

Abstract
Reactive astrogliosis, a complex process characterized by cell hypertrophy and upregulation of components of intermediate filaments, is a common feature in brains of Alzheimer's patients. Reactive astrocytes are found in close association with neuritic plaques; however, the precise role of these glial cells in disease pathogenesis is unknown. In this study, using immunohistochemical techniques and light and electron microscopy, we report that plaque‐associated reactive astrocytes enwrap, engulf and may digest presynaptic dystrophies in the hippocampus of amyloid precursor protein/presenilin‐1 (APP/PS1) mice. Microglia, the brain phagocytic population, was apparently not engaged in this clearance. Phagocytic reactive astrocytes were present in 35% and 67% of amyloid plaques at 6 and 12 months of age, respectively. The proportion of engulfed dystrophic neurites was low, around 7% of total dystrophies around plaques at both ages. This fact, along with the accumulation of dystrophic neurites during disease course, suggests that the efficiency of the astrocyte phagocytic process might be limited or impaired. Reactive astrocytes surrounding and engulfing dystrophic neurites were also detected in the hippocampus of Alzheimer's patients by confocal and ultrastructural analysis. We posit that the phagocytic activity of reactive astrocytes might contribute to clear dysfunctional synapses or synaptic debris, thereby restoring impaired neural circuits and reducing the inflammatory impact of damaged neuronal parts and/or limiting the amyloid pathology. Therefore, potentiation of the phagocytic properties of reactive astrocytes may represent a potential therapy in Alzheimer's disease.
Keywords: Alzheimer's disease, neurodegeneration, neuropathology, reactive astrocyte, synaptopathy
1. INTRODUCTION
Alzheimer's disease (AD) is the most common cause of dementia in elderly individuals and, so far, an effective treatment that prevents onset and disease progression remains elusive. In addition to amyloid plaques and neurofibrillary tangles, the two signature pathological lesions, Alzheimer's brains manifest prominent astrocytic and microglial reactions (De Strooper & Karran, 2016; Heneka et al., 2015; Osborn, Kamphuis, Wadman, & Hol, 2016; Serrano‐Pozo, Muzikansky, et al., 2013; Verkhratsky, Parpura, Pekna, Pekny, & Sofroniew, 2014). Glia‐mediated neuroinflammation has been proposed as a potential mechanism underlying AD pathogenesis and over the past decade activated microglia have been the focus of increasing research (Heneka et al., 2015; Heppner, Ransohoff, & Becher, 2015); however, the precise contribution of reactive astrocytes is still undetermined. Astrocyte reactivity is specifically identified in the vicinity of amyloid plaques and can be monitored by the increased expression of the intermediate filament protein, GFAP, and, morphologically, by the hypertrophy of their main processes (Osborn et al., 2016; Verkhratsky, Zorec, Rodriguez, & Parpura, 2016). Though emerging data support the molecular and functional diversity of astrocytes in adult brain (Haim & Rowitch, 2017; John Lin et al., 2017), little is known about disease‐activated astrocyte diversity. The activation of astrocytes has been often viewed as a neuroinflammatory toxic response in AD; however, this concept is currently changing to the notion of loss‐of‐function (Masgrau, Guaza, Ransohoff, & Galea, 2017) or protection. As an example of the latter, in amyloid precursor protein (APP) mice in which the GFAP and Vimentin genes have been deleted, plaques are more numerous and larger (Kraft et al., 2013), a result that suggest that reactive astrocytes limit the progression of plaques, however the mechanisms underlying these effects are still unclear. Reactive astrocytes could restrict amyloid pathology by phagocytosing and degrading amyloid‐β (Aβ) species (Xiao et al., 2014), since astrocytes have been shown to clear amyloid deposits in vitro (Wyss‐Coray et al., 2003). The discovery that astrocytes express receptors and downstream signaling molecules involved in phagocytic pathways (Cahoy et al., 2008) supports new physiological roles for these cells, including the phagocytosis of synapses, both during development and in the adult brain (Chung et al., 2013). Further supporting a phagocytic profile, a recent study has described different astrocyte subpopulations by their specific gene signatures in adult mouse brain, and, interestingly, some subsets are enriched in genes linked to phagocytosis (John Lin et al., 2017). However, reactive astrocytes might display disease‐specific dysfunctional phenotypes and thus contribute to circuit failure and disease progression.
As far as we know, there is no study showing that astrocytes phagocytose dystrophic neurites or neuronal debris in the context of neurodegenerative pathologies such as AD. In this study, we show that reactive astrocytes surrounding Aβ plaques in the hippocampus of APP/PS1 mice enwrap and engulf axonal/presynaptic dystrophies, probably aiming at eliminating aberrant presynaptic elements. We also detected these phagocytic astrocytes in the hippocampus of AD patients. However, the persistence of dystrophies around plaques with disease progression might result from impaired phagocytic function of astrocytes. Therefore, a therapeutic approach to enhance the efficiency of astrocyte‐mediated clearance activity may contribute to restore neuronal circuits, reduce the Aβ pathology and/or prevent the inflammatory response in AD.
2. MATERIALS AND METHODS
2.1. Transgenic mice
Male transgenic mice expressing both amyloid precursor protein and presenilin‐1 mutations (APP751Swe‐London/PS1M146L) of 4 (n = 8), 6 (n = 8), and 12 (n = 8) months of age were used. These bigenic mice were obtained (see Blanchard et al., 2003) by crossing homozygous PS1 mice (expressing human mutant PS1[M146L] under HMGCoA reductase promoter) to hemizygous APP751SL mice (expressing human mutant APP751 carrying the Swedish [KM670/671NL] and London [V717I] mutations under the control of the Thy1 promoter). We have previously characterized this APP/PS1 transgenic model (Baglietto‐Vargas et al., 2010; Jimenez et al., 2008; Ramos et al., 2006; Sanchez‐Varo et al., 2012; Torres et al., 2012; Trujillo‐Estrada et al., 2014). Age‐matched non‐transgenic mice (WT) of the same genetic background (C57BL:6) were used as controls. All animal experiments were carried out in accordance with the Spanish and the European Union regulations (RD53/2013 and 2010/63/UE), and with the approval of the committee of Animal Research from the University of Malaga (Spain).
2.2. Human samples
Human autopsy specimens (Braak 0, n = 8; Braak II, n = 13, Braak III–IV, n = 10, and Braak V–VI, n = 18) from the medial temporal lobe (hippocampal region) were obtained from the tissue bank Fundacion CIEN (BT‐CIEN; Madrid, Spain) and the Neurological Tissue Bank of IDIBELL‐Hospital of Bellvitge (Barcelona, Spain). The utilization of post mortem human samples was approved by the corresponding biobank ethics committees and the “Comite de Etica de la Investigacion (CEI), Hospital Virgen del Rocio”, Seville, Spain. All the subjects (Braak 0, II, III–IV and V–VI) in this study are identical to those reported by us previously (Sanchez‐Mejias et al., 2016). All cases were scored for Braak tau pathology. Only Braak V–VI cases were clinically classified as demented patients. The number of cases for the different experiments is indicated in the corresponding results section or figure.
2.3. Tissue preparation
For light microscopy studies APP/PS1 and age‐matched WT animals were anesthetized with sodium pentobarbital (60 mg/kg) and transcardially perfused with 0.1 M phosphate‐buffered saline (PBS), followed by 4% paraformaldehyde, 75 mM lysine, 10 mM sodium metaperiodate in 0.1 M phosphate buffer (PB). Fixed and cryoprotected brains were serially sectioned at 40 µm thickness in the coronal plane on a freezing microtome. Human samples were fixed in 4% paraformaldehyde and sectioned at 30 µm thickness on a freezing microtome. For Westerns and quantitative real‐time PCR (qPCR), unfixed and frozen hippocampi from post mortem human samples were used. For transmission electron microscopy (TEM), mice and human sections were postfixed in 1% osmium tetroxide in 0.1 M PB, block stained with uranyl acetate, dehydrated in graded acetone and embedded in Araldite (EMS, USA). Selected areas were cut in ultrathin sections and examined with an electron microscope (JEOL JEM 1400).
2.4. Antibodies
The following primary antibodies were used for this study: anti‐oligomeric Aβ OC rabbit polyclonal (1:5000, Millipore, Burlington, MA); anti‐GFAP (glial fibrillary acidic protein) rabbit polyclonal (1:10000, Dako, Troy, MI); anti‐GFAP chicken polyclonal (1:20000, Millipore); anti‐BLBP (brain lipid‐binding protein) rabbit polyclonal (1:2000, Abcam, Cambridge, MA); anti‐AQP4 (aquaporin 4) rabbit‐polyclonal (1:10000, Sigma, St. Louis, MO); anti‐ALDH1L1 (aldehyde dehydrogenase 1 family member L1; N103/39 clone) mouse monoclonal (1:2000, Millipore); anti‐EAAT2 (excitatory amino acid transporter 2) rabbit polyclonal (1:10000, Abcam); anti‐human APP rabbit polyclonal (1:10000, Sigma); anti‐Iba1 (ionized calcium binding adaptor molecule 1) rabbit polyclonal (1:1000, Wako, Mountain View, CA); anti‐VGluT1 (vesicular glutamate transporter 1) guinea‐pig polyclonal (1:10000, Millipore); anti‐synaptophysin rabbit polyclonal (1:1000, Abcam).
2.5. Light microscopy immunohistochemistry
Free‐floating sections were first treated with 3% H2O2/10% methanol in PBS, pH 7.4 for 20 min to inhibit endogenous peroxidase followed by avidin‐biotin Blocking Kit (Vector Lab, Burlingame, CA) for 30 min to block endogenous avidin, biotin and biotin‐binding proteins. For general antigen retrieval method sections were previously heated at 80°C for 30 min in 50 mM citrate buffer pH 6.0. Sections were immunoreacted with the primary antibody over 24 hr at room temperature. The tissue‐bound primary antibody was detected by the incubation for 1 hr with the corresponding biotinylated secondary antibody (1:500, Vector Laboratories), followed by incubation with streptavidin‐conjugated horseradish peroxidase (1:2000, Sigma‐Aldrich) for 90 min, and finally visualized with 0.05% 3‐3‐diaminobenzidine tetrahydrochloride (Sigma‐Aldrich), 0.01% hydrogen peroxide in PBS and 0.03% nickel ammonium sulfate. The specificity of the immune reactions was controlled by omitting the primary antisera. For double immunofluorescence labeling, sections were first sequentially incubated with the indicated primary antibodies followed by the corresponding Alexa 488/548 secondary antibodies (1:1000, Invitrogen, Carlsbad, CA). GFAP‐, AQP4‐, and EAAT2‐immunolabeled sections were stained with 0.02% thioflavin‐S in 50° ethanol. Double immunofluorescent sections were examined under a confocal laser microscope (Leica SP5 II). For three‐dimensional (3D) reconstructions, serial confocal images of GFAP/APP double labeled sections (acquired in steps of 0.2 µm using a 63× objective) were analyzed with Imaris software (Bitplane), and 3D reconstructions were obtained with the surface plugin.
2.6. Electron microscopy immunogold labeling
50‐µm thick sections from 6 and 12‐month‐old APP/PS1 mice were cryoprotected in a 25% sucrose and 10% glycerol solution, followed by freezing at −80°C in order to increase the antibody binding efficiency. Sections were then incubated 48 hr in primary antibody in a PBS 0.1 M/0.1% sodium azide/2% BSA‐solution at 22°C. The tissue‐bound primary antibody was detected by the incubation with the corresponding 1.4 nm gold‐conjugated secondary antibody (1:100, Nanoprobes, Yaphank, NY) overnight at 22°C. After postfixation with 2% glutaraldehyde and washing with 50 mM sodium citrate, the labeling was enhanced with the HQ Silver Kit (Nanoprobes), and gold toned. Finally, the immunolabeled sections were processed as we previously described by the osmium fixation, dehydration and embedding steps. The primary antibody was omitted in negative control experiments.
2.7. Focused ion beam scanning electron microscopy (FIB/SEM)
Araldite embedded tissue from 12‐month‐old APP/PS1 mice were glued onto a sample stub with carbon adhesive tape in order to facilitate conductivity. Then the sample stub was coated first with iridium and later with platinum for 10 min to allow charge dissipation. With a combined FIB/SEM microscope (Helios Nanolab 650, FEI Company, Hillsboro, OR) we obtained serial TEM images in steps of 30 nm, which allows to represent 3D images from a region of interest.
2.8. Quantitative image analysis
Astroglial loading was defined as the percentage of area stained with anti‐GFAP (total astrocytes) or anti‐BLBP (reactive astrocytes) related to the hippocampal area analyzed. Immunostained sections were examined under a Nikon Eclipse 80i microscope and images were acquired with a Nikon DS‐5M digital camera using the ACT‐2U imaging software (Nikon Corporation, Minato, Tokyo, Japan). The camera settings were adjusted at the start of the experiment and maintained for uniformity. Digital images (4 sections/mouse/age) from APP/PS1 (n = 4/age) and WT mice (n = 4/age) were analyzed using Visilog 6.3 analysis program (Noesis, France). The GFAP‐ or BLBP‐immunopositive signal within the selected brain region (CA1) was converted into 8‐bit gray scale, and immunostained cells were identified by a threshold level mask. The threshold level was maintained throughout the whole image analysis for uniformity. Quantitative comparisons were carried out on sections processed at the same time with same batches of solutions. Plaque loading was defined as the percentage of total CA1 area immunostained with the anti‐Aβ antibody (OC), and the image analysis was carried out as described above. Plaque size and astroglial covered area were measured in TEM images of amyloid plaques (19 plaques/age) from 6 (n = 3) to 12‐month‐old (n = 3) APP/PS1 mice with a JEOL JEM‐1400 electron microscope, analyzed by Visilog 6.3 analysis program (Noesis). This measurement was carried out in the CA1 hippocampal area. The number of total dystrophic neurites around plaque and the number of engulfed dystrophic neurites was measured in the same electron microscopy images in which we previously analyzed plaque size and astroglial covered area.
2.9. Total RNA and protein extraction
Total RNA and proteins were sequentially extracted from cells and human or mouse hippocampal tissue using Tripure Isolation Reagent (Roche, Basel, Switzerland; 100 mg tissue/1 ml Tripure Isolation Reagent) following the manufacturer's recommendations. RNA integrity (RIN) was determined by RNA Nano 6000 (Agilent, Santa Clara, CA). Although no differences between Braak groups were observed, the RIN was lower in human samples compared with transgenic models (RIN: 4.95 ± 1.4 or 8.5 ± 0.5 for human and mouse samples, respectively). RNA was quantified using NanoDrop 2000 spectrophotometer (Thermo Fisher, Waltham, MA). Proteins were quantified using Lowry's method.
2.10. Retrotranscription and qPCR
Retrotanscription and qPCR were performed as described previously (Sanchez‐Mejias et al., 2016). Briefly, retrotranscription (4 g of total RNA as template) was performed with the High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA). For real time qPCR, 40 ng of cDNA were mixed with 2× Taqman Universal Master Mix (Applied Biosystems) and 20× Taqman Gene Expression assay probes. Quantitative PCR reactions (qPCR) were carried out in 96‐well plates using an ABI Prism 7900HT (Applied Biosystems). The cDNA levels of the different samples were determined using GAPDH and β‐actin. Routinely, we used the housekeeper gene, GAPDH, to normalize loading. The amplification of GAPDH was performed in parallel with the gene to be analyzed. Results were expressed using the comparative double‐delta Ct method (2‐ddCt; Bulletin number 2, Applied Biosystems) using Braak 0 as the reference condition.
2.11. Western blots
Western blots were performed as previously described (Sanchez‐Mejias et al., 2016). Proteins (15 μg) were loaded on 4–20% sodium dodecyl sulfate‐Tris‐Glycine‐polyacrylamide gel electrophoresis (Bio‐Rad, Hercules, CA), transferred to nitrocellulose (Optitran, GE Healthcare Life Sciences, Marlborough, MA) and incubated with anti‐GFAP antibody.
2.12. Statistical analysis
Normally distributed data were expressed as the mean ± SD. Mean values were compared using ANOVA followed by Tukey's test (more than two groups) or two‐tailed t test for two group comparisons. Non‐normally distributed data were represented using box‐plots (Sigmaplot) or scatter‐plots with the median and interquartile range (GraphPad), and compared with the Mann–Whitney U test (for two groups comparisons) or Kruskal–Wallis tests (more than two groups) followed by Dunn's post‐hoc test. The significance was set at 95% of confidence. Linear correlations were analyzed using the Spearman test. In all cases, IBM SPSS (v23) Statistics software was used.
3. RESULTS
3.1. Reactive astrocytes are specifically and intimately associated with amyloid plaques in APP/PS1 mouse hippocampus
As revealed by GFAP immunostaining, astrocytes were widely and homogeneously distributed throughout all hippocampal regions in young APP/PS1 mice with none or very low extracellular amyloid deposition (Figure 1a1). However, astroglial reactivity gradually increased with age and amyloid pathology; in fact, astrogliosis was especially patent in areas with abundant amyloid deposits such as the stratum oriens of CA subfields and the hilar region of the dentate gyrus (Figure 1a2,a3). This increase was confirmed by quantification of the GFAP‐positive loading in the CA1 subfield (Figure 1b1). Though WT‐mouse hippocampus displayed also an age‐dependent increase in the GFAP‐positive covered area, the increase was much more pronounced in APP/PS1 mice (2.4 ± 0.6, 2.01 ± 0.23, and 1.68 ± 0.17 fold in 4, 6, and 12 month‐old APP/PS1 compared with age‐matched WT mice; n = 4/age, see Figure 1b1).
Figure 1.
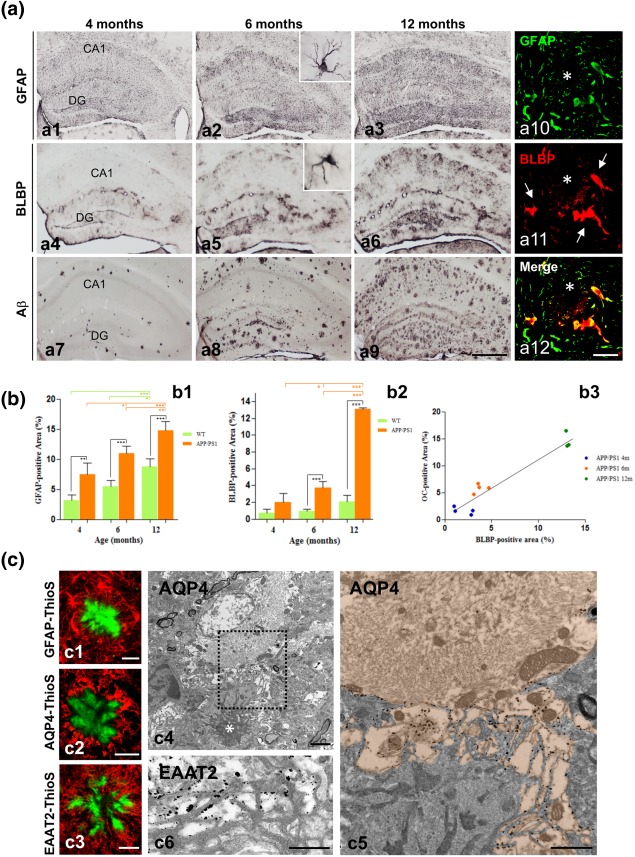
Astrocyte reactivity progresses in parallel to amyloid pathology in the of APP/PS1 hippocampus. (a) Immunostaining for GFAP (a1–a3), BLBP (a4–a6), and OC (a7–a9) in the hippocampus of APP/PS1 at 4, 6, and 12 months of age, immunostained with anti‐GFAP (a1–a3), anti‐BLBP (a4–a6) or anti‐OC (a7–a9) antibodies. Insets in a2 and a5 show details of GFAP‐ and BLBP‐positive astrocytes, respectively, at a higher magnification. Reactive astrocytes are located around amyloid plaques (asterisk) and co‐expressed GFAP and BLBP (arrows; a10–a12) as shown by double immunofluorescence. (b) Image analysis quantification of the GFAP‐positive (b1) and BLBP‐positive (b2) covering areas in the CA1 subfield demonstrates an age‐related increase in the astroglial reactivity, which was significantly higher in the transgenic mice compared to WT mice. BLBP was a more selective marker for reactive astrocytes than GFAP, and the BLPB‐positive area displayed a good correlation with the OC‐positive amyloid load (b3). Data (mean ± SD) from GFAP and BLBP quantification was analyzed by one‐way ANOVA (F(5,18) = 38.18, P = .0001 for GFAP; and F(5,16) = 149.3, P = .0001 for BLBP) followed by Tukey post hoc multiple comparison test. Significance (*P < .05; **P < .01; ***P < .0001) was indicated in the figure. Correlation data between BLBP and OC loading was analyzed by Spearman's Correlation test (r = 0.87; P = .0001). (c) The close relationship between the astrocytic processes and the amyloid fibrils can be observed by confocal microscopy double‐staining for GFAP–Thioflavin S (c1), AQP4–Thioflavin S (c2), or EAAT2–Thioflavin S (c3), as well as by electron microscopy immunogold staining for AQP4 (c4–c5) or EAAT2 (c6); amyloid plaque in c4 is indicated by a white asterisk; Aβ fibrils in c5 and c6 are visualized by their higher electrodensity compared with astroglial (very low electrodense cytoplasm) and microglia (intermediate electrodense cytoplasm). Note the specific gold‐labeling in the astrocytic plasma membrane facing the amyloid fibrils in c5 (the astrocyte cytoplasm is orange colored) and c6. DG, dentate gyrus; CA1, hippocampal CA1 subfield. Scale bars: a1–a9, 500 µm; a10–a12, 25 µm; c1–c3, 20 µm; c4, 2 µm; c5–c6, 1 µm [Color figure can be viewed at wileyonlinelibrary.com]
Figure 2.
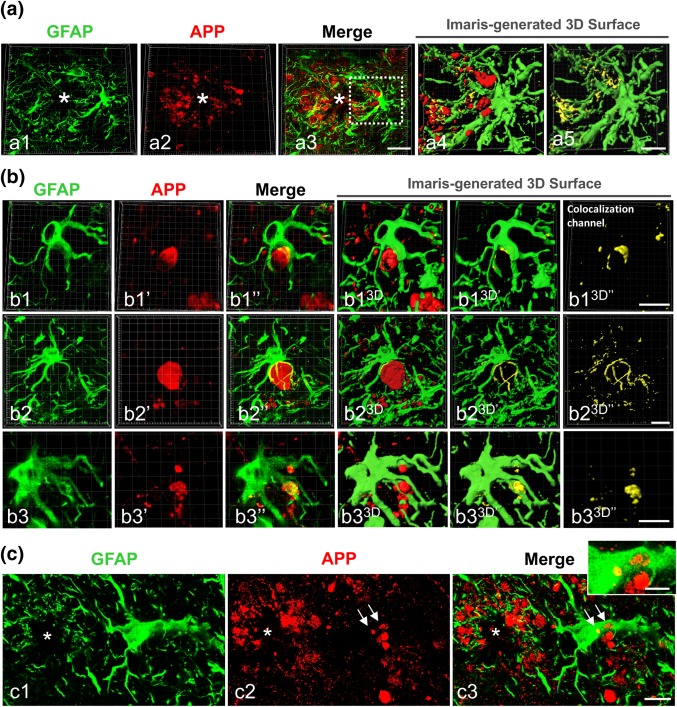
Plaque‐associated reactive astrocytes enwrap axonal dystrophic neurites in APP/PS1 hippocampus. (a) Confocal microscopy of double GFAP/APP immunofluorescence showing plaque (asterisk)‐associated reactive astrocytes making very close contacts with APP‐containing axonal dystrophies (a1–a3). Imaris‐generated 3D surface images (a4 and a5) reveal the tight apposition between the reactive astrocytes and dystrophic neurite membranes (yellow color in the co‐localization channel in a5). (b) Confocal images of double GFAP/APP immunofluorescence showing representative examples of reactive astrocytes with their processes clearly wrapping APP‐positive axonal dystrophies (b1–b3). The corresponding Imaris‐generated 3D reconstructions (b13D–b33D) reveal the extensive apposition surface between the astrocytic processes and the neuronal dystrophies. (c) Confocal images of double GFAP/APP immunofluorescence showing APP‐positive dystrophies (arrows) within a reactive astrocyte in the nearness of an Aβ plaque (star). Scale bars: a1–a3, 20 µm; a4 and a5, b1–b3, and c1–c3, 10 µm; inset, 5 µm [Color figure can be viewed at wileyonlinelibrary.com]
Although most hippocampal astrocytes expressed GFAP‐immunoreactivity, only those located near the Aβ plaques showed enhanced expression of GFAP and displayed a reactive phenotype with hypertrophic morphology and extended processes in and around the plaques. To selectively discriminate reactive from non‐reactive astrocytes, we have used BLBP, a specific marker for reactive astroglia (Gotz, Sirko, Beckers, & Irmler, 2015). Astrocyte‐like neural stem cells in the subgranular zone of dentate gyrus are also positive for this marker. Immunostaining for BLBP (Figure 1a4–a6), and quantification of BLBP‐covered area in the CA1 subfield (Figure 1b2), clearly demonstrated a significant increase in astroglial reactivity with age in APP/PS1 mice (2.6 ± 1.41, 4.06 ± 0.74, and 6.39 ± 0.06 fold at 4, 6, and 12 months compared to age‐matched WT mice; n = 4/genotype/age, see Figure 1b2). As expected, only astrocytes surrounding plaques were double labeled for GFAP and BLBP (Figure 1a10–a12). The regional and temporal pattern for BLBP‐immunoreactivity was coincident with that for amyloid deposition revealed with OC‐immunostaining (see Figure 1a7–a9). Moreover, a good linear correlation (Spearman's correlation coefficient r = 0.87, P < .0001) was found between the astroglial reactivity (BLBP‐positive) and the amyloid load (OC‐immunoreactivity; Figure 1b3).
Figure 4.
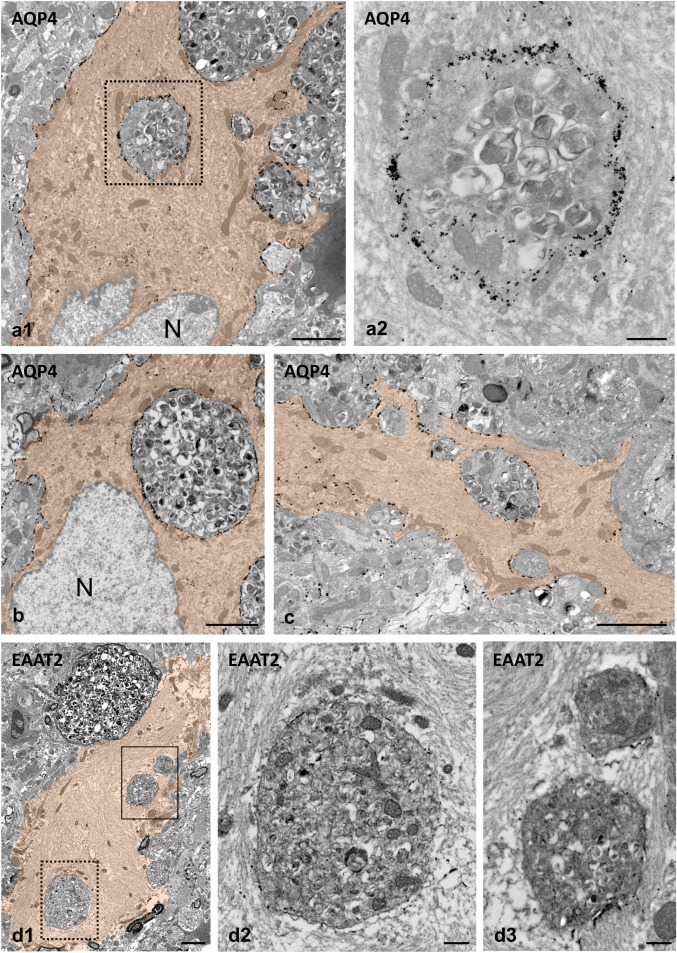
Internalized neuronal dystrophies are enclosed by the plasma membrane of the phagocytic reactive astrocytes. TEM images from AQP4 immunogold‐labeled APP/PS1 mouse hippocampus (a–c). The dystrophies located within the astrocyte cytoplasm (orange colored) are completely enclosed by the reactive astrocyte plasma membrane, which is easily recognized by its specific AQP4 labeling. As can be seen in the enlarged detail (a2), the cytoplasm of a phagocytic astrocyte displays many intermediate filaments and few mitochondria closely associated to the engulfed dystrophy. (d1) Likewise, EAAT2 immunogold‐labeled membranes were detected around engulfed dystrophies within reactive astrocyte (colored in orange); the image shows an example of three engulfed dystrophies (see details in d2 and d3). Astrocyte‐engulfed dystrophies typically contain numerous autophagic vacuoles. N, astrocyte nucleus. Scale bars: a1, b, c, and d1, 2 µm; a2, d2 and d3, 0.5 µm [Color figure can be viewed at wileyonlinelibrary.com]
Figure 7.
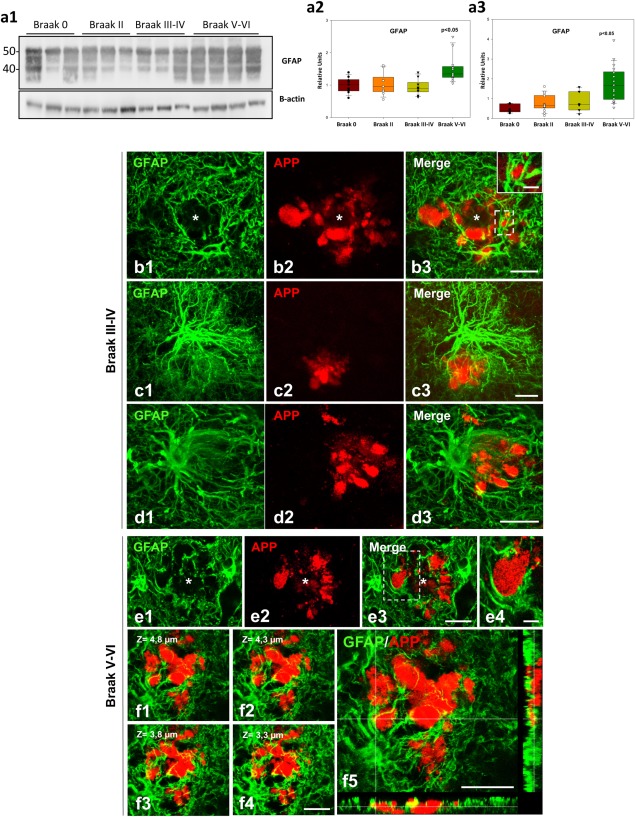
Human reactive astrocytes in Alzheimer's hippocampus envelop the dystrophic neurites that cluster around amyloid plaques. (a1–a3) Marked increase in GFAP protein (a1 and a2) and mRNA (a3) levels in Braak V–VI samples (n = 12–18 for Western blots/qPCR respectively) as compared with controls (Braak 0, n = 6) or earlier tau pathology stages (Braak II, n = 9–13 for Western blots/qPCR respectively; Braak III–IV, n = 9). The data are shown individually (dots) or as box‐plots. Significance (indicated in the figure) was determined by Kruskal–Wallis test followed by Dunn post‐hoc test. (b–f) Confocal microscopy images of double GFAP/APP immunofluorescence showing plaque (asterisk)‐associated reactive astrocytes making very close contacts or encircling APP‐containing dystrophies in the hippocampus of Braak III–IV (b–d) and Braak V–VI (e and f) patients. Single reactive astrocytes are able to cover a cluster of dystrophies (b1–b3 and c1–c3), and the astrocytic processes are directed toward the dystrophies (d3). In Braak V–VI samples, the dystrophic neurites could be seen partially or even broadly covered by astrocytic processes (e1–e4). Serial confocal images (f1–f4) showing a cluster of APP‐positive dystrophies intermingled with astrocyte processes. In f5 is shown an orthogonal plane displaying APP‐positive dystrophies enwrapped by reactive astrocytes. Scale bars: b1–b3, c1–c3, d1–d3, and f1–f5, 20 µm; insets in b3, and e4, 5 µm [Color figure can be viewed at wileyonlinelibrary.com]
Figure 3.
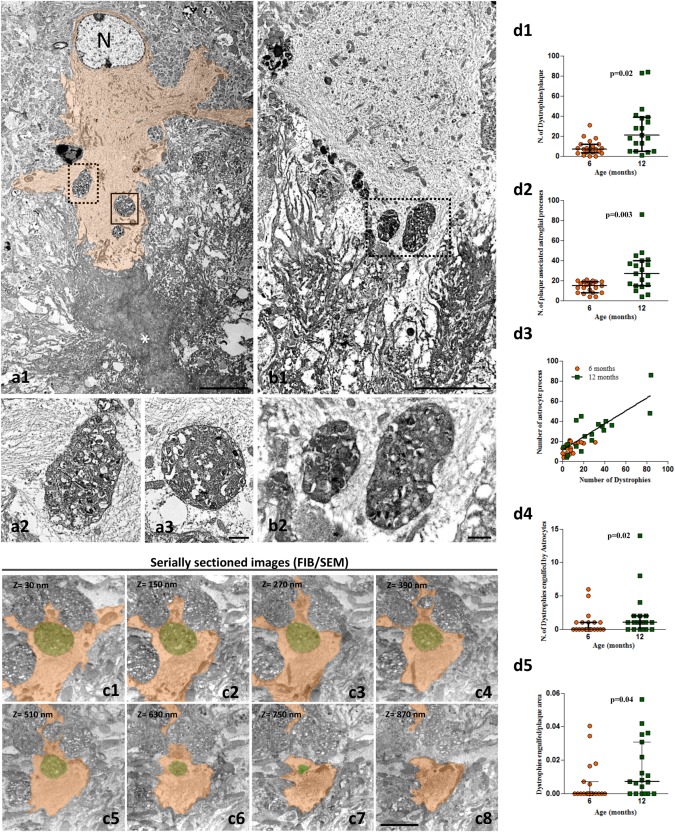
Ultrastructural identification of engulfed dystrophic neurites within the cytoplasm of APP/PS1 reactive astrocytes. TEM images (a1 and b1) showing dystrophic neurites inside plaque‐associated reactive astrocytes (cytoplasm colored in orange in a1; N, astrocyte nucleus). As can be seen in the enlarged images (a2, a3; b2), the engulfed dystrophies are surrounded by the astrocytic cytoplasm completely filled with glial filaments. Representative serial FIB/SEM electron microscopy images (c1–c8) showing an astrocytic process (orange colored) wrapping and engulfing a dystrophic neurite (green colored). Quantitative analysis demonstrates a significant age‐dependent increase in the number of dystrophies associated to plaques (d1), which was paralleled by a significant increment in the number of reactive astrocytic processes contacting the plaque (d2). In consequence, a highly significant linear correlation (d3) between reactive astrocytic processes and the number of dystrophies was also observed (Spearman r = 0.785, P = .0001, n = 40). A significant increase in the number of dystrophic neurites engulfed by astrocytic processes (d4), and in the number of engulfed dystrophies per plaque (d5), is observed at 12 months of age as compared with 6 months. The data (n = 19–21 plaques from three APP/PS1 mice/age) are shown individually (dots) with the median and the interquartile range. Significance was analyzed by Mann–Whitney U comparison test and indicated in the figure. Scale bars: a1 and b1, 5 µm; a2, a3, and b2, 0.5 µm; c1–c8, 3 µm [Color figure can be viewed at wileyonlinelibrary.com]
Thus, amyloid plaques were always surrounded by reactive astrocytes. Double‐staining with thioflavin‐S (for fibrillar Aβ) and anti‐GFAP revealed the intricate relationship between the reactive‐astrocyte processes and the fibrillar plaques (Figure 1c1), which was seen also with other astrocyte markers, such as AQP4 (Figure 1c2) and EAAT2 (Figure 1c3). Immunogold electron microscopy for AQP4 (Figure 1c4,c5) and EAAT2 (Figure 1c6) revealed immunopositive astrocytic processes closely intermixed with Aβ fibres and microglial processes in the periphery of the amyloid plaques, leading to plaques completely wrapped by glial processes. In fact, the percentage of astroglial area surrounding plaques increased with plaque size, showing a highly significant linear correlation (Spearman's correlation coefficient r = 0.745, P = .0001).
Figure 6.
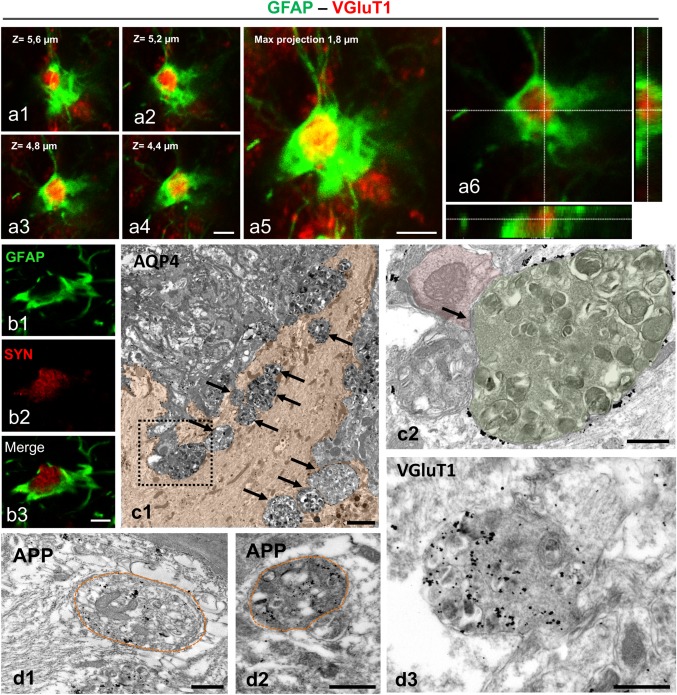
Dystrophies phagocytosed by APP/PS1 reactive astrocytes are of presynaptic nature. Double immunofluorescence labeling for GFAP and VGluT1 (a1–a6) or synaptophysin (b1–b3) confirm that phagocytic reactive astrocytes enwrap presynaptic dystrophies. The orthogonal reconstruction of GFAP/VGluT1 confocal stack images shows that the VGluT1‐positive presynaptic dystrophy is completely enclosed by the astrocyte (a6). TEM image showing an AQP4‐positive reactive astrocyte (cytoplasm colored in orange, c1) containing several dystrophic neurites (arrows). As shown in c2 (enlarged image of the dashed square in c1), a presynaptic dystrophy (green colored) making a synaptic contact (arrow) on a postsynaptic structure (red colored) is surrounded and in close contact with the astrocyte processes (AQP4‐positive). TEM image of immunogold labeled sections shows APP‐positive (d1 and d2) or VGluT1‐positive (d3) neuronal/synaptic structures inside the phagocytic astrocyte cytoplasm. Scale bars: a1–a5, 2 µm; b1–b3, 5 µm; c1, 2 µm; c2 and d1–d3, 0.5 µm [Color figure can be viewed at wileyonlinelibrary.com]
3.2. Reactive astrocytes enwrap and internalize plaque‐associated axonal dystrophic neurites
We have previously showed that almost all amyloid plaques in the APP/PS1 hippocampus are surrounded by dystrophic neurites, and that the number of dystrophies correlates with the plaque size (Sanchez‐Varo et al., 2012). Also, we have reported that dystrophic neurites are APP‐positive and of axonal/synaptic nature (Sanchez‐Varo et al., 2012). However, besides the close spatial relationship between reactive astrocytes and amyloid plaques, there are no studies focusing on astroglia and the dystrophic neurites surrounding neuritic plaques. Since reactive astrocyte processes were better visualized with GFAP marker compared with ALDH1L1 (see Supporting Information, Figure S1), here we analyzed the morphological relationship between GFAP‐reactive astrocytes and APP‐positive aberrant neuronal structures. For that, we have used confocal microscopy and 3D image reconstructions with Imaris software of double GFAP/APP immunolabeling (Figure 2). As shown in Figure 2a, plaque‐associated astrocytic processes were in close proximity to dystrophic neurites (Figure 2a1–a3). The Imaris‐generated 3D reconstructions confirmed the tight interaction between dystrophies and astrocyte processes (Figure 2a4). Moreover, both markers co‐localized in some areas, as shown by the yellow color (Figure 2a5), suggesting close physical contact. Higher magnification images of individual reactive astrocytes (Figure 2b1–b3 are representative images) demonstrated that the astroglial processes enwrap one or several APP‐positive dystrophic neurites. Solid surfaces, best matching the anatomy of astrocytes and dystrophic neurites, were created by 3D reconstructions with Imaris of z‐stacks obtained in the three‐color channels (Fig. 2b1–b33D series). They showed that neuronal dystrophies directly oppose the astrocyte cell surface. The abundance of astrocyte‐dystrophy association per plaque was quantified by confocal imaging. In contrast, most (if not all) plaques presented astrocyte‐dystrophy contacts in 6 and 12‐month‐old APP/PS1 mice (84.74% ± 36.26% and 89.83% ± 30.48%, respectively). In contrast, the confocal analysis showed the presence of APP‐positive dystrophies within reactive astrocytes near amyloid plaques (Figure 2c1–c3), suggesting phagocytosis of dystrophies by astrocytes. To investigate this further we performed ultrastructural analysis (Figure 3). Transmission electron microscopy images revealed the presence of dystrophic neurites (identified by their typical heterogeneous content with autophagic vacuoles) within the cytoplasm of plaque‐associated reactive astrocytes (Figure 3a1–a3,b1,b2). The astrocytic processes containing dystrophies were large and completely filled with intermediate filaments, many of which appeared surrounding and even attached to the outer membrane of the inside‐located dystrophies (Figure 3a3,b2). In contrast, the astrocytic cytoplasm adjacent to the fibrillary Aβ in the periphery of the plaques appeared devoid of filaments and other organelles. To better visualize the spatial relationship of astrocyte‐dystrophies we used FIB/SEM for high‐resolution 3D reconstructions of serially collected transmission electron microscopy images (Figure 3c1–c8 are representative serial images). These serial images confirmed the intracellular location of the dystrophic neurites within reactive astrocyte processes. Quantitative studies using TEM sections revealed, as expected, the existence of an age‐dependent (between 6 and 12 months of age) increase in the plaque area (average 51.42 μm2 (range 5.02–82.67) and 137.68 μm2 (range 6.7–571.30) for 6 and 12 months of age, respectively, n = 20 plaques per age; Mann–Whitney P = .03) and in the number of dystrophic neurites (Mann–Whitney P = .02, Figure 3d1) associated to the Aβ plaques (3.3‐fold between 6 and 12 months of age). This increase was paralleled by a significant increment in the number of astrocytic processes associated to the plaques (13.80 ± 5.49 vs. 29.15 ± 19.17 processes at 6 and 12 months of age respectively, n = 20 per age, Mann–Whitney P = .003, Figure 3d2). In fact, the number of plaque‐associated astrocyte processes significantly correlated with the number of dystrophies (Figure 3d3, Spearman r = 0.785, P = .0001, n = 40) but not with the plaque size (Spearman r = 0.04, P = .39). In consequence, we also observed a significant increase (n = 20 per age, Mann–Whitney P = .02) in the number of dystrophies engulfed by astrocytic processes (Figure 3d4), and in the engulfed dystrophies per plaque (Mann–Whitney P = .04, n = 20 per age, Figure 3d5). It is relevant that the proportion of plaques with associated astrocytic processes containing one or more dystrophies was much higher at 12 months (68.40%) than at 6 months (35%). In spite of this increase, the proportion of dystrophic neurites engulfed by astrocytes with respect to the total number of dystrophies around the plaques was low at both ages (i.e., 6.25% ± 10.14% or 7.72% ± 10.95% of total neurites for 6 or 12‐month‐old APP/PS1 mice).
Figure 5.
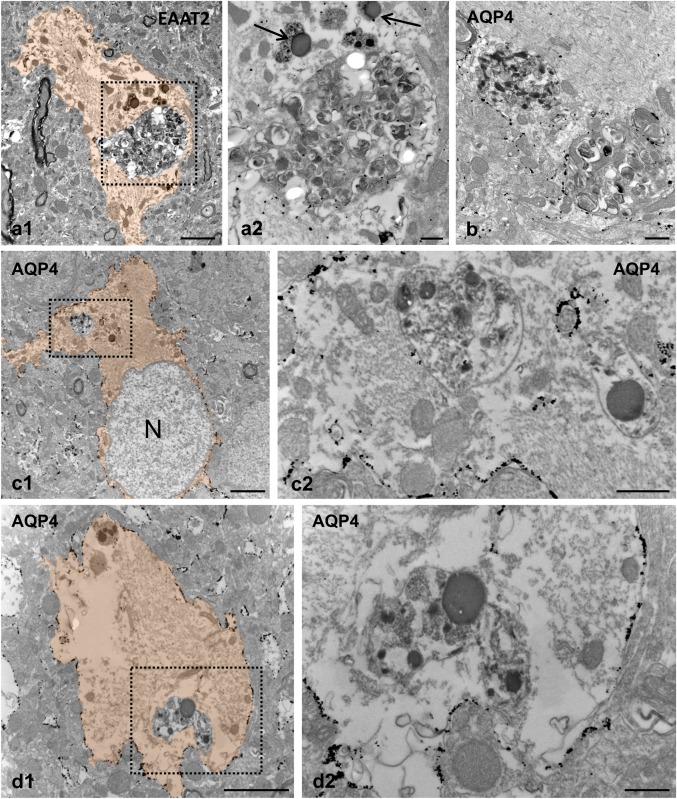
Phagocytic reactive astrocytes degrade engulfed neuronal dystrophies in APP/PS1 hippocampus. TEM images from EAAT2 (a) and AQP4 (b, c, and d) immunogold‐labeled hippocampal APP/PS1 sections showing dystrophic neurites at different stages of digestion. In some cases, the astrocyte membrane around the engulfed dystrophy seemed interrupted or absent (a1, a2 and b), whereas other dystrophies appeared to be in a more advanced degradation process (c and d). a2, c2, and d2 are enlarged views of a1, c1, and d1, respectively. The reactive astrocyte cytoplasm is orange colored in figures a1, c1, and d1; lysosomes are indicated by black arrows. N, astrocyte nucleus. Scale bars: a1, c1, and d1, 2 µm; a2, b, c2, and d2, 0.5 µm [Color figure can be viewed at wileyonlinelibrary.com]
Though microglia, the main phagocytic population within the brain, surrounded amyloid plaques and were also in close contact with dystrophic neurites (see Supporting Information, Figure 2), we could not identify by EM the presence of these abnormal structures within the cytoplasm of these cells. In addition, and in support of the selective clearance of dystrophic neurites by engulfing reactive astrocytes, we never saw other neuronal structures (such as myelinated axons or even myelinated dystrophies) within the cytoplasm of reactive astrocytes.
3.3. Reactive astrocytes engulf and degrade axonal/synaptic dystrophies
Next, we aimed to better visualize the internalization of dystrophies by astrocytes examining by TEM the localization of plasma membrane proteins, AQP4 channel and EAAT2. These proteins are extensively located throughout the plasma membrane of reactive astrocytes in the APP/PS1 mouse hippocampus, and thus allow to dissect the anatomical boundaries of astrocytes (Figures 1 and 3). While AQP4 was mainly located at the astrocyte end‐feet membrane in WT mice hippocampus (Supporting Information, Figure S3), the engulfed dystrophies appeared morphologically intact and surrounded by AQP4‐positive plasma membrane in APP/PS1 mice, as revealed by electron microscopy immunogold labeling for AQP4 (Figure 4 a–c). As mentioned before, the cytoplasm adjacent to the endocytosed dystrophies showed many glial intermediate filaments, and mitochondria frequently appeared closely associated to the phagosome membrane. Similar results were obtained with the antibody for EAAT2 (Figure 4 d1‐3), though the immunostaining intensity was substantially lower than that of AQP4. In most cases, the engulfed dystrophies appeared as double‐membrane‐bound inclusions (astrocyte membrane plus the neuronal dystrophy membrane), but occasionally the membrane of the engulfed material was discontinuous or seemed to be absent (Figure 5a1,a2,b). These TEM images support the existence of astrocyte phagocytosis of pathological neuronal structures. In fact, lysosomes were usually found near the engulfed dystrophies (Figure 5a2,c2,d2). Some of the dystrophies seemed to be in the process of degradation (Figure 5c2,d2), indicating that reactive astrocytes actively engulf and degrade neuronal dystrophies.
To confirm the neuronal/synaptic origin of the engulfed material, we used confocal and TEM immunolabeling with antibodies for neuronal (APP) and synaptic (VGluT1 and synaptophysin) molecules (Figure 6). Double immunofluorescence labelling for GFAP and VGluT1 (Figure 6a1–a6) or synaptophysin (Figure 6b1–b3) confirmed the neuronal and probably synaptic nature of the dystrophies enwrapped by reactive astrocytes. Moreover, TEM analysis identified presynaptic dystrophies surrounded by astrocyte processes (Figure 6c1,c2). In addition, the ultrastructural immunolabeling of the engulfed dystrophies with antibodies for APP (Figure 6d1,d2) or VGluT1 (Figure 6d3) clearly demonstrated positive dystrophies, completely surrounded by the astrocyte cytoplasm, confirming their neuronal and potential presynaptic nature. Furthermore, engulfed dystrophies were significantly (Mann–Whitney, P = .0002) smaller (4.23 μm2 in average; 2.0–9.4 range, n = 28) than those non‐phagocytosed (14.01 μm2 in average; 2.9–50.8 range, n = 28). Altogether, these findings suggest that astrocytes engage in the elimination of presynaptic dystrophies in APP/PS1 mice.
3.4. Astrocyte phagocytosis of neuronal dystrophies is present in human Alzheimer's brains
Astrogliosis is a noticeable event in the hippocampus of Alzheimer's patients (Braak V–VI). The expression of the astrocyte marker GFAP was significantly upregulated in the Braak V–VI samples compared with earlier stages of tau pathology and controls, as measured by Western blot (Figure 7a1,a2) and qPCR (Figure 7a3). Similarly, the expression of vimentin, another astrocyte marker, was also higher in Braak V–VI samples as compared with controls (not shown). This upregulation of GFAP in the hippocampus of Braak V–VI individuals was corroborated by immunostaining and GFAP‐positive astrocyte loading quantification. Reactive astrocytes clustered around amyloid plaques, and the hippocampal area occupied by these astrocytes showed a significant increase in Braak V–VI samples (5.33 ± 2.47 fold, n = 6 for Braak V–VI as compared with Braak II, n = 4, or Braak III–IV, n = 5, samples; Kruskal–Wallis followed by Dunn test, P < .05).
We next examined by confocal microscopy the relationship between dystrophic neurites and astrocytes using double APP–GFAP immunofluorescence. As seen in the APP/PS1 mice, human APP‐positive dystrophic neurites were surrounded by GFAP‐positive astrocyte processes (Figure 7). Similar results were observed using AT8 antibody as another marker for human dystrophic neurites (not shown). The tight astrocyte‐dystrophy association could be seen since initial stages of the amyloid pathology in the hippocampus (Braak III–IV). Reactive astrocytes sent their processes toward the dystrophic neurites forming a net entrapping them (Figure 7b–d). Moreover, in Braak V–VI hippocampus the astrocyte processes were also found in contact with APP‐positive dystrophies, in occasions covering them widely (Figure 7e1–e4,f1–f5); however, the identification of APP‐positive dystrophies inside the astrocyte processes by confocal imaging was difficult. To confirm whether reactive astrocytes in AD Braak V–VI brains phagocytose dystrophic neurites we performed a TEM analysis in post mortem human samples. Similar to APP/PS1 mice, we found engulfed dystrophies within the reactive‐astrocyte cytoplasm (Figure 8a1–a3). The engulfed dystrophic neurites were identified by their high content in autophagy vacuoles, and some even were filled with paired‐helical filaments corroborating the neuronal origin of these structures. However, the limitations of human autopsy brain samples for TEM studies precluded us to further perform subcellular quantitative analysis.
Figure 8.
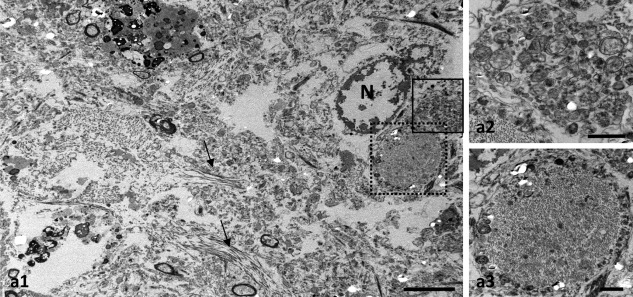
Human reactive astrocytes in Alzheimer's brains phagocytose dystrophic neurites. TEM of post mortem human Braak V–VI tissue showing a reactive astrocyte containing dystrophic neurites (a1). Small bundles of filaments are observed (arrows). Higher magnification views of the squared areas in c showing dystrophic neurites filled with autophagic vesicles (a2) or containing paired‐helical filaments (a3). N, astrocyte nucleus. Scale bars: a1, 5 µm; a2 and a3, 1 µm
4. DISCUSSION
In this study, we provide evidence with light and electron microscopy that amyloid plaque‐associated reactive astrocytes have phagocytic capacity for dystrophic axonal/presynaptic elements in the hippocampus of APP/PS1 mice and in AD brains. Astroglial reactive processes enwrap and internalize axonal/presynaptic dystrophies terminals, which are filled with autophagic vacuoles and surround amyloid plaques. The engulfed neuronal dystrophies are bound for degradation since they were found associated with lysosomal compartments, and partially degraded. To the best of our knowledge, this is the first report of astrocyte‐mediated phagocytic clearance of dystrophic neurites from injured neurons in AD. Below we discuss the relevance of this finding in AD pathogenesis.
As expected (Jimenez et al., 2008; Serrano‐Pozo, Muzikansky, et al., 2013; Serrano‐Pozo, Betensky, Frosch, & Hyman, 2016; Trujillo‐Estrada et al., 2013), the hippocampus of both the transgenic APP/PS1 model and Alzheimer's patients (Braak V–VI) displays significant astrogliosis (measured by GFAP expression and by the area covered by the reactive astrocytes; see also Jimenez et al., 2008), which progresses in parallel to the extracellular amyloid pathology. The increase in astrocytic reactivity involves both morphological (enlarged cell body and processes) and molecular changes (Osborn et al., 2016). In this sense, and in addition to the well‐established increase in GFAP expression, we found that plaque‐associated reactive astrocytes selectively express BLBP, a typical marker for astrocyte‐like neuronal stem cells (Götz & Barde, 2005). This marker has been widely used for studying neuronal progenitors in embryonic and adult brains; however, the expression of BLPB is also a molecular signature for reactive astrocytes in many injury conditions (Robel, Berninger, & Gotz, 2011). We report here for the first time that the extracellular amyloid‐triggered astrogliosis in the brain of a transgenic Alzheimer's model can be monitored by the expression of BLBP. Whether amyloid plaque‐associated reactive astrocytes gain stem cell potential needs to be determined; however, in the APP/PS1 model the proliferation capacity of this reactive population appears very limited if any (Baglietto‐Vargas et al., 2017; Galea et al., 2015; Serrano‐Pozo, Gómez‐Isla, Growdon, Frosch, & Hyman, 2013).
Microglial cells, the professional phagocytes of the brain, were presumably not involved in the clearance of dystrophic neurites in our transgenic model. However, it has been recently reported in another APP/PS1 model, by EM analysis, that activated (dark) microglia is able to encircle synaptic dystrophic elements (Bisht et al., 2016) which might be indicative of their potential phagocytic capacity to eliminate these structures. Therefore, we cannot completely discard a microglial role in the dystrophic clearance but if exists, it must be very infrequent or, on the contrary, a very rapid and highly dynamic process difficult to detect.
We have found that the amount of reactive astrocytic processes correlates with the number of dystrophic neurites around plaques rather than with plaque size, confirming previously reported findings in Alzheimer's brains (Serrano‐Pozo, Muzikansky, et al., 2013). What these morphological analyses indicate is that astrocytes react to damaged neurons, although the functional significance of this response is not known. Our data support the possibility that reactive astrocytes eliminate damaged synapses. The presence of several presynaptic markers at the plaque‐associated dystrophies (this work, Sanchez‐Varo et al., 2012, Trujillo‐Estrada et al., 2014) strongly suggests the presynaptic nature of these pathological structures. Similar findings have been also reported in another APP/PS1 model (Sadleir et al., 2016). However, we cannot rule out the possibility that some of these dystrophies are in fact axonal portions.
Furthermore, our finding of reactive astrocytes actively enwrapping, internalizing and further degrading presynaptic dystrophies in the APP/PS1 mouse brain is an extension of the capacity of astrocytes to phagocytose synapses as a normal process that mediates the refinement of neural circuits in the developing (Chung, Allen, & Eroglu, 2015) and in the adult CNS (Chung et al., 2013), thus suggesting a major role of astrocytes in synaptic pruning in health and disease. Supporting a phagocytic role for reactive astrocytes in AD, it has been suggested that they could mediate Aβ clearance (Osborn et al., 2016) and limit amyloid deposits (Xiao et al., 2014) since these glial cells express many potential phagocytic receptors that bind Aβ (Jones, Minogue, Connor, & Lynch, 2013; Sokolowski & Mandell, 2011). Moreover, astrocytes in culture are highly phagocytic cells (Roldán, Gogg, Ferrini, Schillaci, & De Nicola, 1997; Tansey & Cammer, 1998), and can phagocytose Aβ (Wyss‐Coray et al., 2003) and apoptotic cells (Chang, Barbaro, & Pieper, 2000) and effectively engulf whole dead cells following neural scratch injury in vitro (Lööv, Hillered, Ebendal, & Erlandsson, 2012). Recently, astrocyte phagocytosis of myelin debris has been observed in response to tissue damage (Ponath et al., 2017). Additionally, it has been shown that reactive astrocytes become phagocytic and engulf neuronal cell debris after transient brain ischemia (Morizawa et al., 2017). Moreover, astrocytes degrade mitochondria (transmitophagy) from healthy retinal ganglion cell axons (Davis et al., 2014). Therefore, transcellular degradation of neuronal dystrophies by astrocytes might directly support neuron survival or, alternatively, it might reduce the Aβ release from dystrophic presynaptic elements. In this sense, presynaptic dystrophies are sites of APP accumulation and putative Aβ release (Sanchez‐Varo et al., 2012; Torres et al., 2012) and, as recent studies indicate, the accumulation of BACE1 in presynaptic dystrophies surrounding plaques causes increased cleavage of APP and Aβ generation, which could lead to an exacerbation of amyloid pathology in AD (Sadleir et al., 2016; Torres et al., 2012). Alternatively, insulating dystrophies from the normal tissue might also decrease the probability of possible adverse inflammatory effects. Therefore, the phagocytosis of presynaptic/axonal dystrophies may be a protective function of astrocytes controlling the Aβ production and/or preventing the inflammatory response. However, we cannot discard that healthy synapses, not observed by our EM analysis, were also eliminated by reactive astrocytes contributing then to synaptic loss and cognitive decline. Anyway, further studies are needed for better understanding of the physiological consequences (beneficial or detrimental) of the reactive astrocyte phagocytosis.
Considering recent gene profiling of astrocytes, is it plausible that phagocytic astrocytes are a subtype of the astrocyte population in the brain. The functional and molecular diversity of astrocytes in normal, and especially in diseased brains, remains challenging (Haim & Rowitch, 2017). The extensive array of astrocyte functions supports the existence of different astroglial populations, and, recently, five distinct subpopulations based on molecular profiles have been identified in the adult mouse brain (John Lin et al., 2017). Noteworthy, one of the populations (type C) is highly enriched for genes associated with synaptic activity, and two subpopulations (types B and C) for genes associated with phagocytic capacity. The contribution of the different astrocyte subsets to pathological conditions, and especially to AD, is not known. In fact, that loss‐of‐function of astrocytes may contribute to diseases in a disease‐specific manner and, that therapies should promote their functional recovery are emerging ideas (Khakh et al., 2017; Masgrau et al., 2017).
In line with this idea, we found that the phagocytic activity of reactive astrocytes in APP/PS1 mice did not significantly increase with disease progression. Although the proportion of plaques displaying astrocytic processes containing engulfed dystrophies was greater at 12 months of age (68%) than at 6 months (35%), the number of engulfed dystrophies was relatively low at both ages (around 7% of the total dystrophies that surround plaques). Considering that the amount of plaques and dystrophies significantly increases with age, in parallel with the astrocytic activation, it would be expected that astrocyte‐mediated dystrophy engulfment will increase consequently. Phagocytic activity is a dynamic process, then the proportion of engulfed dystrophies could in fact be low at one time, however the persistence of abundant dystrophic neurites around plaques in older animals highlights the poor efficiency of the astrocyte phagocytosis. Therefore, our data indicate that the phagocytic capacity of reactive astrocytes could be in fact limited, and even impaired, during disease‐course. In contrast, we do not know how efficiently the astrocyte degradation process of these engulfed dystrophies is. In this regard, our TEM studies revealed that the presence of partially degraded material within astrocytes is a less frequent event that the presence of intact dystrophic neurites (surrounded by the astrocyte plasma membrane). It is possible that astrocytes have a low rate of digestion as compared to other typical phagocytes such as microglial cells. In this sense, poor degradation of engulfed dead cells in astrocytes has been reported previously (Lööv et al., 2012). These authors indicate that the slow digestion of the engulfed material in astrocytes is due, at least in part, to the actin‐rings that surround the phagosomes for long periods of time, which physically inhibit the phagolysosome fusion (Lööv, Mitchell, Simonsson, & Erlandsson, 2015). We observed in the APP/PS1 hippocampus numerous instances in which the ingested dystrophies within astrocytes appeared surrounded by intermediate filaments that could cause impairment of lysosome fusion.
Our ultrastructural data in human Braak V–VI samples clearly show that the hippocampal reactive astrocytes exhibit cytoplasmic neurite‐like inclusions containing autophagic vesicles and even paired helical filaments. Therefore, the phagocytosis of dystrophies appears to occur in AD patients as seen in APP/PS1 mice. Supporting a reduced phagocytic capacity of astrocytes in AD brains, a recent study suggests that the susceptibility of APOE4, the strongest genetic risk factor for AD, may arise in part from the defective phagocytic ability of astrocytes (Chung et al., 2016). Therefore, altogether our data support the idea of dysfunctional astroglial cells in the context of AD. In this sense, we have recently reported that microglial cells in the hippocampus of AD brains display a degenerative phenotype (Sanchez‐Mejias et al., 2016). Therefore, the functional impairment of glial cells in AD as a potential driver of disease progression deserves particular consideration.
In summary, our study demonstrates the existence of phagocytic reactive astrocytes which eliminate/isolate presynaptic dystrophies in AD. This phagocytic capacity of astrocytes might result impaired during disease progression. Therefore, exploring how to increase/restore the phagocytic properties of astrocytes could be a novel therapeutic avenue to treat AD.
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article.
Supporting Information
Supporting Information Figure S1
Supporting Information Figure S2
Supporting Information Figure S3
ACKNOWLEDGMENT
This work was supported by La Marató‐TV3 Foundation grants 20141432 (to A. G.), 20141431 (to J. V.), 20141433 (to J. X. C.) and 20141430 (to E. G.); by Fondo de Investigacion Sanitaria (FIS) Instituto de Salud Carlos III (ISCiii) of Spain‐co‐financed by FEDER funds from European Union, through grants PI15/00796 (to A. G.) and PI15/00957 (to J. V.); by CIBERNED PI2015‐2/02 (to A. G., J. V., and J. X. C.); and by Junta de Andalucia, Proyecto de Excelencia (CTS‐2035) (to J. V. and A. G.). We thank to Rocio Romero Pareja from FESEM‐FIB Unit (Bioinnovation Building, University of Malaga, Spain) and Mercedes Aneiros Ferrer for their expert technical assistance.
Gomez‐Arboledas A, Davila JC, Sanchez‐Mejias E, et al. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer's disease. Glia. 2018;66:637–653. https://doi.org/10.1002/glia.23270
Funding information La Marató‐TV3 Foundation, Grant Numbers 20141432 (AG), 20141431 (JV), 20141433 (JXC), 20141430 (EG); Fondo de Investigación Sanitaria (FIS) Instituto de Salud Carlos III (ISCiii) of Spain co‐financed by FEDER funds from European Union, Grant Numbers PI15/00796 (AG) and PI15/00957 (JV); CIBERNED, Grant Number PI2015‐2/02 (AG, JV, JXC); Junta de Andalucía, Proyecto de Excelencia, Grant Number CTS‐2035 (JV, AG)
Contributor Information
Javier Vitorica, Email: vitorica@us.es.
Antonia Gutierrez, Email: agutierrez@uma.es.
REFERENCES
- Baglietto‐Vargas, D. , Moreno‐Gonzalez, I. , Sanchez‐Varo, R. , Jimenez, S. , Trujillo‐Estrada, L. , Sanchez‐Mejias, E. , … Gutierrez, A. (2010). Calretinin interneurons are early targets of extracellular amyloid‐β pathology in PS1/AβPP Alzheimer mice hippocampus. Journal of Alzheimer's Disease, 21(1), 119–132. [DOI] [PubMed] [Google Scholar]
- Baglietto‐Vargas, D. , Sánchez‐Mejias, E. , Navarro, V. , Jimenez, S. , Trujillo‐Estrada, L. , Gómez‐Arboledas, A. , … Gutierrez, A. (2017). Dual roles of Aβ in proliferative processes in an amyloidogenic model of Alzheimer's disease. Scientific Reports, 7(1), 10085 https://doi.org/10.1038/s41598-017-10353-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisht, K. , Sharma, K. P. , Lecours, C. , Sanchez, M. G. , El Hajj, H. , Milior, G. , … Tremblay, M.‐E. (2016). Dark microglia: a new phenotype predominantly associated with pathological states. GLIA, 64, 826–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard, V. , Moussaoui, S. , Czech, C. , Touchet, N. , Bonici, B. , Planche, M. , … Pradier, L. (2003). Time sequence of maturation of dystrophic neurites associated with Aβ deposits in APP/PS1 transgenic mice. Experimental Neurology, 184(1), 247–263. [DOI] [PubMed] [Google Scholar]
- Cahoy, J. D. , Emery, B. , Kaushal, A. , Foo, L. C. , Zamanian, J. L. , Christopherson, K. S. , … Barres, B. A. (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. Journal of Neuroscience, 28(1), 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, G. H. , Barbaro, N. M. , & Pieper, R. O. (2000). Phosphatidylserine‐dependent phagocytosis of apoptotic glioma cells by normal human microglia, astrocytes, and glioma cells. Neuro‐Oncology, 2(3), 174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, W.‐S. , Clarke, L. E. , Wang, G. X. , Stafford, B. K. , Sher, A. , Chakraborty, C. , … Barres, B. A. (2013). Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature, 504(7480), 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, W.‐S. , Verghese, P. B. , Chakraborty, C. , Joung, J. , Hyman, B. T. , Ulrich, J. D. , … Barres, B. A. (2016). Novel allele‐dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proceedings of the National Academy of Sciences, 113(36), 10186–10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, W. S. , Allen, N. J. , & Eroglu, C. (2015). Astrocytes control synapse formation, function, and elimination. Cold Spring Harbor Perspectives in Biology, 7(9), a020370 https://doi.org/10.1101/cshperspect.a020370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, C.‐H. O. , Kim, K.‐Y. , Bushong, E. A. , Mills, E. A. , Boassa, D. , Shih, T. , … Marsh‐Armstrong, N. (2014). Transcellular degradation of axonal mitochondria. Proceedings of the National Academy of Sciences of the United States of America, 111(26), 9633–9638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper, B. , & Karran, E. (2016). The cellular phase of Alzheimer's disease. Cell, 164(4), 603–615. [DOI] [PubMed] [Google Scholar]
- Galea, E. , Morrison, W. , Hudry, E. , Arbel‐Ornath, M. , Bacskai, B. J. , Gómez‐Isla, T. , … Hyman, B. T. (2015). Topological analyses in APP/PS1 mice reveal that astrocytes do not migrate to amyloid‐β plaques. Proceedings of the National Academy of Sciences, 112, 15556–15561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz, M. , & Barde, Y.‐A. (2005). Radial glial cells. Neuron, 46(3), 369–372. [DOI] [PubMed] [Google Scholar]
- Gotz, M. , Sirko, S. , Beckers, J. , & Irmler, M. (2015). Reactive astrocytes as neural stem or progenitor cells: In vivo lineage, in vitro potential, and Genome‐wide expression analysis. GLIA, 63(8), 1452–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haim, L. B. , & Rowitch, D. (2017). Functional diversity of astrocytes in neural circuit regulation. Nature Reviews Neuroscience, 18(1), 31–41. [DOI] [PubMed] [Google Scholar]
- Heneka, M. T. , Carson, M. J. , Khoury, J. E. , Landreth, G. E. , Brosseron, F. , Feinstein, D. L. , … Kummer, M. P. (2015). Neuroinflammation in Alzheimer's disease. The Lancet Neurology, 14(4), 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner, F. L. , Ransohoff, R. M. , & Becher, B. (2015). Immune attack: The role of inflammation in Alzheimer disease. Nature Reviews Neuroscience, 16(6), 358–372. [DOI] [PubMed] [Google Scholar]
- Jimenez, S. , Baglietto‐Vargas, D. , Caballero, C. , Moreno‐Gonzalez, I. , Torres, M. , Sanchez‐Varo, R. , … Vitorica, J. (2008). Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer's disease: Age‐dependent switch in the microglial phenotype from alternative to classic. Journal of Neuroscience, : 28(45), 11650–11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John Lin, C.‐C. , Yu, K. , Hatcher, A. , Huang, T.‐W. , Lee, H. K. , Carlson, J. , … Deneen, B. (2017). Identification of diverse astrocyte populations and their malignant analogs. Nature Neuroscience, 20, 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, R. S. , Minogue, A. M. , Connor, T. J. , & Lynch, M. A. (2013). Amyloid‐β‐induced astrocytic phagocytosis is mediated by CD36, CD47 and RAGE. Journal of Neuroimmune Pharmacology, 8(1), 301–311. [DOI] [PubMed] [Google Scholar]
- Khakh, B. S. , Beaumont, V. , Cachope, R. , Munoz‐Sanjuan, I. , Goldman, S. A. , & Grantyn, R. (2017). Unravelling and exploiting astrocyte dysfunction in Huntington's disease. Trends in Neurosciences, 40(7), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft, A. W. , Hu, X. , Yoon, H. , Yan, P. , Xiao, Q. , Wang, Y. , … Lee, J. M. (2013). Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB Journal, 27(1), 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lööv, C. , Hillered, L. , Ebendal, T. , & Erlandsson, A. (2012). Engulfing astrocytes protect neurons from contact‐induced apoptosis following injury. PLoS ONE, 7(3), e33090 https://doi.org/10.1371/journal.pone.0033090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lööv, C. , Mitchell, C. H. , Simonsson, M. , & Erlandsson, A. (2015). Slow degradation in phagocytic astrocytes can be enhanced by lysosomal acidification. GLIA, 63(11), 1997–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masgrau, R. , Guaza, C. , Ransohoff, R. M. , & Galea, E. (2017). Should we stop saying “glia” and “neuroinflammation”? Trends in Molecular Medicine, 23(6), 486–500. [DOI] [PubMed] [Google Scholar]
- Morizawa, Y. M. , Hirayama, Y. , Ohno, N. , Shibata, S. , Shigetomi, E. , Sui, Y. , … Koizumi, S. (2017). Reactive astrocytes function as phagocytes after brain ischemia via ABCA1‐mediated pathway. Nature Communications, 8(1), 28 https://doi.org/10.1038/s41467-017-00037-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn, L. M. , Kamphuis, W. , Wadman, W. J. , & Hol, E. M. (2016). Astrogliosis: An integral player in the pathogenesis of Alzheimer's disease. Progress in Neurobiology, 144, 121–141. [DOI] [PubMed] [Google Scholar]
- Ponath, G. , Ramanan, S. , Mubarak, M. , Housley, W. , Lee, S. , Sahinkaya, F. R. , … Pitt, D. (2017). Myelin phagocytosis by astrocytes after myelin damage promotes lesion pathology. Brain, 140(2), 399–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos, B. , Baglietto‐Vargas, D. , Rio, J. C. D. , Moreno‐Gonzalez, I. , Santa‐Maria, C. , Jimenez, S. , … Vitorica, J. (2006). Early neuropathology of somatostatin/NPY GABAergic cells in the hippocampus of a PS1/APP transgenic model of Alzheimer's disease. Neurobiology of Aging, 27(11), 1658–1672. [DOI] [PubMed] [Google Scholar]
- Robel, S. , Berninger, B. , & Gotz, M. (2011). The stem cell potential of glia: Lessons from reactive gliosis. Nature Reviews Neuroscience, 12(2), 88–104. [DOI] [PubMed] [Google Scholar]
- Roldán, A. , Gogg, S. , Ferrini, M. , Schillaci, R. , & De Nicola, A. F. (1997). Glucocorticoid regulation of in vitro astrocyte phagocytosis. Biocell, 21(1), 83–89. [PubMed] [Google Scholar]
- Sadleir, K. R. , Kandalepas, P. C. , Buggia‐Prévot, V. , Nicholson, D. A. , Thinakaran, G. , & Vassar, R. (2016). Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Aβ generation in Alzheimer's disease. Acta Neuropathologica, 132(2), 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Mejias, E. , Navarro, V. , Jimenez, S. , Sanchez‐Mico, M. , Sanchez‐Varo, R. , Nuñez‐Diaz, C. , … Vitorica, J. (2016). Soluble phospho‐tau from Alzheimer's disease hippocampus drives microglial degeneration. Acta Neuropathologica, 132(6), 897–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Varo, R. , Trujillo‐Estrada, L. , Sanchez‐Mejias, E. , Torres, M. , Baglietto‐Vargas, D. , Moreno‐Gonzalez, I. , … Gutierrez, A. (2012). Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer's mice hippocampus. Acta Neuropathologica, 123(1), 53–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano‐Pozo, A. , Betensky, R. A. , Frosch, M. P. , & Hyman, B. T. (2016). Plaque‐associated local toxicity increases over the clinical course of Alzheimer disease. American Journal of Pathology, 186(2), 375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano‐Pozo, A. , Gómez‐Isla, T. , Growdon, J. H. , Frosch, M. P. , & Hyman, B. T. (2013). A phenotypic change but not proliferation underlies glial responses in Alzheimer disease. American Journal of Pathology, 182(6), 2332–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano‐Pozo, A. , Muzikansky, A. , Gómez‐Isla, T. , Growdon, J. H. , Betensky, R. A. , Frosch, M. P. , & Hyman, B. T. (2013). Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. Journal of Neuropathology and Experimental Neurology, 72(6), 462–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolowski, J. D. , & Mandell, J. W. (2011). Phagocytic clearance in neurodegeneration. American Journal of Pathology, 178(4), 1416–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey, F. A. , & Cammer, W. (1998). Differential uptake of dextran beads by astrocytes, macrophages and oligodendrocytes in mixed glial‐cell cultures from brains of neonatal rats. Neuroscience Letters, 248(3), 159–162. [DOI] [PubMed] [Google Scholar]
- Torres, M. , Jimenez, S. , Sanchez‐Varo, R. , Navarro, V. , Trujillo‐Estrada, L. , Sanchez‐Mejias, E. , … Vitorica, J. (2012). Defective lysosomal proteolysis and axonal transport are early pathogenic events that worsen with age leading to increased APP metabolism and synaptic Abeta in transgenic APP/PS1 hippocampus. Molecular Neurodegeneration, 7(1), 59 https://doi.org/10.1186/1750-1326-7-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo‐Estrada, L. , Dávila, J. C. , Sánchez‐Mejias, E. , Sánchez‐Varo, R. , Gomez‐Arboledas, A. , Vizuete, M. , … Gutiérrez, A. (2014). Early neuronal loss and axonal/presynaptic damage is associated with accelerated amyloid‐β accumulation in AβPP/PS1 Alzheimer's disease mice subiculum. Journal of Alzheimer's Disease, 42(2), 521–541. [DOI] [PubMed] [Google Scholar]
- Trujillo‐Estrada, L. , Jimenez, S. , De Castro, V. , Torres, M. , Baglietto‐Vargas, D. , Moreno‐Gonzalez, I. , … Vitorica, J. (2013). In vivo modification of Abeta plaque toxicity as a novel neuroprotective lithium‐mediated therapy for Alzheimer's disease pathology. Acta Neuropathologica Communications, 1(1), 73 https://doi.org/10.1186/2051-5960-1-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky, A. , Parpura, V. , Pekna, M. , Pekny, M. , & Sofroniew, M. (2014). Glia in the pathogenesis of neurodegenerative diseases. Biochemical Society Transactions, 42(5), 1291–1301. [DOI] [PubMed] [Google Scholar]
- Verkhratsky, A. , Zorec, R. , Rodriguez, J. J. , & Parpura, V. (2016). Astroglia dynamics in ageing and Alzheimer's disease. Current Opinion in Pharmacology, 26, 74–79. [DOI] [PubMed] [Google Scholar]
- Wyss‐Coray, T. , Loike, J. D. , Brionne, T. C. , Lu, E. , Anankov, R. , Yan, F. , … Husemann, J. (2003). Adult mouse astrocytes degrade amyloid‐beta in vitro and in situ. Nature Medicine, 9(4), 453–457. [DOI] [PubMed] [Google Scholar]
- Xiao, Q. , Yan, P. , Ma, X. , Liu, H. , Perez, R. , Zhu, A. , … Lee, J.‐M. (2014). Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis. Journal of Neuroscience, 34(29), 9607–9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found online in the supporting information tab for this article.
Supporting Information
Supporting Information Figure S1
Supporting Information Figure S2
Supporting Information Figure S3