


Abstract
The elongation of single-stranded DNA repeats at the 3′-ends of chromosomes by telomerase is a key process in maintaining genome integrity in eukaryotes. Abnormal activation of telomerase leads to uncontrolled cell division, whereas its down-regulation is attributed to ageing and several pathologies related to early cell death. Telomerase function is based on the dynamic interactions of its catalytic subunit (TERT) with nucleic acids—telomerase RNA, telomeric DNA and the DNA/RNA heteroduplex. Here, we present the crystallographic and NMR structures of the N-terminal (TEN) domain of TERT from the thermotolerant yeast Hansenula polymorpha and demonstrate the structural conservation of the core motif in evolutionarily divergent organisms. We identify the TEN residues that are involved in interactions with the telomerase RNA and in the recognition of the ‘fork’ at the distal end of the DNA product/RNA template heteroduplex. We propose that the TEN domain assists telomerase biological function and is involved in restricting the size of the heteroduplex during telomere repeat synthesis.
INTRODUCTION
Telomerase is a key player in maintaining chromosome integrity in most eukaryotes. It represents an RNA–protein complex, which extends the 3΄-end of telomeres via the reverse transcription of single-stranded telomeric repeats (1). The enzyme is active in single-cell eukaryotic organisms (yeast, ciliates), whereas in humans, it is expressed only in actively proliferating cells. The lack of telomerase activity in the majority of somatic cells makes the compensation for chromosome shortening after each cell division impossible, leading to cell senescence, and possible cell death (2). Telomerase is reactivated in the majority of cancers, enabling the immortal growth of cancer cells (3,4). Several mutations in telomerase and telomerase-associated proteins are associated with a variety of hereditary diseases (5).
Recent biochemical, genetic and structural studies of telomerase components and telomere/telomerase-associated proteins from different organisms contribute significantly to our understanding of telomeres and telomerase function (6–8). The minimal telomerase catalytic core that is active in vitro consists of telomerase reverse transcriptase (TERT) and telomerase RNA. Despite their low sequence identity among different species, most TERTs contain four structural domains, each with a distinct functional role (Figure 1): the essential N-terminal domain (TEN), the RNA-binding domain (TRBD), the reverse transcriptase domain (RT), and the C-terminal extension (CTE) (9). As yet, only a few structures of TERT domains from different organisms have been determined (10–15). A high-resolution structure of TERT from Tribolium castaneum that naturally lacks the TEN domain (10,13) contains only the conserved telomerase core. This is called the ‘TERT ring’ (Figure 1). Other known structures include the TEN domain from Tetrahymena thermophila (12), the TRBD domain from T. thermophila, Oryzias latipes (15), and Takifugu rubripes (11,14), and the C-terminal domain of human TERT (16). Limited structural data on the organization of the telomerase catalytic core restricts the understanding of telomerase function and the rational drug design of anticancer therapeutics (17).
Figure 1.

Domain structure of telomerase reverse transcriptase (TERT). The TEN (Telomerase Essential N-terminal domain), TRBD (Telomerase RNA-Binding Domain), RT (Reverse Transcriptase) and CTE (C-Terminal Extension) domains are shown as gray boxes. Conserved sequence motifs are shown schematically in dark gray. Two fragments (1–153 and 179–783) of TERT from H. polymorpha discussed in this paper are shadowed in light blue.
The TEN domain is essential for telomerase function, but its role may vary in different species. Several insects with TERT proteins lacking the TEN domain have very low telomerase activity (18). These species have a telomerase-independent mechanism for maintaining telomere length that decreases the demand for telomerase activity to achieve chromosome stability (19). This mechanism was first observed in Drosophila (20). Deletion of the TEN domain from the TERT of the ciliate T. thermophila (ttTERT) causes a complete loss of telomerase activity (21). Several single-residue substitutions in ttTEN affected the initiation and the elongation of nucleotide addition but not the repeat addition processivity (RAP)—an ability to add several telomeric repeats without the dissociation of the DNA product from the enzyme (22). Single molecule-binding studies confirmed that ttTEN binds telomeric DNA substrates and stabilizes short RNA–DNA duplexes in the active site of TERT, thus facilitating telomerase activity (23).
Human TERT (hTERT) does not require its TEN domain for the synthesis of a single repeat (24). However, the deletion of hTEN causes the loss of the RAP. In vivo reconstitution data show that hTEN participates in the RNA-dependent positioning of the DNA substrate and enables processive repeat synthesis by hTERT (24). Co-purification studies demonstrate that hTEN provides a strong contribution to the network of interactions between hTERT and hTR (24). Several mutant variants of hTEN have severe deficiency in enzymatic activity in vitro due to tighter binding and improper positioning of a primer in the active site, or impaired recruitment of dNTP (25,26). In addition, a new DAT region (residues 68–133) in hTEN is dispensable for the activity in vitro but essential for telomerase function in vivo, most probably by providing a stable association between hTERT and telomere-associated proteins (27). Multiple studies implicate that hTEN domain is crucial for the TPP1/POT1-mediated telomerase recruitment to telomeres that facilitates the processivity of telomerase by aiding the translocation step (26–30). Mutant variants of hTEN with replacements of residues 78, 100 and 132 in the DAT region display a drastic reduction of the TPP1/POT1-induced stimulation of the RAP in vitro, or a failure of telomerase to recruit telomeres in vivo, or both, so the DAT region of hTEN is believed to be the site of hTERT-TPP1 interaction (26,27,31).
Based on these observations, hTEN is likely involved in conformational rearrangements during the adaptation of the active site to substrate binding. Mutational analysis showed that the hTEN domain also facilitates the processivity of telomerase through specific interactions between the fragment containing the conserved residue Gly100 and the TPP1 protein.
The functional role of the TEN domain in yeast is less understood, although some interactions with telomeric DNA and the participation in the assembly of the intact TERT ribonucleoprotein were reported (32). The thermotolerant yeast Hansenula polymorpha (also known as Ogataea polymorpha) is an attractive source of macromolecules for structural studies. We previously identified the genes that encode two essential components of the yeast telomerase: TERT and telomerase RNA, which are necessary for telomere maintenance in vitro (33) and in vivo (34). Here, we report on the structures of the hpTERT TEN domain at an atomic level of detail that were obtained in the crystal state using X-ray diffraction and in solution using NMR spectroscopy. We describe the dynamics of the protein backbone and the interactions of hpTEN with the nucleic acid constituents of the telomerase complex. Based on the experimental binding data we also propose an hpTERT model where hpTEN may restrict the size of the heteroduplex during telomere repeat synthesis by forming a steric barrier, in a manner similar to RNA polymerase II and bacterial RNA polymerases.
MATERIALS AND METHODS
Expression of hpTEN protein in E. coli and purification
The wild-type and mutant hpTEN variants, including 15N and 13C-labeled proteins for NMR studies were expressed in E. coli and purified as previously reported (35). l-Selenomethionine-labeled hpTEN, containing a C-terminal His-tag, was expressed in E. coli BL21 (DE3) using SelenoMethionine Medium Base plus Nutrient Mix (Molecular Dimensions, UK) supplemented with L-selenomethionine (50 μg/ml) (SERVA Electrophoresis GmbH, Germany) and kanamycin (100 μg/ml). Cells were grown at 37°C under agitation (210 rpm) until OD600 reached 1.0 (total media volume 4 × 500 ml). The expression was induced with 1 mM IPTG and cells were further incubated at 27°C under agitation (210 rpm) for 16–18 h. Cells were collected by centrifugation at 13 000 g for 15 min at 4°C and stored at –80°C. A pellet obtained from 2 liters of culture was thawed and re-suspended in 35 ml of buffer I (50 mM Hepes, pH 7.0, 100 mM sodium chloride, 10% (v/v) glycerol, 5 mM β-mercaptoethanol and 1 mM PMSF). All purification steps were carried out on ice or at 4°C. Cells were disrupted by sonication (2 × 2 min, 40% amplitude, 3 s on, 7 s off, on ice) and centrifuged at 40 000 g for 30 min at 4°C. The resulting supernatant containing His-tagged l-selenomethionine-labeled protein was filtrated through 0.45 μm filter and purified using affinity chromatography on Ni-NTA agarose (Qiagen, Germany). Ni-NTA agarose (5 ml) was equilibrated with buffer I followed by incubation with the supernatant for 45 min under constant rotation. Contaminants were removed from Ni-NTA agarose by spin-flow chromatography (5 × 6 ml of 20 mM imidazole in buffer I at 4000 g for 5 min at 4°C). The protein was eluted from Ni-NTA agarose by spin-flow chromatography (4 × 2 ml of 320 mM imidazole in buffer I at 4000 rpm for 5 min at 4°C). The His-tag was removed using TEV protease (protease-to-protein ratio 1:100) during dialysis in buffer II (50 mM Hepes pH 7.0, 100 mM sodium chloride, 5 mM β-mercaptoethanol, ∼100:1 v/v) for 16–18 h. Untagged protein was then purified from the His-tagged TEV protease and the hydrolyzed His-tag on Ni-NTA agarose. Ni-NTA agarose (3 ml) was equilibrated with buffer II followed by incubation with the dialyzate for 45 min under constant rotation. The target protein was collected from Ni-NTA agarose by spin-flow chromatography (6 ml of buffer II at 4000 rpm for 5 min at 4°C) and concentrated by ultra-filtration to 2.5 ml (Amicon-Ultra centrifugal filter units, Merck Millipore, Germany). The protein was then further purified by size exclusion chromatography using HiLoad 16/60 Superdex 75 column (GE Healthcare, UK), equilibrated with buffer II. Resulting fractions containing untagged l-selenomethionine-labeled hpTEN were pooled and concentrated to 9 mg/ml by ultra-filtration (Amicon-Ultra centrifugal filter units, Merck Millipore, Germany). Purity of protein fractions was controlled using SDS-PAGE with staining by Instant Blue (Expedeon, UK). Protein concentration was determined using NanoDrop ND-1000 (Thermo Fisher Scientific, USA) at 280 nm (extinction coefficient 29910, Abs 0.1% = 1.61 mg/ml). The protein sample was stored at 4°C for 16–18 h before crystallization. Details of cloning, expression and purification of hpTEN mutant variants are presented in supplementary data.
Construction of truncated variants of hpTEN domain for expression in Hansenula polymorpha
The DNA fragment containing the hpTERT gene, putative promoter and terminator regions was amplified by PCR (primers hpTERTf 5′-aacccgggtactatccagtggtcaaccaaaa-3′ and hpTERTr 5′-aactcgagattctgattggcaaccaggc-3′) and cloned into the SmaI/XhoI sites of the pKAM556 shuttle vector (36). To generate the shuttle vector with the truncated hpTERT gene (coding 179–782 fragment of hpTERT) under control of the hpTERT promoter (pKAM556TERTΔN) the DNA region coding 2–178 amino acids in hpTERT was removed by PCR and self-ligation (primers hpTERTΔNf 5′-atgtctctaatttatgacgttttcaaaacgg-3′ and hpTERTΔNr 5′-aaaaaattcttttcacaaaggatgagctatgatata-3′). To generate the shuttle vector with hpTEN domain under control of the hpTERT promoter (pKAM556TEN) the DNA region coding 179–782 amino acids in hpTERT was removed by PCR and self-ligation (primers hpTERTNf 5′-gaccagcattaaaagtctcgtttctaatc-3′ and hpTERTNr 5′-tgatgacctgcattgcatcttg-3′). The SmaI/XhoI fragment from the vector was cloned into the pKAM556TERTΔN to generate the vector with the N-truncated hpTERT gene and TEN domain gene, each under control of the hpTERT promoter (pKAM556TEN/TERTΔN). Transformation was performed as previously explained (37). Construction of the ΔhpTERT strain (ATCC 26012 leu2 hpTERT::opLEU2) was described earlier (34).
RNA and DNA synthesis
DNA and RNA oligonucleotides were assembled in an MM-12 synthesizer (Bioautomation) with the phosphoramidite method, according to the manufacturer's recommendations at 25 μmol scale. Synthetic procedure is described in detail in supplementary data.
Crystallization of hpTEN proteins
The crystals of native hpTEN and Se-Met variant were grown using vapour diffusion at room temperature. A solution of 9 mg/ml hpTEN in 50 mM bicine, pH 8.25, containing 0.02 mg/ml trypsin was incubated for 30 min prior to crystallization. Precipitating solution contained 1.4 M sodium–potassium–phosphate, 0.15 M sodium citrate and 0.1 M CAPS, pH 10.5. The hanging-drop with a volume of 4 μl contained protein and the precipitant in the 1:1 ratio. The droplets were equilibrated against 0.5 ml reservoir volume. After a few days crystals grew with typical dimensions of 200 × 70 × 70 μm. The crystals were cryoprotected in the precipitating solution additionally containing 25% glycerol (v/v).
X-ray data collection and processing
The SAD data set was collected from a Se-Met hpTEN crystal to 2.2 Å resolution in 7200 images at the EMBL/PETRA III beamline P13 (DESY, Hamburg, Germany) (38) at the radiation wavelength of 0.97858 Å, using 0.1° oscillation, 40 ms exposure, 5% transmission and 50 μm beam size. The crystal belonged to the space group C2221 with unit cell parameters a = 73.3 Å, b = 120.5 Å, c = 123.0 Å. There were three methionines in the 159 residues-long sequences. Significant anomalous signal was present up to 2.6 Å and data to this resolution were used for structure solution. The asymmetric unit contained four hpTEN molecules with 40% solvent content; this was supported by the presence of four pseudo-translation vectors—(1/2, 0, 0), (0, 1/2, 0), (1/4, 1/4, 1/2), (1/4, 3/4, 1/2). The native data were collected at the wavelength of 1.70550 Å to the resolution of 2.25 Å at the same beamline with 10% transmission, but otherwise using the same parameters as for the Se-Met data. The native data belonged to the space group I212121 with unit cell parameters a = 38.2 Å, b = 64.1 Å, c = 126.1 Å, and displayed no pseudo symmetry. The data were indexed using XDS, (39) integrated with XDS (native data) or Dials 1.3.3 (40) (Se-Met data) and merged using the Aimless pipeline (41) from the CCP4 software suite version 7.1 (42).
X-ray structure solution and refinement
In the Se-Met data the anomalous CC1/2 value (43) decreased from 0.9 to 0.3 in the resolution range from 13 to 3 Å. The structure was solved using the Crank2 software pipeline (44). Within the pipeline, SHELXD (45) unambiguously identified 12 Se-sites with the CC and CC-weak values of 0.60 and 0.35, respectively. The correct handedness was determined with MOPRO (46), Solomon (47), Multicomb (48) and REFMAC (49) software. In spite of the low initial value of the overall figure-of-merit of 0.34, after the density modification using Parrot (50), 480 residues (out of 628 expected) were built automatically in 14 fragments using Buccaneer (51) and refined with REFMAC to an R-factor of 0.33. The 14 fragments were combined manually into four chains using Coot (52) and these were used for space group and origin validation (53).
The Se-Met data were in a different space group compared to the native. Therefore a partial model obtained from the SAD solution was used for molecular replacement (54) into the native data limited to 2.4 Å, with Rmeas 0.431 and CC1/2 0.25, 0.99 and 0.87 along a*, b* and c* axes, respectively, in the outer resolution shell, within 2.5 and 2.4 Å. Coot was used for manual corrections. The native model consists of 1017 protein and six solvent atoms treated as one TLS group with an average isotropic equivalent of atomic displacement parameters of 67 Å2. The model was refined using REFMAC to the final R-factor of 0.186 (Rfree 0.224) at 2.4 Å resolution.
Data collection and refinement statistics are summarized in Tables 1 and 2. Overall the electron density maps were of somewhat lower quality than typically expected for 2.4 Å resolution structures, with several blobs and continuous non-interpretable patches of density, possibly owing to disordered regions including short polypeptides produced during crystallization under proteolytic conditions. In particular, the density in the region between His29 and Arg33 could not be interpreted in terms of atomic model but has features suggesting the presence of chain fragments cleaved in this region and overlapping with themselves and, possibly, also with the components of crystallization solution. Similarly, a weak but continuous electron density beyond Thr82 and Asn142 could not be reliably interpreted.
Table 1. X-ray data collection and processing. The numbers for the outer resolution shell are given in parentheses.
| X-ray data set | Native | Se-Met |
|---|---|---|
| Beamline | EMBL/DESY P13 | EMBL/DESY P13 |
| Wavelength (Å) | 1.70550 | 0.97858 |
| Space group | I212121 | C2221 |
| Cell parameters (Å) | a = 38.20 | a = 73.31 |
| b = 64.10 | b = 120.49 | |
| c = 126.06 | c = 122.97 | |
| Monomers in the asymmetric unit | 1 | 4 |
| Wilson plot B-factor (Å2) | 54.6 | 44.0 |
| Resolution range of the data used (Å) | 63.0–2.4 (2.46–2.40) | 63.0–2.6 (2.72–2.60) |
| Number of unique reflections | 6202 (682) | 17 114 (2061) |
| Completeness (%) | 98.0 (98.9) | 99.8 (99.7) |
| Data redundancy | 5.0 (4.7) | 19.3 (20.1) |
| I / σ(I) | 20.6 (4.5) | 14.3 (4.8) |
| CC1/2 | 0.998 (0.971) | 0.999 (0.993) |
| along a* | 1.000 (0.241) | 1.000 (0.864) |
| along b* | 1.000 (0.869) | 1.000 (0.872) |
| along c* | 0.998 (0.997) | 0.999 (0.997) |
| anomalous | 0.782 (0.110) | |
| R meas (%) | 0.053 (0.43) | 0.12 (0.60) |
| R pim (all I+ & I-) | 0.023 (0.194) | 0.028 (0.133) |
Table 2. Refinement of the crystallographic model using the native X-ray data.
| R work | 0.186 |
| R free | 0.224 |
| Fraction of reflections set aside for Rfree monitoring | 0.050 |
| No. of atoms | 1023 |
| Average main chain ADP (Å2) | 64.6 |
| Average side chain ADP (Å2) | 69.9 |
| RMSD from target stereochemistry (target values are given in parentheses) | |
| Bonds (Å): | 0.015 (0.019) |
| Angles (°): | 1.69 (1.96) |
NMR spectroscopy
The NMR samples in concentration of 0.5 mM for 13C,15N-labeled hpTEN and 0.8 mM for 15N-labeled protein were prepared in 90% H2O/10% D2O, 50 mM NaCl, and 20 mM sodium phosphate buffer (pH 7.0). DTT in concentration of 1 mM was added to the final solution to prevent oxidation of free cysteine residue C150. Triple-resonance (1H,13C,15N) spectra were acquired at 303 K on a Bruker Avance III HD 700 MHz spectrometer equipped with a quadruple resonance (1H,13C,15N,31P) CryoProbe. 15N-1H HSQC spectra on 15N-labeled hpTEN in NMR titration experiments by DNA/RNA oligonucleotides were acquired at 303 K on a Bruker Avance 600 MHz spectrometer equipped with a triple resonance (1H,13C,15N) TXI probe. Spectra were processed by NMRPipe (55), and analyzed using NMRFAM-SPARKY (56).
NMR structure determination
Family of NMR structures for the TEN-domain was calculated using conformational restraints for the inter-proton distances and backbone dihedral angles. A set of 199 dihedral angles (Table 3) for the residues located in the well-ordered regions of the protein core, as defined by NMR relaxation experiments and the RCI (Random Coil Index) approach (57), were obtained from the analysis of the 1HN, 1Hα, 15N, 13Cα, 13Cβ and 13C’ chemical shifts using the TALOS+ software (58). 1577 NOEs, used as distance restraints in structure calculation, were obtained from the analysis of 3D 13C–1H and 15N–1H HSQC–NOESY spectra. Inter-proton distance restraints were extracted by the evaluation of the cross-peak intensities in 1H–15N NOESY–HSQC and 1H–13C NOESY–HSQC spectra. NOESY spectra were assigned in a semiautomatic manner: the intra-residue and sequential cross-peaks were assigned manually. The rest of the cross-peaks were assigned using the automatic iterative procedure of spectra assignment / structure calculation implemented in ARIA 2.3 software (59). The automatic assignment and the inter-proton distances provided at the last iteration of the ARIA 2.3 protocol were further manually verified by multiple steps of the structure refinement accomplished using the simulated annealing protocol of the CNS 1.21 software package (60). Structure refinement included a high-temperature torsion-angle molecular dynamics stage followed by a slow-cooling torsion-angle phase, a second slow-cooling phase in Cartesian space and Powell energy minimization. Database values of conformational torsion angle pseudopotentials (61) were implemented during the final cycles of the calculations to improve the quality of protein backbone conformation. Structure refinement was performed until no NOE violations >0.2 Å and no dihedral angle violations higher than 5° occurred. The restraint violations and structure quality were assessed using the CNS tools, Procheck-NMR (62) and in-house software and utilities. At the last iteration of the refinement protocol 100 structures have been calculated using 1577 unambiguous distance and 199 dihedral angle restraints (Table 3). The final family of 20 NMR structures was filtered out in accordance with the lowest-energy criterion. Statistics for the obtained NMR structures is presented in Table 3. Structure visualization and analysis were carried out using PyMOL (Schrödinger LLC).
Table 3. Statistic of the calculated family of NMR structures of the hpTEN.
| A. Restraints used in the structure calculation | ||
| Total NOEs | 1577 | |
| Long range (|i-j| > 4) | 273 | |
| Medium (1< |i-j| ≤ 4) | 191 | |
| Sequential (|i-j| = 1) | 342 | |
| Intraresidue | 771 | |
| Total dihedral angles | 199 | |
| Phi (ϕ) | 98 | |
| Psi (ψ) | 101 | |
| B. Restraint violations and structural statistics (for 25 structures) | ||
| No NOE or dihedral angle violations are above 0.2Å and 10° respectively. | ||
| Average RMSD | <S>a | S rep |
| From experimental restraints | ||
| Distance (Å) | 0.043 ± 0.001 | 0.040 |
| Dihedral (°) | 0.115 ± 0.022 | 0.123 |
| From idealized covalent geometry | ||
| Bonds (Å) | 0.0011 ± 0.0004 | 0.0011 |
| Angles (°) | 0.3065 ± 0.0035 | 0.3090 |
| Impropers (°) | 0.1739 ± 0.0093 | 0.1810 |
| Ramachandran plot statistics | ||
| % of residues in most favorable region of Ramachandran plot | 91.1 | 91.0 |
| % of residues in disallowed region of Ramachandran plot | 0.0 | 0.0 |
| C. Superimposition on the representative structure (A) | ||
| Backbone (C, Cα, N) RMSD of the residues 1–138 | 4.65 ± 1.27 | |
| Backbone (C, Cα, N) RMSD of the protein without flexible loop residues 71–99 | 0.79 ± 0.12 | |
| All heavy-atom RMSD of the protein without flexible loop residues 71–99 | 1.62 ± 0.20 | |
a<S> is the ensemble of 20 final structures; Srep is the representative structure, selected from the final family on the criteria of having the lowest sum of pairwise RMSD for the remaining structures in the family.
NMR relaxation analysis
The 15N relaxation data for protein backbone amide resonances were analyzed using the ‘model-free’ approach of Lipari and Szabo (63). The values of longitudinal relaxation rate constant (R1) were deduced from the collected data as a pseudo-3D experiment with the relaxation delays of 0.1, 0.2, 0.25, 0.3, 0.35, 0.4, 0.45, 0.5, 0.58, 0.64, 0.8, 1.0, 1.3, 1.7, 2.2 and 3.0 s, The values of the transverse relaxation rate constant (R2) were derived from data with relaxation delays of 0, 16.9, 33.9, 50.9, 67.8, 84.8, 101.8, 118.7, 135.7, 152.7, 169.6, 186.6, 203.5, 237.5, 271.4 and 305.3 ms. A 3 s 1H saturation was applied as a relaxation delay for NOE enhancement in the heteronuclear NOE experiment. Values of R1 and R2 with their standard deviations were obtained from non-linear fitting of the integrated peak volumes measured using the nlinLS procedure from the NMRPipe package (55). Estimated standard deviations of the 15N NOE values were calculated using the RMSD noise of the background regions (64) and were further validated and corrected using two independently collected experimental data sets. Experimental values of the relaxation parameters were interpreted using the model-free approach (63), with extensions to include slower internal motions (65) and chemical exchange contributions Rex to the transverse relaxation rate constants (66) under the assumptions of both isotropic and axially symmetric anisotropic rotation using the program RelaxFit (67). The overall correlation time was calculated from the R2/R1 ratios (64) yielding an average value of 10.9 ± 0.7 ns at 303K. The ratio of the principal moments of the axially symmetric diffusion tensor (D║/D┴) was 2.5 ± 0.1. Uncertainties in the calculated parameters S2, Rex and internal motion correlation times were obtained from 1000 cycles of Monte Carlo simulations (68).
NMR titration experiments
Interaction of hpTEN with RNA and/or DNA fragments was studied using NMR titration. 15N–1H HSQC spectra were recorded on the samples of 15N-labeled TEN domain with the increasing content of RNA and/or DNA fragments. The protein concentration in all experiments was between 0.15 and 0.25 mM. The pH of all studied nucleic acid samples was adjusted to be identical to the pH of TEN domain (7.0) by an addition of small aliquots of 10 mM solutions of NaOH or HCl. Aliquots of the samples containing the necessary amount of nucleic acid were preserved by freeze drying. This procedure fixes the protein concentration after dissolution of each consecutive nucleic acid sample in protein solution. DNA-RNA heteroduplexes were prepared using the following annealing protocol: DNA solutions were incubated at 90°C for 5 min, cooled to 70°C, mixed at this temperature with an equimolar amount of RNA component, kept at 70°C for 3 min and then cooled to room temperature. Annealing of the RNA hairpin was carried out at 70°C and under low concentration of RNA (50 μM) to avoid concatemerization. RiboLock RNase inhibitor (Thermo Scientific) in concentration of 700 U/ml was added to the solution containing single strain RNA in order to inhibit RNA cleavage. The 15N–1H HSQC spectrum was recorded for each sample, resonances were assigned and changes of their chemical shifts were analyzed. Values of Kd in case of fast exchange equilibrium between free and complexed protein (Table 4) were obtained by a non-linear fitting of the changes of 1H and 15N chemical shifts in the representative residues (D4, S55, I116, E119).
Table 4. DNA and RNA fragments studied in NMR titration experiments with the hpTEN and their corresponding values of Kd.
| # | Type | RNA and/or DNA fragment | K d, mM |
|---|---|---|---|
| E1 | ssRNA 1 | RNA 5′CGCCACCC3′ | 5.0 ± 2.5 |
| E2 | ssDNA 2 | DNA 5′GTGGCGGGGTGGCG3′ | > 10 |
| E3 | RNA–DNA heteroduplex 1 | DNA 3′GCGGTGGG5′ | 1.4 ± 0.3 |
| RNA 5′CGCCACCC3′ | |||
| E4 | RNA–DNA heteroduplex 2 | DNA 3′GCGGTGGGGCGGTG5′ | 0.9 ± 0.2 |
| RNA 5′CGCCACCCCGCCAC3′ | 1.3 ± 0.3a | ||
| 6.0 ± 0.7b | |||
| E5 | ssRNA 2 | RNA 5′CGCCACCCCGCCAC3′ | 5.0 ± 3.0 |
| E6 | ssDNA 2 | DNA 5′GTGGCGGGGTGGCG3′ | n/a |
| E7 | RNA–DNA fork with inversed polarity | DNA 3′ACTACAGGCGGTG5′ | >10 |
| RNA 5′AUUCAACCGCCAC3′ | |||
| E8 | RNA–DNA fork with native polarity | DNA 3′CGGTGGGCTTTGTC5′ | <0.1 |
| RNA 5′GCCACCCUUCGUCA3′ | |||
| E9 | RNA–DNA half-fork | DNA 3′CGGTGGG5′ | 4.0 ± 2.0 |
| RNA 5′GCCACCCUUCGUCA3′ | |||
| E10 | ssRNA upstream | RNA 5′UUCGUCA3′ | 4.5 ± 2.5 |
| E11 | RNA hairpin |
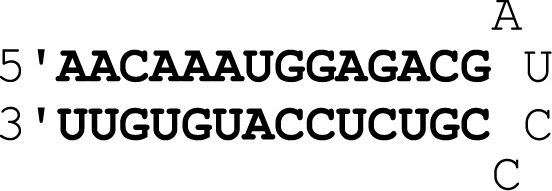
|
1.9 ± 0.4 |
aMeasured by Microscale Thermophoresis (MST) for double mutant N53A/S55A hpTEN.
bMeasured by MST for triple N53A/S55A/R110A and 4-residue N53A/S55A/R110A/E119A mutants of hpTEN.
Microscale thermophoresis measurements, sequence and structure alignment and molecular modeling of hpTERT are described in detail in supplementary data.
RESULTS
Requirement of TEN for in vivo telomerase activity in H. polymorpha
It has been previously shown that the knockout strains of H. polymorpha lacking the genes of TERT (ΔTERT) or telomerase RNA (ΔTER) have the Ever Shorter Telomere (EST) phenotype, which leads to the very rapid onset of senescence (34). Transformation of the ΔTERT strain with a shuttle vector carrying the functional hpTERT gene yields viable yeast colonies (Supplementary Figures S2A, SB). To study the role of hpTEN in H. polymorpha, we expressed a fragment 179–783 of hpTERT lacking TEN under the control of the native hpTERT promoter (Supplementary Figure S2). No colony formation was observed after the introduction of the vector with the TEN-deleted hpTERT into the ΔTERT strain (Supplementary Figure S2C), thus demonstrating the necessity of the N-terminal domain for hpTERT activity and telomerase function in vivo. Even simultaneous separate co-expression of two fragments hpTEN1-153 and hpTERT179-783 in ΔTERT strain of H. polymorpha does not complement the non-viable EST phenotype (Supplementary Figure S2D). This indicates that the integrity of TERT is a key factor for telomerase function and supports the hypothesis of involvement of TEN in the assembly of TERT ribonucleoprotein (69).
Structure of hpTEN determined by X-ray and NMR
The native and Se-Met-labeled forms of hpTEN were crystallized under proteolytic conditions, and the obtained crystals were suitable for X-ray structure determination. The native model was solved using the Se-Met variant and has been refined to an R-factor of 0.186 (Rfree 0.224) at a resolution of 2.4 Å (Tables 1 and 2). The model contains three chain fragments: Met1-His29, Arg33-Thr82 and Arg97-Asn142 (Figure 2A).
Figure 2.

Structure of hpTEN. (A) X-ray structure of hpTEN, rainbow-colored from the N- to the C-terminus (missing protein fragments are represented as dashes); (B) NMR structure of hpTEN (a representative conformer); (C) NMR structure of hpTEN (a stereo view of a family of 20 conformers).
The NMR signal assignments of the hpTEN domain have been reported previously (35). The chemical shift assignments were obtained for 89% of the hpTEN residues. Most of the unassigned residues belong to the fragments Asn70–Leu76 and Lys95–Ala99 and are clustered in the central part of the protein; residues Lys95 and Gly96 are also disordered in the crystal structure. The obtained ensemble of 20 NMR conformers of hpTEN (Figure 2B, Table 3) exhibits a well-structured αβ-core (residues 1–70, 100–138), a flexible central loop (residues 71–99) and an unstructured C-terminus (residues 139–159). The 30–32 and 83–96 regions are present only in the NMR model (α6-helix), and the 71–82 region is better ordered in the crystal structure (α5-helix). The C-terminal residues 143–159 are missing in the crystal structure and are also disordered in the NMR model. The position of a mobile helix, α3, is discernible in crystal and solution structures, with an RMSD of 1.8 Å for its N, C, O and Cα atoms. Helix α4 is longer in the crystal structure, where it precedes the 70–80 coil that is packed along the structured protein core. The residues from the extended part of the helix α4 are unassigned in the NMR spectra due to intense conformational movements. By contrast, helix α2 is longer in the NMR structure since its fragment Gly30–Gly32 is disordered in the crystalline state. The core of the structure contains eight α-helices and three β-strands (Figure 2B). Helix α5 is present in the crystallographic model only, while helix α6 is present only in the solution structure. The spatial position of helix α6 relative to the protein core fluctuates in the NMR model (Figure 2C) but the local structure of this helix is rigid (Supplementary Figure S3). The existence of helix α6 is unambiguously confirmed by the characteristic medium-range NOEs (Supplementary Figure S3A) and the helix-specific values of chemical shifts for 13Cα, 13C', 13Cβ, 1Hα and 15N nuclei (35). The structures of the TEN-domain that were determined independently in the crystal state and in solution are very similar (Figure 2A, B). The protein core, formed by six α-helices (α1, α2, α4, α7, α8 and α9) and three β-strands, has the same structural organization in both structures. The RMSD between the coordinates of the backbone atoms of the residues of the protein core in the X-ray crystallographic and representative NMR structures is 0.80 Å (Supplementary Figure S4). The main differences between the NMR and crystallographic structures are in the flexible parts of the protein, presumably reflecting a dynamic nature of the protein.
The comparison of the structures of hpTEN and ttTEN
The ciliate T. thermophila telomerase (ttTEN, PDB code 2B2A) has been identified as possessing a new protein fold named the TERT essential N-terminal (TEN) domain (12). Despite their low sequence similarity, the structure of hpTEN determined in this study and ttTEN share a common structural core that contains an α/β structure with the central α-helical motif which includes α-helices α7, α8 and α9 (Figure 3). In both structures, helices α8 and α9 are bent at an acute angle (in this paper we will refer to them as a V-motif), with the organism-invariant Gly117 forming the sharpest turn. This V-motif is nearly identical in ttTEN and hpTEN in terms of the main-chain conformations of the involved residues (107–127 in hpTEN, 133–155 in ttTEN). The V-motif sequence contains a highly conserved pattern: hxxhxxxhGxxxhxxhh (where h is a large hydrophobic residue, G is the invariant Gly and x is any residue) (Figure 4). This pattern belongs to the so-called ‘motif T2’ (the most conserved region of the TEN domain), which also contains another conserved block of five residues. These two blocks are separated by a 15–25 residues-long fragment (70). The V-motif is sandwiched between the helix α4 with a long loop on one side and two α-helices (α1, α2) with a β-sheet with three antiparallel β-strands (β1, β2, β3) on the other side (Figure 4B–D).
Figure 3.

The comparison of the structures of hpTEN and ttTEN. (A) The structure of hpTEN (red elements are superimposed for hpTEN and ttTEN (α7–α8–Gly–α9); yellow elements partially correspond, white fragments distinctly differ in two structures or are missing in one of them). (B) Crystal structure of ttTEN (PDB code 2B2A) in the same color scheme. (C) Electrostatic surface potential of the hpTEN. (D) Electrostatic surface potential of the ttTEN. The two views of each protein are related by a 180° rotation about the y-axis. Protein orientation in left view is identical to that shown at panels A or B. Labeled are C-terminal tails.
Figure 4.

Structure-based sequence alignment of the TEN domains. (A) Structure-based sequence alignment of hpTEN (combined NMR and X-ray, residues invisible in X-ray structures are shown in lowercase) with ttTEN (PDB id 2B2A) and hTEN (predicted). Two conserved blocks of sequence motif T2 are shadowed in green, the central loop region is shadowed in orange and conserved residues are shown in bold. Residue numbering corresponds to hpTEN. Numbering of the secondary structure elements correspond to hpTEN and ttTEN. (B–D) Ribbon representation of the structure of hpTEN (B), the predicted structure of hTEN (C) and the structure of ttTEN (D). Structural core elements corresponding to the hpTEN motif α7–α8–Gly–α9 are shown in dark cyan; fragments corresponding to the central loop 71–99 in hpTEN are in orange.
The detailed comparison of the hpTEN and ttTEN structures shows significant differences. Specifically hpTEN lacks 33 residues at the N-terminus and has no equivalent to the α1 and β1 elements that are present in ttTEN (the numbering of helices and strands follows Figure 3B). We note that the lengths of the N-terminal parts of the TEN domains from various species are indeed highly variable, and some of them lack the N-terminal α-helix analogous to that in ttTEN. The loops connecting β2 to α4 in ttTEN and β1 to α4 in hpTEN differ significantly in their lengths and conformations. In addition, the loop in the double turn helix α3 is present in hpTEN but absent in ttTEN. The connection from α4 to the V-motif (Figures 3B and 4B–D) in ttTEN contains a β-hairpin (Thr113-Tyr121) formed by the strands β3 and β4 (Figure 4D). The following chain fragment, up to residue Arg128 at the beginning of helix α7, is missing in the structure of ttTEN (12). The corresponding loop in the structure of hpTEN is longer and contains helix α6 right in the middle (Figure 4B).
Structural alignment of hpTEN and ttTEN fragments of different lengths was conducted using Gesamt (71). This revealed three structurally similar fragments with RMSD on Cα atoms of less than 1.0 Å. The first fragment matches the V-motif (heliсes α6, α7 and α8, hpTEN numbering), while the other two adjoin to the two sides of the V-motif and include α2, β1, β2 and β3 (the second fragment) and a single helix α4 (the third fragment). Their localization and pronounced structural conservation suggests that these three fragments represent the cores of three distinct structural units. The fragments superimpose one-by-one but not all simultaneously. The apparent displacement of these units relative to each other may be of biological significance and also indicates a requirement to have different interaction partners. Indeed, based on the mutagenesis studies and sequence alignments, the previously introduced DAT domain (27) in case of hpTEN, would comprise α4 helix and long linkers connecting α4 to the conserved secondary structural elements β1 and α7 (Figure 2B).
Dynamics of hpTEN backbone
The dynamic properties of the protein backbone at both the ps-ns and ms timescales were examined by analyzing the 15N relaxation data (Supplementary Figure S5). The residues from the unstructured C-terminal region (141–159) have the smallest order parameters (S2), thus indicating the large amplitudes of their motion at the nanosecond timescale (Supplementary Figure S6). The fast motions of the central loop, including the residues from helix α6, are also large in amplitude. The residues from the central flexible loop 71–99 that are close to the junction regions between the loop and the protein core, exhibit motions at the slower ms timescale. Interestingly, most of the unassigned residues belong to these regions. We attribute the motion of these regions to conformational exchange occurring at the ms timescale. Several other protein regions also exhibit conformational rearrangements at the ms timescale. Among these regions are helices α3 and α8, strand β3, and the loops between α2 and β1 (Supplementary Figure S6).
Interaction of TEN with nucleic acid ligands
According to the current understanding of telomerase function, an ‘anchor site’ is needed to handle the 5′-region of single-stranded DNA (about two telomeric repeats) to prevent product release from the active complex (72). Cross-linking data suggest that the anchor site is located either at the TEN domain or at the linker between TEN and TRBD (12,73,74). Several surface-exposed residues have been implicated in DNA binding based on the ttTEN structure from T. thermophila (12). The popular hypothesis that the TEN domain functions as an anchor site for DNA is not supported by the experimental data since the isolated TEN domain shows only weak, if any, affinity to single-stranded DNA fragments (75–78). By contrast, the TERT ring without TEN interacts with single-stranded DNA, although TEN further promotes this binding (77,79). An alternative model proposed that the TEN domain contributes to TERT function by stabilizing the product-template hybrid duplex in the active site rather than by binding the single-stranded DNA in the anchor site (23,79).
In this study, the interaction of hpTEN with RNA and/or DNA fragments mimicking the environment of TERT in the telomerase complex has been investigated using heteronuclear NMR titration experiments. 15N-labeled hpTEN was titrated with an increasing concentration of RNA and/or DNA fragments (Table 4) until the nucleic acids reached a 5–10-fold molar excess over the protein. In this case, the binding of the nucleic acids to 15N-hpTEN can be detected based on changes in the 1H and 15N chemical shifts of the residues that are located near the interaction interface. A single-stranded RNA mimicking TER fragments, as well as several variants of an RNA template/DNA product hybrid duplex, strongly affected the NMR signals of several hpTEN residues, thus indicating specific interactions between these nucleic acids and the protein (Figure 5, Supplementary Figures S7–S16).
Figure 5.

Interaction of hpTEN with RNA and/or DNA fragments. (A) Histograms of 1H and 15N chemical shift changes and the structural location of the TEN domain residues that are most affected by interactions with nucleic acid fragments (NMR titration, Table 4). 15N-labeled TEN was titrated by ssRNA (E1); heteroduplex RNA–DNA (E3); heteroduplex RNA–DNA (E4); RNA upstream fragment (E10); consecutive titration of TEN by ssRNA (E5, black bars) followed by ssDNA (E6, blue bars); half-fork RNA–DNA (E9); RNA hairpin (E11); (h) RNA–DNA fork with inverted orientation (E7); RNA–DNA fork with direct (native) orientation (E8). Blue bars represent the interacting residues of cluster I, red bars represent cluster II. (B and C) Structure of the TEN domain. Residues that interact with the heteroduplex E3 (B, cluster I) or the native fork E8 (С, cluster II) are colored according to the chemical shift perturbation (yellow – no interaction, blue or red – maximum change). Residues that are not observed in 1H–15N HSQC spectra are colored white.
Single-stranded DNA oligonucleotides induced less pronounced changes in the chemical shifts (Supplementary Figure S8a). Mapping the residues affected by oligonucleotide binding onto the structure of hpTEN indicates that they fall into two distinct clusters. The first interface, cluster I (Figure 5B, Supplementary Figures S9-S16), involves residues located in helices α8 and α9 of the structural core (His113–Glu119), which, together with residues from α4 and the loop Asn53–His56, form a groove on the surface of hpTEN (Figure 5B). The second binding interface, cluster II, binds nucleic acids with higher affinity than the first interface. Thus, in the case of binding to cluster II slow exchange is observed between free protein and its complex (Supplementary Figures S16–S18), whereas in the case of cluster I fast exchange equilibrium occurs. Cluster II is built of residues located in α1, α3 and the β-sheet (R2–K11, G30–R39, G79, Ser129–Tyr136) (Figure 5C; Supplementary Figures S16 and S18). This cluster specifically recognizes the oligonucleotide ‘fork’ that is formed at the junction where a heteroduplex splits into single-stranded DNA and RNA (Table 4, E8). It is noteworthy that according to our NMR studies, most of the residues in cluster II do not respond to the binding of RNA or DNA alone, an RNA/DNA duplex, or the fork with reversed polarity (Table 4, E7) where the duplex and single-stranded fragments of the RNA and DNA split in the opposite direction (Figure 5A, Supplementary Figure S15). Interestingly, the residues from clusters I and II either belong to or are immediately adjacent to the cores of two separate structural units, which are highly structurally conserved among hpTEN and ttTEN, and contain conserved sequence patterns shared by TEN domains from a variety of species as discussed below.
In order to further detail the role of the two interfaces in the RNA/DNA binding we produced several hpTEN mutant variants targeting the residues potentially involved in the interaction with nucleic acids. Only surface-exposed residues from cluster I (N53, S55, R110, E119) and cluster II (W36, R131, M132, N134) were chosen for the mutant studies. Our attempts to replace the residues from cluster II failed at the stage of protein expression. This structural element could therefore be deemed important for proper protein folding. The residues in Cluster I were successfully replaced with alanine and the hpTEN variants with double (N53A/S55A), triple (N53A/S55A/R110A) and 4-residue (N53A/S55A/R110A/E119A) mutations were produced. CD spectra of these proteins confirmed that these mutations did not affect the overall structure (Supplementary Figure S19).
Microscale Thermophoresis was used to test the ability of the wild-type hpTEN and produced mutants to interact with the oligonucleotide ligands. This method is based on the use of a covalently linked fluorescent label to one of the binding partners and allows to study molecular interactions with Kd in the range from nM to mM (80). FAM label was linked to the 5′-end of DNA chains of the ligands E4 and E8 (Table 4). Two heteroduplex-containing ligands (heteroduplex E4 and the fork with native polarity E8) were studied, as well as the corresponding ssDNAs. For the interaction with wild-type protein the best affinity was found for the fork E8 (Kd of 0.4±0.2 mM), while heteroduplex E4 and ssDNA E2 were bound with Kd of 0.9±0.2 and 1.0 ± 0.2 mM, respectively. Similar values of Kd were obtained using the fluorescently labeled hpTEN titrated with non-labeled oligonucleotides, so the tighter binding of the fork cannot be attributed to the effects of FAM label. Titration of FAM-labeled oligonucleotides with mutant hpTENs showed that all of them retained the binding ability. Dissociation constant of double mutant was similar to the wild-type hpTEN, whereas triple and 4-residue mutants showed the impaired affinity for the heteroduplex E4 (Supplementary Figure S20A), while the binding of a fork E8 was not affected (Supplementary Figure S20B). These results confirm that the V-motif forms part of the binding interface, and the residues, identified in NMR titration experiments, are directly involved in oligonucleotide binding. At the same time, the binding interface is rather large, and only simultaneous replacements of several residues affect the binding. One can also expect that TEN-oligonucleotide interaction in the assembled telomerase complex would be much stronger than in the described model titration experiments.
The fact that the TEN domain is able to bind the fork between the duplex and the single-stranded parts of the product-template complex indicates a possible involvement of the TEN domain in the restriction of the length of the duplex formed by TERT during telomeric repeat synthesis. Understanding of the detailed structural basis for this specific recognition requires additional studies. Our results support the hypothesis that the TEN domain primarily binds to the RNA strand of a duplex with only a few, if any, contacts to the DNA strand. Some parts of the telomerase RNA may interact with the first binding interface in the TEN domain.
DISCUSSION
Structural conservation of the TEN-core motif
The presented results provide an insight into the shared structural features between TEN domains of TERTs from evolutionary divergent organisms, ciliates (ttTEN) and yeast (hpTEN). Despite the low overall sequence identity, the structural core of hpTEN is topologically similar in these two proteins with the V-motif being the most conserved fragment both in sequence and conformation. Our data demonstrate that the residues in the V-motif are directly involved in binding nucleic acid ligands (cluster I). High sequence similarity of the corresponding region in a wide variety of species implies the structural conservation of this core motif, and supports the notion of functional conservation of TEN domains throughout evolution.
The most noticeable difference between hpTEN and ttTEN in the region close to where the V-motif appears is also functionally relevant. This region, a linker between a well-conserved helix α4 and the V-motif, forms in hpTEN a long loop with the helix α6, while in ttTEN there is a shorter loop with the β-hairpin. From the alignment of the sequences of TEN domains from various species (Supplementary Figure S21), and based on the length of the linker and the presence of the conserved pattern, we conclude that the TEN domains can be classified into three separate groups. The TEN domains from yeast (including hpTEN), animals, and algae belong to a long-linker group (33–39 residues) and contains the conserved pattern NhhххGh (where h is hydrophobic residue, N is a conserved Asn, G is a conserved Gly, and х is any residue). The TEN domains from plants comprise a short-linker group (21 residues) containing most of the residues from the conserved region. The TEN domains from ciliates (including ttTEN) form the third variable-linker group, 22–35 residues without any recognizable common pattern. We therefore propose that the conformations of the linkers in the TEN domains from the long-linker group (including human) are relevant to hpTEN (Figure 4B,C), whereas the β-hairpin-containing TEN domains of ttTEN and other organisms from the variable-linker group are more distant (Figure 4D).
The conformation of the linker should be essential for the function. The linker in hpTEN corresponds to the segment 89–127 of human TEN that comprises most of the DAT region and contains a number of residues crucial for the interaction with TPP1 and the TPP1-mediated recruitment of hTEN to telomeres (26–28,31,81). The yeast homolog of mammalian TPP1, Est3p, is part of a telomerase complex and its direct interaction with the TEN domain of yeast TERT was demonstrated experimentally (82). In ciliates, the TEN-related protein interaction hub is more complex compared to yeast and vertebrates, and this can be the reason for the alternative conformation of the linker in ttTEN compared to other TENs. The structural model of telomerase complex in Tetrahymena thermophila implies the interaction between TEN and three other proteins of the telomerase complex (8,83). ttTEN interacts with the C-terminal OB-fold domain of TEB1 (via β-hairpin-containing linker), N-terminal domain of p50 (via residues R137 and K90 and residues 122–127 disordered in the crystal structure of ttTEN), and probably with TEB2 (via the loop 77–87, also disordered in the crystal structure). p50 is the functional ortholog of TPP1 and Est3p (84), though it has no sequence homology to these proteins and the residues of p50 participating in the interaction with TEN were not yet identified. β-hairpin 112–120 of ttTEN inserts at the interface between p50, Teb1 and Teb2, and therefore plays an important role in controlling the integrity of this interaction hub (83).
Proposed role of the TEN domain in telomerase function
The results obtained here provide important new understanding of the roles of the TEN domain in telomerase function. Our experimental data demonstrate that the hpTEN domain binds to the product-template DNA/RNA duplex at the bifurcation point and to some regions of TER outside the DNA/RNA duplex. Based on this result, we propose a hypothetical model of the full-length hpTERT complex with the product-template hybrid duplex bound in the central pore (Figure 6B). The hpTEN structure is based on the representative NMR conformer reported in this study. The main parts of the models of TERT (the ‘TERT ring’) and the product-template duplex bound in the pore were based on the crystal structure of TERT from T. castaneum (PDB code 3KYL (13)) with sequences of the TER template and telomeric DNA replaced with those of H. polymorpha. The position of hpTEN with respect to the TERT ring is modeled considering the NMR results obtained in this study.
Figure 6.

The proposed role of the TEN domain in the telomerase function. (A) Cartoon of the minimal telomerase complex. (B) Combined 3D hypothetical model of hpTERT (the TERT ring is colored according its TRBD, RT and CTE domains, TEN domain is colored as described in Figure 4C) complexed with the RNA–DNA fork (telomerase RNA in green, telomeric DNA in blue). The view on the left shows TEN domain of hpTERT in the orientation similar to that used in Figures 2A and 3A. The second view, on the right, is related by a 90° rotation about the axis shown.
We propose that the TEN domain is located at the distal (with respect to the active site) end of the central pore of TERT. A similar location was proposed earlier for the model of full-length hTERT where both the N- and C-terminal elements of the TEN domain were placed in the groove of a heteroduplex (85). However, in our model the orientation of the TEN domain differs considerably. We denote the chain fragments immediately before β1 and the loop between β2 and β3 as the determinants for the specific recognition of the DNA–RNA fork (Figure 5C). Therefore, the TEN domain is modeled in the complex, so these fragments are pointing towards the central pore and the fork. In this orientation, the N-terminus of the TEN domain is close enough to interact with the telomerase RNA. The C-terminal tail is disordered in both the crystallographic and NMR structures; however, it can be positioned close to the telomeric DNA to allow their interaction (74).
Due to the second template-like repeat in the TER sequence, the DNA product-TER hybrid duplex could be up to 16 base pairs in length. However, telomerases limit the length of the DNA/RNA heteroduplexes to 5–6 (86) or 7–8 base pairs (87,88) in different organisms. The stepwise unwinding of the product-template hybrid is supposed to coincide with its translocation after every cycle of nucleotide addition. This timing prevents interactions from being too tight and facilitates complete strand separation when the RNA template has to relocate back to the active site (86).
It was proposed that TEN domains could be involved in constraining the length of the product-template heteroduplex (79), but the mechanism of length restriction remains unclear. We want to emphasize a certain functional similarity of TERT to RNA polymerase II (RNApol II). RNApol II also forms a complex with a hybrid duplex (RNA product-DNA template) and keeps its length limited to 8–9 base pairs (89). Protein loops located at the end of the duplex-binding cleft (Supplementary Figure S22) assist in strand separation by (i) providing a steric barrier for the growing duplex and (ii) interacting with the unpaired nucleotides, thus compensating for the loss of enthalpy due to base unpairing (89). In particular, the ‘lid’ element (residues 250–258 in yeast RNApol II, Supplementary Figure S22) is wedged between the DNA and RNA strands and therefore physically restricts the length of the hybrid duplex. In bacterial RNA polymerases, the analogous protein loop of the β' subunit (residues 528–537 in Thermus thermophilus RpoC) not only sterically blocks the growth of the duplex but also directly interacts with its last base pair (90,91). As shown in Supplementary Figure S22, the TERT ring without the TEN domain has no protein fragments outside the duplex-binding pore that are suitably positioned to perform the same role as the ‘lid’ element. However, if the TEN domain is present (Supplementary Figure S22), it is tethered to the TERT ring by a long and flexible linker and can be modeled in a position similar to that of the protein loops of the RNA polymerases involved in strand separation. In the model of hpTERT presented here, chain fragment 131–139 of hpTEN is located directly between the DNA and RNA chains, similar to the position of the ‘lid’ element in RNA polymerases. This fragment contains a conserved sequence pattern: hQhxG (where h is a hydrophobic residue, Q is a conserved Gln, G is an invariant Gly, and x is any residue). This 5-residue block is highly conserved in TEN domains. There are rare exceptions including hpTEN where glutamine is replaced with tyrosine and is included in the ‘motif T2’. This pattern may be related to the ‘lid’ sequence in bacterial RNA polymerases (e.g. VQVDG in RpoC from T. thermophilus), in which the conserved hydrophobic residues stabilize bases in the duplex and upstream RNA (91). This sequence similarity enriches the structural and functional parallels between the TEN domains and RNA polymerases.
Another important issue is the interaction of the TEN domain with telomeric DNA. Based on the proposed model (Figure 6B), the single-stranded DNA, up to the 5′-end of a hybrid duplex, may be sandwiched between the TEN domain and the C-terminal domain of the TERT ring (CTE). There is a hydrophobic patch at the surface of the CTE that is formed by the residues Ile431, Leu434, Ile504, Leu548, and Ile553 that may be involved in the stabilization of the unpaired DNA bases. This hypothesis is consistent with the recently proposed model for the product–template duplex interaction and ssDNA retention by hTERT (92). The C-terminal part of the TEN domain and the disordered linker that is rich in serine, glutamine and lysine residues may assist this interaction with the sugar-phosphate backbone. This scheme is in agreement with previously published results (79).
The presented structural comparison together with the analysis of multiple structure-based sequence alignments, allowed us to define evolutionarily conserved structural elements that may be important for TEN function. These results support the hypothesis that the N-terminal domains of TERT from evolutionarily divergent organisms contain a highly structurally conserved V-motif. The conformation of the central domain region (i.e. residues 71–99, forming flexible linkers, and helix α6, in the case of hpTEN) may vary significantly and thus define the specific features of telomerase function and regulation in different organisms. Based on the local structural differences between the analyzed structures, we consider hpTEN to be an excellent template for modeling the human TEN domain. We propose that the TEN domain is responsible for the restriction of the product-template hybrid length, thus facilitating the strand separation required for RNA translocation and processivity in the addition of telomeric repeats.
ACCESSION NUMBERS
The structural data and experimental restraints used in solution structure calculations have been deposited in the Protein Data Bank under accession number 5LGF. Experimental structure factor amplitudes and the atomic coordinates of the derived model for the reported crystal structure have been deposited in the Protein Data Bank under accession code 5NPT.
Supplementary Material
ACKNOWLEDGEMENTS
Authors are grateful to the Moscow State University (Russia) for the opportunity to use the NMR facilities and the supercomputer SKIF Lomonosov. Authors acknowledge the use of NMR facilities acquired with the support from the Russian Government Program of Competitive Growth of Kazan Federal University. Authors are grateful to Oleg Saveliev and Sofia Mariasina for the expert technical assistance in NMR measurements and Alexander Arutyunyan for technical assistance in CD measurements. The authors also acknowledge the access to the EMBL crystallographic beamline P13 at the PETRA III storage ring at DESY, Hamburg, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Russian Foundation for Basic Research [RFBR-EMBL 15-54-74005 and 17-04-01692_a]; NMR studies were supported by the Russian Science Foundation [14-14-00598]; German Ministry for Science and Education (BMBF) [05K12YE2 and 05K14YEA]. Funding for open access charge: Russian Science Foundation; Russian Foundation for Basic Research.
Conflict of interest statement. None declared.
REFERENCES
- 1. Greider C.W., Blackburn E.H.. Identification of a specific telomere terminal transferase activity in tetrahymena extracts. Cell. 1985; 43:405–413. [DOI] [PubMed] [Google Scholar]
- 2. Schmidt J.C., Cech T.R.. Human telomerase: biogenesis, trafficking, recruitment, and activation. Gene Dev. 2015; 29:1095–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim N.W., Piatyszek M.A., Prowse K.R., Harley C.B., West M.D., Ho P.L., Coviello G.M., Wright W.E., Weinrich S.L., Shay J.W.. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994; 266:2011–2015. [DOI] [PubMed] [Google Scholar]
- 4. Shay J.W. Role of telomeres and telomerase in aging and cancer. Cancer Discov. 2016; 6:584–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Armanios M., Blackburn E.H.. The telomere syndromes. Nat. Rev. Genet. 2012; 13:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Armstrong C.A., Tomita K.. Fundamental mechanisms of telomerase action in yeasts and mammals: understanding telomeres and telomerase in cancer cells. Open Biol. 2017; 7:160338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miracco E.J., Jiang J., Cash D.D., Feigon J.. Progress in structural studies of telomerase. Curr. Opin. Struct. Biol. 2014; 24:115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jiang J., Miracco E.J., Hong K., Eckert B., Chan H., Cash D.D., Min B., Zhou Z.H., Collins K., Feigon J.. The architecture of Tetrahymena telomerase holoenzyme. Nature. 2013; 496:187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wyatt H.D.M., West S.C., Beattie T.L.. InTERTpreting telomerase structure and function. Nucleic Acids Res. 2010; 38:5609–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gillis A.J., Schuller A.P., Skordalakes E.. Structure of the Tribolium castaneum telomerase catalytic subunit TERT. Nature. 2008; 455:633–637. [DOI] [PubMed] [Google Scholar]
- 11. Harkisheimer M., Mason M., Shuvaeva E., Skordalakes E.. A motif in the vertebrate telomerase N-terminal linker of TERT contributes to RNA binding and telomerase activity and processivity. Structure. 2013; 21:1870–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jacobs S.A., Podell E.R., Cech T.R.. Crystal structure of the essential N-terminal domain of telomerase reverse transcriptase. Nat. Struct. Mol. Biol. 2006; 13:218–225. [DOI] [PubMed] [Google Scholar]
- 13. Mitchell M., Gillis A., Futahashi M., Fujiwara H., Skordalakes E.. Structural basis for telomerase catalytic subunit TERT binding to RNA template and telomeric DNA. Nat. Struct. Mol. Biol. 2010; 17:513–518. [DOI] [PubMed] [Google Scholar]
- 14. Rouda S., Skordalakes E.. Structure of the RNA-binding domain of telomerase: implications for RNA recognition and binding. Structure. 2007; 15:1403–1412. [DOI] [PubMed] [Google Scholar]
- 15. Huang J., Brown A.F., Wu J., Xue J., Bley C.J., Rand D.P., Wu L., Zhang R., Chen J.J.L., Lei M.. Structural basis for protein-RNA recognition in telomerase. Nat. Struct. Mol. Biol. 2014; 21:507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hoffman H., Rice C., Skordalakes E.. Structural analysis reveals the deleterious effects of telomerase mutations in bone marrow failure syndromes. J. Biol. Chem. 2017; 292:4593–4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chan H., Wang Y., Feigon J.. Progress in human and Tetrahymena telomerase structure. Annu. Rev. Biophys. 2017; 46:199–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sasaki T., Fujiwara H.. Detection and distribution patterns of telomerase activity in insects. Eur. J. Biochem. 2000; 267:3025–3031. [DOI] [PubMed] [Google Scholar]
- 19. Fujiwara H. Louis EJ, Becker MM. Subtelomeres. 2014; Berlin: Springer, Berlin, Heidelberg; 227–241. [Google Scholar]
- 20. Pardue M.-L., DeBaryshe P.G.. Drosophila telomeres: a variation on the telomerase theme. Fly. 2008; 2:101–110. [DOI] [PubMed] [Google Scholar]
- 21. Jacobs S.A., Podell E.R., Wuttke D.S., Cech T.R.. Soluble domains of telomerase reverse transcriptase identified by high-throughput screening. Protein Sci. 2005; 14:2051–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eckert B., Collins K.. Roles of Telomerase Reverse Transcriptase N-terminal Domain in Assembly and Activity of Tetrahymena Telomerase Holoenzyme. J. Biol. Chem. 2012; 287:12805–12814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Akiyama B.M., Parks J.W., Stone M.D.. The telomerase essential N-terminal domain promotes DNA synthesis by stabilizing short RNA–DNA hybrids. Nucleic Acids Res. 2015; 43:5537–5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Robart A.R., Collins K.. Human telomerase domain interactions capture DNA for TEN domain-dependent processive elongation. Mol. Cell. 2011; 42:308–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jurczyluk J., Nouwens A.S., Holien J.K., Adams T.E., Lovrecz G.O., Parker M.W., Cohen S.B., Bryan T.M.. Direct involvement of the TEN domain at the active site of human telomerase. Nucleic Acids Res. 2011; 39:1774–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stern J.L., Zyner K.G., Pickett H.A., Cohen S.B., Bryan T.M.. Telomerase recruitment requires both TCAB1 and Cajal bodies independently. Mol. Cell. Biol. 2012; 32:2384–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Armbruster B.N., Banik S.S.R., Guo C., Smith A.C., Counter C.M.. N-Terminal domains of the human telomerase catalytic subunit required for enzyme activity in vivo. Mol. Cell. Biol. 2001; 21:7775–7786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zaug A.J., Podell E.R., Nandakumar J., Cech T.R.. Functional interaction between telomere protein TPP1 and telomerase. Gene Dev. 2010; 24:613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang F., Podell E.R., Zaug A.J., Yang Y.T., Baciu P., Cech T.R., Lei M.. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature. 2007; 445:506–510. [DOI] [PubMed] [Google Scholar]
- 30. Latrick C.M., Cech T.R.. POT1–TPP1 enhances telomerase processivity by slowing primer dissociation and aiding translocation. EMBO J. 2010; 29:924–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhong F.L., Batista L.F.Z., Freund A., Pech M.F., Venteicher A.S., Artandi S.E.. TPP1 OB-fold domain controls telomere maintenance by recruiting telomerase to chromosome ends. Cell. 2012; 150:481–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Malyavko A.N., Parfenova Y.Y., Zvereva M.I., Dontsova O.A.. Telomere length regulation in budding yeasts. FEBS Lett. 2014; 588:2530–2536. [DOI] [PubMed] [Google Scholar]
- 33. Smekalova E.M., Petrova O.A., Zvereva M.I., Dontsova O.A.. Hansenula Polymorpha TERT: a telomerase catalytic subunit isolated in recombinant form with limited reverse transcriptase activity. Acta naturae. 2012; 4:70–73. [PMC free article] [PubMed] [Google Scholar]
- 34. Smekalova E.M., Malyavko A.N., Zvereva M.I., Mardanov A.V., Ravin N.V., Skryabin K.G., Westhof E., Dontsova O.A.. Specific features of telomerase RNA from Hansenula polymorpha. RNA. 2013; 19:1563–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Polshakov V.I., Petrova O.A., Parfenova Y.Y., Efimov S.V., Klochkov V.V., Zvereva M.I., Dontsova O.A.. NMR assignments of the N-terminal domain of Ogataea polymorpha telomerase reverse transcriptase. Biomol. NMR Assign. 2016; 10:183–187. [DOI] [PubMed] [Google Scholar]
- 36. Agaphonov M., Romanova N., Choi E.S., Ter-Avanesyan M.. A novel kanamycin/G418 resistance marker for direct selection of transformants in Escherichia coli and different yeast species. Yeast. 2010; 27:189–195. [DOI] [PubMed] [Google Scholar]
- 37. Bogdanova A.I., Agaphonov M.O., Teravanesyan M.D.. Plasmid reorganization during integrative transformation in Hansenula-Polymorpha. Yeast. 1995; 11:343–353. [DOI] [PubMed] [Google Scholar]
- 38. Cianci M., Bourenkov G., Pompidor G., Karpics I., Kallio J., Bento I., Roessle M., Cipriani F., Fiedler S., Schneider T.R.. P13, the EMBL macromolecular crystallography beamline at the low-emittance PETRA III ring for high- and low-energy phasing with variable beam focusing. J. Synchrotron. Radiat. 2017; 24:323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kabsch W. XDS. Acta Crystallogr. D. 2010; 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Waterman D.G., Winter G., Parkhurst J.M., Fuentes-Montero L., Hattne J., Brewster A., Sauter N.K., Evans G., Rosenstrom P.. The DIALS framework for integration software. CCP4 Newslett. Protein Crystallogr. 2013; 49:13–15. [Google Scholar]
- 41. Evans P.R., Murshudov G.N.. How good are my data and what is the resolution. Acta Crystallogr. D. 2013; 69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G.W., McCoy A. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D. 2011; 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Karplus P.A., Diederichs K.. Linking crystallographic model and data quality. Science. 2012; 336:1030–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Skubak P., Pannu N.S.. Automatic protein structure solution from weak X-ray data. Nat. Commun. 2013; 4:2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sheldrick G.M. Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D. 2010; 66:479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guillot B., Viry L., Guillot R., Lecomte C., Jelsch C.. Refinement of proteins at subatomic resolution with MOPRO. J. Appl. Crystallogr. 2001; 34:214–223. [Google Scholar]
- 47. Abrahams J.P., Leslie A.G.W.. Methods used in the structure determination of bovine mitochondrial F-1 ATPase. Acta Crystallogr. D. 1996; 52:30–42. [DOI] [PubMed] [Google Scholar]
- 48. Skubak P., Waterreus W.-J., Pannu N.S.. Multivariate phase combination improves automated crystallographic model building. Acta Crystallogr. D. 2010; 66:783–788. [DOI] [PubMed] [Google Scholar]
- 49. Murshudov G.N., Skubak P., Lebedev A.A., Pannu N.S., Steiner R.A., Nicholls R.A., Winn M.D., Long F., Vagin A.A.. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D. 2011; 67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cowtan K. Recent developments in classical density modification. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D. 2006; 62:1002–1011. [DOI] [PubMed] [Google Scholar]
- 52. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lebedev A.A., Isupov M.N.. Space-group and origin ambiguity in macromolecular structures with pseudo-symmetry and its treatment with the program Zanuda. Acta Crystallogr. D. 2014; 70:2430–2443. [DOI] [PubMed] [Google Scholar]
- 54. Vagin A., Teplyakov A.. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 1997; 30:1022–1025. [Google Scholar]
- 55. Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A.. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995; 6:277–293. [DOI] [PubMed] [Google Scholar]
- 56. Lee W., Tonelli M., Markley J.L.. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics. 2015; 31:1325–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Berjanskii M.V., Wishart D.S.. A Simple Method To Predict Protein Flexibility Using Secondary Chemical Shifts. J. Am. Chem. Soc. 2005; 127:14970–14971. [DOI] [PubMed] [Google Scholar]
- 58. Shen Y., Delaglio F., Cornilescu G., Bax A.. TALOS plus: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR. 2009; 44:213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bardiaux B., Malliavin T., Nilges M.. ARIA for solution and solid-state NMR. Methods Mol. Biol. 2012; 831:453–483. [DOI] [PubMed] [Google Scholar]
- 60. Brunger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S. et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998; 54:905–921. [DOI] [PubMed] [Google Scholar]
- 61. Kuszewski J., Gronenborn A.M., Clore G.M.. Improvements and extensions in the conformational database potential for the refinement of NMR and X-ray structures of proteins and nucleic acids. J. Magn. Reson. 1997; 125:171–177. [DOI] [PubMed] [Google Scholar]
- 62. Laskowski R.A., Rullmannn J.A., MacArthur M.W., Kaptein R., Thornton J.M.. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996; 8:477–486. [DOI] [PubMed] [Google Scholar]
- 63. Lipari G., Szabo A.. Model-Free Approach To The Interpretation Of Nuclear Magnetic-Resonance Relaxation In Macromolecules. 2. Analysis Of Experimental Results. J. Am. Chem. Soc. 1982; 104:4559–4570. [Google Scholar]
- 64. Farrow N.A., Muhandiram R., Singer A.U., Pascal S.M., Kay C.M., Gish G., Shoelson S.E., Pawson T., Forman-Kay J.D., Kay L.E.. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994; 33:5984–6003. [DOI] [PubMed] [Google Scholar]
- 65. Clore G.M., Szabo A., Bax A., Kay L.E., Driscoll P.C., Gronenborn A.M.. Deviations from the simple 2-parameter model-free approach to the interpretation of 15N nuclear magnetic relaxation of proteins. J. Am. Chem. Soc. 1990; 112:4989–4991. [Google Scholar]
- 66. Clore G.M., Driscoll P.C., Wingfield P.T., Gronenborn A.M.. Analysis of the backbone dynamics of interleukin-1 beta using two-dimensional inverse detected heteronuclear 15N-1H NMR spectroscopy. Biochemistry. 1990; 29:7387–7401. [DOI] [PubMed] [Google Scholar]
- 67. Polshakov V.I., Birdsall B., Frenkiel T.A., Gargaro A.R., Feeney J.. Structure and dynamics in solution of the complex of Lactobacillus casei dihydrofolate reductase with the new lipophilic antifolate drug trimetrexate. Protein Sci. 1999; 8:467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tillett M.L., Blackledge M.J., Derrick J.P., Lian L.Y., Norwood T.J.. Overall rotational diffusion and internal mobility in domain II of protein G from Streptococcus determined from N-15 relaxation data. Protein Sci. 2000; 9:1210–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bosoy D., Peng Y., Mian I.S., Lue N.F.. Conserved N-terminal motifs of telomerase reverse transcriptase required for ribonucleoprotein assembly in vivo. J. Biol. Chem. 2003; 278:3882–3890. [DOI] [PubMed] [Google Scholar]
- 70. Lai C.K., Mitchell J.R., Collins K.. RNA Binding Domain of Telomerase Reverse Transcriptase. Mol. Cell Biol. 2001; 21:990–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Krissinel E. Enhanced fold recognition using efficient short fragment clustering. J. Mol. Biochem. 2012; 1:76–85. [PMC free article] [PubMed] [Google Scholar]
- 72. Wallweber G., Gryaznov S., Pongracz K., Pruzan R.. Interaction of human telomerase with its primer substrate. Biochemistry. 2003; 42:589–600. [DOI] [PubMed] [Google Scholar]
- 73. Lue N.F. A physical and functional constituent of telomerase anchor site. J. Biol. Chem. 2005; 280:26586–26591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Romi E., Baran N., Gantman M., Shmoish M., Min B., Collins K., Manor H.. High-resolution physical and functional mapping of the template adjacent DNA binding site in catalytically active telomerase. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:8791–8796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bairley R.C.B., Guillaume G., Vega L.R., Friedman K.L.. A mutation in the catalytic subunit of yeast telomerase alters primer-template alignment while promoting processivity and protein-DNA binding. J. Cell Sci. 2011; 124:4241–4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. O’Connor C.M., Lai C.K., Collins K.. Two purified domains of telomerase reverse transcriptase reconstitute sequence-specific interactions with RNA. J. Biol. Chem. 2005; 280:17533–17539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wyatt H.D.M., Lobb D.A., Beattie T.L.. Characterization of physical and functional anchor site interactions in human telomerase. Mol. Cell Biol. 2007; 27:3226–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Xia J.Q., Peng Y., Mian I.S., Lue N.F.. Identification of functionally important domains in the N-terminal region of telomerase reverse transcriptase. Mol. Cell Biol. 2000; 20:5196–5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wu R.A., Collins K.. Human telomerase specialization for repeat synthesis by unique handling of primer-template duplex. EMBO J. 2014; 33:921–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wienken C.J., Baaske P., Rothbauer U., Braun D., Duhr S.. Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 2010; 1:100. [DOI] [PubMed] [Google Scholar]
- 81. Sealey D.C.F., Zheng L., Taboski M.A.S., Cruickshank J., Ikura M., Harrington L.A.. The N-terminus of hTERT contains a DNA-binding domain and is required for telomerase activity and cellular immortalization. Nucleic Acids Res. 2010; 38:2019–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yen W.F., Chico L., Lei M., Lue N.F.. Telomerase regulatory subunit Est3 in two Candida species physically interacts with the TEN domain of TERT and telomeric DNA. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:20370–20375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jiang J., Chan H., Cash D.D., Miracco E.J., Ogorzalek Loo R.R., Upton H.E., Cascio D., O’Brien Johnson R., Collins K., Loo J.A. et al. Structure of Tetrahymena telomerase reveals previously unknown subunits, functions, and interactions. Science. 2015; 350:aab4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hong K., Upton H., Miracco E.J., Jiang J., Zhou Z.H., Feigon J., Collins K.. Tetrahymena Telomerase Holoenzyme Assembly, Activation, and Inhibition by Domains of the p50 Central Hub. Mol. Cell Biol. 2013; 33:3962–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Steczkiewicz K., Zimmermann M.T., Kurcinski M., Lewis B.A., Dobbs D., Kloczkowski A., Jernigan R.L., Kolinski A., Ginalski K.. Human telomerase model shows the role of the TEN domain in advancing the double helix for the next polymerization step. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:9443–9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Qi X.D., Xie M.Y., Brown A.F., Bley C.J., Podlevsky J.D., Chen J.J.L.. RNA/DNA hybrid binding affinity determines telomerase template-translocation efficiency. EMBO J. 2012; 31:150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Forstemann K., Lingner J.. Telomerase limits the extent of base pairing between template RNA and telomeric DNA. EMBO Rep. 2005; 6:361–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hammond P.W., Cech T.R.. Euplotes telomerase: Evidence for limited base-pairing during primer elongation and dGTP as an effector of translocation. Biochemistry. 1998; 37:5162–5172. [DOI] [PubMed] [Google Scholar]
- 89. Westover K.D., Bushnell D.A., Kornberg R.D.. Structural basis of transcription: Separation of RNA from DNA by RNA polymerase II. Science. 2004; 303:1014–1016. [DOI] [PubMed] [Google Scholar]
- 90. Vassylyev D.G., Vassylyeva M.N., Perederina A., Tahirov T.H., Artsimovitch I.. Structural basis for transcription elongation by bacterial RNA polymerase. Nature. 2007; 448:157–162. [DOI] [PubMed] [Google Scholar]
- 91. Kent T., Kashkina E., Anikin M., Temiakov D.. Maintenance of RNA–DNA Hybrid Length in Bacterial RNA Polymerases. J. Biol. Chem. 2009; 284:13497–13504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wu R.A., Tam J., Collins K.. DNA-binding determinants and cellular thresholds for human telomerase repeat addition processivity. EMBO J. 2017; 36:1908–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.