


Abstract
The Saccharomyces cerevisiae Pif1 protein (ScPif1p) is the prototypical member of the Pif1 family of DNA helicases. ScPif1p is involved in the maintenance of mitochondrial, ribosomal and telomeric DNA and suppresses genome instability at G-quadruplex motifs. Here, we report the crystal structures of a truncated ScPif1p (ScPif1p237−780) in complex with different ssDNAs. Our results have revealed that a yeast-specific insertion domain protruding from the 2B domain folds as a bundle bearing an α-helix, α16. The α16 helix regulates the helicase activities of ScPif1p through interactions with the previously identified loop3. Furthermore, a biologically relevant dimeric structure has been identified, which can be further specifically stabilized by G-quadruplex DNA. Basing on structural analyses and mutational studies with DNA binding and unwinding assays, a potential G-quadruplex DNA binding site in ScPif1p monomers is suggested. Our results also show that ScPif1p uses the Q-motif to preferentially hydrolyze ATP, and a G-rich tract is preferentially recognized by more residues, consistent with previous biochemical observations. These findings provide a structural and mechanistic basis for understanding the multifunctional ScPif1p.
INTRODUCTION
Saccharomyces cerevisiae Pif1 protein (ScPif1p) is the founding member of the Pif1 family helicases conserved from prokaryote to eukaryote. ScPif1p was initially discovered through genetic screening for genes that affect the frequency of mitochondrial DNA recombination between wild-type and mutant strains (1). ScPif1p has both mitochondrial (aa 1−859) and nuclear (aa 40−859) forms due to alternate usages of the first (pif-m1) and the second (pif-m2) AUG sites in the ScPif1p open reading frame (2).
The nuclear form of ScPif1p (Figure 1A and Supplementary Figure S1) plays important roles in many aspects of DNA transactions and genome stabilities (3,4). Genetic characterizations revealed that an overexpression of active ScPif1p results in telomere shortening through a mechanism by which the active telomerase is displaced from DNA ends by ScPif1p (5,6). Furthermore, ScPif1p enhances Okazaki fragment maturation in coordination with the Dna2 helicase/nuclease to process long flaps that cannot be directly cleaved by the FEN1 nuclease (7,8). Nuclear-form ScPif1p is also required for efficient fork arrest at a site called replication fork barrier (RFB) within the ribosomal DNA (rDNA) (9). Furthermore, Rrm3p, which shares highly conserved protein sequences with Pif1p, is needed for timely fork progression through chromosomal sites, including rDNA genes, tRNA genes, centromeres, inactive replication origins, telomeres and subtelomeric regions (10,11). Recently, it has been shown that ScPif1p strongly promotes recombination-specific DNA synthesis during homologous recombination by recruiting polymerase Polδ to the break site and stimulating D-loop migration for conservative DNA replication (12,13). More importantly, ChIP analysis on yeast genome has shown that ScPif1p binds directly to G-rich DNA sequences which potentially form G-quadruplex (G4) structures (9). By mutating potential G4-forming genomic sequences and/or inactivating ScPif1p in cells, Zakian et al. also showed that the potential G4-forming sequences in cells impede DNA replication and lead to chromosomal breakage if Pif1p is impaired (14). Furthermore, it was demonstrated that the G4 DNA-induced damage may lead to new genetic and epigenetic changes in ScPif1p-deficient cells (15).
Figure 1.

Structure of the ScPif1p–DNA–ADP·AlF4 ternary complex. (A) Linear representation of the amino acid sequence of the nuclear-form protein, with labeling of relevant domains or motifs in a color scheme to be used in subsequent figures. (B) Ribbon representation of ScPif1p in complex with ssDNA and ADP·AlF4. (C) Surface representation of the complex structure. (D) Structural alignment of BsPif1 (gray with DNA in hotpink) and ScPif1p (orange with DNA in green). The 2C domain of ScPif1p is shown in yellow. (E) Comparison of the spatial positions and coordinations of ADP·AlF4 in ScPif1p and BsPif1. (F) Stopped-flow kinetic traces for partial duplex DNA unwinding with different NTPs, which were used to determine the unwinding amplitude Am and rate ku from fittings with Equation (2). The stopped-flow assay was performed with 200 nM ScPif1p, 4 nM fluorescently labeled partial duplex DNA (S26ds17) and 1 mM NTP. All stopped-flow kinetic traces in the whole work are averages of over 10 individual traces.
In recent years, much biochemical and biophysical work has been devoted to the understanding of the molecular mechanistic basis of ScPif1p's cellular multifunctions. The nuclear-form ScPif1p is composed of 820 amino acids and displays helicase activity with 5′−3′ polarity in an ATP- and Mg2+-dependent manner (16). The protein is not highly processive and preferentially unwinds RNA/DNA duplex over DNA/DNA duplex (17,18). ScPif1p unwinds duplex DNA with a kinetic step-size of 1 bp and unwinds G4 DNA in a sequential and repetitive unfolding manner (19–21). It is also shown that G-rich and/or G4 motif induces ScPif1p dimerization. Interestingly, ScPif1p-mediated duplex DNA unwinding can be significantly stimulated by an upstream G4 motif at the ss/dsDNA junction through reducing a ‘waiting time’, which is interpreted as the time needed for dimerization of ScPif1p (22,23,24). We and others have recently solved the three-dimensional structures of the ScPif1p homologs from Bacteroides ssp strains (Bacteroides sp. 3_1_23 and sp. 2_1_16) (25,26). However, the structural mechanisms for ScPif1p's recognition of different potential physiological substrates and for the regulation of its enzymatic activities by different substrates remain to be determined.
In the present study, we have solved the crystal structures of the helicase-core region of ScPif1p in complex with a series of ssDNAs and in the presence of ADP·AlF4 or ATPγS. Although the domain folding, the relative domain orientations, the DNA binding arrangements and the overall domain organization resemble that of BsPif1, ScPif1p displays several unique features in its structures and functions.
MATERIALS AND METHODS
Protein expression and purification
The gene (Gene ID = NC_001145) encoding the ScPif1p was truncated from both 5′ and 3′ ends based on the sequences similarities of Pif1 family helicases. The DNA fragment corresponding to amino acids 237−780 was cloned into vector pET15b-SUMO (Takara) and transformed into the C2566H Escherichia coli cells (New England Biolabs). When the culture reached early stationary phase (OD600 = 0.55−0.6) at 37°C, 0.3 mM IPTG was added and the protein expression was induced at 18°C over 16 h. Cells were harvested by centrifugation (4500 g, 4°C, 15 min) and pellets were suspended in lysis buffer (20 mM Hepes, pH 8.0, 500 mM NaCl and 5% glycerol (v/v)). Cells were broken with a French press and then were further sonicated 2−3 times to shear DNA. After centrifugation at 12 000 rpm for 30 min, the supernatants were filtered through a 0.45 μm filter and were loaded onto the Ni-NTA column (QIAGEN) at 4°C. The SUMO-ScPif1p fusion protein was eluted with elution buffer (20 mM Tris-HCl, pH 7.8, 500 mM NaCl, 200 mM Imidazole and 10% glycerol (v/v)). The SUMO tag was cleaved with Sumo protease (Invitrogen, Beijing) and ScPif1p was dialyzed in the lysis buffer at 4°C for 16 h. Protein was reloaded onto the Ni-NTA column (QIAGEN) to remove the SUMO-tag and further purified by cation-exchange chromatography (HiTrap SP, GE Healthcare). The highly purified protein was dialyzed against the storage buffer (20 mM Tris-HCl, pH 7.8, 500 mM NaCl, 1 mM DTT) and concentrated to approximately 12 mg/mL for crystallization. The selenomethionine-substituted protein was expressed using SelenoMet™ Medium kit (Molecular Dimensions) and purified with the same protocol as described above.
DNA substrate preparations
All oligonucleotides used for DNA substrates were synthesized by Sangon Biotech (Shanghai) and listed in Supplementary Table S1. The oligonucleotides used in dynamic light scattering (DLS) assays and small angle X-ray scattering (SAXS) samples were prepared in 10 μM working concentration. The partial duplex DNA S26ds17 used in the stopped-flow assay and G4 DNA used in the binding assay were heated to 95°C in stocking buffer (20 mM Tris-HCl, pH 8.0, 100 mM KCl). The DNA were cooled to room temperature slowly and further purified using monoQ column (GE Healthcare) at a KCl gradient in monoQ buffer (A buffer: 20 mM K2HPO4-KH2PO4, pH 6.8, 100 mM KCl; B buffer: 20 mM K2HPO4-KH2PO4, pH 6.8, 1 M KCl). The oligonucleotides labeled with FAM, HEX or Cy3 were purified by HPLC before being used in DNA binding, ssDNA translocation, DNA unwinding and smFRET assays (Supplementary Table S1).
Analytical size exclusion chromatography
Gel filtration experiments were performed using a 10/30 Superdex 200 GL gel filtration column (GE Healthcare) as described previously (27). Briefly, the column was equilibrated at a flow rate of 0.4 ml/min with 25 mM Tris-HCl (pH 7.5), 200 mM NaCl or 100 mM KCl, 2 mM MgCl2, 2 mM DTT. When specified, 1 mM ADP·AlF4 or ATPγS was added to the elution buffer. About 90 μg of ScPif1p was loaded with a final concentration of 14 μM and absorbance at 280 and 260 nm was recorded. The experiments in the presence of ss-, ds- and G4 DNA were performed by preincubating ScPif1p, DNAs and 1 mM nucleotide for 20 min at 25°C. The calibration graph of log RS versus Kav was constructed using a high and low molecular weight calibration kit from Sigma: cytochrome c (12.4 kDa), carbonic anhydrase (29 kDa), albumin (67 kDa), phosphorylase b (97.4 kDa), and thyroglobulin (669 kDa). Assuming similar shape factors, the plot calibration of log Mw versus Kav allowed the determination, in a first approximation, of the molecular weight of ScPif1p protein.
Crystallization of ScPif1p-nucleic acid complexes
Purified ScPif1p was mixed with the DNA (Supplementary Table S1) at a 1:1.2 molar ratio, yielding a protein-DNA complex (250 μM). Crystallization screening was carried out by vapor diffusion method at 20°C using commercial screening kits (Hampton Research, Molecular Dimensions and Rigaku Reagents), where the ScPif1p-DNA-ADP·AlF4 complex was mixed at a 1:1 ratio with the reservoir solution. Optimized crystallization conditions included 0.1 M Tris-HCl (pH 7.5), 0.2 M Na2SO4 and 15% PEG5000-MME as precipitant. Se-Met-ScPif1p-Poly(T8)-ADP·AlF4 ternary complex was crystallized in 100 mM NaAc-HAc (pH 5.5), 240 mM NaF and 14−16% PEG8000. ADP, AlCl3, and NaF were from Sigma-Aldrich. ADP-AlFx-Mg2+ were prepared by mixing AlCl3 with equimolar amounts of ADP and MgCl2 and a 5-fold molar excess of NaF.
X-ray data collection, phasing and refinement
X-ray diffraction data from native crystals and from a SeMet-substituted protein crystal were collected on BL17U and BL19U1 beamlines at SSRF synchrotron (Shanghai, China) and were processed using the XDS package (28). The ternary complexes containing protein, ssDNA (Poly(G3T5) or Poly(T3G3T2)) and nucleotide ADP·AlF4 crystallized in P212121 or P3121 space groups. The structure in P212121 was solved by SAD using 3.0 Å data collected at the selenium peak wavelength. Thirty four selenium atoms are found with SHELXC/D (29) and phasing was done with AutoSHARP software (30). After solvent flattening, the figure of merit was 0.70 and most of the residues could be built automatically with Buccaneer (31) in the experimental map. The model was then manually built with Coot (32) and refined with Phenix (33) on a native dataset of ScPif1p complexed to ADP·AlF4 and Poly(G3T5) oligonucleotide diffracting to 2.0 Å resolution. This reference structure was used as a model to solve the other structures either in P212121 or P3121 by molecular replacement. Cell parameters and data collection statistics are reported in Supplementary Table S2.
Small-angle X-ray scattering
SAXS experiments were carried out at 20°C with size-exclusion chromatography HPLC coupled to SAXS (SEC-SAXS) data collection on beamline SWING (SOLEIL Synchrotron, France). The samples of protein and protein–DNA complexes at a concentration of 10 mg/ml (injected on HPLC) were measured in SAXS buffer (20 mM Hepes, pH 8.0, 200 mM NaCl or 100 mM KCl, 5% glycerol). Scattering data collection and processing were performed with Foxtrot (34). Additional processing of SEC-SAXS HPLC profile was done with the specific module US-SOMO of Ultrascan2 software (35). Baseline correction of the HPLC profile was applied when necessary and the peaks which were not baseline-resolved were deconvoluted with Gaussian approximation tools included in US-SOMO. SEC-SAXS profiles with treatment of peaks, are shown in Supplementary Figure S6 and Table S3. The peaks were approximated in I(t) dimension as Gaussian functions and SAXS profiles I(q) for the extracted peaks were generated thereafter. I(0) and radius of gyration (Rg) were calculated over the profiles with Guinier approximation. Single peaks were averaged over frames with constant Rg to produce SAXS reference data I(q) for each peak. Pair distance distribution function (PPDF) was calculated using the GNOM5 program from ATSAS 2.8 suite (36). The maximum particle dimension (Dmax) and Rg were extracted from PPDF. Quality of the data was also assessed by Rg calculation from the Guinier fit and from PPDF. Solution ab initio models were determined using MONSA (36) for protein-DNA complexes or DAMMIF for protein or DNA species. Model quality was evaluated further using averaging with DAMAVER (36). Finally, the atomic model profile was calculated, aligned and fitted to the experimental data using CRYSOL and fitted in the SAXS envelope with SUPCOMB (Supplementary Figure S9 and Table S4) (36).
Fluorescence polarization binding assay
The apparent dissociation constants of ScPif1p and the modified proteins were determined with a fluorescence anisotropy assay under equilibrium conditions (27). Protein concentration-dependent changes in fluorescence anisotropy were measured with FAM-labeled DNA using an Infinite F200 instrument (TECAN). Varying amounts of ScPif1p protein were added to a 150 μl aliquot of binding buffer (25 mM Tris-HCl, pH 7.5, 100 mM NaCl or KCl, 2 mM MgCl2 and 2 mM DTT) containing 5 nM FAM-labeled DNA. Each sample was allowed to equilibrate in solution for 2−5 min. Then, the steady-state fluorescence anisotropy (r) was measured. A second reading was taken after 5 min, to ensure that the mixture was well equilibrated and stable. The equilibrium dissociation constants were determined by fitting the binding curves using Equation (1),
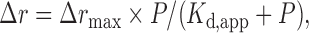 |
(1) |
where Δrmax is the maximal amplitude of the anisotropy (i.e. rmax – rfree,DNA), P is the helicase concentration, and Kd,app is the midpoint of the curve corresponding to the apparent dissociation constant.
Stopped-flow unwinding assay
The stopped-flow assay was carried out according to Duan et al. (23). Briefly, unwinding kinetics were measured in a two-syringe mode, where ScPif1p and fluorescently labelled DNA substrate were pre-incubated at 25°C in syringe 3 for 5 min and the unwinding reaction was initiated by rapid mixing ATP in syringe 4. Each syringe contained unwinding reaction buffer A (25 mM Tris-HCl, pH 7.5, 50 mM NaCl, 2 mM MgCl2, 2 mM DTT). All concentrations listed are after mixing unless noted otherwisely. For converting the output data from volts to percentage unwinding, a calibration experiment was performed in a four-syringe mode, where helicase in syringe 1, hexachlorofluorescein-labeled single-stranded oligonucleotides in syringe 2, and fluorescein-labeled single-stranded oligonucleotides in syringe 3 were incubated in unwinding reaction buffer A, and ATP were in syringe 4. The fluorescent signal of the mixed solution from the four syringes corresponded to 100% unwinding. The standard reaction was usually performed with 4 nM DNA substrates and 200 nM ScPif1p in buffer A.
All stopped-flow kinetic traces were averages of over 10 individual traces. The kinetic traces were analyzed using Bio-Kine (version 4.26, Bio-Logic, France) with the following equation,
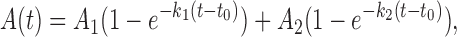 |
(2) |
where A(t) represents the fraction of DNA unwound as a function of time, A1 and A2 are the unwinding amplitudes, k1 and k2 are the unwinding rate constants of the two phases, t0 is the time at which the fraction of DNA unwound starts to rise. From the four parameters obtained through fitting, we can get the total unwinding amplitude Am = A1 + A2 and the initial unwinding rate (i.e., the slope of the kinetic unwinding trace at early times) ku = k1A1 + k2A2.
Stopped-flow translocation assay
This assay was performed essentially according to Liu et al. (27). In brief, translocation experiments in the presence of 1 μM protein trap (dT56 DNA) were performed with final concentrations of 100 nM Cy3-labeled oligonucleotides, 50 nM ScPif1p and 500 μM ATP. Cy3 was excited at 548 nm (2 nm slit width), and its emission was monitored at 568 nm using a high pass filter with 30 nm bandwidth (D568/30, Chroma Technology Co.). All experiments were performed at 25°C in buffer A (25 mM Tris-HCl, pH 7.5, 50 mM NaCl, 2 mM MgCl2 and 2 mM DTT).
Dynamic light scattering assay
DLS measurements were performed using a DynaPro NanoStar instrument (Wyatt Technology Europe GmbH, Germany) equipped with a thermostated cell holder using filtered (0.1 μm filters) solutions in disposable cuvettes (UVette, Eppendorf). The protein concentration was 4.5 μM in a Tris-HCl buffer (50 mM, pH 8.0, 200 mM NaCl, 1 mM DTT) (total volume, 50 μl). The scattered light was collected at an angle of 90°. Recording times were typically between 3 and 5 min (20−30 cycles in average, 10 s in each cycle). The analysis was performed with the Dynamics 7.0 software using regularization methods (Wyatt Technology, France). The molecular weight was calculated from the hydrodynamic radius using the following empirical Equation (3),
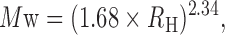 |
(3) |
where Mw and RH represent the molecular weight (in kDa) and the hydrodynamic radius (in nm), respectively.
Single-molecule fluorescence data acquisition
Single-molecule FRET study was carried out with a home-built objective-type total-internal-reflection microscopy and performed according to Zhang et al. (20,24,37). In brief, Streptavidin (10 μg/ml) in a buffer containing 50 mM NaCl and 25 mM Tris-HCl (pH 7.5) was added to the microfluidic chamber made of a polyethylene glycol (PEG)-coated coverslip, and incubated for 10 min. After washing, 50 pM DNAs (smS47ds17 or smS26G4ds17) were added to the chamber and allowed to be immobilized for 10 min. Then free DNA was removed by washing with the reaction buffer. Imaging was initiated before 2.5 nM ScPif1p or mutated ScPif1p (with or without ATP) was flowed into the chamber. We used an exposure time of 100 ms for all single-molecule measurements at a constant temperature of 22°C. A series of movies were recorded with 1 s duration at different times, and the acceptor spots were counted to represent the number of remaining DNA molecules.
Molecular modeling of ScPif1p–G4 complex
The telomeric G4 structure (PDB entry: 3SC8) was docked on ScPif1p with flexible docking using HADDOCK 2.2 Web server (38). G4 structure was modeled at the 3′ end of ssDNA bound to the crystallographic structure of ScPif1p. For HADDOCK calculations, active residues for ScPif1p were chosen as the closest to G4 DNA. Passive residues were automatically defined around the active residues by HADDOCK. All of the bases of the G4 sequence were considered active in the docking. The standard docking procedure was used: from initial 1000 complex structures generated by rigid body docking, the 200 lowest energy structures were further refined in explicit water after semi-flexible simulated annealing. A cluster analysis was performed on the finally docked structures corresponding to the 200 best solutions with lowest intermolecular energies based on a 7.5 Å root-mean-square deviation cut-off criterion. The clusters were ranked according to the averaged Z-score of their top 10 structures.
The best solution was then submitted to 100 ns molecular dynamics simulation with GROMACS using AMBER99SB force fields for protein and DNA (39). Before molecular dynamics, the system was equilibrated in an explicit dodecahedron TIP3 water box at 300 K and 1 bar. Trajectory was analyzed with the tools distributed with GROMACS.
RESULTS
Structure overview
The truncated ScPif1p237−780 displays helicase activity comparable to the nuclear form of ScPif1p (aa 40−859) (Supplementary Figure S2A and B). Therefore, for simplicity, the truncated protein ScPif1p237−780 is mentioned as ScPif1p unless specified otherwisely. The protein was crystallized in complex with ssDNA in the presence of ADP·AlF4 or ATPγS. The ternary complex containing protein, ssDNA (Poly(G3T5)) and nucleotide ADP·AlF4 was crystallized in P212121 space group with two molecules per asymmetric unit (a.u.) related by a non-crystallographic translational symmetry (tNCS). ScPif1p bound with Poly(T8) and ATPγS was also crystallized under similar conditions. Another crystal form in P3121 with two molecules per a.u. was also obtained with ssDNA (poly(T3G3T2)) and ADP·AlF4. Attempts to solve ScPif1p by molecular replacement with BsPif1 or human Pif1 models gave a good solution, but leaving some unsolved missing regions. To remove bias from models, the structure in P212121 was solved by SAD. The asymmetric unit in P212121 contains two independent ScPif1p monomers, related by tNCS that can be superimposed with an r.m.s.d. deviation of 1.503 Å for 496 equivalent α-carbon atoms. From these two molecules in the asymmetric unit and by applying space group symmetries, several types of dimers can be described from the crystal packing. These dimers are associated by different interfaces which all have a buried surface of ∼500 Å2 (Supplementary Figure S3). The structures of these dimers will be described in more detail in another section.
ScPif1p monomer has the structural characteristics of SF1B family helicases with domains 1A (237−306; 326−416), 1B (307−325), 2A (417−478; 699−754), 2B (479−698) and CTD (755−780) (Figure 1A and Supplementary Figure S1) (40). Compared to the previously solved 3D structures of Pif1 family proteins BsPif1 (PDB entry: 5FTE) and human Pif1 (PDB entry: 5FHH) (25,26), ScPif1p displays three striking features (Figure 1B−D): i) the presence of an yeast-specific extra structure protruding from domain 2B, named as domain 2C (aa 544−656); ii) the previously identified loop3 in ScPif1p is significantly shorter than that in BsPif1; iii) while the previously identified domain 1B in BsPif1 (aa 72−88) forms an unstructured loop, that in ScPif1p (aa 307−325) folds into an α-helix (α5) bearing a cluster of positively charged residues. The spatial configuration of α5 helix relative to G4 DNA bears some similarities with a G4 DNA recognition α-helix (RSM) in human RHAU helicase (see late description). The remaining overall structure resembles the BsPif1-Poly(T8)-ADP·AlF4 complex (PDB entry: 5FTE) with an r.m.s.d. calculated on Cα atoms of 1.98 Å (Figure 1D). The conformation of domain 2B and the configuration of ssDNA binding are similar to that observed in the BsPif1-Poly(T8) complex.
Nucleotide binding site
ADP·AlF4 was used to co-crystallize the ternary complex (ScPif1p-ADP·AlF4-ssDNA). AlF4 forms a tetragonal bipyramid that makes it an accurate analog of the transition state of the γ-phosphate of ATP (41). ADP·AlF4 is bound in the cleft between domains 1A and 2A with an hexacoordinated magnesium as found in most DEAD box or DExH proteins (Figure 1E). Resembling the BsPif1 structure (PDB entry: 5FTE), the highly conserved F416 is involved in aromatic stacking of the adenine ring of ADP·AlF4 (Figure 1E). AlF4 is recognized by one uncharged amino acid (Q381) from domain 1A and three basic residues, K264 (in motif I), R417 and R734 (in motifs IV and VI, respectively) from domain 2A (Figure 1E, left panel). ATPγS was built as ADP·AlF4 in the lower-resolution electron density map of crystals of ScPif1p with Poly(T8).
The conserved Q motif of RNA/DNA helicases has been structurally implicated in nucleotide binding and specific recognition of adenine base (42). Although both residues Q11 in BsPif1 and Q241 in ScPif1p are highly conserved, Q11 is too far to engage in hydrogen bonding with adenine base (Figure 1E, right panel) (25). In sharp contrast, the distances between Q241 OE1 and N6, and between Q241 NE2 and N7 are 2.72 and 3.12 Å, respectively, in ScPif1p (Figure 1E, left panel). Thus, the N6 and N7 atoms of adenine base in the ternary complex of ScPif1p are specifically recognized by atoms OE1 and NE2 of Q241, potentially functioning as a canonic Q-motif, which exists also in human Pif1 (Supplementary Figure S4A−C). Figure 1F shows that both the unwinding amplitude and rate with ATP (Am = 0.5 and ku = 1.9 s−1) are significantly higher than that with GTP, UTP and CTP (Am = 0.37 and ku = 0.44 s−1 in average), indicating that ScPif1p uses the Q-motif to more efficiently hydrolyze ATP. In accordance with the above interpretations, the mutant Q241A displays significant reductions in both unwinding amplitude and rate with all the four types of nucleotides. Especially, the ATP-driven helicase activity was reduced to the same level as that of UTP and CTP (Supplementary Figure S4D). These structural and biochemical results, taken together, show that ScPif1p uses the Q-motif to more efficiently hydrolyze ATP, in contrast with BsPif1 which displays general NTPase activities due to its absence of a functional Q-motif (27).
Structural basis for preferential recognition of G-rich tracts
Previous studies have shown that ScPif1p had 5 folds higher binding ability for G-rich over non-G-rich ssDNA (15). We further showed that G4/G-rich sequences may stimulate its duplex DNA unwinding activity (24). To understand the structural basis for specific recognition and binding of G-rich sequences, we co-crystallized ScPif1p in complex with different types of ssDNA oligonucleotides including Poly(T8) and two oligos bearing consecutive G bases at its 5′ or 3′ end. Pure G-rich sequences were not used as they are prone to form G4 structures. Attempts to co-crystallize ScPif1p with Poly(dA) or Poly(dC) were unsuccessful.
Figure 2A (left panel) shows that the 6-base-T binding site traverses along a channel that runs across the top of domains 2A and 1A of ScPif1p in a 5′−3′ direction, as previously observed in BsPif1-ssDNA complex (Figure 2A, right panel). Though the Poly(T8) configurations and the interactions involved in DNA-protein binding in BsPif and ScPif1p are similar, some differences in the sugar-phosphate backbone binding and base recognition can be uncovered. First, while the only phosphate backbone is exclusively bound by residues from domains 1A and 2A in the BsPif1-ssDNA complex, both the phosphate and sugar groups are recognized by residues in domains 1A, 2A and 2B in the ScPif1p-ssDNA complex, in which N533 and N526 interact with the OH groups of phosphates 5 and 6 (Figure 2A and B). In addition, the two highly conserved residues H705 and H303 seem to stack on the deoxyribose rings of nucleotides 2 and 4 (Figure 2A and B). Such interactions are absent in the BsPif1-ssDNA crystal structure (25). Though it is not a stabilizing effect by π-π stacking, this interaction is mainly steric. Thus, these histidine residues may prevent RNA binding through its 2΄ hydroxyl group. These structural features may explain why ScPif1p preferentially loads ssDNA strand over ssRNA strand for translocation (17).
Figure 2.

(A) Detailed views of ssDNA-protein interactions in ScPif1p (left panel) and BsPif1 (right panel). (B) Schematics of ScPif1p's interactions with Poly(T8) (left panel), Poly(G3T5) (middle panel), and Poly(T3G3T2) (right panel). (C) An overlay of spatial conformations of Poly(T8) (show in light gray) and Poly(G3T5) (colored as Figure 1B) with the indicated amino acid side chains involved in ssDNA binding. (D) Representative ssDNA/RNA binding curves determined with ScPif1p (left panel) and mutant H303G/H705G (right panel). The assay was performed according to ‘Materials and Methods’. Inset in the left panel indicates the sequences of ssDNAs used. (E) Representative ssDNA binding curves of ScPif1p determined with Poly(T12) and GR12 in the absence (left panel) and in the presence of 0.1 mM ATPγS (right panel). Inset in the left panel indicates the sequences of ssDNAs used.
The above interpretation is supported by our equilibrium binding assays with the wild-type and a modified version of ScPif1p in which both H303 and H705 were replaced by glycine residues (H303G/H705G). While the wild-type protein displays a stronger binding ability towards Poly(T12) over Poly(U12) (Kd,appDNA = 15.1 nM versus Kd,appRNA = 162.9 nM), the H303G/H705G mutant has a completely inversed DNA/RNA binding property: it almost loses the DNA binding ability (too low to be determined precisely), but gains significant RNA binding activity (Kd,appDNA = ND versus Kd,appRNA = 70.5 nM) (Figure 2D). Second, while both the 5′- and 3′-end bases in the BsPif1-ssDNA complex are free of any contact with amino acids (25), the 5′-end base T1 in the ScPif1p-ssDNA complex is stacked by F723 to form hydrogen bonds between them (Figure 2A and B).
A comparison between the solved structure of ScPif1p in complex with ssDNA bearing 3 consecutive G bases and that with Poly(dT) oligonucleotides (Figure 2A−C) reveals that the residues implicated in interactions with the sugar-phosphate backbones of both oligonucleotides are essentially the same (Figure 2B). Inspection of base recognition shows that the first three consecutive G bases from the 5′ end of the G-rich oligonucleotide are specifically recognized by three additional residues (E724, K387 and K312) while only the first T1 base is recognized in the ScPif1p-Poly(T8) complex (Figure 2A and B). Importantly, while T2 and T3 in the ScPif1p-Poly(T8) complex are free of contact with any residues, G1, G2 and G3 are recognized, respectively, by F723, E724/K387 and K312 in the ScPif1p-ssDNA complex. All these structural differences in nucleotide base-specific recognition may confer ScPif1p a binding preference for G-rich tracts over non-G-rich oligos (15). We further characterized the ScPif1p-mediated DNA binding with fluorescently labeled Poly(T12) and GR12 under equilibrium conditions. The determined dissociation constant (Kd,app) with GR12 (7.8 nM) is about two-fold lower than that with Poly(T12) (15.1 nM) (Figure 2E, left panel). Interestingly, in the presence of ATPγS, the Kd,app value of GR12 is reduced to 6.1 nM and that of Poly(T12) is increased to 33.8 nM (Figure 2E, right panel), indicating that ATP may simultaneously enhance G-rich-sequence and reduce Poly(T12) bindings to ScPif1p, therefore achieving the selective binding of G-rich sequences. The above structural and functional characterizations provide a mechanistic understanding of the specific binding for, and the DNA unwinding-stimulation by G-rich sequences (23,24).
Structural features and functional properties of the yeast-specific insertion domain 2C
In accordance with sequence alignment analysis (Supplementary Figure S1), the structural alignments between BsPif1 and ScPif1p show that the insertion domain 2C is between two β-strands (β12 and β13) (Figure 1A−D), corresponding to the loop 306−309 (EDNV) in BsPif1. Five α-helices (α13−α17) can be built in the electron density map, but some loops connecting these helices are missing (residues 585−588, 605−609, 624−635) (Figure 3A). While α13, α14 and α17 are approximately parallel to each other, α15 is perpendicular to the three α-helices (Figure 3A). The four α-helices are packed as a bundle motif through an elaborate network of hydrophobic and hydrogen-bonding interactions. Furthermore, the spatial conformation of the bundle domain relative to the 2B domain is precisely determined through a cluster of hydrophobic interactions including residues M543 and L672 from domain 2B, and Y551 and Y548 from the 2C bundle (enlarged square in Figure 3A).
Figure 3.

(A) A close-up view of the interactions between domains 2C and 2B. The residues implicated in hydrophobic interactions are boxed and enlarged on the right. (B) Superposition of the 2C bundle motif of ScPif1p and a motif of RecB in RecBCD helicase. (C) The hydrogen bonding between K595 and D677 stabilizes the relative spatial positions of α16 and loop3 in ScPif1p. (D) Summary of α16 sequence deletions and/or mutations. (E) Kinetic unwinding curves of ScPif1p and its variants determined by stopped-flow assay with 4 nM partial duplex DNA (S26ds17) and 200 nM proteins, as described in ‘Materials and Methods’. The determined parameters are summarized in Table 1. (F) Translocation kinetics of the wild-type and mutated ScPif1p.
The SH3 fold of domain 2B in ScPif1p, BsPif1 or human Pif1 displays a striking structural conservation with that of domain 2B of RecD in RecBCD complex (25,26,43). To probe whether the 2C domain protruding from the SH3 fold possesses some potential structural features conserved with other family proteins, we performed a search of structural homologues in the Protein Data Bank with Dali Server (44). A reasonable hit (Z-score 3.1) was obtained with protein RecB determined from the cryoEM structure of RecBCD complex (PDB entry: 5LD2). Sequence 542−583 of domain 2C aligns with sequence 607−646 of RecB with an r.m.s.d. of 2.4 Å over 40 Cα (Figure 3B). This region of RecB is the one which contacts the domain 2B of RecD (homologue of domain 2B in ScPif1p with SH3 fold) in the RecBCD complex (Supplementary Figure S5). More interestingly, a superimposition of ScPif1p with RecD in RecBCD complex by imposing perfect overlapping of the ssDNA configurations reveals an interesting phenomenon: while the SH3 fold in ScPif1p occupies the corresponding position of SH3 fold in RecD, the domain 2C of ScPif1p superimposes with the partial α-helical bundle of RecB that is in contact with the RecD subunit. Thus, the spatial configuration between domains 2B and 2C in ScPif1p mimics the subunit interactions between RecD and RecB in RecBCD (Supplementary Figure S5)(43).
Importantly, α16 of domain 2C protrudes from the bundle domain and further extends above the previously identified loop3 and assumes a parallel conformation relative to loop3 (Figure 3C). Furthermore, it appears that the relative spatial conformation of α16 and loop3 is mainly determined through the interactions between K595 of α16 and D677 of loop3 (Figure 3C).
To determine the functional roles of domain 2C, we first prepared a series of truncation variants in domain 2C (Figure 3D). We observed that total or partial deletion of the 2C bundle leads to protein aggregation and/or degradation, indicating that domain 2C plays an important role in protein folding in addition to its other potential functions. However, we found that the α16 helix can be deleted and is also tolerant for point mutations and/or sequence modifications. Deletion of α16 or alteration of the interaction between K595 of α16 and D677 of loop3 by point mutations (K595A or D677T) resulted in significant reductions in both DNA unwinding rate and amplitude, but no significant reduction in DNA binding (Figure 3E and Table 1), suggesting that α16 is dispensable, but may regulate the unwinding activity through loop3 which has been previously shown to regulate DNA unwinding in BsPif1 (25). Interestingly, replacements of α16 with novel amino acid sequences which allow the formation of either a new α-helix (α16-M1) or an unstructured random coil (α16-M2) led to nearly complete defects in DNA unwinding, although the DNA binding abilities of these two variants were only slightly reduced (Figure 3E and Table 1).
Table 1. DNA unwinding and binding properties of ScPif1p and its variantsa.
| Variant | Helicase activity | DNA binding | |
|---|---|---|---|
| A m | k u (s−1) | K d,app (nM) | |
| ScPif1p | 0.45 ± 0.12 | 1.89 ± 0.14 | 11.2 ± 2.1 |
| D677T | 0.35 ± 0.08 | 0.33 ± 0.11 | 10.5 ± 2.6 |
| K595A | 0.31 ± 0.06 | 0.63 ± 0.20 | 10.2 ± 1.8 |
| Δα16 | 0.38 ± 0.07 | 1.05 ± 0.15 | 11.0 ± 2.2 |
| α16-M1 | 0.02 ± 0.05 | 0.01 ± 0.04 | 15.3 ± 3.1 |
| α16-M2 | 0.06 ± 0.02 | 0.14 ± 0.08 | 15.0 ± 3.7 |
aThe experiments were performed as described under ‘Materials and Methods’. Helicase activities were determined with 4 nM substrate S26ds17 and 200 nM proteins. The apparent dissociation constants were deduced from the titration curves determined with 5 nM fluorescently labeled ssDNA (GR12) and increasing protein concentrations. Values are the averages of at least three experiments and S.E. is reported.
ScPif1p limits yeast telomere lengths by displacing telomerases from telomere ends (6). To address whether domain 2C may regulate the translocation/disruption activities of ScPif1p, we then examined the translocation activities of the two α16 mutants (α16-M1 and α16-M2), in parallel with ScPif1p as a control. Figure 3F shows that, while the fluorescent signal increases and reaches the peak position within 1 s upon addition of ScPif1p, that for α16-M1 and α16-M2 increase only slightly, and then rapidly decrease to very low levels. The amplitude of fluorescent signal of Δα16 is significantly reduced compared with that of ScPif1p. These results, taken together, show that α16 may interact with loop3 to regulate the unwinding and translocation activities of ScPif1p.
G4 motif stabilizes dimer formation
To identify which crystal packing might represent the physiologically relevant dimer in solution, SEC-SAXS experiments were performed under conditions in which ADP·AlF4 was added in all samples measured. The SEC-SAXS data were carefully processed with US-SOMO to correct profiles from baseline drift and to separate the several peaks by deconvolution with Singular Value Decomposition (SVD) and Gaussian tools provided by the software. Elution profiles, SAXS fittings are presented in Supplementary Figures S6 and S9, and Tables S3 and S4. We measured ScPif1p in its apo form and in complex with ssDNA (GR11) under similar conditions as used for protein crystallizations. Both SEC-SAXS profiles have a single peak corresponding to a monomer, but with a major difference in Rg which decreases from 29.3 (for apo form) to 27.5 Å (for ssDNA complex) (Supplementary Figure S6C and D and Table S3). The Krakty plots indicate that the apo form is more elongated compared with the crystal structure (Supplementary Figure S7A and B). The structure of ScPif1p in its apo form was modeled with EOM by flexible fitting of the 2B and 2C domains, which are considered as a rigid domain and can be freely rotated relative to the rest of the structure. Indeed, analysis by EOM indicates a flexibility of the 2B + 2C domain with Rflex = 85.4% (pool 88.5%) and Rσ = 1.68 (Supplementary Figure S8A and B). The fit was then improved from χ2 = 9.83 (crystal structure) to χ2 = 1.16 (Supplementary Figure S9A and Table S4, showing the major conformation of apo ScPif1p). The rotation phenomenon of domain 2B in the apo form was already observed in BsPif1 (25). Interestingly, the same analysis of flexibility of the 2B + 2C domain for ScPif1p bound with ssDNA gives a reduced flexibility with Rflex = 49.1% (pool 89.9%) and Rσ = 0.46 (Supplementary Figure S8C and D), therefore the monomeric ScPif1p in complex with ssDNA becomes more globular than its apo form (Supplementary Figure S7A). The SAXS data of ScPif1p in complex with ssDNA can be reasonably fitted to the monomeric crystal structure with χ2 = 1.23 (Supplementary Figure S9B). Thus, ScPif1p is monomeric both in its apo form and when in complex with ssDNA.
We previously demonstrated a binding cooperativity of the full-length nuclear form of ScPif1p for G4 DNA, but not for ssDNA (23). Here we have further confirmed that the truncated ScPif1p used in this study displays the same cooperative binding to G4 DNA (Supplementary Figure S10A). Interestingly, gel filtration assay shows that G4 motif alone (3G4) or G4 motif bearing a ssDNA (8T3G4) significantly stabilizes the dimer formation (Supplementary Figure S10B). Before measuring SAXS profiles of ScPif1p in complex with 8T3G4 or 3G4, we first characterized the conformations of both G4 DNAs in solution. Uniform parallel G4 DNAs were firstly prepared under molecular crowding conditions and subsequently submitted to SAXS analysis. The registered SAXS profile of 3G4 displays a bell-shaped Kratky plot and can be best fitted according to the available crystal structure (PDB entry: 3SC8), indicating that 3G4 is well folded in solution as expected (Supplementary Figures S7C and D, and S9C). 8T3G4 exhibits some flexibility due to the ssDNA extension at the 5′ end (Supplementary Figures S7C and D, and S9D). Having established that both 8T3G4 and 3G4 were well folded, ScPif1p in complex with 8T3G4 or 3G4 was analyzed by SAXS. The ScPif1p-8T3G4 complex profile shows two peaks, the first one corresponding to a dimer and the second one to a monomer. The measured UV absorbance ratio at 260/280 nm of 0.8 to 0.9 indicates that both peaks contain protein-DNA complexes (Supplementary Figure S6E). The second peak can be modeled according to the monomeric structural model, with 8T3G4 bound, with a good agreement (χ2 = 2.08), which is close to the crystallographic structure (Supplementary Table S3).
From the peak corresponding to the dimer, a clean SAXS profile was generated. ScPif1p-8T3G4 complex was modeled by taking the crystal packing with a minimum buried surface of 450 Å2, and then fitted to SAXS data with Crysol and ranked by χ2. The dimers tested have a χ2 around 5−18 except for one with a better fit, χ2 = 1.66 (Figure 4A and Supplementary Figure S9F−I, and Table S4). According to the best χ2 fit, the more probable dimer in solution is the one in which the two monomers are oriented in a head to tail fashion and interact through helix α2 of domain 1A (273−281) (Figure 4A and B). Furthermore, a G4 motif lies between the dimeric interface in addition to the ssDNA traversing the canonical ssDNA binding channel.
Figure 4.

(A) The SAXS model of ScPif1p dimer stabilized by 8T3G4. (B) Identified crystal structure of dimeric ScPif1p and a close-up view of the interface between the two monomers. (C) Radius distributions of ScPif1p (inset) and the DM mutant obtained from analyses of experimental data in DLS assays. The experiments were performed as described in ‘Materials and Methods’ with 4.5 μM proteins. (D) Individual smFRET traces recorded in experiments performed with a partial duplex DNA (smS47ds17, Supplementary Table S1) or a partial duplex DNA bearing a G4 motif at the ss/dsDNA junction (smS26G4ds17, Supplementary Table S1), in the presence of ScPif1p (upper and middle panels) or the DM mutant (lower panel) in which the dimeric interaction is disrupted. The detailed experimental conditions are described in ‘Materials and Methods’. (E) Histograms of waiting time constructed from about 300 individual records of ScPif1p with smS47ds17 or smS26G4ds17, and DM with smS26G4ds17. They follow an exponential decay with time constants of 26.2, 19.2 and 27.8 s, respectively.
The essential dimeric interactions within the proteins appear to be determined by an indirect interaction between R281 of each monomer, which is stabilized by a phosphate ion (Figure 4A and B). Furthermore, additional residues (E242 and K273) may further strengthen the dimeric interactions. This dimer was also found in P3121 space group which has two molecules in the asymmetric unit. Cell parameters of P3121 and P212121 crystals are unrelated and crystal packings are distinct. In P212121, the dimer involving R281 is between two different molecules of the asymmetric unit related by two-fold non-crystallographic symmetry. However, in P3121 this same dimer is a crystallographic dimer, involving two molecules related by a 2-fold crystallographic symmetry (Supplementary Figure S3C). Because of the different natures of the dimers observed in the crystal structures, it is probably not a crystallographic artifact. Furthermore, the buried surface area between the two monomers is only 542 Å2, suggesting that this dimeric arrangement will not be strongly maintained in solution in the absence of G4 motif.
The SEC-SAXS profile of ScPif1p in complex with 3G4 generated a high molecular-weight peak which can be interpreted as a high-order oligomer peak, and a dimer peak with Mw = 145 kDa (Supplementary Figure S6F). The second peak has a stable radius of gyration which is a homogeneous dimer without detectable monomeric species. The SAXS data obtained with ScPif1p-3G4 DNA yielded the goodness-of-fit χ2 = 1.85 for a dimer model, which is almost identical with the above-mentioned model in Supplementary Figure S9G and J. These results, taken together, not only confirm that G4 motif really stabilizes a stable dimer formation, but also rule out the possibility that the dimerization observed with 8T3G4 is due to the two molecules of ScPif1p being linked by a long ssDNA if 3G4 is partially unfolded.
To confirm the potential protein residues implicated in the dimerization, the interactions inside the dimer were disrupted by triple mutation (E242A/K273A/R281A, named as DM for dimer mutation). We compared the oligomeric states of ScPif1p and DM upon addition of ssDNA, dsDNA and ssDNA linked with a G4 motif by DLS and gel filtration assays. Interestingly, while ScPif1p behaved essentially as a monomer in the absence of DNA and in the presence of ss- and dsDNA (Supplementary Figure S2C−F), only the addition of ssDNA bearing a G4 motif at its 3′ end induced dimeric ScPif1p (∼65% monomers versus 35% dimers) (Figure 4C, inset and Supplementary Figure S10B). In sharp contrast, addition of the same DNA substrate failed to induce dimerization of the DM mutant (92% monomers versus 8−10% dimers) (Figure 4C). We have previously shown that G4 motif greatly stimulates its downstream duplex DNA unwinding by ScPif1p through reducing a so called ‘waiting time’, which is interpreted as the time needed for ScPif1p dimerization (24). We therefore examined the waiting times of the wild-type and the modified ScPif1p (DM) under identical smFRET experimental conditions. We found that while the waiting time was reduced from 26.2 to 19.2 s upon addition of wild-type ScPif1p, that measured with the DM mutant remained unchanged (Figure 4D). More rigorous statistical analyses from 300 individual records further confirmed the above results, indicating that the G4-stimulating effect was significantly compromised due to disruption of the interactions inside the dimer (Figure 4E). The above result has a double significance, it confirms both our present dimeric model and our previous interpretation for G-rich sequence/G4-induced ScPif1p dimerization.
Finally, it should be noted that although it is evident that G4 motif makes a significant contribution to the dimer formation in solution in addition to protein-protein interactions, we cannot give a detailed description of the interactions between the dimer and G4 DNA at the atomic level in the present study, due to the non-availability of crystal structure of ScPif1p-G4 complex.
A potential G4 binding site and identification of residues specifically implicated in G4 binding/unwinding
As shown in the electrostatic potential surface map of ScPif1p in complex with ssDNA, the 3′-end ssDNA is surrounded by a cluster of positively charged residues such as R594/K595 and K321/R324/R326, constituting a positively charged pliers (Figure 5A). Based on SAXS data registered with the ScPif1p-8T3G4 complex and our crystal structure, we further modeled a monomeric ScPif1p in complex with 8T3G4. It appears that the highly negatively charged G4 DNA can be partially clamped into the positively charged pliers and potentially interacts with the above mentioned positive residues, leaving about half solvent-accessible G4 skeleton (Figure 5B). Comparing with our crystal structure of ScPif1p in complex with ssDNA, the bundle motif of domain 2C in the modeled model undergoes a rotation which is accompanied with a re-organization of configuration of the α16 helix relative to loop3 (Figure 5B), making residue E681 of loop3 potentially interact with both the sugar-phosphate and the G base at the 3′ end. Another notable structural feature of the modeled model is the spatial configuration of ScPif1p-specific α5 relative to G4 DNA (Figure 5C). A NMR study has revealed that an α-helix in RHAU helicase (named as RSM) clamps the G4 DNA using three-anchor-point electrostatic interactions between three positively charged amino acids and negatively charged phosphate groups (Figure 5C, left panel). The α5 helix in ScPif1p assumes a similar configuration as RSM relative to G4 DNA, in which at least R324 of ScPif1p occupies the equivalent position of K19 in RSM (Figure 5C, right panel).
Figure 5.

(A) Molecular electrostatic potential map of ScPif1p with the highly positively charged residues indicated in blue. (B) Molecular modeling model of ScPif1p in complex with G4 DNA. The enlarged boxes (left and up) represent the close-up views of the interactions between the positively charged residues and G4 DNA. (C) Structure of Rhau18-G4 DNA complex determined by NMR (left, PDB entry: 2N21) and superposition of the α-helix RSM in Rhau18 and α5 in ScPif1p (right). (D) DNA unwinding activities of ScPif1p and its variants as determined by stopped-flow fluorescence assay. 4 nM partial duplex DNA (S26ds17) (inset) or G4-containing partial duplex DNA (S26G4ds17) and 100 nM protein were used under experimental conditions as described in ‘Materials and Methods’. The parameters derived from these curves are summarized in Table 2.
To confirm whether the above predicted residues in the modeled model are implicated in G4 DNA binding/unwinding, a cluster of amino acids such as R594/K595 in the α16 helix of domain 2C, R324 and R326 in the α5 helix of domain 1B and E681 of loop3 were mutated (Table 2). As controls, residue K363 which is outside of the pliers and residue R323, the configuration of side chain of which is too far to contact with the G4 DNA, were also mutated (Table 2). A fluorescence anisotropy binding assay was used to monitor the equilibrium DNA binding ability of the wild-type and mutated ScPif1p proteins. The apparent dissociation constants (Kd,app) of the proteins were determined with DNA substrates including ssDNA, dsDNA and G4 DNA. The results demonstrated that these mutations do not significantly affect the ss- and dsDNA binding affinities, as revealed by the Kd,app values (Table 2), which indicates that these mutants are properly folded and the mutated residues are not essential for ss- and dsDNA binding. However, the Kd,app values determined with G4 DNA (F-G417) are systematically higher than that determined with ss- and dsDNA, by 2.9−5.7 folds, depending on the types of mutations (Table 2). These results, taken together, strongly suggest that these positively charged residues in the proposed pliers may be indeed specifically implicated in G4 DNA recognition and binding.
Table 2. DNA binding and unwinding properties of ScPif1p and its variantsa.
| Variant | DNA binding | DNA unwinding | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ssDNA (GR12) | dsDNA(dsDNA16) | G4 (F-G417) | Duplex DNA (S26ds17) | G4 DNA (S26G4ds17) | ||||||
| K d,app (nM) | Fold | K d,app (nM) | Fold | K d,app (nM) | Fold | A m | k u (s−1) | A m | k u (s−1) | |
| ScPif1p | 11.2 ± 2.1 | 1 | 81.3 ± 10.3 | 1 | 25.0 ± 5.1 | 1 | 0.41 ± 0.12 | 0.62 ± 0.14 | 0.68 ± 0.11 | 1.39 ± 0.05 |
| R594A/K595A | 10.5 ± 3.5 | 0.94 | 78.9 ± 9.3 | 0.97 | 72.9 ± 4.9 | 2.92 | 0.43 ± 0.11 | 0.16 ± 0.07 | 0.51 ± 0.07 | 0.24 ± 0.08 |
| R324N | 11.4 ± 4.3 | 1.02 | 57.9 ± 1.1 | 0.71 | 87.9 ± 7.1 | 3.51 | 0.34 ± 0.10 | 0.17 ± 0.02 | 0.47 ± 0.09 | 0.25 ± 0.07 |
| R326C | 12.4 ± 2.1 | 1.11 | 78.3 ± 9.6 | 0.96 | 105.0 ± 8.4 | 4.20 | 0.39 ± 0.13 | 0.18 ± 0.02 | 0.50 ± 0.13 | 0.33 ± 0.06 |
| E681G | 12.4 ± 3.9 | 1.10 | 56.10 ± 11.6 | 0.69 | 76.5 ± 10.5 | 3.06 | 0.42 ± 0.04 | 0.51 ± 0.13 | 0.46 ± 0.12 | 0.46 ± 0.03 |
| R323A | 9.9 ± 0.4 | 0.62 | 100.4 ± 5.1 | 1.23 | 25.1 ±1.4 | 1.00 | 0.39 ± 0.13 | 0.30 ± 0.12 | 0.63 ± 0.10 | 0.80 ± 0.11 |
| K363A | 14.4 ± 1.3 | 1.28 | 72.3 ± 8.3 | 0.89 | 43.9 ± 2.2 | 1.76 | 0.38 ± 0.05 | 0.26 ± 0.01 | 0.62 ± 0.08 | 0.77 ± 0.03 |
| DM | 13.38 ± 0.8 | 1.19 | 95.5 ± 8.5 | 1.17 | 23.8 ± 2.5 | 0.98 | 0.45 ± 0.10 | 0.27 ± 0.10 | 0.42 ± 0.09 | 0.23 ± 0.05 |
aThe values were determined according to ‘Materials and Methods’. The DNA substrate sequences are described in Supplementary Table S1. For binding assay, 4 nM fluorescently labelled DNAs were used. Unwinding assays were determined with 4 nM DNA and 100 nM proteins. Unwinding amplitudes and rates were determined from Figure 5D. Values are the averages of at least three experiments and S.E. is reported.
We then measured the G4 DNA unwinding activities of these mutants by a stopped-flow fluorescence assay as we performed previously (23). Significant and specific reductions in G4 DNA unwinding activities of mutants R324N, R326C, R594A/K595A and E681G were observed, while their duplex unwinding activities were almost identical or equivalent to that of the wild-type ScPif1p (Panels 1−4 in Figure 5D and Table 2). Interestingly, both G4 and dsDNA unwinding activities of the control mutants, R323A and K363A, remained unchanged relative to that of the wild-type ScPif1p (Panels 5 and 6 in Figure 5D), as expected. These results, taken together, demonstrate that the identified positively charged cage may be important for G4 DNA recognition, binding and unwinding.
DISCUSSION
Insights into the mechanisms by which Pif1 family helicases perform multiple functions involve answering several fundamental structural questions. First, what is the structural basis for their specific recognition of G-rich tracts and preferential loading and translocating along ssDNA, but not ssRNA strand, to displace telomerases (5,17). Second, what are the structural features and functions of the yeast-specific insertion domain? Third, does ScPif1p form a dimer and what are the structural characters of the dimer? Finally, does ScPif1p possess G4-specific recognition and binding site and use a G4 DNA unwinding mechanism which differs from that for dsDNA unwinding?
Firstly, our structural analysis revealed that, while the sugar-phosphate backbones of both Poly(T8) and Poly(G3T5) nucleotides are bound by the same amino acids from the 1A and 2A domains, the two successive 5′-end G bases (G2 and G3) are preferentially recognized by the additional residues that are not implicated in T-base recognition. Interestingly, the preferential binding of G-rich tracts may be further enhanced by ATP. The potential biological significance of preferential binding of G-rich sequences might be that, ScPif1p may use two mechanisms to efficiently unfold G4 DNA: when G4 DNA is partially unfolded in the G4 DNA binding site (i.e., the electrostatic pliers), the released G bases can then be engaged into the translocation channel; and the more G bases are released from G4 DNA unfolding, the binding and translocation of more ScPif1p molecules occur, and consequently G4 DNA unfolding is further enhanced. More interestingly and importantly, our structures have revealed that both H705 and H303 are implicated in stacking with the deoxyribose rings of nucleotides 2 and 4 and selectively bind to DNA rather than RNA. These structural features together with mutational studies reveal the mechanism by which ScPif1p preferentially loads onto and translocates along ssDNA, but not ssRNA, to efficiently unwind DNA/RNA duplexes and displace telomerases (5,17).
Secondly, the 2C domain, which specifically presents in yeast, but is absent in bacteroides and mammalian Pif1 family helicases, folds as a bundle domain with an extension of the α16 helix. The interactions between α16 and the previously identified loop3 determine the precise spatial conformation of loop3, which is essential for the enzymatic activities, as demonstrated by that alterations of α16 dramatically reduce the unwinding activity of ScPif1p. Furthermore, the domain 2C of ScPif1p can be superimposed with the similar α-helix bundle of RecB subunit in RecBCD helicase with an r.m.s.d. of 2.4 Å over 40 Cα, and the SH3 fold of domain 2B in ScPif1p occupies the equivalent position of the SH3 fold in RecD. Thus, the spatial configurations and interactions between domains 2C and 2B in ScPif1p are reminiscent of that between subunits RecB and RecD in the RecBCD complex, consisting with the observations that the SH3 fold is usually involved in protein-protein interactions (40,45). These structural features of domain 2C may provide a possibility that this domain coordinates ScPif1p's functions through protein-protein interactions in cells.
It appears that many helicases may possess both duplex DNA unwinding and G4 DNA unfolding abilities in vitro. Thus, the challenging questions in studies of G4 DNA unfolding helicases are whether and how some residues, which are different from those implicated in duplex DNA unwinding, are specifically implicated in G4 DNA recognition, binding and unwinding? By a combination of crystal structures, SAXS and biochemical data, and reliable modeling, we have proposed a possible G4 DNA binding site in ScPif1p monomers. In the present work, for the first time to our knowledge, we have identified at least two clusters of residues (R594/K595 in domain 2C, R324/R326 in domain 1B) and one residue (E681 in loop3) of ScPif1p that are specifically involved in G4 DNA unfolding, but not or partially implicated in duplex DNA unwinding. The specific G4 DNA unfolding functions of these residues are fully consistent with their spatial conformations. Firstly, the two clusters of residues are located at the opposite positions of the supposed pliers in which G4 DNA is believed to be clamped. Secondly, a previously identified 13-amino acid α-helix (RSM) in human RHAU helicase clamps the G4 DNA using three-anchor-point electrostatic interactions, which was suggested as a general principle for specific recognition of G4 DNAs (46). The spatial configuration of the α5 helix of domain 1B and G4 DNA is reminiscent of that of RSM and G4 DNA. Especially, R324’s position is equivalent to that of K19 in RSM, providing a comprehensible explanation why R324 plays an important role in G4 DNA unwinding.
Finally, our structures reveal that ScPif1p forms a dimer with a buried surface of only about 550 Å2. The structural features help us to understand why the dimer is not stable without the binding of G4 DNA. Indeed, we have found that only G4 motif stabilizes the dimers that can be identified by DLS, gel filtration and SAXS assays, indicating that both protein-protein and protein-G4 DNA interactions are necessary for stable dimer formation. This is consistent with our previous functional studies that G4 motif greatly stimulates downstream duplex DNA unwinding (23,24). Whether the dimer is stabilized by G4 DNA through a mechanism by which each monomer binds one half of the G4 DNA skeleton as aforementioned remains to be further confirmed. Alternatively, it is also possible that the residues implicated in G4 DNA binding in the monomers may undergo conformational re-organizations to accommodate G4 DNA binding to the dimer. At present, we cannot give a description in great detail about how G4 motif is recognized and bound by the two monomers due to the absence of crystal structure of ScPif1p in complex with G4 motif. In this regard, introducing DM mutation into living cells and analyzing their phenotypes may provide more interesting information for the physiological functions of the dimers.
AVAILABILITY
5O6B: ScPif1p with Poly(G3T5) and ADP·AlF4;
5O6D: ScPif1p with Poly(T8) and ATPγS;
5O6E: ScPif1p with Poly(T3G3T2) and ADP·AlF4.
Supplementary Material
ACKNOWLEDGEMENTS
We thank beamline scientists at BL17U1 of the Shanghai Synchrotron Radiation Facility (China), at BL18U1 and BL19U1 of the National Center for Protein Sciences, Shanghai, for assistance with data collections. We are grateful for access to the SOLEIL (SWING) synchrotron radiation facility for SAXS data collections and acknowledge the support provided by Dr Javier Perez and his team.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [11574252, 31370798, 11774407]; Northwest A&F University Startup Funding [Z101021103 to X.-G.X.]; International Associated Laboratory ‘Helicase-mediated G-quadruplex DNA unwinding and Genome Stability’. Funding for open access charge: National Natural Science Foundation of China [11574252, 31370798]; Northwest A&F University Startup Funding [Z101021103 X.-G.X.]; International Associated Laboratory ‘Helicase-mediated G-quadruplex DNA unwinding and Genome Stability’.
Conflict of interest statement. None declared.
REFERENCES
- 1. Foury F., Kolodynski J.. pif mutation blocks recombination between mitochondrial rho+ and rho- genomes having tandemly arrayed repeat units in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 1983; 80:5345–5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schulz V.P., Zakian V.A.. The saccharomyces PIF1 DNA helicase inhibits telomere elongation and de novo telomere formation. Cell. 1994; 76:145–155. [DOI] [PubMed] [Google Scholar]
- 3. Geronimo C.L., Zakian V.A.. Getting it done at the ends: Pif1 family DNA helicases and telomeres. DNA Repair (Amst.). 2016; 44:151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boule J.B., Zakian V.A.. Roles of Pif1-like helicases in the maintenance of genomic stability. Nucleic Acids Res. 2006; 34:4147–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boule J.B., Vega L.R., Zakian V.A.. The yeast Pif1p helicase removes telomerase from telomeric DNA. Nature. 2005; 438:57–61. [DOI] [PubMed] [Google Scholar]
- 6. Zhou J., Monson E.K., Teng S.C., Schulz V.P., Zakian V.A.. Pif1p helicase, a catalytic inhibitor of telomerase in yeast. Science. 2000; 289:771–774. [DOI] [PubMed] [Google Scholar]
- 7. Pike J.E., Burgers P.M., Campbell J.L., Bambara R.A.. Pif1 helicase lengthens some Okazaki fragment flaps necessitating Dna2 nuclease/helicase action in the two-nuclease processing pathway. J. Biol. Chem. 2009; 284:25170–25180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rossi M.L., Pike J.E., Wang W., Burgers P.M., Campbell J.L., Bambara R.A.. Pif1 helicase directs eukaryotic Okazaki fragments toward the two-nuclease cleavage pathway for primer removal. J. Biol. Chem. 2008; 283:27483–27493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ivessa A.S., Zhou J.Q., Zakian V.A.. The Saccharomyces Pif1p DNA helicase and the highly related Rrm3p have opposite effects on replication fork progression in ribosomal DNA. Cell. 2000; 100:479–489. [DOI] [PubMed] [Google Scholar]
- 10. Ivessa A.S., Zhou J.Q., Schulz V.P., Monson E.K., Zakian V.A.. Saccharomyces Rrm3p, a 5′ to 3′ DNA helicase that promotes replication fork progression through telomeric and subtelomeric DNA. Genes Dev. 2002; 16:1383–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ivessa A.S., Lenzmeier B.A., Bessler J.B., Goudsouzian L.K., Schnakenberg S.L., Zakian V.A.. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol. Cell. 2003; 12:1525–1536. [DOI] [PubMed] [Google Scholar]
- 12. Saini N., Ramakrishnan S., Elango R., Ayyar S., Zhang Y., Deem A., Ira G., Haber J.E., Lobachev K.S., Malkova A.. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature. 2013; 502:389–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wilson M.A., Kwon Y., Xu Y., Chung W.H., Chi P., Niu H., Mayle R., Chen X., Malkova A., Sung P. et al. Pif1 helicase and Poldelta promote recombination-coupled DNA synthesis via bubble migration. Nature. 2013; 502:393–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Paeschke K., Capra J.A., Zakian V.A.. DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell. 2011; 145:678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Paeschke K., Bochman M.L., Garcia P.D., Cejka P., Friedman K.L., Kowalczykowski S.C., Zakian V.A.. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature. 2013; 497:458–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lahaye A., Leterme S., Foury F.. PIF1 DNA helicase from Saccharomyces cerevisiae. Biochemical characterization of the enzyme. J. Biol. Chem. 1993; 268:26155–26161. [PubMed] [Google Scholar]
- 17. Boule J.B., Zakian V.A.. The yeast Pif1p DNA helicase preferentially unwinds RNA DNA substrates. Nucleic Acids Res. 2007; 35:5809–5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li J.H., Lin W.X., Zhang B., Nong D.G., Ju H.P., Ma J.B., Xu C.H., Ye F.F., Xi X.G., Li M. et al. Pif1 is a force-regulated helicase. Nucleic Acids Res. 2016; 44:4330–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ramanagoudr-Bhojappa R., Chib S., Byrd A.K., Aarattuthodiyil S., Pandey M., Patel S.S., Raney K.D.. Yeast Pif1 helicase exhibits a one-base-pair stepping mechanism for unwinding duplex DNA. J. Biol. Chem. 2013; 288:16185–16195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hou X.M., Wu W.Q., Duan X.L., Liu N.N., Li H.H., Fu J., Dou S.X., Li M., Xi X.G.. Molecular mechanism of G-quadruplex unwinding helicase: sequential and repetitive unfolding of G-quadruplex by Pif1 helicase. Biochem. J. 2015; 466:189–199. [DOI] [PubMed] [Google Scholar]
- 21. Zhou R.B., Zhang J.C., Bochman M.L., Zakian V.A., Ha T.. Periodic DNA patrolling underlies diverse functions of Pif1 on R-loops and G-rich DNA. Elife. 2014; 3:e02190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barranco-Medina S., Galletto R.. DNA binding induces dimerization of Saccharomyces cerevisiae Pif1. Biochemistry. 2010; 49:8445–8454. [DOI] [PubMed] [Google Scholar]
- 23. Duan X.L., Liu N.N., Yang Y.T., Li H.H., Li M., Dou S.X., Xi X.G.. G-quadruplexes significantly stimulate Pif1 helicase-catalyzed duplex DNA unwinding. J. Biol. Chem. 2015; 290:7722–7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang B., Wu W.Q., Liu N.N., Duan X.L., Li M., Dou S.X., Hou X.M., Xi X.G.. G-quadruplex and G-rich sequence stimulate Pif1p-catalyzed downstream duplex DNA unwinding through reducing waiting time at ss/dsDNA junction. Nucleic Acids Res. 2016; 44:8385–8394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen W.F., Dai Y.X., Duan X.L., Liu N.N., Shi W., Li N., Li M., Dou S.X., Dong Y.H., Rety S. et al. Crystal structures of the BsPif1 helicase reveal that a major movement of the 2B SH3 domain is required for DNA unwinding. Nucleic Acids Res. 2016; 44:2949–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou X., Ren W., Bharath S.R., Tang X., He Y., Chen C., Liu Z., Li D., Song H.. Structural and functional insights into the unwinding mechanism of Bacteroides sp Pif1. Cell Rep. 2016; 14:2030–2039. [DOI] [PubMed] [Google Scholar]
- 27. Liu N.N., Duan X.L., Ai X., Yang Y.T., Li M., Dou S.X., Rety S., Deprez E., Xi X.G.. The Bacteroides sp. 3_1_23 Pif1 protein is a multifunctional helicase. Nucleic Acids Res. 2015; 43:8942–8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kabsch W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sheldrick G.M. Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vonrhein C., Blanc E., Roversi P., Bricogne G.. Automated structure solution with autoSHARP. Methods Mol. Biol. 2007; 364:215–230. [DOI] [PubMed] [Google Scholar]
- 31. Cowtan K. Completion of autobuilt protein models using a database of protein fragments. Acta Crystallogr. D Biol. Crystallogr. 2012; 68:328–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. David G., Perez J.. Combined sampler robot and high-performance liquid chromatography: a fully automated system for biological small-angle X-ray scattering experiments at the Synchrotron SOLEIL SWING beamline. J. Appl. Crystallogr. 2009; 42:892–900. [Google Scholar]
- 35. Brookes E., Vachette P., Rocco M., Perez J.. US-SOMO HPLC-SAXS module: dealing with capillary fouling and extraction of pure component patterns from poorly resolved SEC-SAXS data. J. Appl. Crystallogr. 2016; 49:1827–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Petoukhov M.V., Franke D., Shkumatov A.V., Tria G., Kikhney A.G., Gajda M., Gorba C., Mertens H.D., Konarev P.V., Svergun D.I.. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012; 45:342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu W.Q., Hou X.M., Li M., Dou S.X., Xi X.G.. BLM unfolds G-quadruplexes in different structural environments through different mechanisms. Nucleic Acids Res. 2015; 43:4614–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Van Zundert G.C., Rodrigues J.P., Trellet M., Schmitz C., Kastritis P.L., Karaca E., Melquiond A.S., van Dijk M., de Vries S.J., Bonvin A.M.. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016; 428:720–725. [DOI] [PubMed] [Google Scholar]
- 39. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E., Berendsen H.J.. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005; 26:1701–1718. [DOI] [PubMed] [Google Scholar]
- 40. Saikrishnan K., Powell B., Cook N.J., Webb M.R., Wigley D.B.. Mechanistic basis of 5′-3′ translocation in SF1B helicases. Cell. 2009; 137:849–859. [DOI] [PubMed] [Google Scholar]
- 41. Xu Y.W., Morera S., Janin J., Cherfils J.. AlF3 mimics the transition state of protein phosphorylation in the crystal structure of nucleoside diphosphate kinase and MgADP. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:3579–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cordin O., Tanner N.K., Doere M., Linder P., Banroques J.. The newly discovered Q motif of DEAD-box RNA helicases regulates RNA-binding and helicase activity. EMBO J. 2004; 23:2478–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wilkinson M., Chaban Y., Wigley D.B.. Mechanism for nuclease regulation in RecBCD. Elife. 2016; 5:e18227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Holm L., Laakso L.M.. Dali server update. Nucleic Acids Res. 2016; 44:W351–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zarrinpar A., Bhattacharyya R.P., Lim W.A.. The structure and function of proline recognition domains. Sci STKE. 2003; 2003:Re8. [DOI] [PubMed] [Google Scholar]
- 46. Heddi B., Cheong V.V., Martadinata H., Phan A.T.. Insights into G-quadruplex specific recognition by the DEAH-box helicase RHAU: Solution structure of a peptide-quadruplex complex. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:9608–9613. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.