

Abstract
Myhre syndrome is a rare autosomal dominant disorder caused by a narrow spectrum of missense mutations in the SMAD4 gene. Typical features of this disorder are distinctive facial appearance, deafness, intellectual disability, cardiovascular abnormalities, short stature, short hands and feet, compact build, joint stiffness, and skeletal anomalies. The clinical features generally appear during childhood and become more evident in older patients. Therefore, the diagnosis of this syndrome in the first years of life is challenging. We report a 2‐year‐old girl diagnosed with Myhre syndrome by whole exome sequencing (WES) that revealed the recurrent p.Ile500Val mutation in the SMAD4 gene. Our patient presented with growth deficiency, dysmorphic features, tetralogy of Fallot, and corectopia (also known as ectopia pupillae). The girl we described is the youngest patient with Myhre syndrome. Moreover, corectopia and tetralogy of Fallot have not been previously reported in this disorder.
Keywords: corectopia, Myhre syndrome, tetralogy of Fallot, WES
1. INTRODUCTION
Myhre syndrome (MIM 139210) is a rare autosomal dominant disorder reported so far in over 60 individuals, mostly males diagnosed in childhood or adolescence (Caputo et al., 2012; Lin et al., 2016). Clinical features of this disorder include poor growth, variable degree of intellectual disability, distinctive dysmorphic features including short palpebral fissures, maxillary hypoplasia, small mouth with thin upper lip and short philtrum, prognathism, and thick skin. Restricted joint mobility and scarring abnormalities are striking features of the disorders (Caputo et al., 2012; Le Goff et al., 2011; Lin et al., 2016). Cardiovascular abnormalities include restrictive cardiomyopathy and pericardial disease although congenital heart defects have also been reported (Lin et al., 2016; Starr et al., 2015). Patients with Myhre syndrome might develop various long‐term and life‐threatening complications including systemic hypertension, cardiomyopathy, pericarditis, laryngo‐tracheal stenosis, and pulmonary insufficiency (Garavelli et al., 2016; Lin et al., 2016; Starr et al., 2015).
Myhre syndrome is caused by recurrent missense heterozygous mutations affecting the Ile500 residue of the SMAD4 gene that encodes a tumor‐suppressor protein affecting the transforming growth factor β (TGF‐β) signaling pathway (Caputo et al., 2012; Le Goff et al., 2011; Piccolo et al., 2014). A mutation in Arg496 has been described in a minority of patients (Caputo et al., 2014; Michot et al., 2014). We report a 2‐year‐old female child harboring a de novo mutation affecting Ile500 who presented with poor growth, dysmorphic features, corectopia (also known as ectopia pupillae), and tetralogy of Fallot (TOF). This case is remarkable for the early diagnosis and the ocular and cardiac abnormalities which have not been previously reported in individuals with Myhre syndrome.
2. CASE REPORT
The child was born to non‐consanguineous parents by ceasarean section after 38 weeks of gestation complicated by intrauterine growth retardation and a prenatal diagnosis of TOF. Her birth weight was 2.3 kg (3th centile). The diagnosis of classic TOF and right aortic arch was confirmed by echocardiogram at birth. She underwent cardiac catheterizations at 3 and 19 days of life for pulmonary balloon dilation that was only partially successful and was followed by 5 mm coronary stent positioning into the right ventricular outflow tract to increase pulmonary blood flow. An aorta of normal caliber was noted during aortic catheterization. At 7 months of age, she underwent complete repair of the TOF with closure of the ventricular septum defect, trans‐anular pulmonary patch, and left pulmonary artery patch repair. At the age of 2 years and 10 months, she underwent another surgical intervention for correction of right ventricular outflow tract obstruction. At chest re‐opening, a diffusely fibrotic scar tissue around the great vessels, on the epicardium, and on the transanular patch was observed.
At the age of 11 months, she was found to have a tense abdomen and an abdominal ultrasound revealed ascites that resolved within approximately 24 hr following treatment with diuretics and a phosphodiesterase III inhibitor (enoximone). Liver and function tests did not reveal any abnormalities and the cause for the ascites remained unclear.
She had feeding difficulties only in the first months of life. Since birth her growth has been deficient with weight, height, and head circumference all below the 5th centile. She had left esotropia, displacement of the eye pupil from its normal and central position (i.e., corectopia) more evident in the left eye, short palpebral fissures, hypertelorism, flat nasal bridge, highly arched palate, and brachydactyly (Figure 1). Her development was slightly delayed: she was able to sit by 11 months and to walk independently at 20 months. By 16 months of age, she said her first words. At her last evaluation at the age of 23 months, her weight was 8.410 kg (<5th centile), height 69.2 cm (<5th centile), and OFC 44.5 cm (<5th centile). The auditory brain steam response (ABR) was normal and the ophthalmology evaluation did not reveal lens dislocation or retinal abnormalities. The array‐CGH showed microdeletions 3q29 of 54 kb (from nucleotide 195,419,168 to 195,472,855 on human reference genome hg19), 4q13.3 of 120 kb (71,162,798–71,283,216), 7q34 of 65 kb (142,825,843–142,890,668), and 15q14 of 71 kb (34,735,949–34,806,953) and a 7q11.21 microduplication of 194 kb (62,460,665–62,654,363) that were all overlapping with copy number variants detected in controls and thus, they were interpreted as non‐pathogenic.
Figure 1.
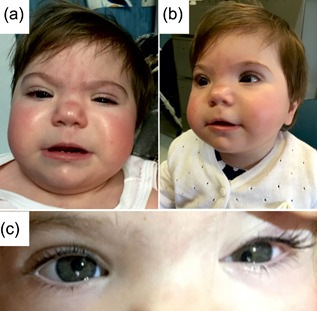
(a) Patient facial features at the ages of 6 months and 2 years. (b) Flat nasal bridge, short palpebral fissures, smooth philtrum, thin upper lip can be noted. (c) Corectopia of the left eye. [Color figure can be viewed at wileyonlinelibrary.com].
2.1. Whole exome sequencing
After informed consent, the child was enrolled in the Telethon Undiagnosed Program (TUDP) and underwent whole exome sequencing (WES). A total of 56,434 high‐quality variants were identified in the proband. Of these variants, 50,825 were single nucleotide variants, 5,609 were indels, 10,625 were predicted to impact a protein sequence, and 510 were also rare (frequency <0.01) according to population database queries. Of these, 14 variants were de novo, 2 were X‐linked, and 461 were homozygous or compound‐heterozygous, considering trio segregation. The subsequent prioritization process was based on the selection of variants with potential loss‐of‐function effect, with higher Combined Annotation Dependent Depletion (CADD) score (Kircher et al., 2014), and involving genes related to the patient phenotype. As top candidate, one de novo (g.chr18:48604676A>G according to GRCh37) variant in SMAD4 (NM_005359.5: c.1498A>G) was identified. This variant causes the p.Ile500Val substitution repeatedly found in Myhre syndrome patients (Caputo et al., 2012; Le Goff et al., 2011). The mutation was confirmed by Sanger sequencing. Both parents did not harbor the mutation. Targeted analysis of the reads corresponding to genes associated to anterior segment dysgenesis (Reis & Semina, 2011) (COL1A1, COL4A, B3GALTL, BMP4, BMP7, CYP1B1, FOX2C1, FOXC2, FOXE3, JAG1, LAMB2, PAX6, PIK3CA, PITX2, PITX3) revealed no variants.
3. DISCUSSION
Previously, the youngest Myhre syndrome patient reported was a 3‐year‐old boy lacking the distinctive findings such as restricted joint movement, compact build and dysmorphic features who was part of a series reporting the results of WES (Need et al., 2012). Early clinical diagnosis of Myhre syndrome in infancy is difficult because several features become evident in late childhood and thus, WES is an effective method for recognition of this disorder.
Ocular involvement with cataracts, strabismus, retinitis pigmentosa, maculopathy, papilledema, and pseudo‐papilledema has been reported in Myhre syndrome (Table 1). However, corectopia has not yet been reported. The lack of variants in known genes associated with anterior segment dysgenesis suggests that the SMAD4 mutation is responsible for the corectopia. Corectopia is generally associated with lens dislocation (Colley et al., 1991), but the child we report herein presented with corectopia without lens dislocation. Although it has not been reported so far in Myhre patients, corectopia with or without lens dislocation has been described in other connective tissue disorders including Marfan syndrome, Weill‐Marchesani syndrome (Colley et al., 1991), and ADAMTSL4‐related disorder (Chandra et al., 2012; Christensen et al., 2010; Sharifi, Tjon‐Fo‐Sang, Cruysberg, & Maat‐Kievit, 2013), which are all characterized by defects of TGF‐β pathway (Table 2). Moreover, corectopia has been reported in Fbn2 and Adamtsl4 null mice showing disruption of extracellular microfibril biogenesis (Collin et al., 2015; Shi, Tu, Mecham, & Bassnett, 2013).
Table 1.
Summary of the ocular findings reported in patients with Myhre syndrome carrying SMAD4 mutation
| Oncular finding | Number of patient with abnormality/total number of patients a |
|---|---|
| Anterior segment defects | |
| Corectopia | 1/56 (2%) |
| Refractory abnormality | 18/56 (32%) |
| Strabismus | 13/56 (23%) |
| Cataract | 6/56 (11%) |
| Posterior segment defects | |
| Pseudopapillema | 3/56 (5%) |
| Papilledema | 1/56 (2%) |
| Retinitis pigmentosa and maculopathy | 2/56 (4%) |
Table 2.
Genetic disorders with corectopia and/or lens dislocation
| Condition | Ectopia lens | Corectopia | Gene defect | Reference |
|---|---|---|---|---|
| Ocular disorders | ||||
| Familial ectopia lentis | + | − | FBN1 | Zhang, Lai, Capasso, Han, and Levin (2015) |
| Rieger anomaly and other anterior segment dysgenesis | − | + | PAX6, PITX2, JAG1 FOXC1, COL4A1, PITX3, BMP4, FOXC2, FOXE3, PRDM5 | Cheong et al. (2016); Micheal et al. (2016); Reis et al. (2011); Reis and Semina (2011); Tümer and Bach‐Holm (2009); Verdin et al. (2014) |
| Connective tissue disorders | ||||
| Marfan syndrome | + | − | FBN1 | Latasiewicz, Fontecilla, Millá, and Sánchez (2016) |
| Beals syndrome | + | − | FBN2 | Viljoen (1994) |
| Ehlers–Danlos syndrome | + | − | COL5A1, COL5A2, COL1A1 | Colley, Lloyd, Ridgway, and Donnai (1991); Sadiq and Vanderveen (2013) |
| ADAMTSL4‐related disorders | + | − | ADAMTSL4 | Christensen, Fiskerstrand, Knappskog, Boman, and Rødahl (2010); Overwater et al. (2017) |
| Weill–Marchesani syndrome | + | + | ADAMTS10, ADAMTS17, LTBP2 | Sadiq and Vanderveen (2013) |
| Knobloch syndrome | + | − | COL18A1, ADAMTS18 | Khan et al. (2012) |
| Inborn errors of metabolism | ||||
| Homocystinuria | + | − | CBS | Burke, O'Keefe, Bowell, and Naughten (1989) |
| Sulfite oxidase deficiency | + | − | SUOX | Shih et al. 1977 |
| Molybdenum cofactor deficiency | + | − | MOCS1 | Lueder and Steiner (1995) |
| Multiple congenital anomalies/malformation syndromes | ||||
| Sturge–Weber syndrome | + | + | GNAQ | Moore, Reck, and Chen (2011); Shields et al. (2015) |
| Traboulsi syndrome | + | − | ASPH | Patel et al. (2014) |
| Spondylometaphyseal dysplasia with cone‐rod dystrophy | + | + | PCYT1A | Yamamoto et al. (2014) |
| Stromme syndrome | − | + | CENPF | Keegan et al. (2004) |
| Microphthalmia/coloboma and skeletal dysplasia syndrome | − | + | MAB21L2 | Deml et al. (2015) |
Cardiovascular anomalies have been reported in approximately 70% of patients with Myhre syndrome (Lin et al., 2016). The most frequent congenital heart defects are large patent ductus arteriosus, atrial and ventricular septal defects, and stenosis of aortic and mitral valves (Lin et al., 2016). Among these defects, TOF has not been reported. Moreover, patients with Myhre syndrome can develop recurrent pericardial effusion, constrictive pericarditis and cardiomyopathy (Lin et al., 2016; Picco et al., 2013). Given the occurrence of such complications, a timely diagnosis of Myhre syndrome is important.
In conclusion, we report a young child with Myhre syndrome due to SMAD4 mutation that presented corectopia and TOF, two features that have not been previously reported in Myhre syndrome patients.
ACKNOWLEDGMENTS
This work was supported by Telethon Foundation, Telethon Undiagnosed Diseases Program (TUDP, GSP15001): S. Banfi, N. Brunetti‐Pierri, A. Bruselles, G. Cappuccio, V. Caputo, R. Castello, G. Chillemi, A. Ciolfi, M. D'Antonio, B. Dallapiccola, M. Dentici, M. Dionisi, G. Esposito, S. Fecarotta, G. Mancano, S. Maitz, L. Monaco, F. Musacchia, M. Mutarelli, V. Nigro, G. Oliva, G. Parenti, M. Pinelli, S. Pizzi, M. Pizzo, F. Radio, E. Rizzi, A. Selicorni, G. Sgroi, M. Tartaglia, A.Torella, R. Turra.
Alagia M, Cappuccio G, Pinelli M, et al. A child with Myhre syndrome presenting with corectopia and tetralogy of Fallot. Am J Med Genet Part A. 2018;176A: 426–430. https://doi.org/10.1002/ajmg.a.38560
Marianna Alagia and Gerarda Cappuccio contributed equally to this work.
REFERENCES
- Burke, J. P. , O'Keefe, M. , Bowell, R. , & Naughten, E. R. (1989). Ocular complications in homocystinuria‐early and late treated. British Journal of Ophthalmology, 73, 427–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo, V. , Bocchinfuso, G. , Castori, M. , Traversa, A. , Pizzuti, A. , Stella, L. , … Tartaglia, M. (2014). Novel SMAD4 mutation causing Myhre syndrome. American Journal Medical Genetics Part A, 164A, 1835–1840. [DOI] [PubMed] [Google Scholar]
- Caputo, V. , Cianetti, L. , Niceta, M. , Carta, C. , Ciolfi, A. , Bocchinfuso, G. , … Tartaglia, M. (2012). A restricted spectrum of mutations in the SMAD4 tumor‐suppressor gene underlies Myhre syndrome. American Journal of Human Genetics, 90, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra, A. , Aragon‐Martin, J. A. , Hughes, K. , Gati, S. , Reddy, M. A. , Deshpande, C. , … Arno, G. (2012). A genotype‐phenotype comparison of ADAMTSL4 and FBN1 in isolated ectopia lentis. Investigative Ophthalmology and Visual Science, 53, 4889–4896. [DOI] [PubMed] [Google Scholar]
- Cheong, S. S. , Hentschel, L. , Davidson, A. E. , Gerrelli, D. , Davie, R. , Rizzo, R. , … Hardcastle, A. J. (2016). Mutations in CPAMD8 cause a unique form of autosomal‐recessive anterior segment dysgenesis. American Journal of Human Genetics, 99, 1338–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen, A. E. , Fiskerstrand, T. , Knappskog, P. M. , Boman, H. , & Rodahl, E. (2010). A novel ADAMTSL4 mutation in autosomal recessive ectopia lentis et pupillae. Investigative Ophthalmology and Visual Science, 51, 6369–6373. [DOI] [PubMed] [Google Scholar]
- Colley, A. , Lloyd, I. C. , Ridgway, A. , & Donnai, D. (1991). Ectopia lentis et pupillae: The genetic aspects and differential diagnosis. Journal of Medical Genetics, 28, 791–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin, G. B. , Hubmacher, D. , Charette, J. R. , Hicks, W. L. , Stone, L. , Yu, M. ,… Nishina, P. M. (2015). Disruption of murine Adamtsl4 results in zonular fiber detachment from the lens and in retinal pigment epithelium dedifferentiation. Human Molecular Genetics, 24, 6958–6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deml, B. , Kariminejad, A. , Borujerdi, R. H. R. , Muheisen, S. , Reis, L. M. , & Semina, E. V. (2015). Mutations in MAB21L2 result in ocular coloboma, microcornea and cataracts. PLOS Genetics, 11, e1005002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdem, H. B., Sahin, I., & Tatar, A. (2017). Myhre syndrome with novel findings: Bilateral congenital cortical cataract, bilateral papilledema, accessory nipple, and adenoid hypertrophy. Clinical Dysmorphology. Published online May 30, 2017. [DOI] [PubMed] [Google Scholar]
- Garavelli, L. , Maini, I. , Baccilieri, F. , Ivanovski, I. , Pollazzon, M. , Rosato, … Tartaglia, M. (2016). Natural history and life‐threatening complications in Myhre syndrome and review of the literature. European Journal of Pediatrics, 175, 1307–1315. [DOI] [PubMed] [Google Scholar]
- Keegan, C. E. , Vilain, E. , Mohammed, M. , Lehoczky, J. , Dobyns, W. B. , Archer, S. M. , & Innis, J. W. (2004). Microcephaly, jejunal atresia, aberrant right bronchus, ocular anomalies, and XY sex reversal. American Journal of Medical Genetics A, 125A, 293–298. [DOI] [PubMed] [Google Scholar]
- Khan, A. O. , Aldahmesh, M. A. , Mohamed, J. Y. , Al‐Mesfer, S. , & Alkuraya, F. S. (2012). The distinct ophthalmic phenotype of Knobloch syndrome in children. British Journal of Ophthalmology, 96, 890–895. [DOI] [PubMed] [Google Scholar]
- Kircher, M. , Witten, D. M. , Jain, P. , O'Roak, B. J. , Cooper, G. M. , & Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latasiewicz, M. , Fontecilla, C. , Millá, E. , & Sánchez, A. (2016). Marfan syndrome: Ocular findings andnovel mutations‐in pursuit of genotype‐phenotype associations. Canadian Journal of Ophthalmology, 51, 113–118. [DOI] [PubMed] [Google Scholar]
- Le Goff, C. , Mahaut, C. , Abhyankar, A. , Le Goff, W. , Serre, V. , Afenjar, A. , & Cormier‐Daire, V. (2011). Mutations at a single codon in Mad homology 2 domain of SMAD4 cause Myhre syndrome. Nature Genetics, 44, 85–88. [DOI] [PubMed] [Google Scholar]
- Lin, A. E. , Michot, C. , Cormier‐Daire, V. , L'Ecuyer, T. J. , Matherne, G. P. , Barnes, B. H. ,… Lindsay, M. E. (2016). Gain‐of‐function mutations in SMAD4 cause a distinctive repertoire of cardiovascular phenotypes in patients with Myhre syndrome. American Journal Medical Genetics Part A, 170A, 2617–2631. [DOI] [PubMed] [Google Scholar]
- Lueder, G. T., & Steiner, R. D. (1995). Ophthalmic abnormalities in molybdenum cofactor deficiency and isolated sulfite oxidase deficiency. Journal of Pediatric Ophthalmology and Strabismus, 32, 334–337. [DOI] [PubMed] [Google Scholar]
- Micheal, S. , Siddiqui, S. N. , Zafar, S. N. , Venselaar, H. , Qamar, R. , Khan, M. I. , & den Hollander, A. I. (2016). Whole exome sequencing identifies a heterozygous missense variant in the PRDM5 gene in a family with Axenfeld‐Rieger syndrome. Neurogenetics, 17, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michot, C. , Le Goff, C. , Mahaut, C. , Afenjar, A. , Brooks, A. S. , Campeau, P. M. , … Cormier‐Daire, V. (2014). Myhre and LAPS syndromes: Clinical and molecular review of 32 patients. European Journal of Human Genetics, 22, 1272–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, D. B. , Reck, S. D. , & Chen, P. P. (2011). Angle closure glaucoma associated with ectopia lentis in a patient with Sturge‐Weber syndrome. Eye (Lond), 25, 1235–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Need, A. C. , Shashi, V. , Hitomi, Y. , Schoch, K. , Shianna, K. V. , McDonald, M. T. , … Goldstein, D. B. (2012). Clinical application of exome sequencing in undiagnosed genetic conditions. Journal of Medical Genetics, 49, 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overwater, E. , Floor, K. , van Beek, D. , de Boer, K. , van Dijk, T. , Hilhorst‐Hofstee, Y. , … Maugeri, A. (2017). NGS panel analysis in 24 ectopia lentis patients; A clinically relevant test with a high diagnostic yield. European Journal of Medical Genetics, 60, 465–473. [DOI] [PubMed] [Google Scholar]
- Patel, N. , Khan, A. O. , Mansour, A. , Mohamed, J. Y. , Al‐Assiri, A. , Haddad, R. , & Alkuraya, F. S. (2014). Mutations in ASPH cause facial dysmorphism, lensdislocation, anterior‐segment abnormalities, and spontaneous filtering blebs, or Traboulsi syndrome. American Journal of Human Genetics, 94, 755–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picco, P. , Naselli, A. , Pala, G. , Marsciani, A. , Buoncompagni, A. , & Martini, A. (2013). Recurrent pericarditis in Myhre syndrome. American Journal Medical Genetics Part A, 161A, 1164–1166. [DOI] [PubMed] [Google Scholar]
- Piccolo, P. , Mithbaokar, P. , Sabatino, V. , Tolmie, J. , Melis, D. , Schiaffino, M. C. , … Brunetti‐Pierri, N. (2014). SMAD4 mutations causing Myhre syndrome result in disorganization of extracellular matrix improved by losartan. European Journal of Human Genetics, 22, 988–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis, L. M. , & Semina, E. V. (2011). Genetics of anterior segment dysgenesis disorders. Current Opinion in Ophthalmology, 22, 314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis, L. M. , Tyler, R. C. , Schilter, K. F. , Abdul‐Rahman, O. , Innis, J. W. , Kozel, B. A. , & Semina, E. V. (2011). BMP4 loss‐of‐function mutations in developmental eye disorders including SHORT syndrome. Human Genetics, 130, 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadiq, M. A. , & Vanderveen, D. (2013). Genetics of ectopia lentis semin. Ophthalmol, 28, 313–320. [DOI] [PubMed] [Google Scholar]
- Sharifi, Y. , Tjon‐Fo‐Sang, M. J. , Cruysberg, J. R. , & Maat‐Kievit, A. J. (2013). Ectopia lentis et pupillae in four generations caused by novel mutations in the ADAMTSL4 gene. British Journal of Ophthalmology, 97, 583–587. [DOI] [PubMed] [Google Scholar]
- Shi, Y. , Tu, Y. , Mecham, R. P. , & Bassnett, S. (2013). Ocular phenotype of Fbn2‐null mice. Investigative Ophthalmology and Visual Science, 54, 7163–7173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields, C. L. , Atalay, H. T. , Wuthisiri, W. , Levin, A. V. , Lally, S. E. , & Shields, J. A. (2015). Sector iris hemangioma in association with diffuse choroidal hemangioma. Official Publication of the American Association for Pediatric Ophthalmology and Strabismus, 19, 83–86. [DOI] [PubMed] [Google Scholar]
- Shih, V. E. , Abroms, I. F. , Johnson, J. L. , Carney, M. , Mandell, R. , Robb, R. M. , … Rajagopalan, K. V. (1977). Sulfite oxidase deficiency. Biochemical and clinical investigations of a hereditary metabolic disorder in sulfur metabolism. The New England Journal of Medicine, 297, 1022–1028. [DOI] [PubMed] [Google Scholar]
- Starr, L. J. , Grange, D. K. , Delaney, J. W. , Yetman, A. T. , Hammel, J. M. , Sanmann, J. N. , … Olney, A. H. (2015). Myhre syndrome: Clinical features and restrictive cardiopulmonary complications. American Journal Medical Genetics Part A, 167A, 2893–2901. [DOI] [PubMed] [Google Scholar]
- Tümer, Z. , & Bach‐Holm, D. (2009). Axenfeld‐Rieger syndrome and spectrum of PITX2 and FOXC1 mutations. European Journal of Human Genetics, 17, 1527–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, G. L. , Baratela, W. A. R. , Almeida, T. F. , Lazar, M. , Afonso, C. L. , Oyamad, M. K. , … Bertola, D. R. (2014). Mutations in PCYT1A cause spondylometaphyseal dysplasia with cone‐rod dystrophy. American Journal of Human Genetics, 94, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viljoen, D. (1994). Congenital contractural arachnodactyly (Beals syndrome). Journal of Medical Genetics, 31, 640–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin, H. , Sorokina, E. A. , Meire, F. , Casteels, I. , de Ravel, T. , Semina, E. V. , & De Baere, E. (2014). Novel and recurrent PITX3 mutations in Belgian families with autosomal dominant congenital cataract and anterior segment dysgenesis have similar phenotypic and functional characteristics. Orphanet Journal of Rare Diseases, 9, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Lai, Y. H. , Capasso, J. E. , Han, S. , & Levin, A. V. (2015). Early onset ectopia lentis due to a FBN1mutation with non‐penetrance. American Journal of Medical Genetics, A167, 1365–1368. [DOI] [PubMed] [Google Scholar]