


Abstract
MicroRNAs (miRNAs) are small endogenous regulatory molecules that modulate gene expression post-transcriptionally. Although differential expression of miRNAs have been implicated in many diseases (including cancers), the underlying mechanisms of action remain unclear. Because each miRNA can target multiple genes, miRNAs may potentially have functional implications for the overall behavior of entire pathways. Here, we investigate the functional consequences of miRNA dysregulation through an integrative analysis of miRNA and mRNA expression data using a novel approach that incorporates pathway information a priori. By searching for miRNA-pathway associations that differ between healthy and tumor tissue, we identify specific relationships at the systems level which are disrupted in cancer. Our approach is motivated by the hypothesis that if an miRNA and pathway are associated, then the expression of the miRNA and the collective behavior of the genes in a pathway will be correlated. As such, we first obtain an expression-based summary of pathway activity using Isomap, a dimension reduction method which can articulate non-linear structure in high-dimensional data. We then search for miRNAs that exhibit differential correlations with the pathway summary between phenotypes as a means of finding aberrant miRNA-pathway coregulation in tumors. We apply our method to cancer data using gene and miRNA expression datasets from The Cancer Genome Atlas and compare ∼105 miRNA-pathway relationships between healthy and tumor samples from four tissues (breast, prostate, lung and liver). Many of the flagged pairs we identify have a biological basis for disruption in cancer.
INTRODUCTION
Cellular functions are carried out by coordinated regulation of genes on a pathway, which facilitate a series of interactions among genes to produce behaviors as diverse as cell metabolism to cell signaling. At the post-transcriptional level, microRNAs (miRNAs, miRs) modulate gene expression by binding to a 6–8 nt target motif of mRNA transcripts, preventing translation and/or inducing degradation of their target genes. Due to the short-binding motif, miRNA targeting is non-specific, such that a single miRNA may target multiple genes and likewise, a single gene may be targeted by multiple miRNAs (1). Currently, it is estimated that ∼103 known miRNAs regulate approximately a third of genes in the genome (2–4). However, not all miRNA–gene relationships are known; studies to predict miRNA targets using sequence matching have had mixed success (5), and the functional consequences of miRNA dysregulation remains an area of active research. It is now thought that the mutliplicity of targets enables miRNAs to exert a cumulative effect at the systems level, by targeting several genes and influencing their downstream interactions. miRNAs have been hypothesized to modulate pathways by regulating targets constituting those pathways (6–10). Such systems-level control may explain the association of aberrant miRNA regulation with multiple diseases, including cancer (11,12), endometriosis (13), inflammation (14) and several others.
High-throughput transcriptomics datasets now enable us to investigate the role of miRNAs in regulating pathway activity by integratively analyzing miRNA and gene expression from the same samples. Such analyses must address the challenges inherent to high-throughput data, including the fact that the number of features typically exceeds the number of samples by orders of magnitude, the data are inherently noisy and many features may be irrelevant to the phenotype of interest. In addition, integrative analyses should account for the multiplicity of interactions that collectively contribute to phenotypic differences. Approaches for integrative miRNA–mRNA analysis generally fall into two categories (15): (i) inferring interacting miR–mRNA pairs from transcriptomic data (e.g. by searching for high correlations (16), using regularized linear regression (17,18) or mutual information (19)); and (ii) combining miRNA and mRNA expression data to identify a signature in the combined feature space that predicts the phenotype of interest (20,21) (e.g. using non-negative matrix factorization (22) or clustering (23) to find combinations of miRNAs and genes that most strongly predict outcomes). A comprehensive review of integrative miRNA–mRNA analysis may be found in (15).
Information about gene interaction networks obtained from pathway databases (such as KEGG (24) or PID (25)) can be used to reduce dimensionality and improve interpretability by focusing on functionally related gene sets. To date, however, most miRNA–mRNA integrative analyses do not explicitly incorporate this information a priori; instead, the interactions and signatures identified in the analysis are tested for overlap with known pathways at the end to lend a systems-level interpretation of the gene-level findings (26,27). Because many pathway analysis approaches (including enrichment methods, such as Gene Set Enrichment Analysis (GSEA) (28)) rely on aggregating single-gene statistics rather than treating the pathway as a whole, they may miss crucial multi-gene interactions, such as the loss of coordinated expression. For example, the relevance of a miRNA that governs the relationship between two genes (such as the amplitude of the oscillation shown in Figure 1) can be missed when considering the target genes in isolation, since neither gene is independently associated with the miRNA.
Figure 1.

An example of two genes cycling out of phase with one another, with the amplitude of the oscillation governed by the expression of a miRNA. The relationship is apparent in the left panel, where the lower values of the miRNA result in a smaller radius in the relationship between gene X and gene Y, yet neither gene X nor gene Y are correlated with the miRNA (right panels, top and bottom).
To overcome this limitation, several groups have proposed schemes to summarize gene expression across the pathway to quantify the overall level of pathway ‘activity’ in each sample (29,30). These approaches apply dimension reduction techniques (such as singular value decomposition [SVD] and principal components analysis [PCA]) to pre-defined gene sets, effectively yielding a single value that encapsulates the coarse coexpression behavior of all the genes in the pathway. In the Pathway Level Analysis of Gene Expression (PLAGE) method (29), SVD was used to obtain a ‘pathway activity level’ quantification based on the expression of genes in the pathway. A similar approach using PCA was employed in the GPC-Score (30) method. A non-linear dimension reduction (NLDR) strategy for pathway summarization was considered in (31), which was shown to more faithfully summarize complex coexpression patterns than linear methods. More recently, the Component Pathway Analysis and Differential Expression Removal (COMPADRE) package (32) presented a framework for pathway summarization using a variety of dimension reduction techniques (including SVD, PCA, ICA, non-negative matrix factorization and non-linear Isomap). The resulting pathway-level quantifications may then be tested for statistical associations with the phenotype, allowing the pathway to be treated as a single functional unit.
Here, we propose a method that identifies miRNAs that differentially regulate the overall activity of pathways by using a pathway summarization technique capable of articulating non-linear and multi-gene effects. Motivated by the observation that NLDR can yield more accurate results when applied to gene expression data (31,33,34), our method uses Isomap (35), an NLDR method, to summarize pathway expression to yield a low-dimensional summary that we call the pathway activity summary (PAS). The PAS provides a faithful ‘snapshot’ of the pathway, a coarse measure of pathway expression in all samples. Our method then computes correlation coefficients between PAS and miRNA expression to identify miRNAs whose expression is associated with the overall activity of the pathway. By comparing class-conditional correlations in cases and controls, we identify miRNA-pathway pairs that appear to have a differential relationship in cases and controls, elucidating the function of the miRNA and its potential mechanistic role in the phenotype of interest.
The approach used here is similar in some respects to our GPC-score method (30), which reveals novel regulatory relationships between genes and pathways. Using PCA for pathway summarization, GPC-score was able to identify differentially regulated gene-pathway pairs and accurately detect the interaction of genes with pathways that were not previously known to include them. The present work augments this prior analysis method in two novel ways. First, by using the non-linear Isomap instead of PCA, we obtain a more faithful summary of pathway activity. Second, by applying the method to miRNA and mRNA data (rather than simply mRNA data), we achieve an integrative analysis of these datasets that can provide insight into the function of miRNAs. We apply this method to miRNA and mRNA expression profiles from four cancers (breast, liver, lung and prostate) using data from The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov/).
Previous analyses have integrated multiple omics platforms to identify specific mechanisms regulating gene expression. Several pipelines have taken into account sample-specific data from TCGA at the transcriptomic, genomic and epigenetic levels and have linked them with cell-generic data from other consortiums (36,37). These studies have identified relationships between expression regulators and genes in some cancer types. Recently, the TCGA Network surveyed miRNAs in the context of expression patterns and clinical outcomes in ovarian cancer, and found widespread impact on gene expression and molecular heterogeneity (38). Our method is also integrative, but novel in that it surveys miRNA regulation in the context of gene expression from a pathway perspective. Importantly, because our approach uses both miRNA and mRNA expression data, it avoids some of the pitfalls that were previously identified (39) with making pathway-level inferences from miRNA data alone.
In this study, we apply our methodology to gene and miRNA expression datasets from TCGA, a freely accessible repository of high dimensional genomic and expression data for several cancers. The datasets include both tumor and adjacent-normal tissue samples across multiple experimental modalities. After identifying class-conditional correlation differences for all possible miRNA-pathway pairs, we assess their significance through permutation testing. We report miRNAs that appear to have pathway-wide effects whose relationships change with the development of cancer, and report results for multiple distinct cancers.
MATERIALS AND METHODS
In order to elucidate the functional role of miRNAs in cancer, we seek to identify miRNAs that appear to influence the overall activity of a pathway, and whose effects on that pathway appear to differ between healthy and tumor tissue. To do so, we first compute an PAS for each sample in each pathway of interest using gene expression data. We then compute, class-conditionally, the correlation between the pathway expression summary and each miRNA in cases and controls to quantify the miRNA-pathway relationship in those tissues, and test whether tumor-normal differences in the miRNA-pathway correlations are statistically significant. We detail the steps of this algorithm below; a summary may be found in Table 1 and Figure 2.
Table 1. Procedure for assessing disrupted pathways regulated by miRNAs.
| miRNA-pathway algorithm | |
|---|---|
| 1. | Subset gene expression data to the pathway genes, forming pathway expression matrix of l genes × N samples. |
| 2. | Apply Isomap to pathway matrix using all samples, obtaining for each sample a PAS value based on the first Isomap coordinate (analogous to using the first principal component from PCA). |
| 3. | Compute the Spearman rank correlation between the miRNA and the PAS in tumor samples, ρ(miR, PAS | T). |
| 4. | Compute the Spearman rank correlation between the miRNA and the PAS in normal samples, ρ(miR, PAS | N). |
| 5. | Compute absolute correlation difference between phenotypes as shown in Equation 1. |
| 6. | Repeat steps 3.–5. using randomly permuted phenotype labels for 105 resamplings to compute the null distribution of Δρ. |
| 7. | Compare the true miRNA-pathway Δρ to the permuted null distribution obtained in step 6. to assess statistical significance of Δρ. |
Figure 2.

Illustration of the algorithm for a particular miRNA-pathway pair. (A) Gene expression data is first subsetted by the genes in a pathway and summarized by Isomap to produce the PAS, a one-dimensional (1D) summary of pathway expression in all samples. (B) PAS and miRNA expression are subsetted by phenotype, and miRNA-pathway correlations are computed for tumor and normal tissue. The difference between correlations gives Δρ. (C) To assess Δρ significance, the Δρ null distribution is estimated by random permutation of the class labels.
Algorithm
To identify miRNAs whose effects across entire systems differ between two conditions, we compute the association of miRNAs with pathways and compare associations between phenotypes by correlating miRNA and pathway gene expression. Because a given pathway may comprise tens to hundreds of genes, we use Isomap (35) to compute a 1D summary of gene expression across the pathway, which we call the PAS. Here, each sample can be be thought of as a point in a high dimensional space whose coordinates correspond to the expression of the genes on that pathway. Because the underlying biology places constraints on the expression of these genes with respect to one another, we make the assumption that the samples lie on a low-dimensional manifold within the gene expression space. Isomap attempts to learn this manifold, yielding a coordinate that articulates the variability among samples; projecting the gene expression data from sample j onto this coordinate obtains the pathway activity score PASj for sample j across the pathway of interest. (The approach is analogous to that of PCA; in contrast to PCA, however, the Isomap coordinate need not be a linear transformation of the gene expression space.) By obtaining PAS values for each sample, we can then compare pathway activity in cases and controls, and test the association of pathway activity with other variables of interest.
Relationships between miRNA expression and pathway activity are then compared between phenotypes as follows. The correlation between the PAS for a pathway and expression for a miRNA is computed class-conditionally, i.e. separately for tumor and normal samples. We then compute the absolute difference of the miRNA-pathway correlation in tumor and normal tissue:
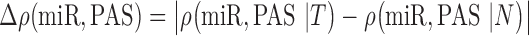 |
(1) |
where, ρ(x, y) is the Spearman rank correlation between x and y, chosen for its insensitivity to outliers, and T and N indicate tumor and normal tissue, respectively. A large correlation difference Δρ between sample classes indicates apparent differential regulation of a pathway by a miRNA. Significance of the correlation difference is assessed by a permutation test, wherein the tumor and normal labels are randomly reassigned and Equation 1 is recomputed to obtain a reference distribution for the miRNA-pathway pair. The steps for the algorithm are listed in Table 1. Figure 2 illustrates the algorithm in visual form.
Implementation
Here we detail the implementation of the algorithm as applied to mRNA and miRNA data from TCGA. Additional details are provided in the Supplementary Data.
Pathway summarization
The goal of pathway summarization is to reduce the dimensionality from that of l genes on the pathway to a single value that encapsulates the pathway activity for each sample. To this end, we define the PAS as the 1D embedding of the pathway mRNA data using Isomap.
The choice to use Isomap for pathway summarization rather than SVD (29) or PCA (30) is motivated by its ability to articulate non-linear geometries in the data. A toy example comparing Isomap to PCA is shown in Figure 3. Here, the data lie on a 2D manifold that is coiled upon itself in three-dimensional space; dimension reduction via Isomap articulates this surface, whereas PCA cannot.
Figure 3.

Swiss roll dimension reduction using PCA and Isomap. The roll is colored from green to red along the roll axis.
For each pathway in the KEGG (24) database, mRNA expression data are subsetted to the genes associated with that pathway to produce pathway-specific matrices. A total of 223 pathways are included in the analysis (after excluding six pathways with fewer than five genes). Expression levels for each gene are scaled to have zero mean and unit variance, allowing features to be measured on the same scale and reducing the disproportionate influence of any outlying samples. Isomap is then applied to the pathway gene expression data, and the projection of the sample on the first Isomap coordinate is used as a measure of the overall activity of the pathway.
An example of the utility of Isomap for summarizing gene expression data is given in Figure 4, where the 39-gene ‘Type I diabetes mellitus pathway’ is summarized by PCA (left) and Isomap (right) for the TCGA breast cancer and normal tissue samples. Because Type I diabetes mellitus has been associated with an increased risk of breast cancer (40,41) and several genes in the pathway are known tumor suppressors and cytokines that are commonly perturbed in tumors, we expect that a low dimensional embedding of the data should enable separation of the tumor and normal samples. However, we observe that this difference is not articulated using PCA; in the left scatterplot matrix of Figure 4, the red and black points overlap. By contrast, the Isomap embedding enables separation of the tumor and normal samples, suggesting that there exists a (non-linear) pattern of gene expression within the pathway that is associated with breast cancer. This example motivates the choice of NLDR as a means of quantifying the overall behavior of a pathway.
Figure 4.

Comparison of gene expression dimension reduction using PCA (left) and Isomap (right) for genes in the Type I diabetes mellitus pathway. Black circles represent TCGA breast cancer tumor tissue and red triangles represent adjacent-normal. Plotted are the projections of the samples in the first four PCA coordinates (left) and first two Isomap coordinates (right). The Isomap embedding enables separation of the tumor and normal samples not achieved by PCA, suggesting that a non-linear pattern of gene expression within the pathway distinguishes tumor and normal samples.
Isomap parameter choice
Isomap has one free parameter, k, which defines the k-nearest neighbors used in reconstructing the local geometry (35). Choosing the optimal value of k is an open question, and different values have the potential to produce different embeddings. We devised a data-driven method for selecting k by employing a comparison between the spectra of PCA and Isomap.
Isomap applies Multidimensional Scaling (MDS) (42) on a distance matrix that approximates geodesic distances, constructed by a k-nearest neighbors search and computing shortest paths. This may be thought of as a localized form of MDS (or, equivalently, PCA (42)), which classically uses distances between all pairs to articulate the global geometry. Like PCA and MDS, Isomap also yields a spectrum of eigenvalues whose magnitude indicates the proportion of variability in the data that is articulated by the corresponding coordinate.
We capitalize on this feature by comparing the spectra of PCA and ISOMAP for different values of k. Spectral comparisons can help find embeddings most different from each other, and may reveal those that articulate manifolds with non-linear structures. In PCA, one chooses the number of components to be retained such that the majority of the variance in the data is captured. A common visualization is the ‘scree plot’ in which the variance for each component (eigenvalues λ1 ≥ λ2, ≥… ≥ λn) is displayed; one looks for an elbow in the spectrum indicating that additional components do not appreciably reduce the residual variance. Mathematically, an elbow at the first component will have a large ratio between the first two eigengaps (i.e. a large change between the first and second eigenvalues, followed by a much smaller change between the second and third), which we call the spectral gap ratio (SGR), 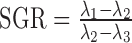 .
.
We choose Isomap k such that it maximizes the SGR ratio between Isomap and PCA, 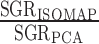 , noting that when k = N − 1 (all data treated as nearest neighbors), Isomap and PCA yield equivalent spectra. The optimal k is guaranteed to yield
, noting that when k = N − 1 (all data treated as nearest neighbors), Isomap and PCA yield equivalent spectra. The optimal k is guaranteed to yield 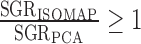 ; that is, it produces an embedding that explains at least as much variance in the first component as PCA. By choosing k to maximize this ratio, we obtain the greatest improvement by Isomap over PCA, which will occur when the data lie on a curved manifold that cannot be articulated by PCA.
; that is, it produces an embedding that explains at least as much variance in the first component as PCA. By choosing k to maximize this ratio, we obtain the greatest improvement by Isomap over PCA, which will occur when the data lie on a curved manifold that cannot be articulated by PCA.
To illustrate our methodology, we apply Isomap to the Swiss roll dataset using different values of k in Figure 5. The ‘optimal’ k (k = 6) produces an embedding that reflects the low-dimensional intrinsic geometry of the roll, the unraveled 2D surface. In comparison, a value that is too small (k = 3) will be sensitive to local distortions, whereas a value that is too large (k = 16) will produce an embedding that poorly learns the intrinsic coordinates. The spectra for all three Isomap embeddings, in addition to the PCA spectrum, are shown in the right-most plot in Figure 5. The green empty circles, corresponding to (k = 6), have the largest 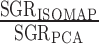 , whereas other k’s have smaller SGR as shown by the red (k = 3) and blue (k = 16) empty circles. The ‘optimal’ k produces a PAS that captures the geodesic of the Swiss roll. We applied this methodology to pathway data such that the PAS best represents the geometry of the data in the high-dimensional space.
, whereas other k’s have smaller SGR as shown by the red (k = 3) and blue (k = 16) empty circles. The ‘optimal’ k produces a PAS that captures the geodesic of the Swiss roll. We applied this methodology to pathway data such that the PAS best represents the geometry of the data in the high-dimensional space.
Figure 5.

2D embedding of the Swiss roll using Isomap for different k values. The bottom right plot shows the spectra using PCA (black dots), and for k = 3 (red), k = 6 (green) and k = 16 (blue) using Isomap. The spectrum at the optimal k, k = 6, is most different from PCA’s spectrum, as computed by the SGR ratio defined in the ‘Materials and Methods’ section.
PAS correlation with miRNAs
Once the PAS is computed, correlations between each pathway’s PAS with each miRNA’s expression are computed class-conditionally. miRNA-pathway correlation differences (Δρ) are computed between tumor and adjacent-normal tissue samples as shown in Equation 1. We emphasize that the PAS is computed class-inclusively (both tumor and adjacent-normal tissue) so that different phenotypes are summarized in context with each other. Thus, we can compare phenotypes on the same scale and quantify their gene expression differences across the pathway. Afterward, we restrict samples to each phenotype and compute their correlation with miRNAs class-conditionally. This enables us to compare how the relationship between a miRNA and a pathway differs in tumor and normal tissue.
The significance of each Δρ is assessed by permutation tests. Each miRNA-pathway pair’s Δρ null distribution is estimated by randomly permuting class labels and recomputing Δρ for 105 resamplings. Within each resampling, the same number of nominal tumor and adjacent-normal samples is preserved. Adjustment for the multiple hypotheses tested is also achieved through permutation (43).
RESULTS
miRNAs with median expression above 0.001 (444 in breast, 455 in liver, 484 in lung and 416 in prostate) and pathways with greater than five genes (223 pathways representing a total of 5869 unique genes) were considered. Each possible miRNA-pathway pair (∼105 pairs) was analyzed for differential association between tumor and adjacent-normal tissue within each organ (breast, prostate, lung and liver) by computing its Δρ and assessing Δρ significance to identify organ-specific relationships between miRNAs and pathways that appear to be strongly altered in tumors. Multiple hypothesis correction was achieved through permutation (43).
We illustrate aberrant miRNA regulation of pathways in tumor tissue by showing sample miRNA versus PAS expression plots which have the most pronounced class-correlation differences (Figure 6). In the plots, tumor samples exhibit distinct trends from adjacent-normal samples for the same miRNA and pathway in the same organ.
Figure 6.

Representative examples of significant miRNA-pathway pairs for all four cancers. miRNA-pathway pairs with the largest Δρ are shown for each cancer (P < 10−5). Tumor samples are represented by red circles and adjacent-normal samples by blue triangles. LOESS curves are overlaid by tissue type (solid line for tumor tissue, dotted line for adjacent-normal tissue) to visualize correlation differences. The number of genes in the pathway which have been used in the computation of the PAS are shown in parenthesis.
In these particular cases, the PAS alone can distinguish phenotypes, as demonstrated by the difference in the location of the tumor and normal samples along the x-axes. However, we emphasize that differential expression within a pathway is unnecessary for achieving significance. Our method also detects aberrant signaling even when no marginal differences can be detected. Figure 7 shows a sample miRNA versus pathway expression plot in prostate cancer with a significant correlation change despite a lack of differential expression across either the PAS or miRNA. Such a pair would not be detected using methods which rely on single gene association statistics, or by looking at the pathway in isolation without the miRNA.
Figure 7.

Example of a miRNA-pathway pair (miRNA ID: hsa-mir-193a, KEGG pathway ID: 04115) with significant Δρ (Δρ = −0.64, P < 10−4) despite no differential expression in prostate cancer. Absence of differential expression is visualized by a rug plot on the top and right. Our method is capable of articulating significant miRNA-pathway coregulation differences regardless of differential expression across either the pathway or miRNA.
Importantly, other evidence from the literature supports the association of this miRNA-pathway pair. The miRNA in Figure 7, hsa-mir-193a, is a tumor suppressor implicated in several cancers whose downregulation has been proposed as a biomarker of oncogenesis (27,44,45). The p53 signaling pathway, a tumor suppressing pathway which responds to cell stress, can activate cell-cycle arrest, senescence or apoptosis. It is known as a prominent regulator which is commonly disrupted in cancer cells (46), and its main tumor protein TP53 is the most mutated gene in cancer. In addition, the p53 pathway contains three genes which are predicted to be targets of hsa-mir-193a (CCND1, SIAH1 and ZMAT3). This example serves to illustrate the capabilities of the method to detect biologically meaningful relationships between miRNA expression and pathway activity.
In the following sections, we list the top 15 pairs with the most pronounced Δρ for each cancer type. The remaining pairs at the same level of significance are listed in the Supplementary Data. Many of the flagged miRNAs and pathways have a biological basis for disruption in cancer.
Breast cancer
Breast cancer pairs with large Δρ are shown in Table 2. Within each pair we include the number of predicted targets of the miRNA on the pathway using TargetScan (47) as well as the target enrichment P-value as calculated by DIANA mirPath v.3 (48). hsa-mir-146b and hsa-mir-135b each regulate multiple pathways class-conditionally and have functional relevance to cancer in the literature. Specifically, hsa-mir-146b is a known tumor suppressor (49,50) that inhibits NF-kB induction of IL-6 to prevent inflammation in breast cells, which chronically leads to oncogenesis. In breast cancer cells, however, promoter methylation decreases hsa-mir-146b expression (51). hsa-mir-135b has previously been associated with several cancer types, including prostate, lung and most prominently colon cancer. In colon cancer, upregulation of hsa-mir-135b promotes cancer progression and activation of hsa-mir-135b is triggered by oncogenic pathways (52). The IL-1R1 pathway, which involves regulation of immune and inflammatory responses, has recently been found to regulate hsa-mir-135b expression in smoke-induced inflammation in lung cells (53).
Table 2. Top breast cancer pairs sorted by the most pronounced Δρ (P < 10−5).
| miRNA | KEGG ID | KEGG name | Δρ | ρ T | ρ N | size | targets | p.DIANA |
|---|---|---|---|---|---|---|---|---|
| hsa-mir-146b | 00330 | Arginine and proline metabolism | 1.15 | 0.44 | −0.71 | 49 (54) | 1 (1) | 0.933 |
| hsa-mir-146b | 05110 | Vibrio cholerae infection | 1.12 | 0.44 | −0.68 | 51 (54) | 0 (0) | 0.933 |
| hsa-mir-146b | 05217 | Basal cell carcinoma | 1.11 | 0.45 | −0.66 | 53 (55) | 1 (1) | 0.933 |
| hsa-mir-146b | 05200 | Pathways in cancer | −1.10 | −0.49 | 0.61 | 316 (326) | 10 (10) | 0.933 |
| hsa-mir-135b | 00980 | Metabolism of xenobiotics by cytochrome P450 | 1.10 | 0.48 | −0.63 | 49 (71) | 0 (0) | 0.685 |
| hsa-mir-146b | 05120 | Epithelial cell signaling in Helicobacter pylori infection | 1.10 | 0.47 | −0.63 | 66 (68) | 0 (0) | 0.962 |
| hsa-mir-135b | 05217 | Basal cell carcinoma | 1.08 | 0.48 | −0.60 | 53 (55) | 5 (5) | 0.517 |
| hsa-mir-146b | 00980 | Metabolism of xenobiotics by cytochrome P450 | 1.08 | 0.46 | −0.61 | 49 (71) | 0 (0) | 1.52e−04 |
| hsa-mir-99a | 00590 | Arachidonic acid metabolism | 1.07 | 0.49 | −0.58 | 48 (59) | 0 (0) | NT |
| hsa-mir-135b | 00520 | Amino sugar and nucleotide sugar metabolism | 1.07 | 0.44 | −0.63 | 47 (48) | 0 (0) | NT |
| hsa-mir-1307 | 00830 | Retinol metabolism | −1.07 | −0.51 | 0.56 | 42 (64) | 0 (0) | NT |
| hsa-mir-1307 | 04976 | Bile secretion | −1.06 | −0.61 | 0.45 | 56 (71) | 0 (0) | 0.046 |
| hsa-mir-135b | 00051 | Fructose and mannose metabolism | 1.06 | 0.44 | −0.63 | 35 (36) | 1 (1) | NT |
| hsa-mir-135b | 05120 | Epithelial cell signaling in helicobacter pylori infection | 1.06 | 0.41 | −0.65 | 66 (68) | 4 (4) | NT |
| hsa-mir-224 | 01040 | Biosynthesis of unsaturated fatty acids | 1.06 | 0.40 | −0.65 | 19 (21) | 0 (0) | NT |
ρ T and ρN are the within-tissue Spearman’s rank correlation for tumor tissue and normal tissue, respectively. A total of 671 tumor samples and 87 normal samples were used to compute correlations. Size denotes the number of genes in the pathway that have been used in the computation of the the PAS. Targets denotes the number of predicted targets of the miRNA on those genes using TargetScan (47). In parenthesis, the total number of genes and targets of the miRNA on the pathway are shown. p.DIANA denotes the miRNA-pathway enrichment P-value as calculated by DIANA mirPath v.3 (48) using TargetScan. NT = no targets found by DIANA on the pathway.
It is notable that several pathways which are listed, including those differentially regulated by hsa-mir-146b and hsa-mir-135b, are inflammatory. Infectious disease pathways, including Vibrio cholerae infection and Epithelial cell signaling in Helicobacter pylori infection, activate proinflammatory responses including the upregulation of various inflammatory cytokines after infection. Cytochrome P450, the main enzyme in Metabolism of xenobiotics by cytochrome P450, is regulated by several inflammatory mediators and its expression and activity is decreased with a host response to inflammation and infection (54).
These miRNAs and pathways are of interest because chronic inflammation is broadly associated with tumorigenesis and cancer. Chronic inflammation has been shown to increase the risk of tumor formation, notably demonstrated in the association between chronic inflammatory bowel disease and colon carcinogenesis. Inflammatory mediators and inflammation in the tumor microenvironment have many cancer-promoting effects including promotion of malignant cells, angiogenesis, subversion of immune responses, metastasis, induction of proneoplastic mutations and altered response to hormones (55–57). Proinflammatory chemokines and cytokines have been found in the tumor microenvironment of many cancers and are typically induced by hypoxic conditions, which are characteristic of tumors (58).
In addition, several metabolic pathways are represented. Arginine and proline metabolism has been known to exhibit changes in cancer (59), and the proline regulatory axis and proline metabolism both undergo alterations that are posited to sustain and promote tumor cell growth (60,61). A plot of hsa-mir-146b differentially regulating Arginine and proline metabolism is shown in Figure 6. Interestingly, two cancer pathways (Pathways in Cancer and Basal Cell Carcinoma) contain the most predicted miRNA targets, including cancer genes NRAS, CCDC6, CSF1R, SMAD4, ITGAV and several others. However, it should be noted that many miRNA-pathway pairs contain no predicted miRNA targets. Sequence matching using TargetScan (47) will fail to capture indirect interactions between miRNAs and pathway genes that may indeed be captured using correlations. For instance, the IL-1R receptor family, which regulates hsa-mir-135b expression (see above), activates cytokines IL-6 and IL-8 which are present or interact with multiple inflammatory pathways in Table 2, even though they are not predicted targets of hsa-mir-135b.
Prostate cancer
hsa-mir-195 is flagged with many pathways in prostate cancer, shown in Table 3. hsa-mir-195 is frequently reported as deleted or downregulated in tumors across multiple cancer types (62–64). In prostate cancer hsa-mir-195 is under-expressed and has been shown to behave as a tumor suppressor by regulating RPS6KB1 (65), BCOX1 (66) and FGF2 (67). hsa-mir-195 itself is part of the hsa-mir-15 family cluster, whose hsa-mir-15a has also been shown to behave as a tumor suppressor by regulating oncogenes BCL2, CCND1 and WNT3 (68). In advanced prostate tumors, hsa-mir-15a is downregulated or deleted and these oncogene levels are markedly increased. Relatedly, the loss of the hsa-mir-15 family in prostate cancer has been found to contribute to metastatic potential including bone lesions (69) (a marker of metastasis). hsa-mir-16, another hsa-mir-15 family member, is flagged with several pathways with comparable P-values in prostate cancer (refer to Supplementary Data), furthering evidence that the hsa-mir-15 family is compromised in prostate cancer.
Table 3. Top prostate cancer pairs sorted by the most pronounced Δρ (P < 10−5).
| miRNA | KEGG ID | KEGG name | Δρ | ρ T | ρ N | size | targets | p.DIANA |
|---|---|---|---|---|---|---|---|---|
| hsa-mir-195 | 04360 | Axon guidance | −0.93 | −0.19 | 0.75 | 128 (129) | 16 (17) | 0.190 |
| hsa-mir-195 | 04510 | Focal adhesion | 0.88 | 0.18 | −0.70 | 194 (200) | 30 (31) | 0.013 |
| hsa-mir-195 | 05218 | Melanoma | 0.88 | 0.18 | −0.70 | 63 (71) | 16 (16) | 0.013 |
| hsa-mir-1307 | 05410 | Hypertrophic cardiomyopathy (HCM) | 0.86 | 0.38 | −0.49 | 75 (83) | 0 (0) | NT |
| hsa-mir-195 | 05217 | Basal cell carcinoma | −0.85 | −0.17 | 0.68 | 54 (55) | 10 (10) | 0.309 |
| hsa-mir-1307 | 05414 | Dilated cardiomyopathy | −0.85 | −0.39 | 0.46 | 81 (90) | 0 (0) | NT |
| hsa-mir-1307 | 04122 | Sulfur relay system | −0.85 | −0.23 | 0.62 | 10 (10) | 0 (0) | NT |
| hsa-mir-200a | 03022 | Basal transcription factors | −0.84 | −0.17 | 0.68 | 35 (36) | 0 (0) | 0.999 |
| hsa-mir-944 | 00982 | Drug metabolism - cytochrome P450 | −0.83 | −0.52 | 0.32 | 55 (73) | 0 (0) | NT |
| hsa-mir-141 | 00592 | alpha-Linolenic acid metabolism | 0.83 | 0.42 | −0.42 | 16 (20) | 0 (0) | NT |
| hsa-mir-195 | 04964 | Proximal tubule bicarbonate reclamation | 0.83 | 0.15 | −0.67 | 22 (23) | 3 (4) | 1 |
| hsa-mir-195 | 04912 | GnRH signaling pathway | 0.83 | 0.13 | −0.70 | 92 (101) | 10 (10) | 0.399 |
| hsa-mir-195 | 05414 | Dilated cardiomyopathy | 0.82 | 0.14 | −0.69 | 81 (90) | 5 (5) | 1 |
| hsa-mir-195 | 04664 | Fc epsilon RI signaling pathway | 0.82 | 0.14 | −0.69 | 70 (79) | 10 (10) | 0.243 |
| hsa-mir-195 | 05100 | Bacterial invasion of epithelial cells | 0.82 | 0.20 | −0.62 | 69 (70) | 5 (5) | 0.271 |
A total of 482 tumor samples and 52 normal samples were used to compute correlations.
Many oncogenes are regulated by hsa-mir-195, including BCL2, CCND1, WNT3, AKT3, CDC42, RAF1 and KRAS that lie on the pathways flagged with hsa-mir-195 in Table 3. These pathways include two cancer pathways (melanoma and basal cell carcinoma), morphological pathways (axon guidance and focal adhesion) and several signaling pathways whose genes are expected to be altered in tumors. Interestingly, most miRNA-pathway pairs in Table 3, and particularly those with hsa-mir-195, exhibit much stronger correlations in normal samples than in tumor samples. These trends may indicate general loss of function in tumorigenesis, in concordance with documented underexpression of hsa-mir-195 in tumors.
Liver cancer
In Table 4, flagged pairs for liver cancer are shown. miRNAs hsa-mir-100, hsa-mir-34a and hsa-mir-210 are represented several times and are each known to be involved in hepatocellular carcinoma. hsa-mir-100 downregulation, concomitant with increased expression of its target PLK1, correlates with poor prognosis and is an early event in hepatocarcinogenesis (70,71). Several studies have shown hsa-mir-34a to be a tumor suppressor that activates apoptosis and cell senescence. In hepatocellular carcinoma, hsa-mir-34a suppresses tumor invasion by modulating c-Met expression and is typically underexpressed (72,73). In addition, hsa-mir-210 upregulation is increased in hypoxic conditions and contributes to metastatic potential in hepatocellular carcinoma (74).
Table 4. Top liver cancer pairs sorted by the most pronounced Δρ (P < 10−5).
| miRNA | KEGG ID | KEGG name | Δρ | ρ T | ρ N | size | targets | p.DIANA |
|---|---|---|---|---|---|---|---|---|
| hsa-mir-100 | 04115 | p53 signaling pathway | 0.85 | 0.38 | −0.47 | 64 (68) | 0 (0) | NT |
| hsa-mir-3607 | 04320 | Dorso-ventral axis formation | 0.82 | 0.26 | −0.56 | 20 (24) | 0 (0) | NT |
| hsa-mir-34a | 04115 | p53 signaling pathway | 0.81 | 0.29 | −0.53 | 64 (68) | 8 (8) | 0.411 |
| hsa-mir-100 | 00360 | Phenylalanine metabolism | 0.81 | 0.53 | −0.28 | 17 (17) | 0 (0) | NT |
| hsa-mir-210 | 03030 | DNA replication | −0.81 | −0.45 | 0.36 | 36 (36) | 0 (0) | NT |
| hsa-mir-210 | 05219 | Bladder cancer | −0.81 | −0.41 | 0.40 | 41 (42) | 0 (0) | NT |
| hsa-mir-210 | 05322 | Systemic lupus erythematosus | −0.81 | −0.35 | 0.46 | 103 (136) | 0 (0) | NT |
| hsa-mir-210 | 04110 | Cell cycle | −0.81 | −0.42 | 0.38 | 117 (124) | 0 (0) | NT |
| hsa-mir-148b | 04614 | Renin-angiotensin system | 0.80 | 0.19 | −0.61 | 14 (17) | 1 (1) | 0.582 |
| hsa-mir-139 | 00053 | Ascorbate and aldarate metabolism | −0.80 | −0.36 | 0.45 | 24 (26) | 0 (0) | NT |
| hsa-mir-34a | 00620 | Pyruvate metabolism | 0.80 | 0.29 | −0.51 | 37 (40) | 1 (1) | 0.999 |
| hsa-mir-34a | 00591 | Linoleic acid metabolism | 0.80 | 0.32 | −0.47 | 23 (30) | 0 (0) | NT |
| hsa-mir-1247 | 00460 | Cyanoamino acid metabolism | −0.80 | −0.46 | 0.34 | 7 (7) | 0 (0) | NT |
| hsa-mir-100 | 00983 | Drug metabolism - other enzymes | 0.79 | 0.56 | −0.24 | 49 (52) | 0 (0) | NT |
| hsa-mir-139 | 00330 | Arginine and proline metabolism | 0.78 | 0.58 | −0.20 | 51 (54) | 2 (2) | NT |
A total of 347 tumor samples and 50 normal samples were used to compute correlations.
hsa-mir-100 and hsa-mir-34a are both found to differentially regulate the p53 signaling pathway in Table 4. This is of interest because TP53 is very commonly implicated in cancer, and in liver cancer, TP53 loss is associated with aggressive carcinomas and restoration of TP53 has been shown to initiate tumor regression (75). Notably, hsa-mir-34a and hsa-mir-34 family members are part of the p53 transcriptional network and are directly regulated by TP53 (76,77). TP53 induces the transcription of the hsa-mir-34 family, which downregulates CDK4 and CDK6 to induce cell cycle arrest and BCL2 to promote apoptosis (78). hsa-mir-34a itself is predicted to directly regulate eight targets on the p53 signaling pathway, including tumor-associated genes CCND1, CCNE2, TP73 and CDK6. In addition, TP53 induces the transcription of other miRNAs (hsa-mir-145, hsa-mir-192/215 and hsa-mir-107) that modulate genes to induce cell cycle arrest, reduce cell proliferation and suppress angiogenesis (78). We illustrate hsa-mir-34a targeting the the p53 signaling pathway in Figure 8, with genes colored by differential expression between tumor and normal tissue in liver cancer. We note this figure does not include connections to hsa-mir-34a since it is only a partial representation of the gene regulatory network. Nevertheless, hsa-mir-34a appears to target genes which have variable degrees of differential expression in liver cancer. Of the genes hsa-mir-34a is predicted to target using TargetScan (47), several (79–83) contain direct literature support, while others lack literature support but are indirectly linked through family members (84) or modulate hsa-mir-34a expression (85). Notably, the bladder cancer pathway is also present in Table 4 and contains TP53 as well as other tumor suppressors and oncogenes that are implicated in multiple cancer pathways.
Figure 8.

The p53 signaling pathway targeted by hsa-mir-34a in liver cancer. Genes in the graph are colored by the degree of differential expression from low to high, from red to white to blue, in tumor versus normal tissue. Genes on the pathway that have not been assayed in TCGA data or used in the computation of the PAS are colored gray. hsa-mir-34a is represented by the orange triangle. Gray edges denote gene-gene interactions. Red edges denote miRNA-gene regulatory relationships between hsa-mir-34a and the genes on the pathway as predicted by TargetScan. Solid red edges contain literature support (79–83). The dotted red edge between hsa-mir-34a and SERPINE1 lacks literature support; its family member hsa-mir-34c targets SERPINE1 (84). The alternating dotted red edge between hsa-mir-34a and TP73 also lacks literature support as predicted by TargetScan. Rather, TP73 has been found to modulate hsa-mir-34a expression by acting on the hsa-mir-34a promotor (85).
Lung cancer
Flagged pairs in lung cancer are shown in Table 5. hsa-mir-141 is represented frequently and is part of a miRNA family containing five members arranged as two clusters, hsa-mir-200a/200b/429 and hsa-mir-141/200c, that is thought to suppress the epithelial to mesenchymal transition (EMT). This is of interest because the EMT is believed to be an important step in metastasis. The EMT is marked by decreased cell adhesions including repression of E-cadherin and increased cell motility. This miRNA family has been observed to play a role in the EMT of many cancer types, including bladder, breast, melanoma, prostate and lung cancer. In lung cancer, it has been shown to suppress the EMT with forced increased expression, while EMT was observed in lung cancer cells with low expression of hsa-mir-200 (86). In addition, hsa-mir-141 has been shown to be a prognostic indicator in lung cancer (87) and promotes proliferation by targeting PHLPP1 and PHLPP2 (88).
Table 5. Top lung cancer pairs sorted by the most pronounced Δρ (P < 10−5).
| miRNA | KEGG ID | KEGG name | Δρ | ρ T | ρ N | size | targets | p.DIANA |
|---|---|---|---|---|---|---|---|---|
| hsa-mir-141 | 04540 | Gap junction | −1.26 | −0.52 | 0.74 | 84 (90) | 8 (8) | 0.357 |
| hsa-mir-141 | 05146 | Amoebiasis | 1.19 | 0.54 | −0.65 | 102 (106) | 5 (5) | 0.958 |
| hsa-mir-203 | 04530 | Tight junction | 1.13 | 0.44 | −0.69 | 121 (132) | 14 (14) | 0.165 |
| hsa-mir-141 | 05100 | Bacterial invasion of epithelial cells | −1.13 | −0.55 | 0.58 | 68 (70) | 5 (5) | 0.471 |
| hsa-mir-141 | 04510 | Focal adhesion | 1.11 | 0.55 | −0.56 | 195 (200) | 8 (8) | 0.958 |
| hsa-mir-141 | 04916 | Melanogenesis | −1.11 | −0.45 | 0.66 | 98 (101) | 5 (5) | 0.745 |
| hsa-mir-141 | 04670 | Leukocyte transendothelial migration | 1.10 | 0.57 | −0.53 | 109 (116) | 2 (2) | 0.999 |
| hsa-mir-141 | 04974 | Protein digestion and absorption | −1.09 | −0.53 | 0.56 | 70 (81) | 3 (3) | 0.999 |
| hsa-mir-150 | 04973 | Carbohydrate digestion and absorption | 1.08 | 0.52 | −0.56 | 35 (44) | 1 (1) | NT |
| hsa-mir-222 | 05146 | Amoebiasis | 1.07 | 0.43 | −0.64 | 102 (106) | 1 (1) | 0.977 |
| hsa-mir-141 | 05200 | Pathways in cancer | 1.06 | 0.53 | −0.53 | 315 (326) | 25 (25) | 0.357 |
| hsa-mir-150 | 04660 | T cell receptor signaling pathway | −1.05 | −0.62 | 0.43 | 103 (108) | 4 (4) | NT |
| hsa-mir-200c | 04540 | Gap junction | −1.05 | −0.39 | 0.66 | 84 (90) | 13 (13) | 0.091 |
| hsa-mir-141 | 04145 | Phagosome | 1.05 | 0.54 | −0.51 | 142 (153) | 6 (6) | 0.924 |
| hsa-mir-141 | 00260 | Glycine, serine and threonine metabolism | −1.04 | −0.44 | 0.60 | 29 (32) | 0 (0) | NT |
A total of 342 tumor samples and 38 normal samples were used to compute correlations.
It is notable that many of the pathways in Table 5 are morphological and dictate cellular processes remodeled during the EMT. Gap junctions, Tight junctions and Focal adhesions all undergo significant changes to decrease cell-cell adhesions and promote invasion. In Table 5, miRNAs hsa-mir-141 and hsa-mir-200c both differentially regulate Gap junctions (hsa-mir-141 versus Gap junction PAS is shown in Figure 6). Diminished Gap junctions or their elimination are seen as important indicators of tumorigenesis (89,90). In addition, many cancer genes targeted by hsa-mir-141 and its family members are on the pathways in Table 5, including SRC, PTEN, GRB2, CDK6, KRAS, DCC and various protein kinases. These genes are reported to play significant roles in multiple cancers in the literature.
Pathway miRNA targets
It is reasonable to ask whether the associations detected between miRNAs and pathways are driven by an abundance of targeted genes on those pathways. Tables 2–5 list the number of genes on the pathway that are targetted by the associated miRNA. As noted above, several pathways do contain multiple targets of a miRNA. However, we detect many more pathways that exhibit a differential association with a miRNA despite the fact that the pathways are not known to contain miRNA targets. To address this question systematically, we tested whether an abundance of miRNA targets in a pathway was predictive of a strong association in the analysis above. Briefly, we were unable to detect any relationship between the strength of the differential miRNA-pathway association and the proportion of miRNA targets on the pathway. Further details may be found in the Supplementary Data.
This is also revealed by comparison of miRNA-pathway associations as predicted by DIANA mirPath v.3 (48) with our results. Briefly, DIANA is a software suite that identifies miRNA regulatory control of functional pathways by looking for targets on annotated pathways, using standard and empirical statistical tests along with database functionalities. In Tables 2–5, DIANA P-values do not appear to correspond with Δρ significance. DIANA is based on predicted miRNA targets within pathways, analogous to our calculations of abundance and enrichment in the Supplementary Data. Both measures are quite distinct from our miRNA-pathway algorithm, which searches for dysregulation by comparing phenotypes using expression data. Thus, target enrichment is a poor predictor of miRNA-pathway dysregulation.
DISCUSSION
We have described a new method for integrating miRNA and gene expression data to elucidate the role of miRNAs in regulating functional pathways and identifying miRNA-pathway pairs whose co-regulation may be disrupted in cancer.
Our approach improves upon other methods that have recently been proposed to study miRNA regulation of pathways in cancer. Many of these approaches rely on miRNA target prediction coupled with enrichment analyses. For instance, Sehgal et al. (91) identified prognostic miRNAs based on survival analysis and then used functional network analysis to identify potential pathways regulated by those miRNAs using gene ontology terms, and Suzuki et al. (92) developed GSEA-FAME (Functional Assignment of miRNAs via Enrichment) to infer miRNA activity from mRNA expression data using enrichment and weighted miRNA–mRNA interaction methods. Both methods have been applied to TCGA data in order to identify biomarkers and interpret miRNA function in cancer. However, functional enrichment has been shown to contain bias (93), and commonly used in silico approaches tend to identify highly related biological processes (39). In addition, these methods typically ignore context dependent changes in miRNA regulation; it is well known that miRNAs exhibit heterogeneous effects across cell, tissue and tumor types.
In contrast, our method does not rely on miRNA target prediction and functional enrichment, avoiding those sources of bias. Rather, our approach is fully data driven, integrating sample-specific miRNA and mRNA expression data for identifying miRNA-pathway regulation. This takes into account any context dependent behavior of miRNAs, since miRNA and mRNA expression are compared using the same biological samples. By summarizing the gene expression behavior on the pathway with the PAS instead of performing enrichment analysis, we capture the overall effect of the miRNA on the pathway, avoiding the bias introduced by correlated genes (93). The use of NLDR to obtain the PAS also enables this method to articulate complex coregulatory dynamics (such as that illustrated in Figure 1). By comparing the miRNA-pathway relationship in tumor tissue to that in adjacent normal tissue, the method is able to identify regulatory relationships which are disrupted in disease. Other methods typically focus only on tumor tissue and therefore cannot distinguish regulation uniquely affected in tumors.
Our pathway summary compresses high-dimensional expression of all constitutive genes using samples of both phenotypes in the same organ. Because it computes the summary collectively, in the context of all other genes and samples, it does not rely on independent statistical associations with the phenotype of interest. Importantly, this approach takes into account systemic effects and has the ability to articulate non-linear geometries, which may separate out phenotypes even if their boundaries are not convex. Class-conditional correlations of the pathway summaries with miRNA expression between phenotypes can identify aberrantly regulated miRNA-pathway relationships even in the absence of differential expression across either the miRNA or pathway. This is in contrast to other approaches which rely on individual differential expression of miRNAs or genes to detect systemic differences across phenotypes. The use of pathways, rather than individual genes, significantly reduces the search space of relevant processes while increasing interpretability.
We integrate sample-specific miRNA and mRNA expression data from TCGA and compare tumor to adjacent-normal tissue samples from breast, prostate, liver and lung cancers. We find that within each cancer type, more miRNA-pathway relationships are aberrantly regulated in tumors than expected by chance. This supports the notion that complex diseases like cancer contain perturbations to entire systems rather than to a few individual genes. Additionally, many of the flagged miRNAs and pathways have a biological basis for disruption in cancer. We find specific relationships related to inflammatory processes, EMT modulation and tumor suppression (p53 signaling) that are highly perturbed in tumors. Comparison of results across cancer types exhibited differences in the miRNA-pathway pairs detected, suggesting that the underlying molecular mechanisms differ across tissues.
Because our method relies on statistical associations of expression data, it does not incorporate known miRNA–gene target relationships a priori. To investigate whether our findings of significant miRNA-pathway paris were driven by an abundance of miRNA targets on the pathway, we tested whether flagged miRNA-pathway pairs were more likely to be enriched with predicted miRNA targets and found poor association in all cancer types (see Supplementary Data). We found no association between the significance of the miRNA-pathway results and the number of miRNA target genes on the pathway, suggesting that indirect coregulation of the miRNA and the pathway genes contributes to our results. Notably, the significance of many miRNA-pathway pairs would be missed using methods that rely on miRNA target lists to identify miRNA-regulated pathways. Other potential artifacts that could influence significance, such as miRNA differential expression and pathway size, also showed little association with our findings (see Supplementary Data), suggesting that these too are not driving our findings. Together, this supports the view that the method is capable of detecting biologically significant miRNA-pathway relationships at the systems level that either cause or emerge from a phenotype change, and which may be missed using other approaches.
We note that our method is limited by its input data. Because we use pathways as annotated systems, the expression of genes in TCGA data that do not map to pathways are disregarded in our analysis, and, in addition, pathway genes which are not assayed are also removed. This limits the amount of genes we can incorporate into our analysis. Our study is also limited by the amount of samples available. Currently, TCGA contains the largest amount of tumor and healthy control samples, although obtaining other comparable datasets would further validate the results of the study. In addition, our method only considers expression data while ignoring other biological factors influencing miRNA regulation of pathways, including the genomic information. Finally, our method only considers one miRNA regulating one pathway at a time, whereas biologically pathways are regulated by hosts of miRNAs simultaneously.
Given miRNA-pathway pairs identified in this study, future work could hone in on these systems to disentangle their causes. For instance, studies could integrate germline or somatic mutations within affected pathways to determine specific miRNA–gene relationships that are compromised in specific cancer types, which may collectively contribute to dysregulation of entire pathways. Other directions could include modifying the algorithm to identify multiple simultaneous miRNAs and pathways in dysregulated subnetworks, rather than a single miRNA, single pathway approach. Currently, TCGA is the largest publicly available consortium of cancer data containing healthy tissue controls, to our knowledge. Increased data collection or applications to other datasets would further validate these results. In addition, experiments could be undertaken to knockdown or upregulate expression patterns of miRNAs and genes in affected miRNA-pathway pairs. These experiments could validate the findings of this study.
Finally, while we apply our algorithm to miRNA and gene expression data in cancer, we note that it can be generalized to other experimental modalities and diseases, provided sufficient data for cases and controls. Future applications could include other regulatory mechanisms such as transcription factors, epigenetic modifications or small molecule inhibitors. In addition, other complex diseases could be investigated that are thought to undergo significant perturbations at the systems level. Identifying altered associations at the systems level helps narrow down the search space for responsible mechanisms that contribute to tumorigenesis.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Sara Solla for helpful suggestions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (5K22CA148779 to R.B.); J.S.McDonnell Foundation (220020394 to R.B.); Northwestern University Data Science Initiative (to G.W. and R.B.). Funding for open access charge: James S. McDonnell Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1. Ambros V. The functions of animal microRNAs. Nature. 2004; 431:350–355. [DOI] [PubMed] [Google Scholar]
- 2. Lewis B.P., Burge C.B., Bartel D.P.. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005; 120:15–20. [DOI] [PubMed] [Google Scholar]
- 3. Griffiths-Jones S., Saini H.K., van Dongen S., Enright A.J.. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2007; 36(Suppl. 1):D154–D158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Friedman R.C., Farh K. K.-H., Burge C.B., Bartel D.P.. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009; 19:92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chi S.W., Hannon G.J., Darnell R.B.. An alternative mode of microRNA target recognition. Nat. Struct. Mol. Biol. 2012; 19:321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cui Q., Yu Z., Purisima E.O., Wang E.. Principles of microRNA regulation of a human cellular signaling network. Mol. Syst. Biol. 2006; 2:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Artmann S., Jung K., Bleckmann A., Beißbarth T.. Detection of simultaneous group effects in microRNA expression and related target gene sets. PLoS One. 2012; 7:e38365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Qiu C., Wang J., Yao P., Wang E., Cui Q.. microRNA evolution in a human transcription factor and microRNA regulatory network. BMC Syst. Biol. 2010; 4:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yoon W.H., Meinhardt H., Montell D.J.. miRNA-mediated feedback inhibition of JAK/STAT morphogen signalling establishes a cell fate threshold. Nat. Cell Biol. 2011; 13:1062–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhu W., Chen Y.-P.P.. Computational developments in microRNA-regulated protein-protein interactions. BMC Syst. Biol. 2014; 8:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Farazi T.A., Spitzer J.I., Morozov P., Tuschl T.. miRNAs in human cancer. J. Pathol. 2011; 223:102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Papagiannakopoulos T., Shapiro A., Kosik K.S.. MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res. 2008; 68:8164–8172. [DOI] [PubMed] [Google Scholar]
- 13. Ohlsson Teague E.M.C., Van der Hoek K.H., Van der Hoek M.B., Perry N., Wagaarachchi P., Robertson S.A., Print C.G., Hull L.M.. MicroRNA-regulated pathways associated with endometriosis. Mol. Endocrinol. 2009; 23:265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ceppi M., Pereira P.M., Dunand-Sauthier I., Barras E., Reith W., Santos M.A., Pierre P.. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:2735–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Walsh C.J., Hu P., Batt J., dos Santos C.C.. Discovering microRNA-regulatory modules in multi-dimensional cancer genomic data: a survey of computational methods. Cancer Inform. 2016; 15(Suppl. 2):25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fu J., Tang W., Du P., Wang G., Chen W., Li J., Zhu Y., Gao J., Cui L.. Identifying microRNA–mRNA regulatory network in colorectal cancer by a combination of expression profile and bioinformatics analysis. BMC Sys. Biol. 2012; 6:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Y., Liang C., Wong K.-C., Luo J., Zhang Z.. Mirsynergy: detecting synergistic miRNA regulatory modules by overlapping neighbourhood expansion. Bioinformatics. 2014; 30:2627–2635. [DOI] [PubMed] [Google Scholar]
- 18. Chen X., Slack F.J., Zhao H.. Joint analysis of expression profiles from multiple cancers improves the identification of microRNA–gene interactions. Bioinformatics. 2013; 29:2137–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Le T.D., Zhang J., Liu L., Li J.. Ensemble methods for miRNA target prediction from expression data. PLoS One. 2015; 10:e0131627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kristensen V.N., Lingjærde O.C., Russnes H.G., Vollan H. K.M., Frigessi A., Børresen-Dale A.-L.. Principles and methods of integrative genomic analyses in cancer. Nat. Rev. Cancer. 2014; 14:299–313. [DOI] [PubMed] [Google Scholar]
- 21. Wei Y. Integrative analyses of cancer data: a review from a statistical perspective. Cancer Inform. 2015; 14(Suppl. 2):173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang Z., Michailidis G.. A non-negative matrix factorization method for detecting modules in heterogeneous omics multi-modal data. Bioinformatics. 2015; 32:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim D., Li R., Dudek S.M., Ritchie M.D.. Predicting censored survival data based on the interactions between meta-dimensional omics data in breast cancer. J. Biomed. Inform. 2015; 56:220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kanehisa M., Goto S.. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000; 28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schaefer C.F., Anthony K., Krupa S., Buchoff J., Day M., Hannay T., Buetow K.H.. PID: the pathway interaction database. Nucleic Acids Res. 2009; 37(Suppl. 1):D674–D679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peng X., Li Y., Walters K.-A., Rosenzweig E.R., Lederer S.L., Aicher L.D., Proll S., Katze M.G.. Computational identification of hepatitis C virus associated microRNA-mRNA regulatory modules in human livers. BMC Genomics. 2009; 10:373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Uhlmann S., Mannsperger H., Zhang J.D., Horvat E.-Á., Schmidt C., Küblbeck M., Henjes F., Ward A., Tschulena U., Zweig K. et al. . Global microRNA level regulation of EGFR-driven cell-cycle protein network in breast cancer. Mol. Syst. Biol. 2012; 8:570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al. . Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tomfohr J., Lu J., Kepler T.B.. Pathway level analysis of gene expression using singular value decomposition. BMC Bioinformatics. 2005; 6:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Braun R., Cope L., Parmigiani G.. Identifying differential correlation in gene/pathway combinations. BMC Bioinformatics. 2008; 9:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Braun R., Leibon G., Pauls S., Rockmore D.. Partition decoupling for multi-gene analysis of gene expression profiling data. BMC Bioinformatics. 2011; 12:497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramos-Rodriguez R.-R., Cuevas-Diaz-Duran R., Falciani F., Tamez-Peña J.-G., Trevino V.. COMPADRE: an R and web resource for pathway activity analysis by component decompositions. Bioinformatics. 2012; 28:2701–2702. [DOI] [PubMed] [Google Scholar]
- 33. Shi J., Luo Z.. Nonlinear dimensionality reduction of gene expression data for visualization and clustering analysis of cancer tissue samples. Comput. Biol. Med. 2010; 40:723–732. [DOI] [PubMed] [Google Scholar]
- 34. Kim M.H., Seo H.J., Joung J.-G., Kim J.H.. Comprehensive evaluation of matrix factorization methods for the analysis of DNA microarray gene expression data. BMC Bioinformatics. 2011; 12:S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tenenbaum J.B., De Silva V., Langford J.C.. A global geometric framework for nonlinear dimensionality reduction. Science. 2000; 290:2319–2323. [DOI] [PubMed] [Google Scholar]
- 36. Li Y., Liang M., Zhang Z.. Regression analysis of combined gene expression regulation in acute myeloid leukemia. PLoS Comput. Biol. 2014; 10:e1003908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Krasnov G.S., Dmitriev A.A., Melnikova N.V., Zaretsky A.R., Nasedkina T.V., Zasedatelev A.S., Senchenko V.N., Kudryavtseva A.V.. CrossHub: a tool for multi-way analysis of the Cancer Genome Atlas (TCGA) in the context of gene expression regulation mechanisms. Nucleic Acids Res. 2016; 44:e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Creighton C.J., Hernandez-Herrera A., Jacobsen A., Levine D.A., Mankoo P., Schultz N., Du Y., Zhang Y., Larsson E., Sheridan R. et al. . Integrated analyses of microRNAs demonstrate their widespread influence on gene expression in high-grade serous ovarian carcinoma. PLoS One. 2012; 7:e34546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Godard P., van Eyll J.. Pathway analysis from lists of microRNAs: common pitfalls and alternative strategy. Nucleic Acids Res. 2015; 43:3490–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wolf I., Sadetzki S., Catane R., Karasik A., Kaufman B.. Diabetes mellitus and breast cancer. Lancet Oncol. 2005; 6:103–111. [DOI] [PubMed] [Google Scholar]
- 41. Larsson S.C., Mantzoros C.S., Wolk A.. Diabetes mellitus and risk of breast cancer: a meta-analysis. Int. J. Cancer. 2007; 121:856–862. [DOI] [PubMed] [Google Scholar]
- 42. Cox T.F., Cox M.A.. Multidimensional Scaling, Second Edition. 2000; NY: CRC Press. [Google Scholar]
- 43. Sham P.C., Purcell S.M.. Statistical power and significance testing in large-scale genetic studies. Nat. Rev. Genet. 2014; 15:335–346. [DOI] [PubMed] [Google Scholar]
- 44. Liang H., Liu M., Yan X., Zhou Y., Wang W., Wang X., Fu Z., Wang N., Zhang S., Wang Y. et al. . miR-193a-3p functions as a tumor suppressor in lung cancer by down-regulating ERBB4. J. Biol. Chem. 2015; 290:926–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang P., Ji D.-B., Han H.-B., Shi Y.-F., Du C.-Z., Gu J.. Downregulation of miR-193a-5p correlates with lymph node metastasis and poor prognosis in colorectal cancer. World J. Gastroenterol. 2014; 20:12241–12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sherr C.J., McCormick F.. The RB and p53 pathways in cancer. Cancer Cell. 2002; 2:103–112. [DOI] [PubMed] [Google Scholar]
- 47. Lewis B.P., Shih I.-h., Jones-Rhoades M.W., Bartel D.P., Burge C.B.. Prediction of mammalian microRNA targets. Cell. 2003; 115:787–798. [DOI] [PubMed] [Google Scholar]
- 48. Vlachos I.S., Zagganas K., Paraskevopoulou M.D., Georgakilas G., Karagkouni D., Vergoulis T., Dalamagas T., Hatzigeorgiou A.G.. DIANA-miRPath v3.0: deciphering microRNA function with experimental support. Nucleic Acids Res. 2015; 43:460–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Garcia A.I., Buisson M., Bertrand P., Rimokh R., Rouleau E., Lopez B.S., Lidereau R., Mikaélian I., Mazoyer S.. Down-regulation of BRCA1 expression by miR-146a and miR-146b-5p in triple negative sporadic breast cancers. EMBO Mol. Med. 2011; 3:279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bhaumik D., Scott G., Schokrpur S., Patil C., Campisi J., Benz C.. Expression of microRNA-146 suppresses NF-κB activity with reduction of metastatic potential in breast cancer cells. Oncogene. 2008; 27:5643–5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xiang M., Birkbak N.J., Vafaizadeh V., Walker S.R., Yeh J.E., Liu S., Kroll Y., Boldin M., Taganov K., Groner B. et al. . STAT3 induction of miR-146b forms a feedback loop to inhibit the NF-κB to IL-6 signaling axis and STAT3-driven cancer phenotypes. Sci. Signal. 2013; 7:ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Valeri N., Braconi C., Gasparini P., Murgia C., Lampis A., Paulus-Hock V., Hart J.R., Ueno L., Grivennikov S.I., Lovat F. et al. . MicroRNA-135b promotes cancer progression by acting as a downstream effector of oncogenic pathways in colon cancer. Cancer Cell. 2014; 25:469–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Halappanavar S., Nikota J., Wu D., Williams A., Yauk C.L., Stampfli M.. IL-1 receptor regulates microRNA-135b expression in a negative feedback mechanism during cigarette smoke-induced inflammation. J. Immunol. 2013; 190:3679–3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Morgan E.T. Regulation of cytochrome p450 by inflammatory mediators: why and how. Drug Metab. Dispos. 2001; 29:207–212. [PubMed] [Google Scholar]
- 55. Shacter E., Weitzman S.A.. Chronic inflammation and cancer. Oncology. 2002; 16:217–226. [PubMed] [Google Scholar]
- 56. Mantovani A., Allavena P., Sica A., Balkwill F.. Cancer-related inflammation. Nature. 2008; 454:436–444. [DOI] [PubMed] [Google Scholar]
- 57. Colotta F., Allavena P., Sica A., Garlanda C., Mantovani A.. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009; 30:1073–1081. [DOI] [PubMed] [Google Scholar]
- 58. Balkwill F., Mantovani A.. Inflammation and cancer: back to Virchow. Lancet. 2001; 357:539–545. [DOI] [PubMed] [Google Scholar]
- 59. Catchpole G., Platzer A., Weikert C., Kempkensteffen C., Johannsen M., Krause H., Jung K., Miller K., Willmitzer L., Selbig J. et al. . Metabolic profiling reveals key metabolic features of renal cell carcinoma. J. Cell. Mol. Med. 2011; 15:109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Phang J.M., Liu W., Hancock C., Christian K.J.. The proline regulatory axis and cancer. Front. Oncol. 2012; 2:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Phang J.M., Liu W., Hancock C.N., Fischer J.W.. Proline metabolism and cancer: emerging links to glutamine and collagen. Curr. Opin. Clin. Nutr. Metab. Care. 2015; 18:71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Calin G.A., Dumitru C.D., Shimizu M., Bichi R., Zupo S., Noch E., Aldler H., Rattan S., Keating M., Rai K. et al. . Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:15524–15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li D., Zhao Y., Liu C., Chen X., Qi Y., Jiang Y., Zou C., Zhang X., Liu S., Wang X. et al. . Analysis of miR-195 and miR-497 expression, regulation and role in breast cancer. Clin. Cancer Res. 2011; 17:1722–1730. [DOI] [PubMed] [Google Scholar]
- 64. Deng H., Guo Y., Song H., Xiao B., Sun W., Liu Z., Yu X., Xia T., Cui L., Guo J.. MicroRNA-195 and microRNA-378 mediate tumor growth suppression by epigenetical regulation in gastric cancer. Gene. 2013; 518:351–359. [DOI] [PubMed] [Google Scholar]
- 65. Cai C., Chen Q.-B., Han Z.-D., Zhang Y.-Q., He H.-C., Chen J.-H., Chen Y.-R., Yang S.-B., Wu Y.-D., Zeng Y.-R. et al. . miR-195 inhibits tumor progression by targeting RPS6KB1 in human prostate cancer. Clin. Cancer Res. 2015; 21:4922–4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Guo J., Wang M., Liu X.. MicroRNA-195 suppresses tumor cell proliferation and metastasis by directly targeting BCOX1 in prostate carcinoma. J. Exp. Clin. Cancer Res. 2015; 34:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liu C., Guan H., Wang Y., Chen M., Xu B., Zhang L., Lu K., Tao T., Zhang X., Huang Y.. miR-195 inhibits EMT by targeting FGF2 in prostate cancer cells. PLoS One. 2015; 10:e0144073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bonci D., Coppola V., Musumeci M., Addario A., Giuffrida R., Memeo L., D’Urso L., Pagliuca A., Biffoni M., Labbaye C. et al. . The miR-15a–miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat. Med. 2008; 14:1271–1277. [DOI] [PubMed] [Google Scholar]
- 69. Bonci D., Coppola V., Patrizii M., Addario A., Cannistraci A., Francescangeli F., Pecci R., Muto G., Collura D., Bedini R. et al. . A microRNA code for prostate cancer metastasis. Oncogene. 2016; 35:1180–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Petrelli A., Perra A., Schernhuber K., Cargnelutti M., Salvi A., Migliore C., Ghiso E., Benetti A., Barlati S., Ledda-Columbano G. et al. . Sequential analysis of multistage hepatocarcinogenesis reveals that miR-100 and PLK1 dysregulation is an early event maintained along tumor progression. Oncogene. 2012; 31:4517–4526. [DOI] [PubMed] [Google Scholar]
- 71. Chen P., Zhao X., Ma L.. Downregulation of microRNA-100 correlates with tumor progression and poor prognosis in hepatocellular carcinoma. Mol. Cell. Biochem. 2013; 383:49–58. [DOI] [PubMed] [Google Scholar]
- 72. Dang Y., Luo D., Rong M., Chen G.. Underexpression of miR-34a in hepatocellular carcinoma and its contribution towards enhancement of proliferating inhibitory effects of agents targeting c-MET. PLoS One. 2013; 8:e61054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Li N., Fu H., Tie Y., Hu Z., Kong W., Wu Y., Zheng X.. miR-34a inhibits migration and invasion by down-regulation of c-Met expression in human hepatocellular carcinoma cells. Cancer Lett. 2009; 275:44–53. [DOI] [PubMed] [Google Scholar]
- 74. Ying Q., Liang L., Guo W., Zha R., Tian Q., Huang S., Yao J., Ding J., Bao M., Ge C. et al. . Hypoxia-inducible microRNA-210 augments the metastatic potential of tumor cells by targeting vacuole membrane protein 1 in hepatocellular carcinoma. Hepatology. 2011; 54:2064–2075. [DOI] [PubMed] [Google Scholar]
- 75. Xue W., Zender L., Miething C., Dickins R.A., Hernando E., Krizhanovsky V., Cordon-Cardo C., Lowe S.W.. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007; 445:656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bommer G.T., Gerin I., Feng Y., Kaczorowski A.J., Kuick R., Love R.E., Zhai Y., Giordano T.J., Qin Z.S., Moore B.B. et al. . p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol. 2007; 17:1298–1307. [DOI] [PubMed] [Google Scholar]
- 77. Chang T.-C., Wentzel E.A., Kent O.A., Ramachandran K., Mullendore M., Lee K.H., Feldmann G., Yamakuchi M., Ferlito M., Lowenstein C.J. et al. . Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell. 2007; 26:745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Feng Z., Zhang C., Wu R., Hu W.. Tumor suppressor p53 meets microRNAs. J. Mol. Cell Biol. 2011; 3:44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sun F., Fu H., Liu Q., Tie Y., Zhu J., Xing R., Sun Z., Zheng X.. Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett. 2008; 582:1564–1568. [DOI] [PubMed] [Google Scholar]
- 80. Mandke P., Wyatt N., Fraser J., Bates B., Berberich S.J., Markey M.P.. MicroRNA-34a modulates MDM4 expression via a target site in the open reading frame. PLoS One. 2012; 7:e42034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. He L., He X., Lim L.P., De Stanchina E., Xuan Z., Liang Y., Xue W., Zender L., Magnus J., Ridzon D. et al. . A microRNA component of the p53 tumour suppressor network. Nature. 2007; 447:1130–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bukeirat M., Sarkar S.N., Hu H., Quintana D.D., Simpkins J.W., Ren X.. MiR-34a regulates blood–brain barrier permeability and mitochondrial function by targeting cytochrome c. J Cereb. Blood Flow Metab. 2016; 36:387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yin H., Zhang S., Sun Y., Li S., Ning Y., Dong Y., Shang Y., Bai C.. MicroRNA-34/449 targets IGFBP-3 and attenuates airway remodeling by suppressing Nur77-mediated autophagy. Cell Death Dis. 2017; 8:e2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Francis S. M.S., Davidson M.R., Tan M.E., Wright C.M., Clarke B.E., Duhig E.E., Bowman R.V., Hayward N.K., Fong K.M., Yang I.A.. MicroRNA-34c is associated with emphysema severity and modulates SERPINE1 expression. BMC Genomics. 2014; 15:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Agostini M., Tucci P., Killick R., Candi E., Sayan B.S., di Val Cervo P.R., Nicotera P., McKeon F., Knight R.A., Mak T.W. et al. . Neuronal differentiation by TAp73 is mediated by microRNA-34a regulation of synaptic protein targets. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:21093–21098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gibbons D.L., Lin W., Creighton C.J., Rizvi Z.H., Gregory P.A., Goodall G.J., Thilaganathan N., Du L., Zhang Y., Pertsemlidis A. et al. . Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009; 23:2140–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zhang X., Li P., Rong M., He R., Hou X., Xie Y., Chen G.. MicroRNA-141 is a biomarker for progression of squamous cell carcinoma and adenocarcinoma of the lung: clinical analysis of 125 patients. Tohoku J. Exp. Med. 2015; 235:161–169. [DOI] [PubMed] [Google Scholar]
- 88. Mei Z., He Y., Feng J., Shi J., Du Y., Qian L., Huang Q., Jie Z.. MicroRNA-141 promotes the proliferation of non-small cell lung cancer cells by regulating expression of PHLPP1 and PHLPP2. FEBS Lett. 2014; 588:3055–3061. [DOI] [PubMed] [Google Scholar]
- 89. Eghbali B., Kessler J., Reid L., Roy C., Spray D.. Involvement of gap junctions in tumorigenesis: transfection of tumor cells with connexin 32 cDNA retards growth in vivo. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:10701–10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Leithe E., Sirnes S., Omori Y., Rivedal E.. Downregulation of gap junctions in cancer cells. Crit. Rev. Oncog. 2006; 12:225–256. [DOI] [PubMed] [Google Scholar]
- 91. Sehgal V., Seviour E.G., Moss T.J., Mills G.B., Azencott R., Ram P.T.. Robust selection algorithm (RSA) for multi-omic biomarker discovery; integration with functional network analysis to identify mirna regulated pathways in multiple cancers. PLoS One. 2015; 10:e0140072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Suzuki H.I., Mihira H., Watabe T., Sugimoto K., Miyazono K.. Widespread inference of weighted microRNA-mediated gene regulation in cancer transcriptome analysis. Nucleic Acids Res. 2013; 41:e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bleazard T., Lamb J.A., Griffiths-Jones S.. Bias in microRNA functional enrichment analysis. Bioinformatics. 2015; 31:1592–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.