

Abstract
Glecaprevir (nonstructural protein 3/4A protease inhibitor) and pibrentasvir (nonstructural protein 5A inhibitor) (G/P), a coformulated once‐daily, all oral, ribavirin (RBV)‐free, direct‐acting antiviral regimen, was evaluated for safety and efficacy in hepatitis C virus genotype 2 (GT2)–infected Japanese patients, including those with compensated cirrhosis. CERTAIN‐2 is a phase 3, open‐label, multicenter study assessing the safety and efficacy of G/P (300/120 mg) once daily in treatment‐naive and interferon ± RBV treatment–experienced Japanese patients without cirrhosis but with GT2 infection. Patients were randomized 2:1 to receive 8 weeks of G/P (arm A) or 12 weeks of sofosbuvir (400 mg once daily) + RBV (600‐1000 mg weight‐based, twice daily) (arm B). The primary endpoint was noninferiority of G/P compared to sofosbuvir + RBV by assessing sustained virologic response at posttreatment week 12 (SVR12) among patients in the intent‐to‐treat population. SVR12 was also assessed in treatment‐naive and interferon ± RBV treatment‐experienced patients with GT2 infection and compensated cirrhosis who received G/P for 12 weeks in the CERTAIN‐1 study. A total of 136 patients were enrolled in CERTAIN‐2. SVR12 was achieved by 88/90 (97.8%) patients in arm A and 43/46 (93.5%) patients in arm B. No patient in arm A experienced virologic failure, while 2 did in arm B. The primary endpoint was achieved. In CERTAIN‐1, 100% (18/18) of GT2‐infected patients with compensated cirrhosis achieved SVR12. Treatment‐emergent serious adverse events were experienced by 2 patients without cirrhosis in each arm and no patient with cirrhosis. Conclusion: The results demonstrate high efficacy and favorable tolerability of G/P in GT2‐infected Japanese patients. (Hepatology 2018;67:505‐513).
Abbreviations
- AE
adverse event
- CI
confidence interval
- DAA
direct‐acting antiviral
- GLE
glecaprevir
- G/P
pangenotypic regimen of GLE and PIB
- GT2
genotype 2
- HCV
hepatitis C virus
- IFN
interferon
- ITT
intention‐to‐treat
- mITT
modified ITT
- NS
nonstructural protein
- peg
pegylated
- PIB
pibrentasvir
- RBV
ribavirin
- SOF
sofosbuvir
- SVR
sustained virologic response
- SVR12
SVR at 12 weeks
Approximately 1.5 million individuals in Japan have chronic hepatitis C virus (HCV) infection,1, 2, 3, 4 30% with genotype 2 (GT2).5, 6 The Japanese HCV patient population tends to be older and at a higher risk for developing hepatocellular carcinoma than non‐Japanese patients.7, 8, 9 The burden of HCV infection in Japan is expected to rise due to disease progression combined with an aging population.10
Sofosbuvir (SOF), a nucleoside direct‐acting antiviral agent (DAA), was demonstrated to have a high rate of sustained virologic response (SVR) in Japanese patients with chronic GT2 HCV infection when coadministered with ribavirin (RBV).11 The two‐DAA regimen ombitasvir/paritaprevir/ritonavir (ritonavir is a pharmacoenhancer with no anti‐HCV activity) coadministered with RBV also achieved high SVR rates in HCV GT2‐infected Japanese patients without cirrhosis.12 The Japan Society of Hepatology currently recommends SOF + RBV for 12 weeks for HCV GT2‐infected patients with or without compensated cirrhosis or ombitasvir/paritaprevir/ritonavir + RBV for 16 weeks for HCV GT2a‐infected patients without cirrhosis as a first line of treatment.11, 13 As RBV is known to have adverse events (AEs) related to decreases in hemoglobin and elevations of indirect bilirubin, its elimination from HCV regimens can improve tolerability. Furthermore, SOF is contraindicated in Japan in patients with severe renal impairment (creatinine clearance <30 mL/minute), while RBV is contraindicated in moderate and severe renal impairment (creatinine clearance ≤50 mL/minute).11, 14 Therefore, no DAA treatment regimens are approved in Japan for HCV GT2‐infected patients with advanced chronic kidney disease.
Glecaprevir (GLE, formerly ABT‐493, identified by AbbVie and Enanta), a nonstructural protein (NS) 3/4A protease inhibitor, coformulated with pibrentasvir (PIB, formerly ABT‐530), an NS5A inhibitor, is currently being evaluated as a pangenotypic regimen (G/P) for the treatment of patients with HCV infection including those with compensated cirrhosis, as well as other patient subpopulations that have been previously considered difficult to treat. Preclinical and previous clinical studies have demonstrated that this combination regimen has a high barrier to resistance and potency against common NS3 and NS5A polymorphisms.15 High efficacy of G/P in various patient populations over short treatment durations compared to currently recommended treatments has been demonstrated outside of Japan.16 Here, we report the safety and efficacy of once‐daily G/P administered for 8 weeks compared to SOF + RBV administered for 12 weeks in DAA‐naive GT2 HCV‐infected Japanese patients without cirrhosis and in a separate, small cohort of DAA‐naive Japanese patients with GT2 and compensated cirrhosis who received G/P for 12 weeks.
Participants and Methods
STUDY DESIGN
CERTAIN‐1 (NCT02707952) and CERTAIN‐2 (NCT02723084) are phase 3, open‐label, multicenter studies assessing the safety and efficacy of G/P (300/120 mg once daily) in Japanese patients with HCV infection. The same dose has been used in various studies assessing the efficacy and safety of G/P outside Japan.16 Patients enrolled in CERTAIN‐2 had GT2 HCV infection without cirrhosis and were randomized 2:1 to receive 8 weeks of treatment with G/P (arm A) or 12 weeks of treatment with SOF (400 mg once daily) + RBV (600‐1,000 mg weight‐based, divided, twice daily) (arm B). The randomization was stratified by prior interferon (IFN)/pegylated (peg) IFN experience (naive versus experienced) and HCV RNA viral load (<6 million IU/mL or ≥6 million IU/mL). A cohort of patients enrolled in substudy 2 of CERTAIN‐1 (arm C) who had HCV GT2 infection and compensated cirrhosis were assigned to treatment with G/P for 12 weeks. Prior to study initiation, the number of GT2 patients with cirrhosis to be enrolled in CERTAIN‐1 substudy 2 was predetermined to be 15 based on the relative distribution of GT2 infection with cirrhosis in Japan. Patients were followed for 24 weeks after the final dose of study drug. Figure 1 shows the study design.
Figure 1.
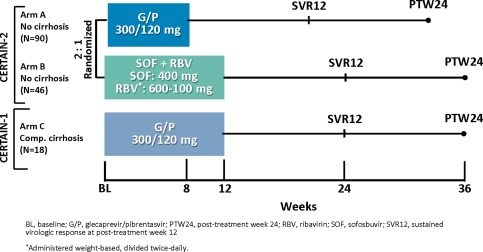
Study design for GT2‐infected DAA‐naive patients enrolled in the CERTAIN‐2 study and a subset of patients (GT2‐infected patients with compensated cirrhosis) enrolled in the CERTAIN‐1 arm C study.
All patients provided written, informed consent to participate, and the study was consistent with the ethical guidelines of the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice Guidelines. The study was approved by an institutional review board of each study site prior to the initiation of any screening or study‐specific procedures. Supporting Fig. S1 shows patient disposition.
PATIENTS
Patients were screened from February 22, 2016, to July 15, 2016, at 56 study sites in Japan. Adults ≥18 years of age with GT2 HCV infection without cirrhosis were eligible for enrollment in CERTAIN‐2, and those with GT2 HCV infection and compensated cirrhosis were eligible for enrollment in CERTAIN‐1 substudy 2. Patients were required to be either HCV treatment‐naive or to have failed prior IFN/pegIFN ± RBV therapy, be DAA treatment‐naive, test positive for anti‐HCV antibody, have a plasma HCV RNA load >1,000 IU/mL at the time of screening, and have a laboratory result indicating HCV infection with GT2 only.
Patients enrolled in CERTAIN‐2 were required to demonstrate absence of cirrhosis with one of the following criteria: a liver biopsy demonstrating absence of cirrhosis (e.g., METAVIR score of ≤3 or Ishak score of ≤4), a FibroScan score <12.5 kPa or a FibroTest score ≤0.72, and aspartate aminotransferase to platelet ratio index ≤2 or screening discriminant score (z) <0. Patients with compensated cirrhosis (Child‐Pugh A) enrolled in CERTAIN‐1 substudy 2 were required to have one of the following: a liver biopsy with a METAVIR (or equivalent) fibrosis score >3 or Ishak fibrosis score >4, a FibroTest score ≥0.73 with an aminotransferase to platelet ratio index >2, a FibroScan score ≥14.6 kPa, or z >0. Absence of hepatocellular carcinoma was confirmed with a negative ultrasound, computed tomographic scan, or magnetic resonance imaging scan within 3 months prior to screening or a negative ultrasound at screening. HCV genotype was assessed with the Versant HCV Genotype Inno LiPA Assay, version 2.0 or higher; if the Versant assay failed to provide the result, a Sanger sequencing assay of the NS5B region was used.
Patients were excluded if they had a positive test result for hepatitis B surface antigen or anti–human immunodeficiency virus antibody; estimated glomerular filtration rate <30 mL/minute/1.73 m2 (CERTAIN‐1 substudy 2) or CrCl ≤50 mL/min (CERTAIN‐2); any current or past clinical evidence of Child‐Pugh B or C classification; clinical history of decompensated liver disease such as ascites, hepatic encephalopathy, or variceal bleeding; any cause of liver disease other than HCV infection; any clinically significant abnormalities or comorbidities that made the patient an unsuitable study candidate in the opinion of the investigator; or abnormal screening laboratory results as listed in Supporting Table S1.
STUDY ASSESSMENT
Virologic response was assessed using serum HCV RNA concentration with a lower limit of quantitation of 15 IU/mL. Samples were collected at the screening visit; day 1 visit; treatment weeks 1, 2, 4, and 8 (and week 12 for arm B patients and patients with compensated cirrhosis); and posttreatment weeks 2, 4, 8, 12, and 24. The primary efficacy endpoint for GT2 HCV‐infected patients without cirrhosis was to demonstrate noninferiority of 8 weeks of G/P compared to 12 weeks of SOF + RBV in achieving SVR at 12 weeks (SVR12) among patients in the intent‐to‐treat (ITT) population, defined as those who received at least one dose of study drug. For GT2‐infected patients with compensated cirrhosis, SVR12 was assessed in the ITT population. Additional secondary endpoints for all patients reported here were the percentage of patients with on‐treatment virologic failure and posttreatment relapse.
Next‐generation sequencing was conducted on HCV NS3 and NS5A genes from samples collected from all patients at baseline, and presence of HCV baseline polymorphisms was evaluated using a 15% detection threshold. Blood samples for pharmacokinetic assessment of the study drugs were collected from patients during each study visit (G/P, n = 90 for patients without cirrhosis and n = 18 for patients with compensated cirrhosis; SOF + RBV, n = 46 patients without cirrhosis). Patients consenting to intensive pharmacokinetic sampling had samples drawn at the study day 1 (at 2, 4, and 6 hours postdose) and at the week 4 visit at hour 0 (before study drug administration) and 2 and 4 hours postdose. Plasma concentrations for GLE, PIB, SOF, GS‐331007 (primary metabolite of SOF), and RBV were summarized as steady‐state trough levels (Ctrough) based on binning of pharmacokinetic samples that fall in the interval of 22‐26 hours after dosing. Plasma concentrations were determined using a validated liquid chromatography assay.
AEs and laboratory tests were assessed and recorded at each visit throughout the treatment period and 24 weeks posttreatment for safety evaluations. From the time of study drug administration until 30 days following discontinuation of study treatment, all treatment‐emergent AEs were collected, whether solicited or spontaneously reported by the patient. After 30 days following completion of study treatment and throughout the posttreatment period, only spontaneously reported serious AEs were collected. All AEs were graded according to Common Terminology Criteria for Adverse Events, version 4.0.
STATISTICAL ANALYSIS
The primary analysis was conducted after all enrolled patients completed the posttreatment week 12 visit or prematurely discontinued the study. Efficacy, safety, and demographic analyses were performed on all patients in the ITT population. For the primary efficacy endpoint in CERTAIN‐2, the difference in SVR12 rate between arm A and arm B and the two‐sided 95% confidence interval (CI) were computed using the normal approximation to the binomial distribution. Noninferiority was considered established if the lower bound of the CI for this difference was above −10%. SVR12 was also assessed in the modified ITT (mITT) population that excluded patients who did not achieve SVR12 due to reasons other than virologic failure. The number and percentage of patients meeting the primary and secondary endpoints were determined for each endpoint, and a two‐sided 95% CI was computed for the percentage. Safety analyses compared the rate of AEs and laboratory abnormalities between treatment groups (arm A versus arm B) with the use of Fisher's exact test.
Results
BASELINE PATIENT DEMOGRAPHICS AND CHARACTERISTICS
A total of 136 patients with HCV GT2 infection without cirrhosis were randomized to arms A (8 weeks with G/P, n = 90) and B (12 weeks of SOF + RBV, n = 46) in CERTAIN‐2. Of those patients, 53% and 54% were female in arms A and B, respectively, and 17% in each arm were treatment‐experienced with IFN/pegIFN ± RBV. The median age was 57 and 58 in arms A and B, respectively. The mean HCV RNA at baseline was 6.0 ± 0.8 log10 IU/mL and 6.1 ± 0.8 log10 IU/mL, respectively.
A total of 18 patients with GT2 HCV infection and compensated cirrhosis were enrolled in CERTAIN‐1 substudy 2. Of the patients with cirrhosis, 61% were female, and 39% were treatment‐experienced with IFN/pegIFN ± RBV, with a median age of 70. The mean HCV RNA at baseline was 5.3 ± 1.0 log10 IU/mL. Detailed patient demographic and baseline characteristics are shown in Table 1.
Table 1.
Baseline Demographics and Disease Characteristics of GT2 HCV‐Infected Patients Without Cirrhosis Enrolled in Arms A and B of CERTAIN‐2 and GT2 HCV‐Infected Patients With Compensated Cirrhosis Enrolled in Arm C of CERTAIN‐1 Substudy 2
| Characteristic |
CERTAIN‐2 Without Cirrhosis |
CERTAIN‐1 Substudy 2 Compensated Cirrhosis |
|
|---|---|---|---|
|
Arm A G/P 8 weeks (n = 90) |
Arm B SOF + RBV 12 weeks (n = 46) |
Arm C G/P 12 weeks (n = 18) |
|
| Female, n (%) | 48 (53) | 25 (54) | 11 (61) |
| Age, median (range), years | 57 (26‐83) | 58 (21‐84) | 70 (49‐85) |
| Age, n (%) | |||
| ≥65 | 29 (32) | 17 (37) | 13 (72) |
| ≥75 | 10 (11) | 8 (17) | 6 (33) |
| BMI, mean ± SD (kg/m2) | 22.9 ± 3.3 | 23.0 ± 4.2a | 22.2 ± 3.5 |
| IL28B non‐CC genotype, n (%) | 23 (26) | 9 (20) | 3 (17) |
| Treatment‐experienced (IFN‐based with or without RBV), n (%) | 15 (17) | 8 (17) | 7 (39) |
| HCV subtypeb | |||
| 2a, n (%) | 65 (72) | 30 (65) | 10 (56) |
| 2b, n (%) | 25 (28) | 16 (35) | 8 (44) |
| HCV RNA, mean ± SD, log10 IU/mL | 6.0 ± 0.8 | 6.1 ± 0.8 | 5.3 ± 1.0 |
| FIB‐4 index, median (range) | 1.6 (0.6‐7.7) | 2.1 (0.6‐7.3) | 5.1 (1.6‐17.0) |
| Proton pump inhibitor use, n (%) | 12 (13) | 2 (4) | 7 (39) |
| Liver protectant use, n (%) | 15 (17) | 12 (26) | 10 (56) |
| Calcium channel blockers use, n (%) | 23 (26) | 11 (24) | 6 (33) |
n = 45.
HCV subtype determined by phylogenetic analysis of baseline NS3/4A and/or NS5A sequence.
Abbreviations: BMI, body mass index; IL28B, interleukin 28B; SD, standard deviation.
EFFICACY OUTCOMES
A total of 97.8% (88/90) of patients enrolled in arm A achieved SVR12; no virologic failures occurred, resulting in an SVR12 rate of 100% in the mITT population. One patient not achieving SVR12 was lost to follow‐up after achieving SVR at 4 weeks, while the other prematurely discontinued study drug due to an AE of grade 2 nausea and vomiting after 18 days of treatment. SVR12 was achieved by 93.5% (43/46; 95% CI, 92.5%‐97.8) of patients enrolled in arm B; virologic relapse occurred in 2 patients by posttreatment week 12, resulting in an SVR12 rate of 95.6% in the mITT population; the third patient not achieving SVR12 prematurely discontinued study drug due to an AE of grade 1 malaise. The CERTAIN‐2 study met the primary endpoint, demonstrating that G/P for 8 weeks was noninferior to SOF + RBV for 12 weeks as the lower bound of the 95% CI for the difference in SVR12 between arms A and B (4.3; 95% CI, –3.5 to 12.1) was above the predefined threshold of –10%.
All (18/18, 100%) HCV GT2‐infected patients with compensated cirrhosis enrolled in arm C of CERTAIN‐1 substudy 2 and treated with G/P for 12 weeks achieved SVR12. Figure 2 shows the SVR12 rates and 95% CIs for all cohorts in the ITT and mITT populations.
Figure 2.
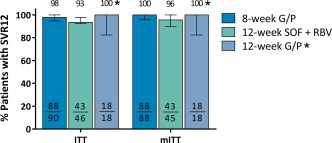
SVR12 rates for each arm in the ITT and mITT populations. Error bars represent the 95% CIs. Arm A: 8‐week G/P treatment; arm B: 12‐week SOF + RBV treatment; arm C: 12‐week G/P treatment. *GT2 patients with compensated cirrhosis from CERTAIN‐1 substudy 2.
IMPACT OF BASELINE POLYMORPHISM
Prevalence of baseline polymorphisms relative to a reference sequence was determined using a 15% detection threshold at amino acid positions 155, 156, and 168 in NS3 and 24, 28, 30, 31, 58, 92, and 93 in NS5A for patients in arms A and C. The most common polymorphism in HCV GT2‐infected patients was the NS5A L31M polymorphism, occurring at a prevalence rate of 94% and 80% in patients without cirrhosis and with compensated cirrhosis, respectively. The most common polymorphisms in HCV GT2b‐infected patients were M31I/L/V in NS5A, occurring at a combined prevalence rate of 18% in patients without cirrhosis, and NS5A L28F, occurring at a prevalence rate of 30% in patients with compensated cirrhosis. There were no virologic failures among GT2‐infected, DAA‐naive patients treated with G/P; therefore, baseline polymorphisms did not have an impact on treatment outcome. Prevalence of baseline polymorphisms is shown in Supporting Table S2.
PHARMACOKINETIC RESULTS
Following administration of G/P, GLE and PIB plasma concentrations attained steady state by the week 1 visit and remained constant throughout the treatment period (weeks 1‐8 for DAA‐naïve, GT2‐infected patients without cirrhosis and weeks 1‐12 for DAA‐naïve, GT2‐infected patients with cirrhosis). No apparent drug accumulation was observed. GLE plasma concentrations were higher in GT2 HCV‐infected, DAA‐naive patients with compensated cirrhosis compared to patients without cirrhosis, while PIB plasma concentrations were comparable between patients with and without cirrhosis.
SAFETY OUTCOMES
Treatment‐emergent AEs were experienced by 48% and 76% of patients in arm A (administered G/P for 8 weeks) and arm B (administered SOF + RBV for 12 weeks) of CERTAIN‐2, respectively, with 18% and 50% of patients having AEs assessed as study drug–related. The lower rates of AEs in arm A compared to arm B were statistically significant (P = 0.002 and P < 0.001 for all AEs and drug‐related AEs, respectively). AEs that occurred at a statistically significantly lower rate in arm A than arm B (P < 0.05) were anemia, blood bilirubin increase, and hyperuricemia. Two serious AEs (2%) occurred in arm A, spontaneous pneumothorax in 1 patient and unstable angina in 1 other, neither of which was assessed by the investigator as being related to study drug. Two serious AEs (4%) occurred in arm B, pneumonia and Castleman disease, the former assessed as not study drug–related and the latter as study drug–related. One patient (1%) in arm A discontinued study drug due to a grade 2 AE (nausea and vomiting) after 18 days of treatment, and 1 patient (2%) in arm B discontinued treatment due to an AE (malaise) after 12 days of treatment and withdrew consent; both AEs were assessed as study drug–related by the investigator, and neither patient achieved SVR12. No AEs occurred with a frequency >10% in arm A. AEs with a frequency >10% in arm B of CERTAIN‐2 included anemia (35%) and increased blood bilirubin (15%).
AEs were experienced by 67% of GT2‐infected patients with compensated cirrhosis in CERTAIN‐1 substudy 2, and 39% patients had events that were assessed as study drug–related by the investigator; no serious AEs occurred. One patient (6%) discontinued study drug due to a grade 2 drug‐related AE (drug eruption, on day 12, characterized as a patchy purpuric rash and eczema), which resolved on day 29; the patient discontinued study drug on day 14 and achieved SVR12. AEs with a frequency >10% were pruritus (22%), nasopharyngitis (11%), and increased blood bilirubin (11%). No drug‐related liver injury or liver decompensation events occurred in any arm. Table 2 provides a list of AEs in the safety population.
Table 2.
Treatment‐Emergent AEs
| Event |
CERTAIN‐2 Without Cirrhosis |
CERTAIN‐1 Substudy 2 Compensated Cirrhosis |
|
|---|---|---|---|
|
Arm A G/P (n = 90), n (%) |
Arm B SOF + RBV (n = 46), n (%) |
Arm C G/P (n = 18), n (%) |
|
| Any AEa | 43 (48) | 35 (76) | 12 (67) |
| Any drug‐related AEb | 16 (18) | 23 (50) | 7 (39) |
| Any serious AE | 2 (2)c | 2 (4)d | 0 |
| Any study drug–related serious AE | 0 | 1 (2) | 0 |
| Any AE leading to discontinuation of study drug | 1 (1.1)e | 1 (2.2)f | 1 (6) |
| Any AE leading to interruption of study drug | 0 | 2 (4.3)g | 0 |
| Common AEs (occurring in ≥5% and ≥2 patients in any group) | |||
| Anemiah | 0 | 16 (35) | 1 (6) |
| Pruritus | 3 (3) | 2 (4) | 4 (22) |
| Blood bilirubin increasedi | 1 (1) | 7 (15) | 2 (11) |
| Nasopharyngitis | 9 (10) | 5 (11) | 2 (11) |
| Malaise | 5 (6) | 4 (9) | 1 (6) |
| Headache | 6 (7) | 1 (2) | 0 |
| Nausea | 3 (3) | 3 (7) | 0 |
| Stomatitis | 1 (1) | 3 (7) | 0 |
| Hyperuricemiaj | 0 | 3 (7) | 0 |
Difference between arm A and arm B is statistically significant (P = 0.002).
Difference between arm A and arm B is statistically significant (P < 0.001).
Spontaneous pneumothorax and unstable angina.
Pneumonia and Castleman disease, the former assessed as not drug‐related and the latter as drug‐related.
Nausea and vomiting.
Malaise.
Pneumonia and anemia, the latter assessed as study drug–related.
Difference between arm A and arm B is statistically significant (P < 0.001).
Difference between arm A and arm B is statistically significant (P = 0.002).
Difference between arm A and arm B is statistically significant (P = 0.037).
Grade ≥3 laboratory abnormalities were rare across all treatment arms. In arm A patients, no grade ≥3 abnormalities occurred in hemoglobin, alanine aminotransferase, or aspartate aminotransferase levels. One patient with compensated cirrhosis treated with G/P (arm C) had a grade 3 elevation in total bilirubin (predominantly indirect bilirubin) at week 1, which had returned to baseline (grade 2) on the day 15 visit, with no concomitant alanine aminotransferase elevation. One patient in arm B treated with SOF + RBV had a grade 3 elevation in total bilirubin level lasting for approximately 58 days. The rates of postbaseline grade ≥2 levels in hemoglobin and total bilirubin were statistically significantly lower in patients in arm A compared to arm B (Table 3).
Table 3.
Key Laboratory Abnormalities (Worsening From Baseline)
| Laboratory Abnormalities |
CERTAIN‐2 Without Cirrhosis |
CERTAIN‐1 Substudy 2 Compensated Cirrhosis |
|
|---|---|---|---|
|
Arm A G/P (n = 90), n (%) |
Arm B SOF + RBV (n = 46), n (%) |
Arm C G/P (n = 18), n (%) |
|
| Hemoglobin | |||
| Grade 2 (8‐10 g/dL)a | 1 (1) | 4 (9) | 2 (11) |
| Grade ≥3 (<8 g/dL) | 0 | 1 (2) | 0 |
| Alanine aminotransferase | |||
| Grade 2 (>3‐5 × ULN) | 0 | 0 | 0 |
| Grade ≥3 (>5 × ULN) | 0 | 0 | 0 |
| Aspartate aminotransferase | |||
| Grade 2 (>3‐5 × ULN) | 0 | 0 | 1 (6) |
| Grade ≥3 (>5 × ULN) | 0 | 0 | 0 |
| Total bilirubin | |||
| Grade 2 (>1.5‐3 × ULN)b | 4 (4) | 9 (22) | 2 (11) |
| Grade ≥3 (>3 × ULN) | 0 | 1 (2) | 1 (6) |
Difference in grade ≥2 between arms A and B was statistically significant (P = 0.017).
Difference in grade ≥2 between arms A and B was statistically significant (P = 0.005).
Abbreviation: ULN, upper limit of the normal range.
Discussion
Once‐daily 8‐week treatment with G/P was noninferior to 12‐week treatment with SOF + RBV in DAA‐naive HCV GT2‐infected patients without cirrhosis, as demonstrated by the primary endpoint being met in the CERTAIN‐2 study. SVR12 was achieved in 97.8% of Japanese patients with HCV GT2 infection without cirrhosis treated with G/P for 8 weeks; no virologic failures occurred. Additionally, 100% of patients with HCV GT2 infection and compensated cirrhosis in CERTAIN‐1 substudy 2 (arm C) achieved SVR12 following 12‐week treatment with G/P. The patient population treated with G/P included patient subgroups who have been previously considered difficult to treat such as those with a high baseline viral load (i.e., ≥6 million IU/mL; n = 6), those with non‐CC interleukin‐28 genotype (n = 26), those who failed to achieve SVR with prior IFN‐based treatment (n = 22), and those with NS3 and/or NS5A baseline polymorphisms (n = 83). High prevalence rates of baseline polymorphisms were detected in patients with HCV GT2a infection with no impact on treatment outcome as no virologic failures occurred in patients treated with G/P.
G/P was well tolerated in patients without cirrhosis and in those with compensated cirrhosis with no drug‐related serious AEs reported; no clinically significant laboratory abnormalities in hemoglobin, alanine aminotransferase, or aspartate aminotransferase levels; and only one patient experiencing a grade ≥3 transient bilirubin elevation. Two patients (<2%) discontinued study drug due to an AE. In patients without cirrhosis, no AEs occurred with a frequency >10%. Three AEs, pruritis (22%), increased blood bilirubin (11%), and nasopharyngitis (11%), occurred with a frequency >10% in the 18 patients with compensated cirrhosis. Additionally, no patient experienced laboratory abnormalities indicating liver disease progression, no patient experienced hepatic decompensation, and no cases occurred that were consistent with drug‐induced liver injury.
Currently, no HCV treatment for GT2 is approved in Japan for a duration of less than 12 weeks or without RBV. The elimination of RBV from DAA regimens can improve tolerability and reduce rates of treatment discontinuation due to AEs as RBV is associated with decreases in hemoglobin and elevations of indirect bilirubin.17 In the CERTAIN‐2 study, 35% of patients receiving SOF + RBV had anemia and 15% had elevations in blood bilirubin levels compared to no cases of anemia in patients treated with G/P and 1% with elevations in blood bilirubin level. The reduced treatment duration of 8 weeks for DAA‐naive, GT2‐infected patients without cirrhosis compared to the currently recommended treatment for Japanese patients (12 weeks of SOF + RBV for HCV GT2‐infected patients or 16 weeks of ombitasvir/paritaprevir/ritonavir + RBV for HCV GT2‐infected patients) is a significant reduction in treatment duration that, in addition to simplifying treatment, can help reduce the burden of managing concomitant medications, particularly in Japan's HCV patient population where about 70% are older than 60 years.18 A total of 39% and 15% of patients treated with G/P and reported on here were older than 65 and 75 years of age, respectively; 23% used liver protectants; 27% used calcium channel blockers; and 18% used proton‐pump inhibitors—and none of these patients experienced virologic failure. As no virologic failures occurred in patients with HCV GT2a (n = 75) or GT2b (n = 33) infection, these results indicate that HCV GT2 subtyping may not be required with G/P. The high efficacy and favorable tolerability observed with G/P in this study were similar to those observed in Japanese patients with GT1 HCV infection treated with G/P for 8 weeks19 and are consistent with studies of G/P conducted outside Japan.16
Limitations of this study include an open label design and the lack of an active comparator for patients with compensated cirrhosis. The use of objective, laboratory‐based efficacy endpoints and laboratory assessments for safety, however, mitigate these limitations. The relatively small number of patients in certain subgroups included in this study such as GT2 HCV‐infected patients with cirrhosis, patients with prior treatment experience, patients with a viral load ≥6 million log10 IU/mL, and patients with the interleukin‐28 non‐CC genotype precludes drawing further conclusions. However, high SVR12 rates have been observed in those subpopulations with relatively large numbers of patients in studies outside Japan.16, 20 An additional limitation is the use of SOF + RBV as an active comparator because other DAA regimens are approved to treat GT2 patients in regions other than Japan. Twelve‐week treatment with either SOF/ledipasvir or SOF/velpatasvir demonstrates high SVR12 rates with favorable safety profiles in GT2 patients outside Japan.21, 22, 23 However, while SOF/ledipasvir is approved to treat GT1 infections in Japan, neither of these regimens is currently approved or recommended for treatment of GT2 in Japan. Therefore, SOF + RBV was chosen as the comparator for this study because it is the standard of care for GT2 infection in Japan.
In summary, a high SVR12 rate was achieved with 8 weeks of G/P in treatment‐naive and IFN ± RBV treatment–experienced (DAA‐naive) patients with GT2 HCV infection without cirrhosis; no virologic failures occurred. Noninferiority of the 8‐week treatment with G/P compared to the 12‐week treatment with SOF + RBV was achieved. A 100% SVR12 rate was achieved in the 18 patients with GT2 HCV infection and compensated cirrhosis following 12 weeks of treatment with G/P; however, the size of this patient subgroup precludes drawing further conclusions. The regimen was well tolerated. These findings suggest that G/P may provide an all‐oral, IFN‐free and RBV‐free treatment option with high efficacy and favorable tolerability for GT2 HCV‐infected patients, largely independent of baseline patient demographics and disease characteristics, and a shorter treatment duration for GT2 HCV‐infected patients without compensated cirrhosis than currently recommended regimens.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.29510/suppinfo.
Supporting Information 1
Acknowledgment
We thank the patients, their families, and study investigators and coordinators. Medical writing support was provided by Maher Quraan, Ph.D., and Daniel O'Brien, Ph.D., both from AbbVie.
Potential conflict of interest: Dr. Chayama is on the speakers' bureau and received grants from MSD, AbbVie, Ajinomoto, Bristol‐Myers Squibb, Chugai, Sumitomo Dainippon, and Mitsubishi Tanabe. He is on the speakers' bureau for Abbott, Astellas, and Gilead. He received grants from Otsuka, Roche, Janssen, EA, and Toray. Dr. Atarashi received grants from AbbVie. Dr. Atsukawa is on the speakers' bureau and received grants from AbbVie. Dr. Kumada is on the speakers' bureau for AbbVie, MSD, Gilead, Bristol‐Myers Squibb, and Sumitomo Dainippon. Dr. Nakamuta is on the speakers' bureau and received grants from Bristol‐Myers Squibb and AbbVie. He received grants from MSD. Dr. Osaki is on the speakers' bureau for AbbVie, MSD, and Gilead. Dr. Sato received grants from AbbVie and MSD. Dr. Suzuki is on the speakers' bureau for Bristol‐Myers Squibb, AbbVie, and Gilead. Dr. Takaguchi is on the speakers' bureau for Bristol‐Myers Squibb, AbbVie, and AstraZeneca. Dr. Toyoda is on the speakers' bureau for AbbVie and Bristol‐Myers Squibb. Dr. Burroughs is employed by and owns stock in AbbVie. She owns stock in Merck. Dr. Kato is employed by and owns stock in AbbVie. Dr. Redman is employed by and owns stock in AbbVie. Dr. Pilot‐Matias is employed by and owns stock in AbbVie. Dr. Oberoi is employed by and owns stock in AbbVie. Dr. Fu is employed by and owns stock in AbbVie. Dr. Pugatch is employed by and owns stock in AbbVie. Dr. Alves is employed by and owns stock in AbbVie.
Supported by AbbVie (NCT02723084 and NCT02707952).
REFERENCES
- 1. Bennett H, Waser N, Johnston K, Kao JH, Lim YS, Duan ZP, et al. A review of the burden of hepatitis C virus infection in China, Japan, South Korea and Taiwan. Hepatol Int 2015;9:378‐390. [DOI] [PubMed] [Google Scholar]
- 2. Sievert W, Altraif I, Razavi HA, Abdo A, Ahmed EA, Alomair A, et al. A systematic review of hepatitis C virus epidemiology in Asia, Australia and Egypt. Liver Int 2011;31(Suppl. 2):61‐80. [DOI] [PubMed] [Google Scholar]
- 3. Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age‐specific antibody to HCV seroprevalence. Hepatology 2013;57:1333‐1342. [DOI] [PubMed] [Google Scholar]
- 4. Liu GG, DiBonaventura M, Yuan Y, Wagner JS, L'Italien GJ, Langley P, et al. The burden of illness for patients with viral hepatitis C: evidence from a national survey in Japan. Value Health 2012;15:S65‐S71. [DOI] [PubMed] [Google Scholar]
- 5. Yu ML, Chuang WL. Treatment of chronic hepatitis C in Asia: when East meets West. J Gastroenterol Hepatol 2009;24:336‐345. [DOI] [PubMed] [Google Scholar]
- 6. Messina JP, Humphreys I, Flaxman A, Brown A, Cooke GS, Pybus OG, et al. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 2015;61:77‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tanaka J, Kumagai J, Katayama K, Komiya Y, Mizui M, Yamanaka R, et al. Sex‐ and age‐specific carriers of hepatitis B and C viruses in Japan estimated by the prevalence in the 3,485,648 first‐time blood donors during 1995‐2000. Intervirology 2004;47:32‐40. [DOI] [PubMed] [Google Scholar]
- 8. Mizokami M, Tanaka Y, Miyakawa Y. Spread times of hepatitis C virus estimated by the molecular clock differ among Japan, the United States and Egypt in reflection of their distinct socioeconomic backgrounds. Intervirology 2006;49:28‐36. [DOI] [PubMed] [Google Scholar]
- 9. Chung H, Ueda T, Kudo M. Changing trends in hepatitis C infection over the past 50 years in Japan. Intervirology 2010;53:39‐43. [DOI] [PubMed] [Google Scholar]
- 10. Chayama K, Hayes CN, Ohishi W, Kawakami Y. Treatment of chronic hepatitis C virus infection in Japan: update on therapy and guidelines. J Gastroenterol 2013;48:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Omata M, Nishiguchi S, Ueno Y, Mochizuki H, Izumi N, Ikeda F, et al. Sofosbuvir plus ribavirin in Japanese patients with chronic genotype 2 HCV infection: an open‐label, phase 3 trial. J Viral Hepat 2014;21:762‐768. [DOI] [PubMed] [Google Scholar]
- 12. Sato K, Chayama K, Alves K, Toyoda H, Suzuki F, Kato K, et al. Randomized phase 3 trial of ombitasvir/paritaprevir/ritonavir and ribavirin for hepatitis C virus genotype 2‐infected Japanese patients. Adv Ther 2017;34:1449‐1465. [DOI] [PubMed] [Google Scholar]
- 13. Japan Society of Hepatology . Guidelines for the management of hepatitis C virus infection. Edition 5.4, published April 2017.
- 14. Sovaldi tablets [package insert]. Gilead, Tokyo, Japan. Version 7, published March 2017.
- 15. Ng TI, Krishnan P, Pilot‐Matias T, Kati W, Schnell GB, Beyer J, et al. In vitro antiviral activity and resistance profile of the next‐generation hepatitis C virus NS5A inhibitor pibrentasvir. Antimicrob Agents Chemother 2017;61:e02558‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Puoti M, Foster G, Wang S, Mutimer D, Gane E, Moreno C, et al. High SVR rates with eight and twelve weeks of pangenotypic glecaprevir/pibrentasvir: integrated efficacy and safety analysis of genotype 1‐6 patients without cirrhosis. J Hepatol 2017;66(Suppl.):S721. [Google Scholar]
- 17. Banerjee D, Reddy KR. Review article: safety and tolerability of direct‐acting anti‐viral agents in the new era of hepatitis C therapy. Aliment Pharmacol Ther 2016;43:674‐696. [DOI] [PubMed] [Google Scholar]
- 18. Kurosaki M. Treatment strategy for eldery patients with chronic hepatitis C. Igakunoayumi 2014;249:247‐252. (in Japanese). [Google Scholar]
- 19. Chayama K, Suzuki F, Karino Y, Kawakami Y, Sato K, Atarashi T, et al. CERTAIN‐1: efficacy and safety of glecaprevir/pibrentasvir in Japanese patients with chronic genotype 1 hepatitis C virus infection with and without cirrhosis. J Hepatol 2017;66(Suppl.):S527. [Google Scholar]
- 20. Forns X, Lee S, Valdes J, Lens S, Ghalib R, Aguilar H, et al. EXPEDITION‐I: efficacy and safety of glecaprevir/pibrentasvir in adults with chronic hepatitis C virus genotype 1, 2, 4, 5 or 6 infection and compensated cirrhosis. J Hepatol 2017;66(Suppl.):S3‐S4. [DOI] [PubMed] [Google Scholar]
- 21. Feld JJ, Jacobson IM, Hezode C, Asselah T, Ruane PJ, Gruener N, et al. Sofosbuvir and velpatasvir for HCV genotype 1, 2, 4, 5, and 6 infection. N Engl J Med 2015;373:2599‐2607. [DOI] [PubMed] [Google Scholar]
- 22. Asselah T, Boyer N, Saadoun D, Martinot‐Peignoux M, Marcellin P. Direct‐acting antivirals for the treatment of hepatitis C virus infection: optimizing current IFN‐free treatment and future perspectives. Liver Int 2016;36(Suppl. 1):47‐57. [DOI] [PubMed] [Google Scholar]
- 23. Gane EJ, Hyland RH, Yang Y, Svarovskaia E, Stamm LM, Brainard DM, et al. Efficacy of ledipasvir plus sofosbuvir for 8 or 12 weeks in patients with hepatitis C virus genotype 2 infection. Gastroenterology 2017;152:1366‐1371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.29510/suppinfo.
Supporting Information 1