

Abstract
A photoswitchable arylazopyrazole (AAP) derivative binds with cucurbit[8]uril (CB[8]) and methylviologen (MV2+) to form a 1:1:1 heteroternary host–guest complex with a binding constant of K a=2×103 m −1. The excellent photoswitching properties of AAP are preserved in the inclusion complex. Irradiation with light of a wavelength of 365 and 520 nm leads to quantitative E‐ to Z‐ isomerization and vice versa, respectively. Formation of the Z‐isomer leads to dissociation of the complex as evidenced using 1H NMR spectroscopy. AAP derivatives are then used to immobilize bioactive molecules and photorelease them on demand. When Arg‐Gly‐Asp‐AAP (AAP–RGD) peptides are attached to surface bound CB[8]/MV2+ complexes, cells adhere and can be released upon irradiation. The heteroternary host–guest system offers highly reversible binding properties due to efficient photoswitching and these properties are attractive for designing smart surfaces.
Keywords: arylazopyrazoles, cucurbit[8]uril, host–guest systems, photo-responsive, stimuli-responsive
External control of bioactivity on biointerfaces and coatings has attracted considerable interest across bioanalytical and biomedical applications.1, 2, 3, 4 In supramolecular chemistry, an important focus lies on the use of light to exploit and direct reversible control over the state of molecular assemblies and complex structures because light does not require additional components and light can be applied with a very high degree of spatio‐temporal control.5, 6, 7 Shinkai et al. proposed to use light for manipulating self‐assembled structures. Their self‐complementary azobenzene derivative underwent cyclic oligomerization in E‐form and lead to intramolecular cyclization when irradiated with UV light and subsequent switching to Z‐isomer.8 Feringa and co‐workers used dithienylcyclopentene photochromic switches to tune the viscosity of solutions.8, 9 Azobenzene isomerization processes have successfully been employed to enable a large functional change in biomolecules and ligands in a number of instances.10, 11 Inclusion of azobenzene derivatives as guests in macrocyclic hosts such as cyclodextrins (CD) and cucurbiturils (CB) can give rise to photosensitive host–guest complexation to regulate recognition and function.12, 13, 14, 15, 16, 17, 18 Photosensitive host–guest complexation is currently intensively explored on surfaces and is promising to come yet a step closer to mimic natural cell‐extracellular matrix (ECM) interactions on surfaces.1 Zhang et al. prepared β‐CD surfaces modified with azobenzene‐containing propyltriethoxysilane guests to tune the wettability properties of surfaces.19 We have demonstrated the possibility of photospecific protein assembly via azobenzene‐functionalized ligands on β‐CD surfaces.20 Gong and co‐workers designed α‐CD self‐assembled monolayers (SAM) to immobilize azobenzene‐modified cell adhesive RGD peptides and subsequently control cell attachment and release on this surface with UV light.19
Cucurbit[8]uril‐mediated host–guest heteroternary complexes including photoactive azobenzenes have been used in photomodulation of the assembly of (bio)molecules on surfaces.22, 23, 24, 25, 26, 27, 28, 29 For example, Scherman et al. reported the photoinduced disassembly of raspberry‐shaped colloids, representing these systems as useful tools for cargo delivery.22 We have recently assembled these photoresponsive azobenzene‐containing heteroternary complexes onto chips and employed them to attract and release proteins, viruses and bacteria by photoisomerization.23, 24 Interestingly, when these heteroternary complexes have been constructed with both redox‐ and light‐responsive elements multi stimuli‐responsivity becomes available.25
Azobenzenes have a thermodynamically stable E‐isomer and a metastable Z‐isomer and they can be switched from E to Z with UV irradiation (light of wavelength λ≈360 nm) and back from Z to E with visible irradiation (λ≈460 nm).26 Unfortunately, the thermodynamic stability of the Z‐isomer is low and the overlapping absorbance bands lead to incomplete photoswitching with a photostationary state (PSS) of about 70–80 %.6, 12, 27 Increasing the half‐life time, while retaining good addressability has been a major challenge and led to the design of new derivatives, such as o‐methoxy and o‐fluoro azobenzenes or bridged azobenzenes.28, 29
Alternatively, recent literature documents excellent switching efficiencies and improved half‐life times of Z‐isomers in the case one aryl ring in the azobenzene system is exchanged with a five‐membered nitrogen based heteroaromatic ring, so‐called azoheteroaryl photoswitches.30, 31, 32 For example, N‐substituted arylazopyrazoles (AAP) show half‐life times of 10 to 1000 days at 25 °C.31 PSS for both isomers are ≥98 % upon irradiation with λ=365 nm for E to Z isomerization while employing λ=520 nm leads to Z to E isomerization.30 Ravoo and co‐workers have recently shown that AAPs form photoresponsive inclusion complexes with β‐CD.33 Light‐responsive switching of these β‐CD‐AAP host–guest inclusion complexes occurred more efficient and with a superior thermal half‐life time of the Z‐isomer compared to commonly used azobenzenes.33 Incorporation of the AAP in CD vesicles and nanoparticles revealed excellent photoresponsive aggregation and dispersion.33 Since heterocyclic azo‐compounds such as AAPs are relatively unknown in host–guest chemistry and its surface‐related applications, we describe the novel use of AAP as potent photoswitchable ligand for heteroternary CB[8]‐based inclusion complexes in solution and on surfaces (Figure 1). We show the potential for fabricating photosensitive bioactive surfaces using an AAP‐modified integrin binding Arg‐Gly‐Asp (RGD) peptide. Our work represents an alternative to the use of photolabile caging groups on RGD motifs for the controlled attachment of cells.34
Figure 1.
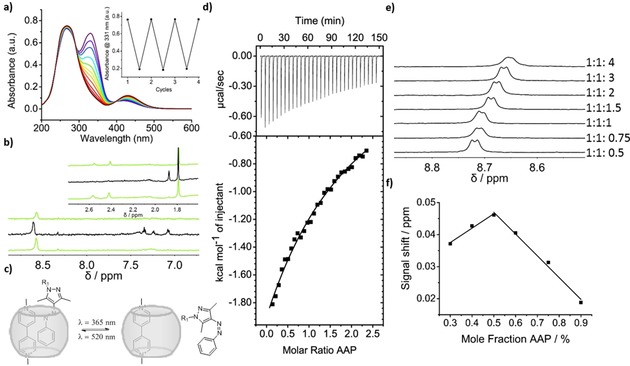
a) UV/Vis spectrum of the photo‐isomerization of AAP in the presence of CB[8]/paraquat (1:1:1) at 100 μm in water with corresponding switching cycles (see text for details). b) 1H NMR spectrum of the re‐isomerization of a 1:1:1 mixture irradiated with λ=520, 365 and 520 nm for 10 min ((E‐Z‐E) from top to bottom) at 100 μm. c) Scheme of heteroternary inclusion complex formation and dissociation (R1: CH2CONH‐(OCH2CH2)4‐OH). d) Isothermal calorimetry of CB[8]/paraquat with E‐AAP and the corresponding 1:1 fit. e) 1H NMR titration with 100 μm CB[8]/paraquat and E‐AAP. f) Job plot based on a 1H NMR titration.
To verify CB[8]‐mediated supramolecular complexation of AAP (Figure 1 c, see Supporting Information for synthetic details and characterization of AAP), a solution of E‐AAP (1 mm) was titrated to a 1:1 solution (0.1 mm) of CB[8] and paraquat while monitoring the change in heat using isothermal titration calorimetry (ITC, Figure 1 d). An exothermic, 1:1 binding event was observed and a binding constant between CB[8]/paraquat and E‐AAP was determined to be K a=2.5×103 m −1 (Figure 1 d). Additionally, AAP concentrations in a range of 0.05 to 0.4 mm where titrated to a 0.1 mm solution of CB[8]/paraquat in a 1H NMR titration (Figure 1 e, full spectra are given in Figure S2a). On the basis of the downfield shift of the aromatic signals of paraquat, a binding constant of K a=2.1×103 m −1 was obtained. A 1:1 binding stoichiometry was verified using a Job plot (Figure 1 f, 1H NMR spectra are given in Figure S2b).35 Upon increasing the AAP concentration the aromatic signals of paraquat as well as both methyl signals of the pyrazole unit of AAP shifted upfield. These observations are in agreement with CB[8] guest complexation and show that paraquat is interacting with the AAP moiety inside the CB[8] cavity.25 The ternary complex was also identified using ESI‐ToF mass spectrometry, which showed a signal at m/z 974.4 corresponding to a doubly charged heteroternary complex (see Supporting Information Figure S1).
To investigate the photoisomerization of AAP in the presence of CB[8]/paraquat, 1H NMR and UV/Vis spectroscopy measurements were performed (Figure 1 a, b). Upon irradiation with UV light (λ=365 nm) the AAP shows the characteristic changes in absorbance, which remained detectable when complexed. A significant decrease and a 10 nm blue‐shifted π–π* absorbance band at λ=328 nm concomitant with an increase of and a 10 nm red‐shifted n–π* band at λ=430 nm confirmed the isomerization from the E to Z‐isomer.33 Moreover, Z to E isomerization occurred upon irradiation with light of λ=520 nm, optically similar as observed in the case of non‐complexed AAP. Alternating the wavelength of irradiation between λ=520 nm and λ=365 nm and following the maximum absorption of E‐AAP at λ=311 nm showed that AAP stably and near‐quantitatively switched between the two isomers (Figure 1 a, inset). Re‐isomerization was also characterized using 1H NMR (Figure 1 b, full spectra are given in Figure S2c). The methyl signals of E‐AAP at δ=2.55 nm and 2.41 nm reversibly shifted to δ=1.87 nm and 1.54 nm, due to formation of the Z‐isomer. Comparing the spectra of the E and Z‐isomer in the aromatic region around δ=7.3 ppm, the signals of AAP sharpened in the Z‐state, indicating a disassembly of the 1:1:1 complex (Figure 1 b). These photo‐isomerization properties of AAP, that is, the separated excitation of the different states, are advantageous for molecular switches and photoresponsive materials and are an improvement when compared to the photo‐isomerization properties of azobenzenes. In addition, these results demonstrate that the photo‐isomerization properties of AAP are unaffected when complexed with CB[8] and paraquat.
Having established that a heteroternary, photoswitchable complex forms in aqueous solution, we prepared self‐assembled monolayers (SAMs) that are modified with AAP conjugated ternary complexes (Figure 2). To introduce specific cell interactions an integrin‐specific binding peptide RGD was attached to the AAP moiety (see Supporting Information for synthetic details). In short, the carboxylic acid functionality of AAP was modified with a tetraethylene glycol spacer bearing an azide moiety. This AAP–azide derivative was suitable for strain‐induced, metal‐free cycloaddition with a purified bicyclononyne‐RGD conjugate. The AAP–RGD peptide binds to CB[8] entities through inclusion of the hydrophobic E‐AAP moiety and to cells via the integrin‐binding peptide RGD.
Figure 2.
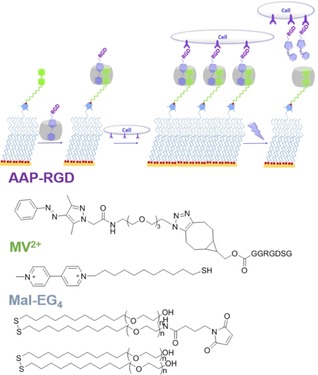
Assembly of heteroternary complex on antifouling SAMs. After cell adhesion, UV irradiation releases AAP–RGD and cells.
SAMs were then prepared on gold sensors for surface plasmon resonance (SPR) and quartz crystal microbalance with dissipation monitoring (QCM‐D) with a background layer of antifouling oligo(ethylene glycol) alkanethiols consisting of 1 or 0.1 % maleimide groups (Mal‐EG4, see Supporting Information for details).36 Thiolated methyl viologen (MV2+) was conjugated to the maleimide groups and acted as the first guest for CB[8] to bind the macrocycle to the surface (for clarity only explicitly shown in QCM‐D plot, Figure 3 b). SPR spectroscopy and QCM‐D measurements confirmed that the formed monolayer is efficiently binding CB[8] and AAP (Figure 3 a,b and Figure S3). Contact angle measurements verified the assembly of the surface (Figure S4). A concentration series of AAP over a range of 0.1–1 mm was performed at a flow rate of 100 μL min−1, in the presence of 50 μm CB[8], and followed by SPR (Figure 3 a) and QCM‐D (Figure 3 b). The binding constant of the binary CB[8]/MV complex has been estimated K=1.3×105 m −1.24 As we performed the binding studies at 50 μm of CB[8], nearly 80 % of CB[8] is expected bound to the MV2+‐surface. The binding constant of AAP to the binary complex of CB[8]/MV2+ is estimated to be K a=1.9×103 m −1 (K d=518 μm, SPR) and K a=3.5×103 m −1 (K d=283 μm, QCM‐D) (Figure 3 d). These values are in good agreement with the binding constants determined in solution using ITC and 1H NMR. Control experiments confirmed negligible nonspecific interactions of AAP–RGD and RGD with other surface components while when the smaller host CB[7] was used, AAP showed no binding (Figure S3). Therefore, we conclude that the selective formation of the AAP consisting heteroternary complex on the surface occurred as envisioned.
Figure 3.
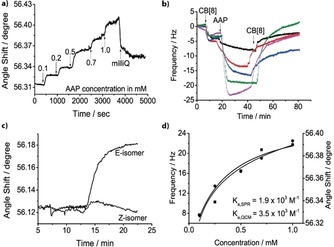
Concentration‐dependent E‐AAP assembly to a MV2+/CB[8] surface studied by a) SPR and b) QCM‐D. c) SPR response of flowing E‐ and Z‐AAP over MV2+/CB[8] SAMs. d) Change in SPR angle shift (square) and QCM‐D frequency (circle) vs. E‐AAP concentration (1:1 Langmuir fit is shown).
Subsequently, binding of E‐ and Z‐AAP isomers to the surface were studied using SPR (Figure 3 c). A significant change in the SPR angle appears when flowing an irradiated (with λ=520 nm) solution of 1 mm E‐AAP over a surface of CB[8]/MV2+ (Figure 3 c). This change was absent when the solution was irradiated with UV light (λ=365 nm) indicating that the binding of the Z‐isomer to the CB[8]/MV2+ surface is negligible (Figure 3 c). The binding of AAP to the surface was also photomodulated in flow (Figure S3c).
Based on the results that we formed CB[8]‐mediated heteroternary N‐substituted arylazopyrazole complexes on the surface, we then performed a set of experiments to demonstrate that these responsive supramolecular layers can be used for the photocontrolled cell adhesion. These experiments were performed on monolayers presenting the AAP–RGD complexes using the above‐mentioned assembly strategy. The supramolecular substrates were seeded with mouse myoblast C2C12 cells for 1 h in cell culture medium. Control surfaces without RGD, bearing just MV2+ or CB[8]/MV2+, showed limited cell adhesion, whereas CB[8]‐mediated AAP–RGD containing surfaces showed increased number of adhered cells (an overview of images is shown in Tables S1 and S2). In addition, adhered cells on AAP–RGD surfaces were more elongated and covered a larger area compared to all control surfaces of MV2+ and CB[8]/MV2+ (Figure 4 a). Staining in green of focal adhesion protein vinculin revealed well‐formed focal adhesions at the ends of the red‐stained actin only on the CB[8]‐mediated AAP–RGD presenting SAMs. This result indicates that efficient integrin‐mediated adhesion only occurred on CB[8]‐mediated AAP–RGD surfaces (inset Figure 4 a). Similar results were visible on SAMs using 0.1 % maleimide groups (Table S1). We note that specific integrin mediated cell adhesion occurred despite the modest binding constant of AAP–RGD to the binary complex. Presumably the weakly bound AAP–RGD molecules remain close to the surface available for rebinding as we have observed before in a cell force microscopy study.36
Figure 4.
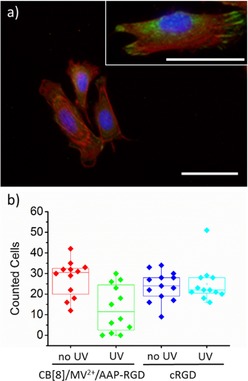
a) Fluorescence microscopy image of fixed C2C12 cells seeded for 1 h on CB[8]/MV2+/AAP–RGD SAMs. Cells were stained for nucleus (blue), actin (red) and vinculin (green), scale bar 100 μm. Inset is a magnified image of same surface, scale bar 50 μm. b) Quantitative analysis of C2C12 cells before (no UV) and after (UV) irradiation of λ=365 nm of the CB[8]/MV2+/AAP–RGD and cRGD SAMs.
Having established that specific cell adhesion occurs on self‐assembled CB[8]/MV2+/AAP–RGD monolayers, we evaluated cell detachment from the surfaces induced by photoswitching of AAP–RGD. As control surfaces we used a maleimide–SAM to which a cysteine‐capped RGD peptide was coupled (cRGD SAM).36 After C2C12 cells were seeded for 1 h, parts of these surfaces were irradiated for 10 min with UV light and dipped once in PBS. Cells were imaged (Table S3) and counted before and after irradiation on at least 10 spots on the entire surfaces. Significantly less cells were counted on the supramolecular AAP–RGD surface after partly irradiation while this was not the case when partly irradiation was performed on the control surfaces (Figure 4 b). This result leads to the conclusion that AAP–RGD is switchable on surfaces. Shortening the irradiation time to 1 min removed similar amounts of cells from the supramolecular surface while longer irradiation did not improve the results.
In conclusion, we have reported a novel CB[8]‐mediated photoresponsive heteroternary complex consisting of N‐substituted arylazopyrazole compounds. Complexation of the heteroternary complex on the surface has been studied using SPR and QCM‐D and yields binding constants that are similar to the values we found in solution studies using ITC and 1H NMR. We applied this new type of photoresponsive complexes for fabricating bioactive surfaces. Cells adhered to the supramolecularly immobilized arylazopyrazole‐modified RGD peptide and were removed by irradiation with UV light. This type of arylazopyrazoles with improved photoswitching behavior when compared to the commonly applied azobenzene derivatives are of interest for constructing supramolecular assemblies in solution and on surfaces. The design of dynamic reversible bioactive surfaces opens possibilities for novel innovative schemes including multi‐responsive bioactivity.25, 37
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. J. Voskuhl for providing an intermediate compound. This work was supported by the Netherlands Organization for Scientific Research (NWO) (NWO‐VIDI 723.012.106 to P.J.).
M. Wiemann, R. Niebuhr, A. Juan, E. Cavatorta, B. J. Ravoo, P. Jonkheijm, Chem. Eur. J. 2018, 24, 813.
Contributor Information
Prof. Bart Jan Ravoo, Email: b.j.ravoo@uni-muenster.de.
Prof. Pascal Jonkheijm, Email: p.jonkheijm@utwente.nl.
References
- 1. Brinkmann J., Cavatorta E., Sankaran S., Schmidt B., van Weerd J., Jonkheijm P., Chem. Soc. Rev. 2014, 43, 4449. [DOI] [PubMed] [Google Scholar]
- 2. Yang H., Yuan B., Zhang X., Scherman O. A., Acc. Chem. Res. 2014, 47, 2106. [DOI] [PubMed] [Google Scholar]
- 3. Chilkoti A., Hubbell J. A., MRS Bull. 2005, 30, 175. [Google Scholar]
- 4. Love J. C., Estroff L. A., Kriebel J. K., Nuzzo R. G., Whitesides G. M., Chem. Rev. 2005, 105, 1103. [DOI] [PubMed] [Google Scholar]
- 5. Feringa B. L., van Delden R. A., Koumura N., Geertsema E. M., Chem. Rev. 2000, 100, 1789. [DOI] [PubMed] [Google Scholar]
- 6. Yagai S., Kitamura A., Chem. Soc. Rev. 2008, 37, 1520. [DOI] [PubMed] [Google Scholar]
- 7. Uhlenheuer D. A., Petkau K., Brunsveld L., Chem. Soc. Rev. 2010, 39, 2817. [DOI] [PubMed] [Google Scholar]
- 8. Seiji S., Tohru Y., Kiminori M., Osamu M., Bull. Chem. Soc. Jpn. 1987, 60, 1819. [Google Scholar]
- 9. Lucas L. N., van Esch J., Kellogg R. M., Feringa B. L., Chem. Commun. 2001, 0, 759. [Google Scholar]
- 10. Sadovski O., Beharry A. A., Zhang F., Woolley G. A., Angew. Chem. Int. Ed. 2009, 48, 1484; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 1512. [Google Scholar]
- 11. Liu D., Xie Y., Shao H., Jiang X., Angew. Chem. Int. Ed. 2009, 48, 4406; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 4470. [Google Scholar]
- 12. Wu J., Isaacs L., Chem. Eur. J. 2009, 15, 11675. [DOI] [PubMed] [Google Scholar]
- 13. Yamaguchi H., Kobayashi Y., Kobayashi R., Takashima Y., Hashidzume A., Harada A., Nat. Commun. 2012, 3, 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stoffelen C., Voskuhl J., Jonkheijm P., Huskens J., Angew. Chem. Int. Ed. 2014, 53, 3400; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3468. [Google Scholar]
- 15. Blass J., Bozna B. L., Albrecht M., Krings J. A., Ravoo B. J., Wenz G., Bennewitz R., Chem. Commun. 2015, 51, 1830. [DOI] [PubMed] [Google Scholar]
- 16. del Barrio J., Horton P. N., Lairez D., Lloyd G. O., Toprakcioglu C., Scherman O. A., J. Am. Chem. Soc. 2013, 135, 11760. [DOI] [PubMed] [Google Scholar]
- 17. Yu Z., Zhang J., Coulston R. J., Parker R. M., Biedermann F., Liu X., Scherman O. A., Abell C., Chem. Sci. 2015, 6, 4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim Y., Ko Y. H., Jung M., Selvapalam N., Kim K., Photochem. Photobiol. Sci. 2011, 10, 1415. [DOI] [PubMed] [Google Scholar]
- 19. Shen Q., Liu L., Zhang W., Langmuir 2014, 30, 9361. [DOI] [PubMed] [Google Scholar]
- 20. Voskuhl J., Sankaran S., Jonkheijm P., Chem. Commun. 2014, 50, 15144. [DOI] [PubMed] [Google Scholar]
- 21. Gong Y. H., Li C., Yang J., Wang H. Y., Zhuo R. X., Zhang X. Z., Macromolecules 2011, 44, 7499. [Google Scholar]
- 22. Lan Y., Wu Y., Karas A., Scherman O. A., Angew. Chem. Int. Ed. 2014, 53, 2166; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2198. [Google Scholar]
- 23. Sankaran S., van Weerd J., Voskuhl J., Karperien M., Jonkheijm P., Small 2015, 11, 6187. [DOI] [PubMed] [Google Scholar]
- 24. Weineisen N. L., Hommersom C. A., Voskuhl J., Sankaran S., Depauw A. M. A., Katsonis N., Jonkheijm P., Cornelissen J. J. L. M., Chem. Commun. 2017, 53, 1896. [DOI] [PubMed] [Google Scholar]
- 25. Tian F., Jiao D., Biedermann F., Scherman O. A., Nat. Commun. 2012, 3, 1207. [DOI] [PubMed] [Google Scholar]
- 26. Fenske T., Korth H.-G., Mohr A., Schmuck C., Chem. Eur. J. 2012, 18, 738. [DOI] [PubMed] [Google Scholar]
- 27. Nalluri S. K. M., Voskuhl J., Bultema J. B., Boekema E. J., Ravoo B. J., Angew. Chem. Int. Ed. 2011, 50, 9747; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9921. [Google Scholar]
- 28. Bléger D. J., Schwarz J., Brouwer A. M., Hecht S., J. Am. Chem. Soc. 2012, 134, 20597. [DOI] [PubMed] [Google Scholar]
- 29. Siewertsen R., Neumann H., Buchheim-Stehn B., Herges R., Näther C., Renth F., Temps F., J. Am. Chem. Soc. 2009, 131, 15594. [DOI] [PubMed] [Google Scholar]
- 30. Calbo J., Weston C. E., White A. J. P., Rzepa H. S., Contreras-García J., Fuchter M. J., J. Am. Chem. Soc. 2017, 139, 1261. [DOI] [PubMed] [Google Scholar]
- 31. Weston C. E., Richardson R. D., Haycock P. R., White A. J. P., Fuchter M. J., J. Am. Chem. Soc. 2014, 136, 11878. [DOI] [PubMed] [Google Scholar]
- 32. Travieso-Puente R., Budzak S., Chen J., Stacko P., Jastrzebski J. T. B. H., Jacquemin D., Otten E., J. Am. Chem. Soc. 2017, 139, 3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stricker L., Fritz E. C., Peterlechner M., Doltsinis N. L., Ravoo B. J., J. Am. Chem. Soc. 2016, 138, 4547. [DOI] [PubMed] [Google Scholar]
- 34.
- 34a. Lee T. T., García J. R., Paez J. I., Singh A., Phelps E. A., Weis S., Shafiq Z., Shekaran A., del Campo A., García A. J., Nat. Mater. 2014, 14, 352; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34b. Petersen S., Alonso J. M., Specht A., Duodo P., Goeldner M., Del Campo A., Angew. Chem. Int. Ed. 2008, 47, 3192; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3236. [Google Scholar]
- 35. Renny J. S., Tomasevich L. L., Tallmadge E. H., Collum D. B., Angew. Chem. Int. Ed. 2013, 52, 11998; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12218. [Google Scholar]
- 36. Sankaran S., Jaatinen L., Brinkmann J., Zambelli T., Vörös J., Jonkheijm P., ACS Nano 2017, 11, 3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. An Q., Brinkmann J., Huskens J., Krabbenborg S., deBoer J., Jonkheijm P., Angew. Chem. Int. Ed. 2012, 51, 12233; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12399. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary