

Abstract
Reported herein is a photochemical cascade process that combines the excited‐state and ground‐state reactivity of chiral organocatalytic intermediates. This strategy directly converts racemic cyclopropanols and α,β‐unsaturated aldehydes into stereochemically dense cyclopentanols with exquisite stereoselectivity. Mechanistic investigations have enabled elucidating the origin of the stereoconvergence, which is governed by a kinetic resolution process.
Keywords: cascade reactions, kinetic resolution, organocatalysis, photochemistry, radicals
Cascade reactions are valuable tools for streamlining the synthesis of structurally complex chiral molecules in a single operation and from readily available substrates.1 Their combination with asymmetric aminocatalysis2 has recently led to innovative techniques for the one‐step enantioselective preparation of stereochemically dense molecules.3 The iminium ion–enamine activation sequence depicted in Figure 1 a was crucial to fully harnessing the synthetic potential of organocascade catalysis.4 The domino process is initiated by the conjugated addition of a nucleophile to the electrophilic iminium ion intermediate I, generated from enals 1 and the chiral amine catalyst, followed by α‐functionalization of the resulting electron‐rich enamine II with an electrophile. In this well‐defined sequence, the chiral catalyst has an active role in both bond‐forming steps. This strategy, which relies on the established ground‐state polar reactivity of intermediates I and II, has reached high levels of efficiency, as seen in applications for the total synthesis of natural products.5
Figure 1.
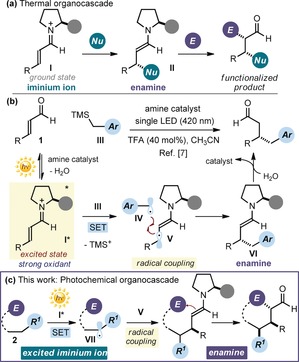
a) The established strategy for designing organocascade reactions based on the ground‐state reactivity of iminium ions and enamines. b) The previous study demonstrating that light excitation turns iminium ions I into chiral oxidants, and the resulting SET‐based radical mechanism for the enantioselective β‐alkylation of enals. c) Proposed approach for a photochemical organocascade reaction proceeding through an excited iminium ion/ground‐state enamine sequence, where a stereocontrolled radical pathway is combined with two‐electron‐pair reactivity. The gray circle represents the chiral fragment of the aminocatalyst scaffold. E=electrophile, Nu=nucleophile, SET=single electron transfer.
Recently, our laboratories found that the synthetic potential of aminocatalytic intermediates is not limited to the ground‐state domain, but can be expanded by exploiting their photochemical activity.6 For example, the photoexcitation of iminium ions can switch on novel catalytic functions that are unavailable to ground‐state reactivity.7 Specifically, selective excitation with a violet light emitting diode (LED) brings an iminium ion I into an electronically excited state (I* in Figure 1 b). This turns an electrophilic species into a strong oxidant, which could trigger the formation of benzylic radicals IV through single electron transfer (SET) oxidative cleavage of the silicon–carbon bond within benzyl trimethylsilane derivatives III. Importantly, compounds III are non‐nucleophilic substrates that are recalcitrant to classical ground‐state conjugate addition manifolds. The subsequent stereoselective coupling between IV and the chiral β‐enaminyl radical V, emerging from the SET, leads to the enamine derivative VI, which affords the final β‐benzylation product after hydrolysis. We reasoned that if the ground‐state nucleophilic reactivity of intermediate VI could be exploited to trigger a subsequent process, this might form a basis with which to implement a photochemical enantioselective cascade process. A crucial step would be to identify a suitable radical precursor 2 that, upon SET oxidation from the excited iminium ion I*, could generate an intermediate VII with ambivalent reactivity (Figure 1 c). Initially, VII should behave as a radical to then unveil, after stereocontrolled radical coupling governed by V, an electrophilic reactive center amenable to an enamine‐mediated cyclization. Herein, we detail the successful realization of this idea, which allowed us to expand the potential of organocascade catalysis by including photochemical reactivity8 as a new design principle for enantioselective domino reactions.
Figure 2 details our design plan for combining the photoexcitation of iminium ions with the ground‐state reactivity of enamines. A central element of our approach was to identify a suitable radical precursor. We envisioned that cyclopropanols of type 2 could be suitable as, upon SET oxidation from I*, unstable oxycyclopropyl radical cations VIII would be generated.9 Because of the release of strain energy, these intermediates have a strong tendency to undergo rapid ring opening to afford the β‐keto radical cations IX. Intermediates IX have the required ambivalent reactivity, possessing both radical and electrophilic character. The stereocontrolled radical coupling with the chiral β‐enaminyl radical V would initially occur to set the first stereogenic center. The resulting ground‐state enamine intermediate X would then be well‐poised to promote an aldol cyclization step by reacting with the newly formed electrophilic ketone, eventually affording the cyclopentanols 3 with three contiguous stereocenters. The feasibility of our plan was corroborated by previous studies by Mariano and co‐workers, who exploited the photoactivity of preformed cyclic non‐conjugated iminium ions to oxidize cyclopropanols of type 2 in the 1980s.10
Figure 2.
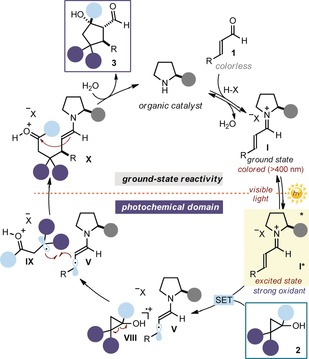
Mechanistic proposal for the excited iminium ion/enamine cascade sequence. Central to this study is the ability of the excited iminium ion I*, acting as an oxidant, to drive the generation of intermediate IX, which displays radical and electrophilic behavior.
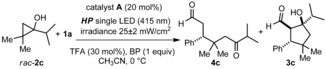
We selected cinnamaldehyde (1 a) as the model substrate while using the gem‐difluorinated diarylprolinol silyl ether catalyst A 7 to promote the formation of the chiral iminium ion I a (Table 1). The experiments were conducted in CH3CN under irradiation with a single high power (HP) LED (λ max=415 nm) with an irradiance of 25 mW cm−2, as controlled by an external power supply (for full details of the illumination set‐up, see the Supporting Information, Figure S1). The racemic cyclopropanol 2 a was selected as the radical precursor (E ox (2 a .+/2 a)=+1.66 V versus Ag/Ag+ in CH3CN). The excited iminium ion has a reduction potential (E* red (I a*/I a .−)) as high as +2.4 V (vs. Ag/Ag+ in CH3CN), as estimated from electrochemical and spectroscopic measurements.7 The SET oxidation of 2 a is therefore thermodynamically feasible. This reasoning was confirmed experimentally, as the cyclopentanol product 3 a was generated with high stereoselectivity (8:1 d.r., 95 % ee for the major diastereoisomer), albeit in moderate chemical yield (53 % yield, entry 1), when performing the reaction at ambient temperature. During control experiments, no product formation was detected in the absence of amine catalyst A or light (entry 2), demonstrating that the photoexcitation of the chiral iminium ion I a, which absorbs up to 440 nm, is essential to promote the cascade reaction. Lowering the temperature to 0 °C slightly increased the reaction yield (65 %; entry 3). We next found that the addition of 1,1′‐biphenyl (BP), commonly used as a redox mediator,11 positively influenced the reactivity,12 without affecting the stereoselectivity (entry 4). As a catalytic amount of biphenyl slightly decreased the overall efficiency, we used the conditions described in Table 1, entry 4 to demonstrate the generality of the photochemical organocascade process.
Table 1.
Optimization studies.[a]
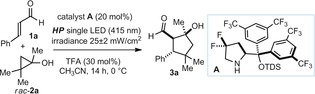
| Entry | Additive | Yield[b] [%] | d.r.[c] | ee [d] [%] |
|---|---|---|---|---|
| 1[e] | – | 53 | 8:1 | 95 |
| 2[e,f] | – | 0 | – | – |
| 3 | – | 65 | 8:1 | 97 |
| 4 | BP (1 equiv) | 88 | 8:1 | 97 |
| 5 | BP (0.2 equiv) | 75 | 8:1 | 97 |
[a] Reactions performed at 0 °C on a 0.1 mmol scale using 2 equiv of 1 a under illumination with a single high‐power (HP) LED (λ max=415 nm) with an irradiance of 25 mW cm−2. [b] Yield of 3 a isolated as a mixture of diastereomers. [c] Diastereomeric ratio inferred by 1H NMR analysis of the crude mixture. [d] Enantiomeric excess of 3 a determined by UPC2 analysis on a chiral stationary phase. [e] Performed at ambient temperature. [f] In the dark. BP=1,1′‐biphenyl, TDS=thexyldimethylsilyl, TFA=trifluoroacetic acid.
As highlighted in Figure 3, there appears to be significant tolerance towards structural and electronic variations of both substrates, enabling access to a variety of complex cyclopentanols 3 with three stereocenters. Cyclopropanols 2 bearing linear and branched alkyl (adducts 3 a–3 f), benzyl (3 g), and heterocyclic (3 h, 3 i) substituents all reacted to give the products with good yields and exquisite selectivity. The method also tolerates the presence of a cyclopropyl ring (adduct 3 d), a valuable fragment that frequently appears in complex small molecules with drug‐like properties.13 Spirocyclic compounds could also be effectively synthesized (products 3 e and 3 f). In terms of the scope with respect to α,β‐unsaturated aldehydes 1, different substitution patterns at the β‐aromatic moiety were tolerated well, regardless of their electronic and steric properties and position on the aryl ring (products 3 j–3 p). As a limitation of the method, the presence of a β‐alkyl fragment in 1 completely inhibited the reaction. In addition, the dialkyl substitution pattern on the cyclopropanol 2 was necessary to facilitate the cyclization, owing to the Thorpe–Ingold effect, and the formation of the cascade adduct 3 (see Section D3 in the Supporting Information for details). Crystals from compound 3 q were suitable for X‐ray crystallographic analysis,14 which established the relative and absolute configurations of the three stereogenic centers.
Figure 3.
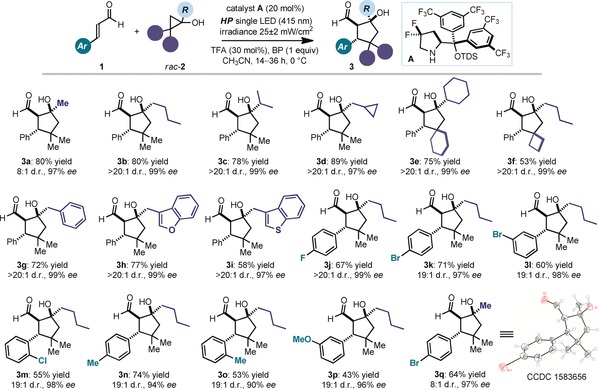
Survey of the cyclopropanols and α,β‐unsaturated aldehydes that can participate in the reaction. Reactions performed on 0.1 mmol scale using 2 equiv of 1. Yields and ee values of the isolated products are indicated below each entry (average of two runs per substrate). For all entries, yields refer to diastereomerically pure products. The d.r. values refer to the overall cascade reaction, and were inferred by 1H NMR analysis of the crude mixture. The ee values were determined by UPC2 analysis on a chiral stationary phase. BP=1,1′‐biphenyl, TDS=thexyldimethylsilyl, TFA=trifluoroacetic acid.
The results in Figure 3 indicate that the cascade reactions provide the cyclopentanol adducts 3 essentially with perfect selectivity, since a single stereoisomer out of the eight possible isomers is generally formed. It is well‐established that the combination of multiple asymmetric catalytic transformations in a cascade sequence imparts increased enantiomeric excess to the final product compared to the corresponding discrete transformations.15 However, the asymmetric amplification observed during successive cycles of a cascade comes at the expense of the diastereoselectivity. In general, the products are generated with high optical purity but a moderate d.r. As the very high diastereoselectivity of the photochemical cascade process was incongruent with this general behavior, we performed control experiments to elucidate the origin of the stereoselectivity.
We studied the reaction of 1 a with the racemic substrate 2 c because the enamine‐mediated cyclization step was relatively slow to allow for isolation of the non‐cyclized open adduct 4 c (Table 2). Thus we could monitor the yield and the enantiomeric excess of both cyclopentanol 3 c and its predecessor 4 c during the progression of the photochemical cascade reaction.
Table 2.
Understanding the origin of the stereoselectivity.[a]
| Entry | Time | 4 c | 3 c | |||||
|---|---|---|---|---|---|---|---|---|
| [h] | Yield [%] | ee [%] | Yield [%] | ee [%] | ||||
| 1 | 3 | 40 | 92 | 16 | 99 | |||
| 2 | 7 | 37 | 90 | 30 | 99 | |||
| 3 | 14 | 29 | 85 | 61 | 99 | |||
| 4 | 36 | 8 | 39 | 78 | 99 | |||
[a] Reactions performed on a 0.1 mmol scale using 2 equiv of 1 a. Yields of 3 c and 4 c determined by 1H NMR analysis of the crude reaction mixture using trichloroethylene as the internal standard. The ee values were determined by UPC2 analysis on a chiral stationary phase.
After 3 hours, the cyclic adduct 3 c, arising from the photocatalytic iminium ion/enamine cascade sequence, was formed in 16 % yield and 99 % ee. The open product 4 c, emerging from the light‐triggered radical coupling event, was generated in 40 % yield and 92 % ee. The increased enantiopurity of 3 c with respect to 4 c demonstrates that the chiral secondary amine catalyst A controls both steps of the cascade process. We also observed that the optical purity of the open product 4 c decreased with the progression of 3 c formation (entries 2–4). This behavior is consonant with a kinetic resolution regime governing the enamine‐mediated cyclization, where the chiral catalyst A selects the major enantiomer of 4 c for selective cyclization to afford 3 c as a single stereoisomer, while the minor enantiomer of 4 c remains essentially unreacted. This scenario was confirmed by reacting independently prepared racemic open adduct 4 c with the enantiopure catalyst A in the absence of light irradiation (Scheme 1). After 2 hours, a single diastereomer of the cascade adduct 3 c was formed in 31 % yield and 88 % ee, while the unreacted 4 c was enantioenriched (47 % ee). In consonance with the proposed path, unreacted 4 c had the opposite absolute configuration than in the experiment depicted in Table 2.
Scheme 1.
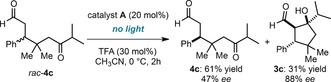
Demonstration that a kinetic resolution pathway governs the thermal enamine‐mediated step of the cascade.
Overall, these studies suggest that the two stereocontrolled steps of the cascade reaction operate sequentially to drive the formation of the cyclopentanol product 3 essentially as a single stereoisomer. The enamine‐mediated aldol reaction magnifies the original stereoselectivity of the photochemical step (about 92 % ee, as inferred from entry 1 in Table 2), selecting exclusively the major enantiomer of the intermediary open adduct 4 for cyclization.
In summary, we have developed an enantioselective cascade process that combines the excited‐state and ground‐state reactivity of chiral organocatalytic intermediates. This transformation demonstrates the possibility of effectively merging a stereocontrolled radical pattern with a classical ionic process in a cascade sequence. Further studies are ongoing to develop photochemical radical cascade processes16 to rapidly generate structural and stereochemical complexity from simple starting materials.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support was provided by the Generalitat de Catalunya (CERCA Program), Agencia Estatal de Investigación (AEI; CTQ2016‐75520‐P and Severo Ochoa Excellence Accreditation 2014–2018, SEV‐2013‐0319), and the European Research Council (ERC 681840—CATA‐LUX). G.M. thanks ICIQ‐LMU (SEV‐2013‐0319) for a predoctoral fellowship.
Ł. Woźniak, G. Magagnano, P. Melchiorre, Angew. Chem. Int. Ed. 2018, 57, 1068.
Contributor Information
Dr. Łukasz Woźniak, http://www.iciq.org/research/research group/prof‐paolo‐melchiorre/
Prof. Dr. Paolo Melchiorre, Email: pmelchiorre@iciq.es.
References
- 1.
- 1a. Nicolaou K. C., Edmonds D. J., Bulger P. G., Angew. Chem. Int. Ed. 2006, 45, 7134–7186; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 7292–7344; [Google Scholar]
- 1b. Tietze L. F., Brasche G., Gericke K. M. in Domino Reactions in Organic Synthesis, Wiley-VCH, Weinheim, 2006; [Google Scholar]
- 1c. Xu P.-F., Wang W. in Catalytic Cascade Reactions, Wiley-VCH, Weinheim, 2013. [Google Scholar]
- 2.
- 2a. C. F. Barbas III , Angew. Chem. Int. Ed. 2008, 47, 42–47; [Google Scholar]; Angew. Chem. 2008, 120, 44–50; [Google Scholar]
- 2b. List B., Angew. Chem. Int. Ed. 2010, 49, 1730–1734; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 1774–1779; [Google Scholar]
- 2c. Lelais G., MacMillan D. W. C., Aldrichimica Acta 2006, 39, 79–87. [Google Scholar]
- 3.For reviews, see:
- 3a. Enders D., Grondal C., Hüttl M. R. M., Angew. Chem. Int. Ed. 2007, 46, 1570–1581; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 1590–1601; [Google Scholar]
- 3b. Pellissier H., Adv. Synth. Catal. 2012, 354, 237–294; [Google Scholar]
- 3c. Hepburn H. B., Dell′Amico L., Melchiorre P., Chem. Rec. 2016, 16, 1787–1806. [DOI] [PubMed] [Google Scholar]
- 4.For the pioneering studies that established the possibility of integrating the ground-state reactivity of iminium ions and enamines into elaborate cascade sequences, see:
- 4a. Huang Y., Walji A. M., Larsen C. H., MacMillan D. W. C., J. Am. Chem. Soc. 2005, 127, 15051–15053; [DOI] [PubMed] [Google Scholar]
- 4b. Marigo M., Schulte T., Franzen J., Jørgensen K. A., J. Am. Chem. Soc. 2005, 127, 15710–15711; for another landmark report, see: [DOI] [PubMed] [Google Scholar]
- 4c. Enders D., Hüttl M. R. M., Grondal C., Raabe G., Nature 2006, 441, 861–864. [DOI] [PubMed] [Google Scholar]
- 5.For a review on the impact of organocascade catalysis in total synthesis, see:
- 5a. Grondal C., Jeanty M., Enders D., Nat. Chem. 2010, 2, 167–178; for a selected example, see: [DOI] [PubMed] [Google Scholar]
- 5b. Jones S. P., Simmons B., Mastracchio A., MacMillan D. W. C., Nature 2011, 475, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Arceo E., Jurberg I. D., Álvarez-Fernández A., Melchiorre P., Nat. Chem. 2013, 5, 750–756; [DOI] [PubMed] [Google Scholar]
- 6b. Woźniak Ł., Murphy J. J., Melchiorre P., J. Am. Chem. Soc. 2015, 137, 5678–5681; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Silvi M., Arceo E., Jurberg I. D., Cassani C., Melchiorre P., J. Am. Chem. Soc. 2015, 137, 6120–6123; [DOI] [PubMed] [Google Scholar]
- 6d. Bahamonde A., Melchiorre P., J. Am. Chem. Soc. 2016, 138, 8019–8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Silvi M., Verrier C., Rey Y., Buzzetti L., Melchiorre P., Nat. Chem. 2017, 9, 868–873. [DOI] [PubMed] [Google Scholar]
- 8.For an example of a non-stereoselective cascade process where the different photochemical activities of a single light-absorbing catalyst were combined, see:
- 8a. Metternich J. B., Gilmour R., J. Am. Chem. Soc. 2016, 138, 1040–1045; for a review on enantioselective catalytic photochemical reactions, see: [DOI] [PubMed] [Google Scholar]
- 8b. Brimioulle R., Lenhart D., Maturi M. M., Bach T., Angew. Chem. Int. Ed. 2015, 54, 3872–3890; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3944–3963. [Google Scholar]
- 9.For a review on the chemistry of cyclopropanols, see:
- 9a. Kulinkovich O. G., Chem. Rev. 2003, 103, 2597–2632; for selected examples of oxidative functionalization, see: [DOI] [PubMed] [Google Scholar]
- 9b. Iwasawa N., Hayakawa S., Isobe K., Narasaka K., Chem. Lett. 1991, 20, 1193–1196; [Google Scholar]
- 9c. Abe M., Oku A., J. Org. Chem. 1995, 60, 3065–3073; [Google Scholar]
- 9d. Abe M., Nojima M., Oku A., Tetrahedron Lett. 1996, 37, 1833–1836; [Google Scholar]
- 9e. Rinderhagen H., Waske P. A., Mattay J., Tetrahedron 2006, 62, 6589–6593; [Google Scholar]
- 9f. Bloom S., Bume D. D., Pitts C. R., Lectka T., Chem. Eur. J. 2015, 21, 8060–8063. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Stavinoha J., Bay E., Leone A., Mariano P. S., Tetrahedron Lett. 1980, 21, 3455–3458; [Google Scholar]
- 10b. Mariano P. S., Stavinoha J., Bay E., Tetrahedron 1981, 37, 3385–3395. [Google Scholar]
- 11.
- 11a. Mizuno K., Kamiyama K., Ichinose N., Otsuji N., Tetrahedron 1985, 41, 2207–2214; [Google Scholar]
- 11b. Mangion D., Arnold D. R., Acc. Chem. Res. 2002, 35, 297–304; [DOI] [PubMed] [Google Scholar]
- 11c. Skubi K. L., Blum T. R., Yoon T. P., Chem. Rev. 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Redox mediators are generally used to facilitate exergonic redox processes that are kinetically slow, or to mitigate the occurrence of a back-electron transfer. In this system, a possible alternative role of 1,1′-biphenyl (BP) is to prevent the SET oxidation of the secondary amine catalyst A, which leads to decomposition (E ox (BP.+/BP)=+1.90 V, E ox (A .+/A)=+2.20 V vs. Ag/Ag+ in CH3CN). In consonance with this scenario, preliminary kinetic measurements indicate that the presence of BP does not influence the initial rate of the cascade process. In contrast, a large amount of catalyst A remains at the end of the reaction when adding BP while catalyst degradation is much more significant in the absence of the redox mediator.
- 13. Talele T. T., J. Med. Chem. 2016, 59, 8712–8756. [DOI] [PubMed] [Google Scholar]
- 14. CCDC 1583656 (3 q) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- 15.The Horeau principle provides the mathematical foundation for rationalizing the enantioenrichment observed during successive cycles of an enantioselective catalytic cascade process; see: Vigneron F. J. P., Dhaenens M., Horeau A., Tetrahedron 1973, 29, 1055–1059. [Google Scholar]
- 16. Plesniak M. P., Huang H.-M., Procter D. J., Nat. Rev. Chem. 2017, 1, 0077. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary