODUCTION (TO DNA NANOTECHNOLOGY)
DNA has proved to be versatile polymer for the directed self-assembly of custom 2D and 3D objects, arrays and devices bearing features on the nanometer scale (1,2). The high fidelity and programmability of Watson–Crick (W–C) base pairing allows double-helical domains to be used reliably as secondary structural elements (Figure 1). As a consequence, engineering with DNA has required the design of motifs that obey the inherent sequence and geometrical properties of individual W–C duplexes, as well as the necessity for certain solution and assembly conditions. Various topologies are made available by exploiting the natural B-form helical pitch (ca. 10.5 base pairs/turn), the 5′-3′ polarity of strands, and the positioning of strand crossovers between neighbouring helices (Figure 1A). Sheets of helices can be generated by positioning crossovers a whole integer of half-helical turns apart (e.g. Figure 1B and C) (3,4). Such sheets can be stacked into hexagonal (5,6) or square lattices (7) by introducing multiple crossovers between many or all of the helices, whilst sheets or individual helices can be programmed to project at various angles by shifting the register of crossovers along adjacent inter-helical interfaces (e.g. Figure 1D and E) (8–10). Features may also be introduced by varying the length of adjacent domains and/or forcing deviations to the natural twist density (11,12). Structures can be assembled through the hybridisation of a few short oligonucleotides (3,5,8,9) or by the folding of a long single-stranded scaffold (usually the 7249 nucleotide genome of the M13mp18 virus) by multiple short strands (‘staples’), an approached termed DNA origami (4,6,7,10–12). The size of these structures, often referred to as tiles, can be extended by over five orders of magnitude through the coaxial assembly of blunt (13) or sticky-ended segments (14–16) and the concomitant formation of duplexes containing discontinuities (‘nicks’) in their phosphodiester backbones (e.g. Figure 1B, D and E). The stability of such multi-stranded structures is improved by the presence of various counter ions that screen the high degree of negative charge repulsion (e.g. Na+ or Mg2+) (17) and may also take part in the folding process (18). In addition, sequence symmetry minimization (19), computer aided design (20), and the control of annealing protocols (21) can be exploited to limit the yield of undesired complexes, such as those formed through unintentional base pair mismatches. A growing number of applications have been mooted for these designer complexes, including their use in diagnostics, detection and therapy, as nanomechanical devices, as well as for the precise positioning of non-nucleic acid components in 2D and 3D space (1,2).
Figure 1.

The double-helix as a secondary structural element: Structural motifs assembled through single (A), double (B), and multiple (C–E) strand crossovers between adjacent double-helical domains. The size of these structures can be extended by the appropriate positioning of complementary single-stranded overhangs (sticky-ends): (B) 2D array generated from ca. 106 copies of a double-crossover molecule; (D) tetrahedron assembled from four copies of a three-point star motif; and (E) 3D crystal formed from ca. 1012 copies of a tensegrity triangle motif. The single-layer structures shown in (B) and (C) were imaged by atomic force microscopy; the tetrahedral structure shown in (D) was reconstructed from cryo-electron microscopy analysis; whilst the crystal shown in (E) was imaged by light microscopy and its underlying DNA structure later solved by X-ray diffraction analysis. The zig-zag lines in (A) represent half-helical turns and arrows reflect the 5′-3′ polarity of strands. Where possible non-crossover strands are shown in gold and cylinders denote the double-helical regions within each motif. Adapted from (4,9,14,15) with permission.
Clearly the success of DNA nanotechnology is testament to, and based upon, the sequence and structural features of the double-helix first described by Watson and Crick over 60 years ago (22). However DNA is known to adopt a variety of other helical forms involving specific non-canonical base-base interactions (23). The sequence, structural and assembly requirements that differentiate these complexes from their duplex counterparts has introduced a novel paradigm into the process of engineering with DNA (24). Perhaps the most exploited of these structures is the triple-helix: a three-stranded complex generated by the binding of a third nucleic acid strand within the duplex major groove (Figure 2). Triplex motifs that exploit the duplex-to-triplex transition have been incorporated into structures designed for dynamic and/or structural applications (25), whilst the sequence-specific recognition of DNA by triplex-forming oligonucleotides has been used for the targeted introduction of non-nucleic acid components within a DNA scaffold (26). Here, we review the properties of triplexes that have proved useful for engineering DNA nanostructures with an emphasis on the applications that hitherto have not been possible by duplex formation alone.
Figure 2.

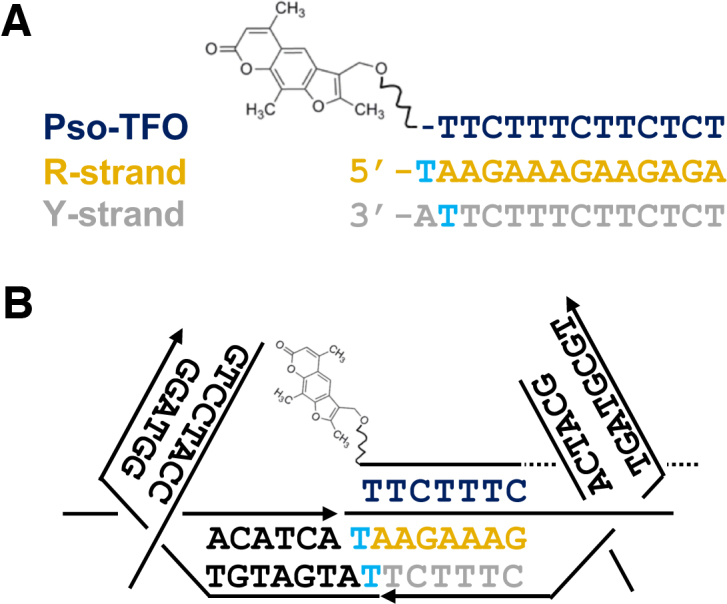
The parallel triple-helix: (A) NMR structure of a parallel triplex formed by the binding of a third strand within the major groove of a polypurine-polypyrimdine duplex (PDB code: 13DX). (B) Chemical structures of parallel T-AT and C+-GC triplets. The notation X-RY refers to a triplet in which the third strand base (X) binds to a purine (R) and pyrimidine (Y) base pair of its target duplex. (C) Sequence of a typical 13-mer triplex that we have used in our own work, also shown in a zig-zag format. The third strand (X-strand) is shown in dark blue and the duplex oligopurine (R-strand) and oligopyrimidine (Y-strand) strands in orange and grey, respectively. Where possible zig-zag diagrams, strand colourings and X, R and Y strand labels remain constant throughout the text.
SEQUENCE, STRUCTURAL AND ASSEMBLY REQUIREMENTS OF TRIPLEXES
Nucleic acid triplexes were first observed experimental 60 years ago by Rich and coworkers upon mixing the polyribonucleotides polyU and polyA in a 2:1 ratio (27). Frank-Kamenetskii's group later showed that a homopurine-homopyrimidine mirror repeat within a supercoiled plasmid was capable of forming an intramolecular triplex under low pH conditions (28,29), implicating a physiological role for these complexes in gene regulation, as well as the cause of genome instability (30). Around this time, the Dervan and Hélène laboratories realised that the formation of an intermolecular triplex by a synthetic oligonucleotide (31,32) could provide a means to target unique genomic sequences and allow the modulation of specific genes (33). However, since these seminal studies there has been limited evidence for the formation of triplexes within genomic DNA, and interest in their use for gene-targeting has dwindled, perhaps due to the growing success of other gene-targeting methodologies, such as zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs). Nevertheless, this work serendipitously laid the foundation for the application of triplexes to DNA nanotechnology.
Triplex formation can occur at almost any given oligopurine-oligopyrimidine duplex sequence and such sequences may be present or easily embedded within a DNA nanostructure with little effect on its overall topology. Binding of the third strand is asymmetric within the major groove, with the third strand bases forming Hoogsteen or reverse Hoogsteen hydrogen bonds with the ‘central’ oligopurine-containing strand of the target duplex (Figure 2A). Pyrimidine-containing strands bind in a parallel orientation with respect to the central strand, with thymine and protonated cytosine recognising AT and GC base pairs, generating T-AT and C+-GC base triplets, respectively (Figure 2B) (v). Purine-containing strands bind in an antiparallel orientation, with adenine and guanine recognising AT and GC base pairs, generating A-AT and G-GC base triplets, respectively (34,35). The notation X-RY used here refers to a triplet in which the third strand base X interacts with the duplex base pair RY, forming hydrogen bonds to base R. Triplex formation at mixed sequence targets is also possible (i.e. with oligopurine sequences containing pyrimidine interruptions) by using third strands containing base or nucleoside analogues (36,37). Triplexes can be generated intramolecularly, through the association of a single-stranded region of the same duplex that folds back on itself, or intermolecularly, through the association of a triplex-forming oligonucleotide (TFO) or sequence (TFS) with a separate duplex. Importantly, intramolecular and intermolecular motifs are compatible with duplex regions assembled by crossover strand exchange and is discussed in a later section.
Both parallel and antiparallel motifs can be exploited in the design of DNA nanostructures but in practise the parallel motif has been more widely adopted due to the following reasons: Firstly, parallel triplexes are more stable than their antiparallel counterparts since the T-AT and C+-GC triplets formed in this motif are structurally isomorphic; that is, if the C-1′ atoms of their W–C base pairs are superimposed, the positions of the C-1′ atoms of the third strand are almost identical. This minimises backbone distortion of both the third strand and duplex between adjacent triplets, leading to only a slight perturbation of the underlying duplex structure. NMR structures have suggested only a slight widening of the major groove upon triplex formation, resulting in a helix that is slightly more A than B-like (38,39). As a consequence third strands composed of ribonucleotides or other nucleotides that possess an N-type sugar pucker are slightly more stable than their deoxynucleotide equivalents. Secondly, the formation of the parallel motif is dependent on low pH conditions (pH < 6.0) necessary for imino protonation of the N3 position of cytosine and the formation of a second hydrogen bond with the N7 position of guanine (Figure 2B). Although this may seem to be a limitation, the presence of the positive charge acts to increase triplex stability by screening the charge repulsion between the three polyanionic strands (40). However, runs of contiguous cytosine residues are destabilising due to electrostatic repulsion between residues (41). The most stable triplexes are therefore composed of regions containing separated C+-GC and T-AT triplets and an example sequence often used in our own work is shown in Figure 2C. Importantly, the pH dependence of the parallel motif is also a useful property that can be used to fine-tune the binding and/or removal of the third strand by adjusting the solution pH, most frequently between a pH of 5.0 and 7.0, respectively. In general, the underlying duplex regions within a nanostructure are much less affected by this change in pH. This pH dependence can also be adjusted or removed by using various cytosine mimics that allow stable triplex formation at a variety of pH values (37). Thirdly, and lastly, the antiparallel motif requires the use of G-rich oligonucleotides and is hampered by the tendency of such purine-rich strands to adopt other non-canonical structures, such as G-quadruplexes and GA-duplexes, that compete with triplex formation. Consequently the studies described in the remainder of this article deal solely with triplexes generated through the parallel binding motif using pyrimidine-rich third strands.
Under low pH conditions the stability of a parallel triplex can be greater than its underlying duplex, i.e. the affinity of a third strand for its target duplex is greater than the affinity of a duplex strand for its W–C partner (42). For example, the melting temperature (Tm) determined for the dissociation of the 13-mer third strand shown in Figure 2C is 65°C, whilst the Tm for underlying duplex is 62°C (experiments undertaken in pH 5.0 tris-acetate buffer containing 15 mM magnesium acetate; unpublished observation). The selectivity of triplex formation is also similar to that of a W–C duplex; single base mismatches between the third strand and duplex results in a typical free energy change of around 3 kcal mol−1 (43–46). The extent of destabilization is dependent on the nature and position of the mismatch and central mismatches are more destabilising than terminal ones since they disrupt the cooperative interaction between neighbouring triplets (44,46). Nevertheless, triplex formation is still possible with mismatches in the third strand (e.g. by forming G-TA and T-CG triplets) and it is also possible to generate triplexes with mismatched base pairs within the duplex (47). The affinity of the third strand can be adjusted by altering its length, by incorporating stabilizing nucleoside analogues (37), through the addition (48) or conjugation of triplex-stabilizing ligands to the third strand (49), and as with other multi-stranded structures, by increasing the counterion concentration, of which Mg2+ is the most effective for stabilizing parallel triplexes (50). Consequently, there is a substantial repertoire of duplex and third strand sequences that can be introduced into DNA nanostructures depending on the proposed application.
Lastly, the kinetics of triplex formation are considerably different from their duplex counterparts. The rate of triplex formation is about three orders of magnitude slower than duplex formation with reported association rate constants of ca. 103 M−1 s−1 (41,44,51–53). Binding of the third strand is thought to proceed via a nucleation-zipper mechanism, dependent on the formation of a quasi-stable intermediate consisting of a few productive triplets, before a ‘zippering’ of the remainder of the strand around the duplex (44). The apparent association rate therefore decreases with temperature, as lower temperatures stabilise this transient intermediate (41,44,51). For example, it has been shown that a 10°C reduction in temperature leads to roughly a 2-fold increase in TFO association rate (51). Although such slow association kinetics might seem to be a limitation, it is a useful property that can be exploited for the ‘one-pot’ assembly of triplex-based nanostructures, since it allows duplex regions within the structure to first form before binding of the third strand, thereby reducing its influence on the annealing process. The rate of triplex dissociation is also slow, with reports suggesting half-lives of between 30 minutes and several days (41,44,51), and can be increased using stabilizing nucleoside analogues (52,53).
RECONFIGURABLE STRUCTURES BASED ON THE DUPLEX-TO-TRIPLEX TRANSITION
One of the major goals of DNA nanotechnology is the construction of switchable structures capable of occupying two or more distinct structural states with time (1,54). The first duplex-based device that possessed such qualities exploited the structural transition of right-handed B-DNA to left-handed Z-DNA promoted by the addition of hexamminecobalt(III) chloride to the sample solution (55). Not long after, this was extended to a pair of ‘molecular tweezers’ that could be reconfigured through a strand-displacement reaction; a robust process by which two strands with partial or full complementarity hybridize to each other, displacing one or more pre-hybridized strands in the process (56). Similar strategies have been exploited for the design of triplex-based devices that exploit the reversibility of the duplex-to-triplex transition, either by pH change, the use of triplex-stabilizing ligands, or through a strand displacement process. Such devices have been designed for sensing solution pH, for directing chemical reactions, for capturing and/or releasing substrates, for strand displacement circuits, as well as to aid in the hierarchical assembly and/or dissociation of extended DNA structures and heterogeneous complexes.
For sensing solution pH
The majority of triplex-based devices exploit the pH dependence of parallel triplex formation with pyrimidine-rich oligonucleotides (57,58). In general, the input ‘fuel’ used in these systems is H+ and OH− ions introduced by the addition of either HCl or NaOH to the sample solution. Unlike devices based on duplex strand-displacement, that generate duplexes as so-called ‘waste’, the products of the reaction are simply H2O and NaCl. In addition, the gradual increase of ionic strength due to the accumulating salt is not expected to change the electrostatic potential of the DNA structure, and consequently its performance, until the salt reaches molar concentration. Moreover, the diffusion rate of ions (milliseconds) is much faster than oligonucleotides (seconds) used for strand-displacement reactions, and subsequently, allows faster cycling times between states.
The first demonstration of a DNA device that exploited this pH dependence was reported by Mao and coworkers just over 10 years ago (57). Their device was based loosely on the molecular tweezers first described by Yurke et al. (56). The device consists of three oligonucleotide strands and operates through the reversible formation and dissociation of an intramolecular triplex generated within the device (Figure 3A(i)). At pH 8.0 the three strands forms an ‘open’ complex consisting of three duplexes and a single-stranded triplex-forming sequence that adopts a random coil conformation. Upon lowering to pH 5.0 the single-stranded section folds back with the adjacent duplex, forming a triplex, and results in a more compact ‘closed’ structure. The structural change was demonstrated by comparing the mobility of the two complexes by non-denaturing gel electrophoresis and by the incorporation of fluorescent dyes into the system. In the fluorescent experiments, two pH-independent dyes were attached to the opposing duplexes so that in the open state the dyes were far apart and not capable of fluorescence resonance energy transfer (FRET). By contrast, in the closed state the two dyes were in close proximity and a fluorescence signal could be monitored (Figure 3A(i)). Experiments revealed that the conformational change occurred in seconds but the cycling efficiency deteriorated over 16 cycles (Figure 3A(ii)). It was suggested this decrease was due to photobleaching of the fluorescence dyes, or as a consequence of diluting the sample volume by the addition of the acid or base that reduced the effective concentration and signal of the complexes.
Figure 3.

Triplex-based devices that respond to pH change: (A) (i) pH-dependent device based on molecular tweezers. The complex contains an appropriately positioned FRET pair (F1 and F2) that allows the opening and closure of the device to be monitored upon pH change; (ii) FRET data generated through the repeated pH cycling of such a device. Adapted from (57) with permission. (B) (i) pH-dependent device based on a simple intramolecular triplex. The complex contains an appropriately positioned fluorophore (F) and quencher (Q) that allows the opening and closure of the device to be monitored upon pH change. (ii) Varying the relative T-AT and C+-GC content of such a device allows the sensing of different pH values. Adapted from (60) with permission. (C) (i) pH-dependent complex composed of an intramolecular triplex and graphene oxide (GO) used to monitor pH changes associated with apoptosis in living cells. At high pH, the triplex-forming sequence interacts with GO, whilst at low pH triplex formation prevents this interaction. Since GO quenches fluorescence a fluorophore attached to the triplex allows association, and hence the pH of the solution, to be monitored directly. (ii) Fluorescence images of living cells transfected with the GO-device. Adapted from (61) with permission.
A further device was later developed by the Samori laboratory that was based on a much simpler architecture but similar operation style (58). The device consists of two partially complementary DNA strands with a single-stranded region capable of forming an intramolecular triplex with the adjacent duplex region due to the presence of an unstructured 5-nucleotide loop (Figure 3B(i)). In addition to FRET and gel electrophoresis based measurements the system was characterized by ultraviolet (UV) melting and circular dichroism (CD). At low pH, the latter resulted in a spectrum with a negative peak around 215 nm indicative of triplex formation. By employing a polymer statistics model it was estimated that the positioning of the two termini within the triplex differed by ∼6 nm during each cycle. Varying the pH of the solution allowed the device to cycle between its two states without deterioration over time with cycling occurring with a time frame of milliseconds. However, it was later observed that the expected FRET efficiency never exceeded 90%, suggesting that the number of devices that were closed in solution was lower than expected (59). This was attributed to intermolecular interactions between devices but was overcome at low oligonucleotide concentrations (picomolar) by tethering the device to a solid support.
One of the drawbacks of using such devices as pH sensors is that they are restricted to sensing over a short range, typically between 1.5 and 2.0 pH units. To overcome this issue Ricci's group exploited the simple notion that the pH window for a duplex-to-triplex transition will depend on the relative C+-GC and T-AT content of the triplex (60). For example, the opening of a switch containing mainly T-AT triplets will be triggered at a more basic pH (9.0–11.0) due to deprotonation of thymine (pKa ∼ 10). In contrast, the opening of a switch containing mainly C+-GC triplets will be triggered at a more acidic pH (5.0–7.0) due to deprotonation of cytosine (pKa ∼ 5) (Figure 3B(ii)). Using the simple intramolecular system described above (58) the authors demonstrated that a range of 5.5 pH units could be measured by using two or more switches in solution, each triggered over a different pH range. The authors proposed that such devices would be useful for real time sensing of cellular extracts, in in vivo cells, or in other media where pH changes represent an important input both in healthy and pathological biological pathways. Indeed a triplex-based sensor based on the above design has been used to monitor pH changes associated with apoptosis in living cells to aid in the diagnosis of cancer (61). This was achieved by exploiting the fact that single-stranded regions of a DNA molecule can bind to graphene oxide (GO) whereas duplex or triplex regions do not. The sensor was composed of a double-stranded hairpin tailed by a triplex-forming sequence that at pH 8.0 is capable of interacting with the GO, whilst at pH 5.0 triplex formation prevents this interaction (Figure 3C(i)). Since GO is capable of quenching fluorescence the attachment of a fluorescent dye to one of the duplex strands of the sensor allows the duplex-to-triplex transition to be monitored by a change in fluorescence intensity. The authors preformed the complex at pH 8.0 before transfection into Ramos (lymphoma) cells and demonstrated successful translocation across the cell membrane. Addition of vincristine sulfate to the cells, which induces apoptosis and intracellular acidification, resulted in triplex formation within the device and its release from the GO. An increase in fluorescence signal was then observed and localised by confocal microscopy (Figure 3C(ii)).
For directing chemical reactions
Duplex-to-triplex transitions have also been exploited for directing specific chemical reactions with a triplex structure. This was first elegantly demonstrated by Mao and coworkers who used a system that utilises this conformational change to direct amide bond formation (amine acylation) between a carboxylic acid group and one of two identical amines positioned on different strands within the device (Figure 4A) (62). In the presence of a condensation agent the reaction was directed to one of the two amines by the association and dissociation of the third strand due to a change of the solution pH. Moreover, the efficiency of the reaction was high, with yields of 88% and 67% for the reactions at pH 8.0 (reaction 1) and pH 5.0 (reaction 2), respectively (Figure 4A). Such an approach could be useful in synthetic chemistry where protection-deprotection strategies are difficult or expensive. Indeed a similar triplex-based device has been integrated into a microfluidic chip, which allowed electronic control over local pH cycling and switching, and consequently the reactions, within the device (63). The authors demonstrated rapid control over a DNA ligation reaction between disulfide linkages within the triplex complexes and suggest wider applications of the device in biotechnology, in DNA computations and control of self-assembly.
Figure 4.

Triplex-directed chemical reactions: (A) Directing amide bond formation by triplex formation. A carboxylic acid group attached to the triplex-forming sequence is positioned adjacent to a terminal (amine 1) or central amine (amine 2) upon duplex and triplex formation, respectively. The reaction is initiated by the addition of a condensation agent. Adapted from (62) with permission. (B) Control of copper-catalysed alkyne-azide cycloaddition reactions. (i) Upon duplex formation the reaction leads to the linkage of the two duplex strands (reaction 1). (ii) Subsequent addition of a triplex binder, which promotes triplex formation, leads to the linkage of the third strand to the pre-linked duplex (reaction 2). Adapted from (64) with permission.
It is also possible to control duplex-to-triplex transitions by using small-molecules that selectively stabilise triplex over duplex DNA. Such small-molecules are usually composed of aromatic rings for stacking between the base triplets and may also incorporate a positive charge to partially alleviate the charge repulsion between the three polyanionic strands (48). Gothelf and co-workers were the first to demonstrate such control over a dual copper-catalysed alkyne–azide cycloaddition CuAAC (‘click’) reaction using such a strategy (Figure 4B) (64). Their system was composed of an intermolecular triplex that contained azides on both the X- and R-strands and a dual alkyne modification on the Y-strand. In the absence of ligand the reaction between the R- and Y-strands would proceed (i.e. between duplex strands), with a triazole linkage formed between the azide on the R-strand and one of the two alkynes positioned on the Y-strand. Upon addition of the ligand, which promoted triplex formation, a second triazole linkage was formed between the azide on the third strand and the unreacted second of the two alkynes positioned on the Y-strand. The product of the reaction was therefore a dual-linked product connecting the three oligonucleotides. The yield of the reaction was 90% and 80% for the duplex and triplex systems, respectively. The authors suggest that the rate of the triplex-directed reaction could be controlled by using different triplex-stabilizing ligands, each with different binding affinities for a triplex. For example, the rate of the reaction could be increased by the addition of a strong triplex binder, such as naphthylquinoline, or slowed down by the addition of a weaker binder, such as coralyne. Interestingly, the authors suggest that the product of their reaction; a three-way branched structure, could itself be used as the basis for novel DNA architectures.
For capturing and/or releasing substrates
The ability to switch between duplex and triplex states has been used for the opening and closing of DNA structures of various architectures and offers the ability to catch, encapsulate and/or release different substrates. This was first demonstrated by Deng et al. who designed a pair of molecular tweezers containing a triplex-forming region that allows the capture of a DNA target (Figure 5A) (65). The tweezers were loosely based on a double-crossover (DX) molecule first described by the Seeman laboratory with the ‘closed’ device, containing its captured target, designed to emulate a fully formed DX structure (e.g. Figure 1B). The ‘open’ device was first assembled from four oligonucleotide strands; two non-crossover strands that extend the length of each helix and two strands that hold the molecule together by forming a single crossover on just one side of the molecule. Introduction of the target strand at pH 5.0 then led to its binding by triplex formation to specific duplex regions assembled at the centre of the molecule. A DNA set (or locker) strand was then employed to capture the strand by closing the tweezers through the formation of a single-stranded second crossover on the other side of the molecule. The pH was then switched to 5.7, and at this increased pH the tweezers still firmly held the target via the closing action and some remaining Hoogsteen bonding with the target. The locker strand contained a single-stranded toehold that could be used to release the DNA target though strand displacement and the opening of the molecule. Since the device is based on a DX molecule (3) it offers the intriguing possibility of its interfacing within a 2D DX-array (e.g. Figure 1B) (14). Not only might this be used to visualize its action but might be capable of inducing molecular motion within the structure.
Figure 5.

Triplex-based devices capable of capturing and releasing molecules: (A) Control of a tweezer-like DX device that captures/releases single-stranded DNA. The captured strand binds through triplex formation and is subsequently held in place by the addition of toe-hold containing strand that forms the second crossover of the molecule. Upon increasing the pH, the oligonucleotide remains trapped, and is only released by removal of the toe-hold containing strand by addition of its W–C complement. Adapted from (65) with permission. (B) Control of a clamp-like device that detects ATP. A triplex generated within the molecule by the addition of its R-strand brings into close proximity two halves of a split aptamer capable of binding ATP. Adapted from (67) with permission.
A simpler structure designed by Ricci and co-workers was a clamp-like probe that worked by the formation of an intermolecular triplex with the target molecule being a single-strand of DNA (66). The clamp was composed of two recognition elements separated by an unstructured 10-nucleotide loop. The first recognition element was a polypyrimidine sequence designed to bind to the single-stranded polypurine target sequence by W–C base pairing, whilst the second was a polypyrimidine sequence that bound this duplex by Hoogsteen base pairing. The formation of this triplex conformation led to the closure of the switch. Compared to the simple interaction of two W–C duplex strands the inclusion of the third strand increased not only the affinity of the probe but also improved its discrimination efficiency towards a single-base pair mismatch by 1.2 ± 0.2 kcal mol−1. The authors have since exploited such a clamp as a means to regulate various target-responsive nucleic acid aptamers evolved to detect either adenosine triphosphate (ATP), cocaine, or gentamicin; an antibiotic used to treat various bacterial infections (Figure 5B) (67). Each of the aptamers was split into two segments (split aptamers) with one segment being coupled to one end of the clamp oligonucleotide, and the other segment coupled to the other end of the oligonucleotide. In the absence of triplex formation the two halves are kept far apart and are not capable of binding their target molecule. By contrast, addition of the central oligonucleotide strand (DNA activator) generated a triplex and brings the two halves of the split aptamer into close proximity, allowing binding of the target molecule. In order to monitor this process, the split aptamer was labelled with a fluorophore and a quencher at each end which allowed the detection of the target to be observed by a fluorescence change. It was shown that the efficiency with which the nanoswitch binds to ATP could be varied by 2–3 orders of magnitude (nM – uM KD) by simply varying the concentration of the DNA activator and obeyed a model for simple allosteric activation, often seen with biological molecules such as protein receptors. Moreover, regulation could be further tuned by removing the DNA activator through pH change or strand displacement, as well as by using more than one DNA activator in tandem.
More recently, Willner et al. have described the assembly of triplex-based microcapsules capable of releasing CdSe/ZnS quantum dots (QD) (68). To assemble the capsules CaCO3 microparticles were first loaded with the QDs and subsequently coated with a positively charged polyallylamine hydrocholoride (PAH) polyelectrolyte. This allowed the layer-by-layer deposition of a nucleic acid shell through the intermolecular hybridization of a number of strands designed to interact through the formation of W–C duplexes. The CaCO3 core was then dissolved by the addition of ethylenediaminetetraacetic acid (EDTA) freeing the encapsulated QDs within the capsule. Importantly, the oligonucleotides that make up the shell were designed in such a fashion so as to form intramolecular triplexes upon lowering the pH. This in turn disrupts their interaction with one another and allows the release of the QDs. Assembly of the capsules and the release of the QDs was monitored due to the intrinsic florescence of the encapsulated QDs and the authors showed that varying the relative T-AT and C+-GC content allowed the release of the QDs at various pH values. The size of the microcapsules was calculated to be 3.5 ± 0.8 μm and are likely to find use in imaging and other studies, but are perhaps too big to offer cellular delivery of drugs and proteins.
For strand displacement reactions
Duplex strand displacement is often initiated at a single-stranded domain (a ‘toehold’) complementary to an ‘invading’ oligonucleotide and progresses through a branch migration process. The reaction proceeds because more base pairs are formed by hybridisation of the toehold containing-strand to the invading strand than when bound to its original partner. The simplest means to displace a third strand from its duplex partner is achieved by the addition of an excess of its W–C complement (e.g. Figure 6A(i)). A toehold-mediated strategy is also possible by the addition of a single stranded domain to the end of the third strand (i.e. Figure 6A(ii)). The former is kinetically controlled and has been used to determine the dissociation kinetics of triplex formation (52,53,69), whilst the latter is thermodynamically controlled and can be adjusted in much the same way that has been demonstrated for duplex strand displacement reactions, e.g. by adjusting the base sequence and/or length of the toehold (unpublished observation). Importantly, and in both cases, the structure and stability of the underlying duplex region within the nanostructure is likely to remain unaffected.
Figure 6.

Triplex-mediated strand displacement reactions: (A) (i) Mechanism of third strand displacement by addition of its W–C complement; (ii) the addition of a toehold to the end of the third strand can be used to enhance the reaction. (B) Mechanism of duplex strand displacement using a short triplex domain as a toehold. Third strand binding positions strand S2 adjacent to identical strand S and as a result leads to its gradual displacement from the duplex by branch migration. Adapted from (70) with permission. (C) (i) OH−-activated duplex strand displacement. At low pH, binding of a third strand blocks access of the invading strand to the duplex toehold and prevents strand displacement, whilst at high pH the third strand dissociates and the displacement reaction can proceed. (ii) H+-activated duplex strand displacement. At low pH triplex formation with a clamp-like oligonucleotide containing both Y2- and X-strands leads to the displacement of identical strand Y. Adapted from (71) with permission.
Triplex formation has also been used to direct duplex strand displacement (i.e. removal of one of the two strands of the underlying W–C duplex) and was first demonstrated by Mao and co-workers (Figure 6B) (70). In their system the duplex target was composed of a template strand with a hairpin region at one end of the molecule and a shorter W–C complement designed to bind to the remaining single-stranded region, generating a continuous duplex with a nick site where the two strands abut. To displace the shorter strand a third oligonucleotide was designed that first binds to the hairpin region of the molecule by triplex formation, whilst simultaneously positioning in close proximity a segment of the oligonucleotide identical in sequence to the shorter strand. Consequently, binding of the third strand leads to the displacement of the shorter strand from the duplex. The first two steps are reversible, but the last step is essentially irreversible, thus driving the overall reaction to completion. Interestingly, the authors have used this approach to detect the transient formation of a cytosine-containing triplex at neutral pH, which is not possible using other techniques. This is a simple strategy that could be further controlled by adjusting the pH, and hence the stability and kinetics of the triplex region formed. Following this Ricci's group developed two different approaches that exploit the pH dependence of parallel triplexes as a means to activate or inhibit toehold-based duplex strand displacement reactions (Figure 6C) (71). The first approach exploited the formation of a triplex at low pH to physically prevent the displacement reaction from occurring due to steric hindrance between the bound TFO and the invading strand (Figure 6C(i)). The second approach exploited a clamp-like invading strand that can initiate strand displacement directly, but only upon formation of a triplex through a decrease in pH. This was achieved by limiting the length of the Y-region of the strand so it would not lead to duplex formation alone and required binding of the third strand in tandem to generate a stable complex (Figure 6C(ii)). Both strategies were demonstrated using a second cascade where the released strand disrupted a duplex containing a fluorophore and quencher, and thus led to an increase in fluorescence signal. Because triplex stability can be tuned at different pHs the authors suggest the gradual inhibition/activation of the strand displacement process could be achieved by small changes to the solution's pH.
For hierarchical assembly and/or dissociation
Extended DNA structures
Ricci and co-workers have exploited the ability to activate or inhibit duplex strand displacement by triplex formation as a means to control the assembly of DNA concatamers generated through a hybridisation chain reaction (HCR) (72). HCR is a process through which two metastable duplex hairpins react with each other in the presence of a triggering single strand. The addition of the initiator opens a hairpin of one species, exposing a new single-stranded region that opens a hairpin of the second species. This in turn exposes a single-stranded region identical to the original initiator. The resulting chain reaction leads to the formation of a nicked duplex that can grow until the hairpin supply is exhausted. The group adapted this approach by redesigning one of the two hairpin species (HP1) to include a nine nucleotide tail that at low pH can form a triplex with the terminal portion of the duplex (Figure 7A(i)). The structure acts as a molecular trap sequestering a toehold domain found in the single-stranded loop connecting the triplex-forming tail to the hairpin, and in doing so, prevents the binding of the initiator strand and the start of polymerization. By increasing the pH, the third strand dissociates, and the HCR reaction proceeds with the second hairpin species (HP2) generating a concatemer of the two hairpin duplexes (Figure 7A(ii)). A second strategy was also utilised that allowed the reaction to be activated at acidic pH’s. In this instance a triplex-forming sequence attached to the hairpin is used to stabilise, not prevent, the interaction of the initiator strand with a shorter toehold sequence, which under normal conditions is not long enough to allow initiator nucleation and an efficient HCR. The same group have also developed a triplex-based strand displacement circuit as a means to control the assembly of double-crossover tile arrays (Figure 7B) (73). The circuit consists of a single-stranded catalyst that binds to a pH-dependent substrate leading to the release of a deprotector strand (Figure 7B(i) and (ii)), which in turn activates a downstream self-assembling reaction by irreversibly associating with a protected tile (Figure 7B(iii)). The result is a reactive tile that assembles into lattices similar to those shown in Figure 1B. The pH-dependent substrate is formed using a clamp-like triplex-forming strand that physically inhibits strand displacement with the catalyst until the pH of the solution is increased and the third strand dissociates (Figure 7B(i)). The authors suggest that such systems could offer better spatiotemporal control over the self-assembly processes of DNA-based nanostructures.
Figure 7.

Hierarchical formation of extended DNA complexes through the control of strand displacement reactions. (A) Formation of a DNA concatemer at basic pH. The system is composed of two metastable hairpin species that react with each other in the presence of an initiator strand. The initiator binds to a toehold region on the hairpin of one species (HP1) and through strand displacement exposes a new single-stranded region that opens the hairpin of the second species (HP2), generating a concatemer of the two duplexes. (i) At low pH the binding of the initiator to HP1 is inhibited due to the formation of a triplex which sequesters the toehold portion of the molecule. (ii) Increasing the pH leads to the dissociation of the third strand, which allows initiator binding, and subsequent polymerisation of the two hairpin species. Adapted from (72) with permission. (B) Formation of double-crossover lattices at basic pH. The system is composed of a triplex-based strand displacement circuit that activates a downstream self-assembling reaction: the formation of a DX array similar in design to the one shown in Figure 1B. (i) The circuit is initiated by the binding of a catalyst strand to a pH-dependent substrate. At low pH, triplex formation prevents this interaction, whilst at high pH the reaction can proceed. (ii) Catalyst binding releases a deprotector strand through strand displacement. (ii) The deprotector strand then associates with a protected tile that is made reactive through the displacement of protecting strands that cover the sticky-ends of the molecule, resulting in array formation. Adapted from (73) with permission.
Using a different approach Willner's group have demonstrated the controlled cyclic assembly and separation of hexagonal origami dimer and trimer systems based on the pH-dependent duplex-to-triplex transition (74). To do this, the edge of one of the origami tiles (Tile 1) was designed with a nucleic acid tether that, at neutral pH, consisted of a CG-duplex and a single-stranded overhang capable of binding through W–C pairing to a single-stranded tether attached to a different origami tile (Tile 2) (Figure 8A). Upon lowering the pH the first single-stranded domain of tile 1 preferentially forms an intramolecular triplex and disrupts its association with tile 2, separating the origami complex into its constituent monomers. The system was reversibly reconfigured by increasing the pH back to neutral conditions, allowing the two origami monomers to once again associate. A second strategy was also developed based on the formation of an intermolecular triplex containing predominantly T-AT triplets that allowed the structure to be cycled through higher pH values (pH 7.0–9.0), with the origami tiles dissociating at the higher pH. Both strategies were then coupled to create origami trimers that can dissociate into two different dimers by an appropriate pH change: tile 3 contained single stranded extensions that are complementary to those in tile 4, but folds back on itself to form an intramolecular C+-GC triplex at pH 4.5, whilst tiles 4 and 5 were connected by a T-AT triplex bridge that dissociates at pH 9.5. Thus by cycling the pH from 7.0 to 4.5 the trimer dissociates to form a dimer of tiles 4–5, while cycling the pH from 7.0 to 9.5 the trimer dissociates to form a dimer of tiles 3–4. In both cases, the trimer could be reassembled by reverting back to the original pH of 7.0.
Figure 8.
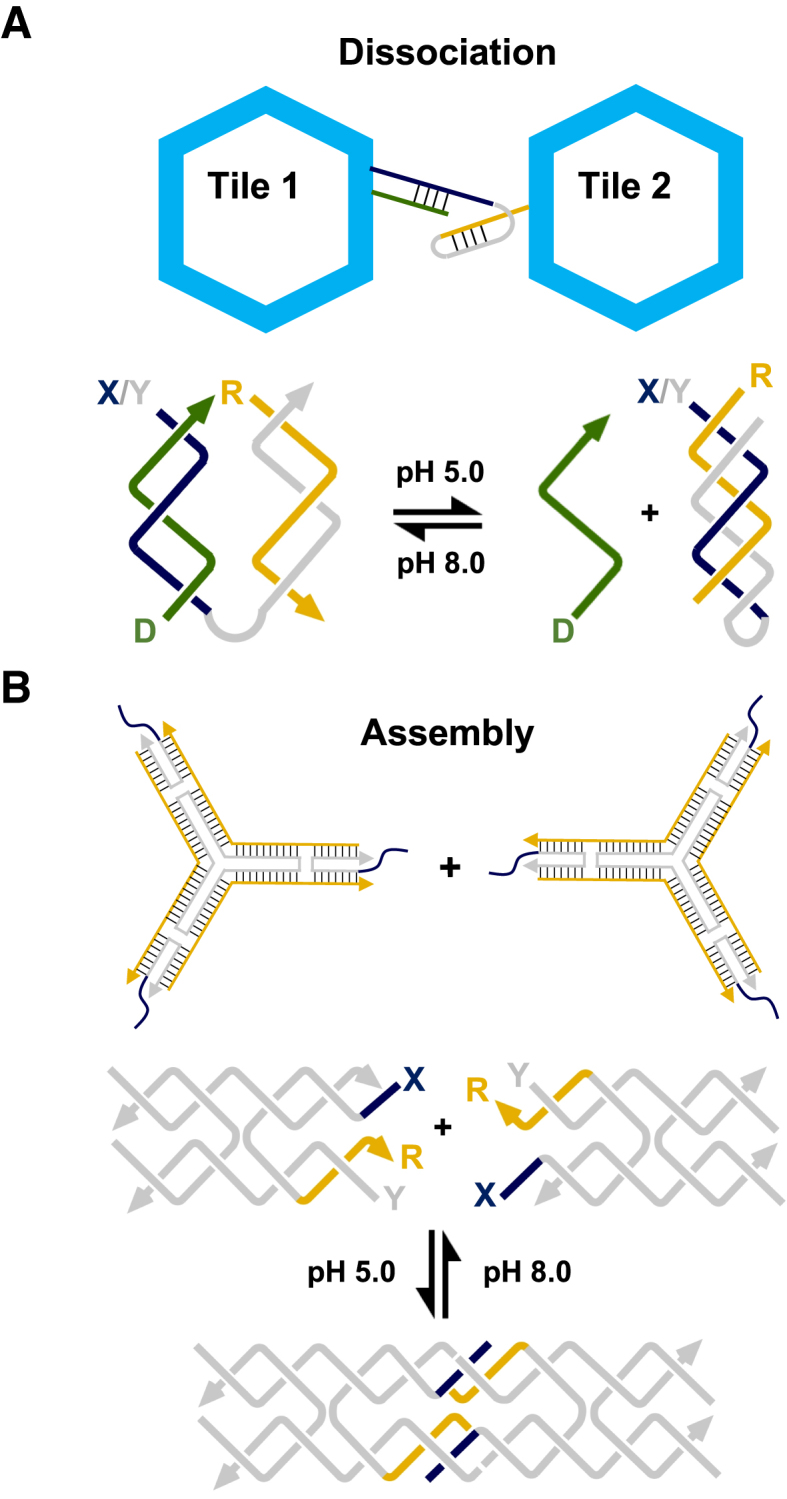
Hierarchical assembly and/or dissociation of extended DNA complexes through pH change: (A) Triplex motif used for reconfiguring the interactions of hexagonal origami tiles. At high pH, the strands form two inter-linked duplexes between tile 1 and tile 2, whilst at low pH one strand of one of the duplex partners folds back and forms an intramolecular triplex leading to the dissociation of the two tiles. Adapted from (74) with permission. (B) Triplex motif used for reconfiguring the interaction of a three-point star motif into a DNA tetrahedron similar to the one shown in Figure 1D. At low pH, the two triplex-modified sticky-ends interact, whilst at high pH they do not. Adapted from (75) with permission.
Triplex formation has also been exploited by Mao and coworkers to isothermally assemble and dissociate a DNA tetrahedron in response to pH change (75). The tetrahedron was based on the authors previously designed structure that assembles from four copies of a three-point star motif through sticky-end cohesion (Figure 1D) (9). In this instance, a single-stranded triplex-forming sequence was introduced alongside the sticky-ends of the molecule that was capable of triplex formation across the helices of the individual motifs (Figure 8B). While the sticky-ends bring together the motifs to direct the overall polyhedral geometry they were designed to be intrinsically unstable in the absence of the triplex forming sequence which provides extra cohesion strength under the right conditions. At pH 5.0, the tetrahedron formed from the three-point star motifs and when the pH was increased, it dissociated into the component motifs. The assembly/disassembly process could also be cycled by changing the pH. This was an interesting study that offers the ability to encapsulate and/or trigger the release of molecular cargo based on a pH change.
Heterogeneous complexes
Triplex formation has also been exploited as a means to aggregate/dissociate nanoparticle (NP) clusters and assemblies, in particular, those composed of gold (AuNPs) and silver nanoparticles (AgNPs). Since the optical and fluorescent properties of such NPs is dependent on their inter-particle distance the ability to reconfigure their arrangement (i.e. through pH change) provides a way to study and/or exploit these properties. The first reversible system was developed by the Choi laboratory (76). It was composed of two sets of oligonucleotide-modified AuNPs; the first contained an oligonucleotide designed to fold into a hairpin duplex, whilst the second contained an oligonucleotide capable of binding to this duplex through triplex formation at low pH (Figure 9A). Since the thiol conjugation process leads to the attachment of multiple oligonucleotides per gold particle, triplex formation between many or all of the strands led to the generation of an extended 3D network with the NPs in a closer proximity than when free in solution. Successful clustering of the complexes was observed as a red to reddish-purple colour change of the sample solution, and characterized by transmission electron microscopy (TEM) (Figure 9A). Further studies have been undertaken to improve control over the average inter-particle separation in such complexes (77,78). Mao et al. have also demonstrated the specific expansion/contraction of AuNP aggregates using a different strategy (79). Two sets of AuNPs were functionalized with oligonucleotides that associated to form a duplex at one end whilst leaving a flanking single-stranded region at the other. The design was such that the single-stranded region could fold back on the duplex formed between the particles and generate a triplex at low pH. Cycling the pH led to the expansion and contraction of the nanoparticles which was observed as a change to the AuNP plasmon resonance peak from 524 to 533 nm, respectively. Interestingly, various groups have exploited such systems as a colorimetric assay for screening potential triplex-binding molecules (80,81). Such agents are of interest since they could be used to stabilize putative triplexes formed in genomic DNA that might modulate the expression of specific genes.
Figure 9.

Hierarchical assembly/dissociation of heterogeneous complexes: (A) pH-dependent aggregation of gold nanoparticles. One set of gold nanoparticles is functionalized with a third strand, whilst the second set is functionalized with its duplex partner. Dissociation/assembly of the two sets of gold particles into a 3D network is controlled through pH change. Adapted from (76) with permission. (B) Small-molecule induced aggregation of single-walled carbon nanotubes. Each nanotube is functionalised with one or more polyT-polyA duplexes capable of repartitioning into a polyT-polyA-polyT triplex upon addition of a triplex-stabilizing ligand, such as coralyne. Adapted from (84) with permission; (C) pH-responsive hydrogels. The acrylamide chains are functionalized with a single DNA strand capable of forming either an intermolecular duplex at high pH, or intramolecular triplex at low pH leading to the association and dissociation of the copolymer chains, respectively. Adapted from (85) with permission.
A different approach for the directed assembly of NPs clusters has been demonstrated by Qu and co-workers (82) who exploited the observation that a silver ion (Ag(I)) can specifically displace the N3 proton of a cytosine within a C+-GC triplet removing its pH dependency (83). Inspired by this unique feature, they demonstrated the formation of homogenous Ag2 clusters by reducing Ag(I) ions present at a specific C-GC site within a triplex by Tollens chemistry. Moreover, this was extended to multiple locations within the structure by the inclusion of several C-GC triplets. The authors suggest that since the fluorescence properties of Ag2 are different to Ag this approach would be useful for the template-directed synthesis of fluorescent Ag2 clusters.
Triplex formation has also been used in the self-assembly of single-walled carbon nanotubes (SWCNTs) through exploiting triplex-stabilising ligands (84). One set of SWCNTs was tagged with single-stranded polyT whilst another set was tagged with polyA. In the presence of a triplex-inducer coralyne, the dT22–dA22 duplex was induced to form a dT22–dA22–dT22 triplex, resulting in the aggregation of SWCNTs (Figure 9B). This aggregation occurs only in the presence of the triplex-inducer and can thus be used in the construction of multifunctional architectures for electrical and biosensing applications. Lastly, Willner et al. have recently applied triplex formation to the preparation of pH-responsive DNA hydrogels (85). In one of their systems double-helices were used as bridging units between acrylamide chains that formed an acrylamide gel (Figure 9C). The design of the duplex regions was such that, upon lowering of the pH, the duplex units would generate intramolecular triplexes, thereby preventing their association, and resulted in dissolution of the gel into its liquid phase. In another system the acrylamide chains were held together by the intermolecular association of a triplex composed predominantly of T-AT triplets. This time the dissolution of the gel occurred at pH 10.0 due to the separation of the triplex bridging units upon thymine deprotonation. The authors demonstrated that both hydrogel systems could undergo reversible and cyclic hydrogel/solution transitions by subjecting the systems to appropriate pH values. In addition, the triplex-stabilizing ligand coralyne could be used within the hydrogel to increase its stiffness. By combining both systems the authors have also demonstrated the creation of shape-memory DNA hydrogels capable of varying their states based on pH change (86).
FUNCTIONALIZED STRUCTURES BASED ON TRIPLEX SEQUENCE ADDRESSABILITY
Many of the proposed applications that have been mooted for DNA nanostructures will first require the attachment of biological and/or chemical components to precise regions along the DNA scaffold. For example, the incorporation of a protein within the structure would impart such properties as molecular recognition (e.g. antibodies), catalytic turnover (e.g. enzymes) and/or energy conversion (e.g. photosynthetic proteins). In addition, it provides a means to characterise such molecules by various imaging and structural analysis techniques. The method most frequently employed for incorporating such components has relied on their attachment to one or more of the oligonucleotide strands that make up part of the nanostructure itself, or where applicable, their recruitment to a reactive group incorporated in the same manner (14). However, many components or reactive groups will not tolerate the high temperatures and slow annealing steps required for structure assembly, and unwanted side interactions may also disrupt the folding pathway and/or decrease the stability of the underlying complex. In addition, components are usually attached to the oligonucleotide terminus since the introduction of internal modifications is synthetically more tedious and the incorporation of multiple reactive groups within the same oligonucleotide can lead to cross-reactivity during the conjugation step (e.g. thiols). This not only restricts the positioning and number of components that can be introduced, but also limits the ability to ligate the oligonucleotide ends, which has been used to improve the structural integrity of a nanostructure (87). One approach used to circumvent these problems is to introduce single-stranded overhangs that protrude beyond or above the nanostructure surface. Conjugation of a component to an oligonucleotide designed to hybridize to these regions can be used to target the structure after the assembly process (88). But this is again limited by the necessity of positioning the overhangs at the oligonucleotide termini and is not applicable to nanostructures that exhibit covalently closed topologies. A more useful strategy is to exploit the sequence addressability of the double-helical regions of a nanostructure using a programmable DNA recognition agent such as triplex-forming oligonucleotides (26). Attachment of a component or reactive group to the end of the TFO will then lead to its targeted introduction to sequences either present or embedded within the nanostructure through design. A major benefit of using TFOs over other DNA recognition agents is their compatibility with a variety of conjugation strategies developed for the attachment of components to an oligonucleotide, such as covalent copper and copper-free ‘click’ strategies, maleimide and amino chemistries, as well as non-covalent NTA:His-Tag and biotin:streptavidin interactions, to name a few. Moreover, the binding and/or removal of the TFO can be controlled in a similar fashion to the devices described above (i.e. by pH change, triplex-stabilising ligands, strand displacement etc.). Consequently, the triplex approach to DNA recognition has been used for the scaffolding of components in 2D and 3D, as well as a means to direct the positioning of molecules that can chemically modify the underlying DNA.
For scaffolding of non-nucleic acid components
The original motivation for engineering objects and lattices from DNA was the use of these structures as scaffolds for the spatial organisation of non-nucleic acid molecules in 2D and 3D space (89). Such heterogeneous complexes could be used to study the structure and interactions of appended molecules, for the manipulation of biological or chemical cascades, to construct optical and electronic devices, as well as for nanoscale patterning and lithographic applications.
Arrangements in 2D
The Norden and Brown groups were the first to examine the binding of a TFO to a specific double-helical region within a DNA nanostructure (90). The structure was composed of two adjacent hexagonal units (analogous to naphthalene) assembled from ten unique three-way branched oligonucleotides, and in principle, each of the eleven 10-mer double-helical edges could be addressed by a different TFO (Figure 10A). The authors suggest that such a device could be used for information storage with an extremely dense information content since its overall area is just 10 × 20 nm2. Binding to one of the helical edges was demonstrated by FRET between a dye located at the TFO termini (F1) and a second dye located on the underlying nanostructure (F2) and as expected could be reversed by adjusting the solution pH. Cycling was achieved at pH values higher than described above, between pH values of 6.3 and 7.5, since the TFO was composed of the stabilising nucleoside analogues 2′-aminoethoxy-T and 2′-aminothoxy-2-amino-pyridine, which offer enhanced recognition of AT and GC base pairs, respectively (37).
Figure 10.

Triplex-directed targeting of DNA nanostructures: Triplex sequence addressability of various DNA architectures. (A) Targeting of an individual sequence within a hexagonal array generated from three-way branched oligonucleotides. The complex contains an appropriately positioned FRET pair (F1 and F2) that allows the association and dissociation of the TFO to be monitored by pH change. Adapted from (90) with permission. (B) Triplex-directed scaffolding of a double-crossover tile and array with a streptavidin proteins. Adapted from (91) with permission. (C) Targeting of an individual sequence positioned within a DNA origami frame. Binding of the TFO results in the association of the two duplexes running along the centre of the frame into an X-shaped structure that is visualised by AFM. Adapted from (93) with permission. (D) Triplex- directed scaffolding of a tensegrity triangle crystal with a cyanine dye. Adapted from (94) with permission.
More recently we have extended this strategy to the targeting of a more complex structure assembled by crossover strand exchange; a double-crossover tile and array (Figure 1B) (91). Before these studies we were concerned that the densely packed nature of helices and close proximity of crossovers may have occluded binding of the oligonucleotide. An AB-type array was chosen since it allowed the interaction of the TFO to be examined first with individual tiles (i.e. Tile A alone) by simple electrophoretic, thermal denaturation and enzymatic protection assays, followed by direction visualisation of its interactions with the extended tile assemblies by AFM (i.e. both Tile A and B in an AB-type array) (Figure 10B). These studies revealed that the TFO was capable of interacting specifically with the tile and array and that binding sites were accessible at oligopurine sequences located within the crossover and non-crossover strands, and intriguingly across a region that spanned the junction itself. The latter is possible since binding of a TFO within the major groove is asymmetric, with the oligonucleotide recognising only the oligopurine sequence of the duplex, which was located along the non-crossover strand of the junction. To our knowledge, this was the first example of a triple-helical crossover junction and gives rise to the possibility of designing structures based on this motif. More importantly, these studies also demonstrated that a bound TFO was capable of recruiting a protein (streptavidin) to a DNA nanostructure through its interaction with a biotin group attached to the end of the oligonucleotide. Through design this led to the periodic positioning of the protein on the array with a repeat spacing of 32 nm in the direction that the tiles cohere (yellow spheres; Figure 10B). Other spacing should be possible by adjustment to tile design. For example, the smallest theoretical stable DX tile is generated with a separation of 16 base pairs between helix ends (i.e. one half and one whole turn between crossovers within or between tiles, respectively) and the positioning of single binding sites in adjacent tiles would yield a repeat spacing of 5.5 nm. By exploiting tiles of the AB-, ABC- and ABCD-type systems, where only tile A contains a binding site, would increase the repeat spacing to 11, 16.5 and 22 nm, respectively (92).
The Sugiyama and Endo groups have also examined triplex formation within the context of a DNA origami structure (93). The goal of this study was not to demonstrate its targeted modification of the structure per se but to visualise the process of triplex formation itself. To do so, the groups designed a DNA origami frame composed of two adjacent but separated double-helices running through the centre of the frame (Figure 10C). These were designed in such a fashion that a triplex was formed through the association of the two single-stranded regions within the helices upon addition of either its R-strand or third strand. In doing so the two helices are brought together and generate an X-shaped structure in the centre of the frame which could be imaged by AFM. The formation of both parallel and antiparallel triplexes was observed, with the former, as expected, dependent on the pH of the solution. In an elegant adaption to this study the authors also monitored the association of the TFO in real-time using high speed AFM. To do so the TFO was modified with a photocaged group (N3-6-nitropiperonyloxymethyl thymidine) that prevents TFO binding until exposure with UV light. Under their experimental conditions triplex formation was observed to occur in a time frame of seconds.
Arrangements in 3D
One of the most exciting structures to have been developed in recent years is the tensegrity triangle crystal described by Mao and Seeman (8,15). The tensegrity triangle is a robust motif consisting of three double-helices directed along linearly independent vectors (8). By tailing the helices with sticky-ends, each triangle can associate with six others, along three different directions, yielding macroscopic DNA crystals (Figure 1E) (15). Crystals have been assembled from triangles containing from two to four helical turns per edge and offer rhombohedral cavities with dimensions that exceed 1000 nm3. It therefore seems plausible that they will be able to host a variety of components, ranging from small-molecules, to nanoclusters, to larger macromolecules such as proteins. Consequently, we have been investigating the triplex addressability of the tensegrity triangle crystal for the scaffolding of molecules in 3D space (Figure 10D). Since triangles can be assembled with and without three-fold rotational symmetry we have demonstrated the selective targeting of triangles with binding sites embedded in either one or three of the double-helices, respectively (94,95). In both cases the TFO-bound triangles were capable of self-assembled crystal growth and grew to the expected size and morphology. Interestingly, we have also demonstrated one-pot assembly of the triplex-modified crystal by exploiting the slow association rate of the TFO; annealing the triangle at a fast rate before the crystallisation step allowed the double-helical regions of the triangle to first form before TFO binding and did not seem to adversely affect crystal growth or morphology. Moreover, these studies have shown that TFOs can be used to incorporate non-nucleic components within the asymmetric unit cell of the crystal (i.e. one third of the triangle). For example the attachment of a cyanine dye to the end of the TFO led to the expected change in colour of the crystal (e.g. attachment of Cy3 resulted in a red crystal; Figure 10D). These experiments were undertaken using unmodified and modified TFOs containing the nucleoside analogues 2′-aminoethoxy-T and 2′-aminoethoxy-C that help to alleviate the pH dependence of triplex formation and allowed modification of the crystal at neutral pH. The triangle system we used here contained three helical turns per edge and the targeting of a single component to each helix within the crystal dictates its positioning with sub-nanometer precision; each component is separated by ca. 10.5 nm along the helix axis between tiles and 5.8 nm through 3D space within the same tile (i.e. between the 5’ ends of each TFO). Since a typical 100 μm crystal contains an estimated 1012 unit cells, full occupancy of each binding site within the crystal would result in the incorporation of the same number of periodically repeating components within the crystal, resulting in a local concentration of ca. 10 mM. Other spacings should also be possible by using triangles with two or four helical turns per edge (15), or alternatively, by using AB-type crystal systems with TFO binding sites positioned on every other tile in the 3D lattice (96). Such modified crystals are likely to offer applications that include the organisation of nanoelectronics, the manipulation of biological or chemical cascades, and the structure determination of periodically positioned molecules by X-ray diffraction analysis.
For improving structure stability
One of the major drawbacks of generating nanostructures through W–C hybridization is that it involves reversible non-covalent interactions, limiting these structures to applications at relatively low temperatures, under conditions that promote stable hydrogen bond formation (i.e. specific ionic conditions, pH, etc.). This is particularly problematic for extended tile-based structures held together by short sticky-ends, such as the DX and tensegrity triangle systems described above (14,15). The latter is also hampered by the necessity for an increased ionic strength during crystallization; removal of the crystals into a physiological, or other relevant buffer, is not possible, since it lowers their stability, resulting in crystal dissolution. The simplest means to overcome this problem is to increase the stability of the duplex regions formed by sticky-end cohesion by the binding of a third strand across these sites (i.e. TFO binding across the staggered nick sites in the duplex). Triplex formation is also likely to increase the torsional rigidity (97) of the underlying duplex. Mao and co-workers exploited this approach to increase the stability of a tensegrity triangle crystal by the inclusion of an appropriate site across the 2-nucleotide sticky-ends between the tiles of the crystal (98). In the absence of TFO the DNA crystals were only stable in solutions of high ionic strength (e.g. >1.2 M (NH4)2SO4), whilst in its presence the crystals were stable at ionic strengths as low as that of 0.02 M solution of (NH4)2SO4. More importantly, the strategy can be undertaken post crystal assembly by soaking the crystals with the TFO, minimizing any influence on the crystal assembly process.
We have developed a different strategy for improving nanostructure stability that involves directing cross-linking reactions to the underlying DNA using the photo-cross-linking agent 4,5,8-trimethylpsoralen (psoralen) (95,99). Intercalation of free psoralen at TpA steps leads to a 2+2 cycloaddition reaction with the adjacent thymidines upon UV exposure, thereby cross-linking the two duplex strands. Indeed experiments have shown that cross-linking can increase the thermal stability of nanostructures assembled by DNA origami (100). However, multiple intercalation events are not tolerated well by smaller motifs, such as DX or tensegrity triangle motif since it will unwind the DNA and disrupt the precise crossover positioning required for tile and/or lattice assembly. We have therefore demonstrated that TFOs can be used to overcome this limitation by directing the specific photo-cross-linking of psoralen to unique loci within a nanostructure by its attachment to the end of the oligonucleotide. This was achieved by embedding an appropriate oligopurine–oligopyrimidine sequence with an adjacent TpA step located at the 5′-end of the TFO target sequence (Figure 11A). Positioning the sequence across regions that span the sticky-ends between tiles of both DX and tensegrity triangle systems (e.g. Figure 11B) resulted in their cross-linking upon UV exposure (95,99). The latter was particular advantageous since it led to an increase in the thermal stability of the crystal; targeting just one of the three helices led to an increase in the melting temperature of the crystal by ca. 8°C. Importantly, UV exposure did not seem to disrupt crystal assembly. Crystal stability might be improved further by designing a system that cross-links the intermolecular contacts at each end of all three helices within the triangle. Such covalently closed crystals may prove useful for applications that entail removal of the crystals from their mother liquor. These studies also highlight that triplex formation could be used to direct any reactive group compatible with oligonucleotide synthesis to unique locations within a DNA nanostructure, for example a DNA cleavage agent (31,32). One can envisage that by exploiting a ‘tool box’ of such small molecule–oligonucleotide conjugates it will be possible to reconfigure a variety of DNA nanostructures into topologies previously unattainable by W–C hybridization alone.
Figure 11.
Triplex-directed modification of DNA nanostructures: (A) triplex-directed intercalation and photo-cross-linking of a tethered psoralen molecule to a TpA step introduced adjacent to a TFO target sequence. UV exposure results in a 2+2 cycloaddition reaction with the adjacent thymidines (shown in light blue), thereby cross-linking the two strands. (B) Appropriate targeting of a TpA step and target sequence embedded between adjacent tiles of a tensegrity triangle crystal allows cross-linking across the sticky-ends and increases crystal stability. Adapted from (95) with permission.
OUTLOOK: A THIRD STRAND FOR DNA NANOTECHNOLOGY?
Triplexes have found most use in oligonucleotide- and tile-based structures since oligopurine–oligopyrimidine target sequences can easily be embedded within the double-helical regions of the structure with little influence on nanostructure topology. This is in contrast to their incorporation within structures assembled by the DNA origami approach which is restricted by the limited number of naturally present target sequences located within the M13mp18 scaffold: simple sequence analysis reveals three potential oligopurine sequences greater than ten nucleotides in length, and around 13 that contain one or two pyrimidine interruptions. One means to increase the number of suitable sequences would be to use third strands containing base analogues designed to recognise pyrimidine bases (36,37). It should also be possible to remove problematic pyrimidines within the scaffold by standard cloning or mutagenesis procedures or alternatively, within the staple strands by generating mismatched base pairs, i.e. by generating AG, GG, AA or GA mismatches. The latter would substantially increase the number of target sequences with only a slight loss in stability of the origami (47). Another strategy would be to include appropriate binding sites within hairpin or dumbbell duplexes projected above the nanostructure surface (4). The number of possible binding sites would then depend on the number of projections per staple, as well as the total number of modified staples (>250 sites) required for origami assembly.
There is a variety of base, sugar and backbone modifications that can be used to improve the triplex-forming properties of oligonucleotides, such as their binding affinity, pH dependence and kinetics, and the catalogue of such modifications compatible with standard phosphoramidate chemistry is continually expanding (36,37). The use of such modifications offers the ability to fine-tune the binding of the third strand in a manner that has already been seen with duplex modifications (101,102). One interesting class of oligonucleotide modifications that could be applied to the field is peptide nucleic acids (PNA), where the phosphate backbone is replaced with uncharged repeating (2-aminoethyl)glycine units to which nucleobases are linked by methylene bridges (103,104). PNA can be programmed to interact with DNA via standard triplex formation (103), but more intriguingly, can also interact with duplexes through strand displacement and P-loop formation. In this instance two pyrimidine-containing strands of PNA interact with the purine-containing strand of the target duplex, generating a local triplex (104). The resultant triplex is much more stable than the equivalent DNA triplex on account of the lower charge repulsion and may reduce the concentration of counterions required to stabilise such structures.
FUNDING
Funding for open access charge: This work was supported by BBSRC grant BB/H019219/1 to D.A.R.
Conflict of interest statement. None declared.
REFERENCES
- 1. Seeman N.C. Nanomaterials based on DNA. Annu. Rev. Biochem. 2010; 79:65–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Linko V., Dietz H.. The enabled state of DNA nanotechnology. Curr. Opin. Biotechnol. 2013; 24:1–7. [DOI] [PubMed] [Google Scholar]
- 3. Fu T.-J., Seeman N.C.. DNA double-crossover molecules. Biochemistry. 1993; 32:3211–3220. [DOI] [PubMed] [Google Scholar]
- 4. Rothemund P.W.K. Folding DNA to create nanoscale shapes and patterns. Nature. 2006; 440:297–302. [DOI] [PubMed] [Google Scholar]
- 5. Kuzuya A., Wang R., Sha R., Seeman N.C.. Six-helix and eight-helix DNA nanotubes assembled from half-tubes. Nano Lett. 2007; 7:1757–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Douglas S.M., Dietz H., Liedl T., Graf F., Shih W.M.. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature. 2009; 459:414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ke Y., Douglas S.M., Liu M., Sharma J., Cheng A., Leung A., Liu Y., Shih W.M., Yan H.. Multilayer DNA origami packed on a square lattice. J. Am. Chem. Soc. 2009; 131:15903–15908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu D, Wang M., Deng Z., Walulu R., Mao C.. Tensegrity: construction of rigid DNA triangles with flexible four-arm junctions. J. Am. Chem. Soc. 2004; 126:2324–2325. [DOI] [PubMed] [Google Scholar]
- 9. He Y., Ye T., Zhang C., Ribbe A.E., Jiang W., Mao C.. Hierarchical self-assembly of DNA into symmetric supramolecular polyhedra. Nature. 2008; 452:198–199. [DOI] [PubMed] [Google Scholar]
- 10. Ke Y., Sharma J., Liu M., Jahn K., Liu Y., Yan H.. Scaffold DNA origami of a DNA tetrahedron molecular container. Nano Lett. 2009; 9:2445–2447. [DOI] [PubMed] [Google Scholar]
- 11. Dietz H., Douglas S.M., Shih W.M.. Folding DNA into twisted and curved nanoscale shapes. Science. 2009; 325:725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Han D., Pal S., Nangreave J., Deng Z., Liu Y., Yan H.. DNA origami with complex curvatures in three-dimensional space. Science. 2011; 332:342–346. [DOI] [PubMed] [Google Scholar]
- 13. Woo S., Rothemund P.W.K.. Programmable molecular recognition based on the geometry of DNA nanostructures. Nat. Chem. 2011; 3:620–627. [DOI] [PubMed] [Google Scholar]
- 14. Winfree E., Liu F., Wenzler L.A., Seeman N.C.. Design and self-assembly of two-dimensional DNA crystals. Nature. 1998; 394:539–544. [DOI] [PubMed] [Google Scholar]
- 15. Zheng J., Birktoft J.J., Chen Y., Wang T., Sha R., Constantinou P.E., Ginell S.L., Mao C., Seeman N.C.. From molecular to macroscopic via the rational design of a self- assembled DNA crystal. Nature. 2009; 461:74–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu W., Zhong H., Wang R., Seeman N.C.. Crystalline two-dimensional DNA origami arrays. Angew. Chem. Int. Ed. Engl. 2011; 50:264–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martin T.G., Dietz H.. Magnesium-free self-assembly of multi-layer DNA objects. Nat. Commun. 2012; 3:1103–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Duckett D.R., Murchie A.I.H., Lilley D.M.J.. The role of metal ions in the conformation of the four-way junction. EMBO J. 1990; 9:583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Seeman N.C. De novo design of sequences for nucleic acid structural engineering. J. Biomol. Struct. Dyn. 1990; 8:573–581. [DOI] [PubMed] [Google Scholar]
- 20. Douglas S.M., Marblestone A.H., Teerapittayanon S., Vazquez A., Church G.M., Shih W.M.. Rapid prototyping of 3D DNA-origami shapes with caDNAno. Nucleic Acids Res. 2009; 37:5001–5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sobczak J.P., Martin T.G., Gerling T., Dietz H.. Rapid folding of DNA into nanoscale shapes at constant temperature. Science. 2012; 338:1458–1461. [DOI] [PubMed] [Google Scholar]
- 22. Watson J.D., Crick F.H.C.. Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid. Nature. 1953; 171:737–738. [DOI] [PubMed] [Google Scholar]
- 23. Rich A. DNA comes in many forms. Gene. 1993; 135:99–109. [DOI] [PubMed] [Google Scholar]
- 24. Yatsunyk L.A., Mendoza O., Mergny J.L.. “Nano-oddities”: unusual nucleic assemblies for DNA based nanostructures and nanodevices. Acc. Chem. Res. 2014; 47:1836–1844. [DOI] [PubMed] [Google Scholar]
- 25. Hu Y., Cecconello A., Idili A., Ricci F., Willner I.. Triplex DNA nanostructures: From basic properties to applications. Angew. Chem. Int. Ed. 2017; 56:15210–15233. [DOI] [PubMed] [Google Scholar]
- 26. Rusling D.A., Fox K.R.. Sequence-specific recognition of DNA nanostructures. Methods. 2014; 67:123–133. [DOI] [PubMed] [Google Scholar]
- 27. Felsenfeld G., Davies D.R., Rich A.. Formation of a three-stranded polynucleotide molecule. J. Am. Chem. Soc. 1957; 79:2023–2024. [Google Scholar]
- 28. Lyamichev V.I., Mirkin S.M., Frank-Kamenetskii M.D. A pH-dependent structural transition in the homopurine-homopyrimidine tract in superhelical DNA. J. Mol. Biol. Struct. Dyn. 1985; 3:327–338. [DOI] [PubMed] [Google Scholar]
- 29. Mirkin S.M., Lyamichev V.I., Drushlyak K.N., Dobrynin V.N., Filipov S.A., Frank- Kamenetskii. DNA H form requires a homopurine-homopyrimidine mirror repeat. Nature. 1987; 330:495–497. [DOI] [PubMed] [Google Scholar]
- 30. Jain A., Wang G., Vasquez K.M.. DNA triple helices: biological consequences and therapeutic potential. Biochemie. 2008; 90:117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moser H.E., Dervan P.B.. Sequence-specific cleavage of double-helical DNA by triple helix formation. Science. 1987; 238:645–650. [DOI] [PubMed] [Google Scholar]
- 32. Le Doan T., Perrouault L., Praseuth D., Habhoub N., Decout J.L., Thuong N.T., Lhomme J., Hélène C.. Sequence-specific recognition, photocrosslinking and cleavage of the DNA double helix by an oligo-(alpha)-thymidylate covalently linked to an azidoproflavine derivative. Nucleic Acids Res. 1987; 15:7749–7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rusling D.A., Broughton-Head V.J., Brown T., Fox K.R.. Towards the targeted modulation of gene expression by modified triplex-forming oligonucleotides. Curr. Chem. Biol. 2008; 2:1–10. [Google Scholar]
- 34. Beal P.A., Dervan P.B.. Second structural motif for recognition of DNA by oligonucleotide-directed triple-helix formation. Science. 1991; 251:1360–1363. [DOI] [PubMed] [Google Scholar]
- 35. Durland R.H., Kessler D.J., Gunnell S., Duvic M., Pettitt B.M., Hogan M.E.. Binding of triple-helix forming oligonucleotides to sites in gene promoters. Biochemistry. 1991; 30:9246–9255. [DOI] [PubMed] [Google Scholar]
- 36. Rusling D.A., Powers V.E.C., Ranasinghe R.T., Wang Y., Osborne S.D., Brown T., Fox K.R.. Four base recognition by triplex-forming oligonucleotides at physiological pH. Nucleic Acids Res. 2005; 33:3025–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rusling D.A, Brown T., Fox K.R.. Waring M. DNA recognition by triplex formation. Sequence-Specific DNA Binding Agents. 2006; RSC Publishing; 1–28. [Google Scholar]
- 38. Koshlap K.M., Gillespie P., Dervan P.B., Feigon J.. Solution structure of an intramolecular DNA triplex containing an N7-glycosylated guanine which mimics protonated cytosine. Biochemistry. 1993; 9:7908–7909. [DOI] [PubMed] [Google Scholar]
- 39. Asensio J.L., Carr R., Brown T., Lane A.N.. Solution conformation of a parallel DNA triple helix containing 5’ and 3’ triplex-duplex junctions. J. Am. Chem. Soc. 1999; 121:11063–11070. [DOI] [PubMed] [Google Scholar]
- 40. Lee J.S., Woodsworth M.L., Latimer L.J.P., Morgan A.R.. Poly(pyrimidine).poly(purine) synthetic DNAs containing 5-methylcytosine from stable triplexes at neutral pH. Nucleic Acids Res. 1984; 12:6603–6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. James P.J., Brown T., Fox K.R.. Thermodynamic and kinetic stability of intermolecular triple helices containing different portions of C+-GC and T-AT triplets. Nucleic Acids Res. 2003; 31:5598–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rusling D.A., Rachwal P.A., Brown T., Fox K.R.. The stability of triplex DNA is affected by the stability of the underlying duplex. Biophys. Chem. 2009; 145:105–110. [DOI] [PubMed] [Google Scholar]
- 43. Roberts R., Crothers D.. Specificity and stringency in DNA triplex formation. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:9397–9401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rougée M., Faucon B., Mergny J.L., Barcelo F., Giovannangeli C., Garestier T., Thoung N.T., Hélène C.. Kinetics and thermodynamics of triple-helix formation; effects of ionic strength and mismatches. Biochemistry. 1992; 31:9269–9278. [DOI] [PubMed] [Google Scholar]
- 45. Best G.C., Dervan P.B.. Energetics of formation of sixteen triple helical complexes which vary at a single position within a pyrimidine motif. J. Am. Chem. Soc. 1995; 117:1187–1193. [Google Scholar]
- 46. Mergny J.L., Sun J.S., Rougée M., Montenay-Garester T., Barcelo F., Chomilier J., Hélène C.. Sequence-specificity in triple-helix formation: experimental and theoretical studies of the effect of mismatches on triplex stability. Biochemistry. 1991; 30:9791–9798. [DOI] [PubMed] [Google Scholar]
- 47. Rusling D.A, Brown T., Fox K.R.. DNA triple-helix formation at target sites containing duplex mismatches. Biophys. Chem. 2006; 123:134–140. [DOI] [PubMed] [Google Scholar]
- 48. Darby R.A.J., Fox K.R. Demeunynck M, Bailly C, Wilson WD. Triple helix-specific ligands. Interaction of Small Molecules with DNA and RNA: from Synthesis to Nucleic Acid Complexes. 2002; WILEY-VCH Verlag GmbH; 360–383. [Google Scholar]
- 49. Rusling D.A., Fox K.R. Fox KR, Brown T. Small-molecule oligonucleotide conjugates. DNA Conjugates and Sensors. 2002; RSC Publishing; 103–117. [Google Scholar]
- 50. Kohwi Y., Kohwi-Shigematsu T.. Magnesium ion-dependent triple-helix structure formed by homopurine-homopyrimdine sequences in supercoiled DNA. Proc. Natl. Acad. Sci. U.S.A. 1998; 85:3781–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sarai A., Sugiura S., Torigoe H., Shindo H.. Thermodynamic and kinetic analysis of DNA triplex formation: application of filter binding assay. J. Biomol. Struct. Dyn. 1993; 11:245–252. [DOI] [PubMed] [Google Scholar]
- 52. Rusling D.A., Broughton-Head V.J., Tuck A., Khairallah H., Osborne S.D., Brown T., Fox K.R.. Kinetic studies on the formation of DNA triplexes containing the nucleoside analogue 2’-O-(2-aminoethyl)-5-(3-amino-1-propynyl)uridine. Org. Biomol. Chem. 2008; 6:122–129. [DOI] [PubMed] [Google Scholar]
- 53. Rusling D.A., Peng G., Srinivasan N., Fox K.R., Brown T.. DNA triplex formation with 5-dimethylaminopropargyl deoxyuridine. Nucleic Acids Res. 2009; 37:1288–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang Y., Seelig G.. Dynamic DNA nanotechnology using strand-displacement reactions. Nat. Chem. 2011; 3:103–113. [DOI] [PubMed] [Google Scholar]
- 55. Mao C., Sun W., Shen Z., Seeman N.C.. A nanomechanical device based on the B-Z transition of DNA. Nature. 1999; 397:144–146. [DOI] [PubMed] [Google Scholar]
- 56. Yurke B., Turberfield A.J., Mills A.P., Simmel F.C., Neumann J.L.. A DNA-fuelled molecular machine made of DNA. Nature. 2000; 406:605–608. [DOI] [PubMed] [Google Scholar]
- 57. Chen Y., Lee S-H., Mao C.. A DNA nanomachine based on a duplex-triplex transition. Angew. Chem. Int. Ed. 2004; 43:5335–5338. [DOI] [PubMed] [Google Scholar]
- 58. Brucale M., Zuccheri G., Samori B.. The dynamic properties of an intramolecular transition from DNA duplex to cytosine-motif triplex. Org. Biomol. Chem. 2005; 3:575–577. [DOI] [PubMed] [Google Scholar]
- 59. Kolaric B., Sliwa M., Brucale M., Vallée R.A.L., Zuccheri G., Samori B., Hofkens J., De Schryver F.C.. Single molecule fluorescence spectroscopy of pH sensitive oligonucleotide switches. Photochem. Photobiol. Sci. 2007; 6:614–618. [DOI] [PubMed] [Google Scholar]
- 60. Idili A., Vallée-Bélisle A., Ricci F.. Programmable pH-triggered DNA nanoswitches. J. Am. Chem. Soc. 2014; 136:5836–5839. [DOI] [PubMed] [Google Scholar]
- 61. Li X.M., Song J., Cheng T., Fu P.Y.. A duplex-triplex nucleic acid nanomachine that probes pH changes inside living cells during apoptosis. Anal. Bioanal. Chem. 2013; 405:5993–5999. [DOI] [PubMed] [Google Scholar]
- 62. Chen Y., Mao C.. Reprogramming DNA-directed chemical reactions on the basis of a DNA conformational change. J. Am. Chem. Soc. 2004; 126:13240–13241. [DOI] [PubMed] [Google Scholar]
- 63. Minero G.A.S., Wagler P.F., Oughli A.A., McCaskill J.S.. Electronic pH switching of DNA triplex reactions. RSC Adv. 2015; 5:27313–27325. [Google Scholar]
- 64. Jacobsen M.F., Ravnsbaek J.B., Gothelf K.V.. Small molecule induced control in duplex and triplex DNA-directed chemical reactions. Org. Biomol. Chem. 2010; 8:50–52. [DOI] [PubMed] [Google Scholar]
- 65. Han X., Zhou Z., Yang F., Deng Z.. Catch and release: DNA tweezers that can capture, hold, and release an object under control. J. Am. Chem. Soc. 2008; 130:14414–14415. [DOI] [PubMed] [Google Scholar]
- 66. Idili A., Plaxco K.W., Valleé-Bélisle A., Ricci F.. Thermodynamic basis for engineering high-affinity, high-specificity binding-induced DNA clamp nanoswitches. ACS Nano. 2013; 7:10863–10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Grosso E.D., Idili A., Porchetta A., Ricci F.. A modular clamp-like mechanism to regulate the activity of nucleic-acid target-responsive nanoswitches with external activators. Nanoscale. 2016; 8:18057–18061. [DOI] [PubMed] [Google Scholar]
- 68. Liao W.C., Riutin M., Parak W.J., Wilner I.. Programmed responsive microcapsules for the controlled release of CdSe/ZnS quantum dots. ACS Nano. 2016; 10:8683–8689. [DOI] [PubMed] [Google Scholar]
- 69. Hampsire A.J., Rusling D.A., Broughton-Head V.J., Fox K.R.. Footprinting: a method for determining the selectivity, affinity and kinetics of DNA-binding ligands. Methods. 2007; 42:128–140. [DOI] [PubMed] [Google Scholar]
- 70. Liu Z., Mao C.. Reporting transient molecular events by DNA strand displacement. Chem. Commun. 2014; 50:8239–8241. [DOI] [PubMed] [Google Scholar]
- 71. Zhao B., Porchetta A., Idili A., Castronovo M., Fan C., Ricci F.. Rational design of pH-controlled DNA strand displacement. J. Am. Chem. Soc. 2015; 15:5539–5544. [DOI] [PubMed] [Google Scholar]
- 72. Idili A., Porchetta A., Amodio A., Vallee-Belise A., Ricci F. Controlling hybridisation chain reactions with pH. Nano Lett. 2015; 15:5539–5544. [DOI] [PubMed] [Google Scholar]
- 73. Amodio A., Adedeji A.F., Castronovo M., Franco E., Ricci F.. pH-controlled assembly of DNA tiles. J. Am. Chem. Soc. 2016; 138:12735–12738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wu N., Willner I.. pH-stimulated reconfiguration and structural isomerization of origami dimer and trimer systems. Nano Lett. 2016; 16:6650–6655. [DOI] [PubMed] [Google Scholar]
- 75. Liu Z., Li Y., Tian C., Mao C.. A smart DNA tetrahedron that isothermally assembles or dissociates in response to the solution pH value changes. Biomacromolecules. 2013; 14:1711–1714. [DOI] [PubMed] [Google Scholar]
- 76. Jung Y.H., Lee K-B., Kim Y.G., Choi I.S.. Proton-fueled, reversible assembly of gold nanoparticles by controlled triplex formation. Angew. Chem. Int. Ed. 2006; 45:5960–5963. [DOI] [PubMed] [Google Scholar]
- 77. Yan H.L., Xiong C., Yuan H., Zeng Z.X., Ling L.S.. Spacer control the dynamic of triplex formation between oligonucleotide-modified gold nanoparticles. J. Phys. Chem. C. 2009; 113:17326–17331. [Google Scholar]
- 78. Guerrini L., Mckenzie F., Wark A.W., Faulds K., Graham D.. Tuning the interparticle distance in nanoparticle assemblies in suspension via DNA-triplex formation: correlation between plasmonic and surface-enhanced Raman scattering responses. Chem. Sci. 2012; 3:2262–2268. [Google Scholar]
- 79. Chen Y., Mao C.. pH-induced reversible expansion/contraction of gold nanoparticle aggregates. Small. 2008; 4:2191–2194. [DOI] [PubMed] [Google Scholar]
- 80. Han M.S., Lytton-Jean A.K.R., Mirkin C.A.. A gold nanoparticle based approach for screening triplex DNA binders. J. Am. Chem. Soc. 2006; 128:4954–4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Xiong C., Wu C., Zhang H., Ling L.. Gold nanoparticles-based colorimetric investigation of triplex formation under weak alkali pH environment with the aid of Ag+. Spectrochim. Acta A. 2011; 79:956–961. [DOI] [PubMed] [Google Scholar]
- 82. Feng L., Huang Z., Ren J., Qu X.. Toward site-specific, homogeneous and highly stable fluorescent silver nanocluster fabrication on triplex DNA scaffolds. Nucleic Acids Res. 2012; 40:e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ihara T., Ishii T., Araki N., Wilson A.W., Jyo A.. Silver ion unusually stabilizes the structure of a parallel-motif DNA triplex. J. Am. Chem. Soc. 2009; 131:3826–3827. [DOI] [PubMed] [Google Scholar]
- 84. Zhao C., Qu K., Xu C., Ren J., Qu X.. Triplex inducer-directed self-assembly of single-walled carbon nanotubes: a triplex DNA-based approach for controlled manipulation of nanostructures. Nucleic Acids Res. 2011; 39:3939–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ren J., Hu Y., Lu C-H., Guo W., Aleman-Garcia M.A., Ricci F., Willner I.. pH-responsive and switchable triplex-based hydrogels. Chem. Sci. 2015; 6:4190–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hu Y., Guo W., Kahn J.S., Aleman-Garcia M.A., Willner I.. A shape memory DNA-based hydrogel exhibiting two internal memories. Angew. Chem. Int. Ed. 2016; 55:4210–4214. [DOI] [PubMed] [Google Scholar]
- 87. Goodman R.P., Schaap I.A., Tardin C.F., Erben C.M., Berry R.M., Schmidt C.F., Turberfield A.J.. Rapid chiral assembly of rigid building blocks for molecular nanofabrication. Science. 2005; 310:1661–1665. [DOI] [PubMed] [Google Scholar]
- 88. Lund K., Liu Y., Lindsay S., Yan H.. Self-assembling a molecular peg board. J. Am. Chem. Soc. 2005; 127:17606–17607. [DOI] [PubMed] [Google Scholar]
- 89. Seeman N.C. Nucleic acid junctions and lattices. J. Theor. Biol. 1982; 99:237–247. [DOI] [PubMed] [Google Scholar]
- 90. Tumpane J., Kumar R., Lundberg E.P., Sandin P., Gale N., Nandhakumar I.S., Albinsson B., Lincoln P., Wilhelmsson L.M., Brown T., Norden B.. Triplex addressability as a basis for functional DNA nanostructures. Nano Lett. 2007; 7:3832–3839. [DOI] [PubMed] [Google Scholar]
- 91. Rusling D.A., Nandhakumar I.S., Brown T., Fox K.R.. Triplex-directed recognition of a DNA nanostructure assembled by crossover strand exchange. ACS Nano. 2012; 6:3604–3613. [DOI] [PubMed] [Google Scholar]
- 92. Furong L., Sha R., Seeman N.C.. Modifying the surface features of two-dimensional DNA crystals. J. Am. Chem. Soc. 1999; 121:917–922. [Google Scholar]
- 93. Yamagata Y., Emura T., Hidaka K., Sugiyama H., Endo M.. Triple helix formation in a topologically controlled DNA nanosystem. Chem. Eur. J. 2016; 22:5494–5498. [DOI] [PubMed] [Google Scholar]
- 94. Rusling D.A., Chandrasekaran A.R., Ohayon Y.P., Brown T., Fox K.R, Sha R., Mao C., Seeman N.C.. Functionalizing designer DNA crystals with a triple-helical veneer. Angew. Chem. Int. Ed. 2014; 3:3979–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Abdallah H.O., Ohayon Y.P., Chandrasekaran A.R., Sha R., Fox K.R., Brown T., Rusling D.A., Mao C., Seeman N.C.. Stabilisation of self-assembled DNA crystals by triplex-directed photo-cross-linking. Chem. Commun. 2016; 52:8014–8017. [DOI] [PubMed] [Google Scholar]
- 96. Wang T., Sha R., Birktoft J, Zheng J., Mao C., Seeman N.C.. A DNA crystal designed to contain two molecules per asymmetric unit. J. Am. Chem. Soc. 2010; 132:15471–15473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Barone F., Chirico G., Matzeu M., Mazzei F., Pedone F.. Triple helix DNA oligomer melting measured by fluorescence polarization spectroscopy. Eur. Biophys. J. 1998; 27:137–146. [DOI] [PubMed] [Google Scholar]
- 98. Zhao J., Chandrasekaran A.R., Li Q., Li X., Sha R., Seeman N.C., Mao C.. Post-assembly stabilization of rationally designed DNA crystals. Angew. Chem. Int. Ed. 2015; 54:9936–9939. [DOI] [PubMed] [Google Scholar]
- 99. Rusling D.A., Nandhakumar I.S., Brown T., Fox K.R.. Triplex-directed covalent cross-linking of a DNA nanostructure. Chem. Commun. 2012; 48:9592–9594. [DOI] [PubMed] [Google Scholar]
- 100. Rajendran A., Endo M., Katsuda Y., Hidaka K., Sugiyama H.. Photo-cross-linking-assisted thermal stability of DNA origami structures and its application for higher- temperature self-assembly. J. Am. Chem. Soc. 2011; 133:14488–14491. [DOI] [PubMed] [Google Scholar]
- 101. Lukeman P.S, Mittal A.C., Seeman N.C.. Two dimensional PNA/DNA arrays: estimating the helicity of unusual nucleic acid polymers. Chem. Commun. 2004; 1694–1695. [DOI] [PubMed] [Google Scholar]
- 102. Rinker S., Liu Y., Yan H.. Two dimensional LNA/DNA arrays: estimating the helicty of LNA/DNA hybrid duplex. Chem. Commun. 2006; 2675–2677. [DOI] [PubMed] [Google Scholar]
- 103. Nielsen P.E., Egholm R.H., Berg R.H., Buchardt O.. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science. 1991; 254:1497–1500. [DOI] [PubMed] [Google Scholar]
- 104. Wittung P., Nielsen P., Norden B.. Extended DNA-recognition repetoire of peptide nucleic acid (PNA): PNA ds-DNA triplex formed with cytosine-rich homopyrimidine PNA. Biochemistry. 1997; 36:7973–7979. [DOI] [PubMed] [Google Scholar]