

Abstract
Scope
Metabolic programming can occur not only in the perinatal period, but also post‐weaning. This study aims to assess whether fructose, in comparison to glucose, in the post‐weaning diet programs body weight, adiposity, glucose tolerance, metabolic flexibility, and health at adult age.
Methods and results
Three‐week‐old male and female C57BL6/JRccHsd mice are given an intervention diet with 32 energy percent (en%) glucose or fructose for only 3 weeks. Next, all animals are switched to the same 40 en% high fat diet for 9 weeks. Neither body weight nor adiposity differs significantly between the animals fed with glucose or fructose diets at any point during the study in both sexes. Glucose tolerance in adulthood is not affected by the post‐weaning diet, nor are activity, energy expenditure, and metabolic flexibility, as measured by indirect calorimetry. At the end of the study, only in females fasting serum insulin levels and HOMA‐IR index are lower in post‐weaning fructose versus glucose diet (p = 0.02), without differences in pancreatic β‐cell mass.
Conclusions
Our present findings indicate no adverse programming of body weight, adiposity, glucose tolerance, and metabolic flexibility by dietary (solid) fructose in comparison to glucose in the post‐weaning diet in mice.
Keywords: carbohydrates, indirect calorimetry, metabolic programming, metabolism, monosaccharides
1. Introduction
Obesity is considered a major public health problem. In 2014, worldwide over 600 million adults and 41 million children younger than five years of age were obese (whttp://www.who.int/mediacentre/factsheets/fs311/en/). Excessive intake of high energy foods and low physical activity are considered the most important factors contributing to the high global obesity prevalence. Furthermore, it has been shown that early life nutrition influences the susceptibility of an individual to obesity in later life, and also diabetes type II and cardiovascular diseases.1, 2, 3 This is “nutritional programming”, defined as the process whereby “a stimulus or insult (a nutrient) operating at a critical or sensitive period of development results in a long‐standing or life‐long effect on the structure or function of the organism”.4 It is of imminent importance for the development of preventive strategies to understand how early life diet affects later life health.
Focus in nutritional programming research has mainly been on caloric or protein restriction of mothers (in the perinatal period). However, evidence suggests that also carbohydrates can have programming effects. Supplementing maternal diet during gestation and lactation with sucrose reduced insulin sensitivity in the adult offspring in rats.5, 6 In mice, providing dams before and throughout gestation and lactation with sucrose led to hyperinsulinemia and decreased glucose tolerance in female offspring when adult, while food intake was increased in both sexes.7 Moreover, insulin levels were higher in weanlings of rat dams whose drinking water was supplemented with fructose, compared to weanlings of dams who received only tap water; glucose in drinking water of the dams did not affect insulin levels in offspring at weaning.8 Programming effects of carbohydrates have also been observed when exposure was only during lactation.9, 10 Formula feeding with a high‐carbohydrate formula (56 energy percent (en%) carbohydrates) increased β‐cell mass, hyperinsulinemia, and body weight (BW) in male rats, compared to rats reared by a dam on natural rat milk (≈8 en% carbohydrates).10 Similarly, in female pups, artificial rearing with high carbohydrate formula led to higher food intake, BW, and insulin levels in adulthood.9 These studies show that carbohydrates in the perinatal period can program metabolic health.
Moreover, also the period directly following lactation, the post‐weaning period, can be considered as a critical window of development, impacting lifelong metabolic health, although this is much less investigated. For example, post‐weaning dietary lipids arachidonic acid/docosahexaenoic acid supplementation prevented excessive BW gain in later life, and affected lipid metabolism beneficially.11 Similarly, the structure of lipid droplets (size and phospholipid coating) in the post‐weaning diet also affected body composition (BC) and metabolic profile.12 Little is known on post‐weaning programming by sugars, although sucrose (in comparison to starch) in weaning diet of rats was shown to increase circulating insulin and cholesterol levels at later life, when rats were switched to the starch diet.13
Sugars are a major component of the diet, in particular in infants and children. Overall, sugar energy intake in children represents 25% of total energy intake, being relatively higher than in adults with about 20 en% intake.14 High sugar diets are causally related to obesity, and consumption of sugar‐sweetened beverages is causally related to diabetes mellitus type II.15 Because of this high intake of simple carbohydrates and their role in the development of obesity, it is important to study their potential for nutritional programming in the post‐lactation period. While the amount of sugar in diets have been causally related to metabolic health, the effects of type of sugar are less clear.15 It has been postulated that especially high fructose consumption leads to higher adiposity and increased risk of metabolic syndrome.16, 17 However, this theory is under debate, as it is considered that fructose may not contribute more to metabolic syndrome than glucose.18, 19, 20
Underlying the debate on the different risks of glucose and fructose is the knowledge that the metabolism and intestinal uptake of the two monosaccharides is different. Circulating glucose is selectively taken up in various peripheral organs, while the majority of fructose is metabolized in the liver. There, fructose enters glycolysis, bypassing the rate limiting and tightly controlled step by phosphofructokinase. Part of glucose is also metabolized in the liver, yet at high concentrations of intracellular ATP inhibiting phosphofructokinase, glucose goes into glycogenesis to be stored.21 Moreover, glucose causes an insulin response in the blood, while fructose does not.22
In summary, young children are exposed to high dosages of simple sugars. As early childhood is considered a critical period of development, and glucose and fructose are metabolized differently, their respective effects on lifelong metabolic function and health might be different. Therefore, the aim of this study was to investigate whether fructose in comparison to glucose in the post‐weaning diet programs (negatively) for bodyweight, adiposity, glucose tolerance, and metabolic flexibility at adult age.
2. Experimental Section
2.1. Animals and Experimental Setup
All experimental procedures were approved by the Animal Experimental Committee (DEC 2014085, Wageningen) and complied with the principles of good laboratory animal care following the EU‐directive for the protection of animals used for scientific purposes (2010/63/EU). All experiments were carried out at controlled laboratory conditions (23 ±1 °C; 12:12 light dark cycle) with ad libitum access to food, unless stated otherwise.
Male and female C57BL/6JRccHsd mice (Harlan Laboratories BV, Horst, The Netherlands) were time mated and kept on a semi‐synthetic purified low fat breeding diet (Research Diet Services BV, Wijk bij Duurstede, The Netherlands) (see Supporting Information S1). One or 2 d after birth, litters were standardized to six pups per nest with at least two pups of each sex. Pups were used for the intervention study; see Figure 1A for an overview of the setup. Pups (males: n = 12 per group, females n = 14 per group) were weaned three weeks after birth, stratified by body weight (BW), housed individually and placed on the post‐weaning intervention diet for three weeks. The post‐weaning diets (Research Diet Services) contained 20 en% protein, 16 en% fat, and 64 en% carbohydrates of which 32 en% starch and 32 en% monosaccharides, being either glucose or fructose. At 6 weeks of age, all animals were switched to a high fat (HF) diet with 40 en% fat (for details on the dietary composition see Supporting Information S1). GLU and FRU are used to refer to animals on the glucose and fructose diets before diet switch, HF is added for the period after the diet switch (i.e. GLU‐HF and FRU‐HF). Food was refreshed every week. Food intake was determined by subtracting the weight of remaining pellets from the weight of the pellets provided. BW was measured weekly throughout the study. BC—lean mass (LM) and fat mass (FM)—was measured using the EchoMRI 100V (EchoMedical Systems, Houston, TX, USA). BC measurements were carried out weekly during the post‐weaning intervention period, and bi‐weekly once animals were on HF.
Figure 1.
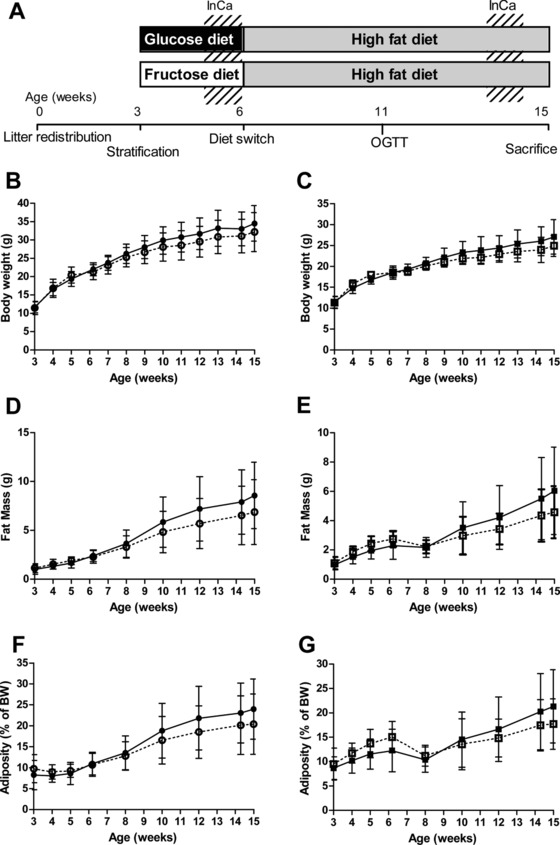
Study setup, body weight (BW) and body composition development over time. A) Schematic overview of the study setup. Briefly, litters were standardized to six pups per nest. In week 3, pups were stratified by BW and placed on a glucose or fructose diet (animals in these groups are GLU or FRU). In week 6, all animals were switched to the same high fat diet (HF; animals are referred to as GLU‐HF or FRU‐HF). Animals were tested in the indirect calorimetry (InCa) system in week 5 and week 14, an oral glucose tolerance test (OGTT) was performed in week 11. Animals were sacrificed in week 15. B) BW over the intervention period (week 3–6) and HF period (week 6–15) for males and C) females, D) fat mass (FM) for males and E) females, F) adiposity for males and G) females. Black symbols and lines represent GLU until week 6 and GLU‐HF from week 7 till week 15; open symbols and dotted lines represent FRU groups until week 6 and FRU‐HF groups from week 7 till week 15. Values represent mean± SD, n = 12 for males, n = 14 for females.
2.2. Diets
Diets were an adaptation from the BIOCLAIMS diet,23 adjusted for the fat content as recommended by AIN‐93 for growing and lactating animals.24 Moreover, all fructose was omitted from the breeding diet, to prevent contact with fructose in dams and offspring before feeding the post‐weaning diet.
2.3. Indirect Calorimetry (InCa)
A subset of the mice was placed into the Phenomaster LabMaster Metabolism Research Platform (TSE systems GmbH, Bad Homburg, Germany) for measurements of activity, energy expenditure (EE), respiratory exchange ratio (RER), and food intake (FI) as described previously.25 The animals were placed in the InCa at the end of the monosaccharide diet intervention, and again at the end of the HF period (Figure 1A). Measurements consisted of 24 h adaptation, 24 h recording of basal conditions, and a fasting‐refeeding challenge. The fasting‐refeeding challenge assesses metabolic flexibility in vivo, by determining an individual's ability to switch from fat oxidation to carbohydrate oxidation after a period of fasting, as determined by the RER. The challenge consisted of restricted feeding of own diet (1.5 g for low fat diets, 1.1 g for HF) 2 h before dark phase started. The next morning, fasting state was verified by low RER. The animals were refed ad libitum with the low fat glucose diet (Supporting Information S1), 1 h before dark phase on that day (25 h after the restricted feeding). The animals were taken out of the InCa 15 h after the refeeding, during the next light phase. BW and BC were determined before and after the InCa measurements. Animals that did not go into the InCa system, were fasted and refed according to the same procedure to ensure identical treatment.
2.4. Oral Glucose Tolerance Test (OGTT)
In week 11, OGTT analyses were performed as previously published,26 with minor modifications. Briefly, food was removed 1 h after start of the light phase. Mice remained without food for the following 5 h, after which blood glucose was measured via a tail cut with the Freestyle blood glucose system (Abbott Diabetes Care, Hoofddorp, The Netherlands) and 2 g glucose kg–1 BW was given by oral gavage. Fifteen, 30, 45, 60, 90, and 120 min after glucose administration, glucose concentration was determined (Freestyle). Glucose tolerance was analyzed with time course data and incremental area under the curve (iAUC).
2.5. Sacrifice
In week 15, animals were fasted for 2–5 h at the start of the light period, and sacrificed by decapitation to prevent effects of anesthesia on metabolic parameters including glucose levels.27 Animals were sacrificed in random order (males, females, and treatments randomized). A drop of blood was used to measure blood glucose levels (Freestyle), the rest of the blood was collected in MiniCollect® serum tubes (Greiner Bio one B.V., Alphen aan de Rijn, The Netherlands), and centrifuged for 10 min at 3000 × g and 4 °C to obtain serum. Serum samples were aliquoted and stored at –80 °C. Liver and gonadal fat pads were collected, weighed, and snap frozen in liquid nitrogen. Mesenteric fat and pancreas were excised at once, separated based on density by placing it in PBS, cut and weighed. Pancreata were fixated in 4% PAF overnight, and embedded in paraffin.
2.6. Serum Measurements
Serum leptin and adiponectin were measured using Bio‐Plex Pro mouse diabetes assays (Bio‐Rad laboratories, Veenendaal, the Netherlands) in accordance with the manufacturer's instructions. Samples were tested in duplicate; leptin samples were diluted ten times, adiponectin samples were diluted 1600 times. Insulin levels were measured using an Ultra‐Sensitive Mouse Insulin ELISA Kit (ChrystalChem, Downers Grove, Illinois, United States) according to the manufacturer's instructions. Samples were tested in duplicate and averaged. HOMA‐IR was calculated with the formula HOMA‐IR = (glucose)* (insulin)/14.1, according to van Dijk et al.,28 where 14.1 is a C57BL/6J mice adjusted factor (instead of 22.5 used for humans).
2.7. Liver Triglycerides
Liver triglyceride (TG) levels were determined using the Liquicolor kit (Human, Wiesbaden, Germany). Briefly, liver tissue (20mg mL–1) was placed in 10 mm Tris, 2 mm EDTA, 250 mm sucrose buffer, pH 7.5, and homogenized by sonication. Protein content of the liver homogenates was measured using a DC protein Assay (Biorad) according to the manufacturer's instructions. Samples were tested in triplicate, 100 μL of reagent was added to 4 μL sample.
2.8. Immunohistochemistry of the pancreas
Paraffin‐embedded pancreata were cut into sections of 5 μm. Five sections per sample, spaced at least 200 μm apart, were mounted on Superfrost plus slides (Menzel, Braunschweig, Germany) for immunohistochemistry, as published.29 In detail, sections were deparaffinized, rehydrated, and incubated in 1% H202 in methanol for 20 minutes. After microwave antigen retrieval in sodium citrate buffer, sections were blocked with 5% goat serum in PBS for 30 min. Sections were incubated at 4 °C overnight with primary rabbit anti‐insulin antibody (Cell signaling, Technology, Leiden, The Netherlands; 1:500). Negative controls were incubated with rabbit IgG solution (Vector Laboratories, Burlingame, California, United states). Sections were incubated for 60 min at room temperature with goat anti‐rabbit biotinylated antibody (Vector Laboratories; 1:200) followed by 60 min incubation with Vectastain Elite ABC kit (Vector Laboratories; 1:2000). For visualization, a 3‐3′diaminobenzidine kit (Immpact DAB, Vector Laboratories; 1:200) was used. Sections were counterstained with Hematoxylin QS (Vector laboratories). Specific staining was absent in controls. Pictures were taken with a Leica DM6 microscope (Leica Microsystems, Wetzlar, Germany) and merged to overview pictures of whole sections with LasX pc software (Leica). Pancreatic β‐cell area was determined with the ROI manager in ImageJ‐software, version 1.51f (http://rsbweb.nih.gov/ij/). DAB‐positive areas were manually encircled, and expressed as percentage of automatically calculated total area. β‐cell mass was estimated by multiplying the percentage of insulin positive surface area by the pancreas weight.
2.9. Hardness
Hardness of dietary pellets was measured using a Kahl device, as described.23
2.10. Statistics
Adiposity was calculated by dividing EchoMRI‐determined FM by BW. Data was analyzed in GraphPad Prism, version 5.04 (GraphPad Software Inc., San Diego, CA, USA). Data were checked for normality by D'Agostino and Pearson omnibus normality test. Normally distributed data were compared with an independent Students’ t‐test, with Welch's correction when applicable. Not normally distributed data were log (or square root) transformed and rechecked for normality. BW, FM, LM, and adiposity were checked by two‐way ANOVA. Findings were compared within sex. All data are shown as mean ± SD. Significance was defined at p < 0.05.
3. Results
BW did not significantly differ between the animals fed with fructose or glucose enriched diets in the 3 week post‐weaning period, nor during the following HF period (Figure 1B for males, Figure 1C for females). These animals are referred to as GLU or FRU when on the intervention diets, or as GLU‐HF and FRU‐HF during the high fat feeding period. FM was not significantly different at any point in males (Figure 1D) nor females (Figure 1E), although there was a trend for higher FM in FRU females in week 6 (p = 0.058, Table 1). However, the interaction post‐weaning diet × time was significant by two‐way ANOVA for BW in males and females, and for FM in females. Similarly, adiposity was not significantly affected by the post‐weaning diet (Figure 1F for males and 1G for females), nor during the HF period. In females, the interaction post‐weaning diet × time was significant. Lean mass was not different between groups at any point in the study (data not shown). Thus, body composition parameters did not differ at any specific time point, but the two‐way ANOVA analysis indicated a lower FM for the FRU‐HF females.
Table 1.
Body weight (BW), fat mass (FM) and cumulative food intake after the 3 weeks intervention and subsequent 9 weeks high fat (HF) diet
| Cumulative food intake intervention (g) | BW after intervention (g) | FM after intervention (g) | Cumulative food intake HF(g) | BW after HF period (g) | FM after HF period (g) | ||
|---|---|---|---|---|---|---|---|
| GLU Males | 63 ± 3 | 22.0 ± 1.7 | 2.4 ± 0.6 | GLU‐HF Males | 172 ± 13 | 34.5 ± 4.9 | 8.6 ± 3.4 |
| FRU Males | 71 ± 8a) | 21.2 ± 2.0 | 2.3 ± 0.7 | FRU‐HF Males | 171 ± 20 | 32.2 ± 5.3 | 6.9 ± 3.3 |
| GLU Females | 59 ± 5 | 18.5 ± 1.6 | 2.3 ± 0.9 | GLU‐HF Females | 162 ± 15 | 27.1 ± 4.1 | 6.0 ± 3.0 |
| FRU Females | 69 ± 7b) | 18.5 ± 1.2 | 2.8 ± 0.5 | FRU‐HF Females | 154 ± 18 | 24.9 ± 2.5 | 4.6 ± 1.8 |
Values represent mean ± SD, n = 12 for males, n = 14 for females.
a)Differs significantly between GLU males and FRU males (p < 0.01).
b)Differs significantly between GLU females and FRU females (p < 0.001).
Food intake measurements during the intervention seem to suggest a higher intake of the fructose intervention diet in both sexes (Table 1). However, the fructose intervention diet was more brittle and had a wetter appearance than the glucose intervention diet. This was reflected in the hardness of the diets, being 24.5 ± 3.0 kgf for glucose intervention diet and 4.1 ± 0.5 kgf for the fructose diet. Moreover, spillage of diet seemed to occur more often in the fructose group, thus likely overestimating food intake in the FRU animals. Interestingly, during the measurement in the InCa in week 5, no differences in 24 h FI were found between GLU and FRU (Table 2). Food intake of the HF was not affected by the post‐weaning intervention in regular measurements (Table 1) or during the 24 h basal measurement in the InCa (Table 2).
Table 2.
Energy expenditure, activity, and food intake during the 24 hour basal indirect calorimetry (InCa) measurements
| Average EE (KJ h–1) | Total activity (counts) | Cumulative FI (g) | |
|---|---|---|---|
| Week 5: | |||
| GLU Males | 1.84 ± 0.13 | 4.48 × 104 ± 7.28 × 103 | 4.14 ± 0.57 |
| FRU Males | 1.81 ± 0.08 | 4.23 × 104 ± 2.92 × 103 | 3.30 ± 1.36 |
| Week 14: | |||
| GLU‐HF Males | 2.19 ± 0.06 | 2.97 × 104 ± 7.28 × 103 | 3.44 ± 0.57 |
| FRU‐HF Males | 2.16 ± 0.19 | 3.28 × 104 ± 7.00 × 103 | 3.29 ± 0.44 |
| Week 5: | |||
| GLU Females | 1.73 ± 0.13 | 5.98 × 104 ± 8.05 × 103 | 3.57 ± 0.79 |
| FRU Females | 1.70 ± 0.15 | 5.61 × 104 ± 1.13 × 104 | 3.36 ± 0.69 |
| Week 14: | |||
| GLU‐HF Females | 1.95 ± 0.19 | 3.92 × 104 ± 1.08 × 104 | 3.09 ± 0.70 |
| FRU‐HF Females | 1.98 ± 0.20 | 4.39 × 104 ± 1.17 × 104 | 3.65 ± 0.74 |
Results are given for 24 h basal measurements. Energy expenditure (EE) is given as the average per hour of the 24 h measurements. Activity and food intake (FI) are cumulative over the 24 h. No significant differences were observed between dietary groups.
Energy expenditure (EE) and activity were not significantly different between GLU or FRU males, nor between FLU‐HF and FRU‐HF males (Table 2). For the females, also no difference in EE or activity between GLU and FRU was found, nor for GLU‐HF versus FRU‐HF, although in week 14 EE was higher and activity was lower compared to week 5 (see Table 2), likely due to increased BW and FM. RER values during 24 h basal measurements were not different between GLU males and FRU males (Figure 2A). During the fasting–refeeding measurements, RER values in fasted state were similar (Figure 2B and C), while RER was higher in the 2 h following the refeeding challenge in GLU compared to FRU males (Figure 2B and C). GLU males ate more in the two hours following the refeeding than the FRU males (Figure 2D). The GLU‐HF and FRU‐HF males did not differ in RER during the 24 h basal measurements (Figure 2E) or during the fasting–refeeding challenge (Figure 2F and G). Food intake in the 2 h after the refeeding was not different for GLU‐HF and FRU‐HF males (Figure 2H). As expected, RER showed a circadian rhythm over 24 h (Figure 2A), which was dampened when animals were fed the HF diet (Figure 2E). For females, no difference in RER during the basal measurements nor during the fasting–refeeding challenges were found between GLU and FRU animals in week 5 (Figure 3A–D), nor for GLU‐HF and FRU‐HF animals in week 14 (Figure 3E–G). Thus, energy metabolism parameters were not significantly different between the diets.
Figure 2.
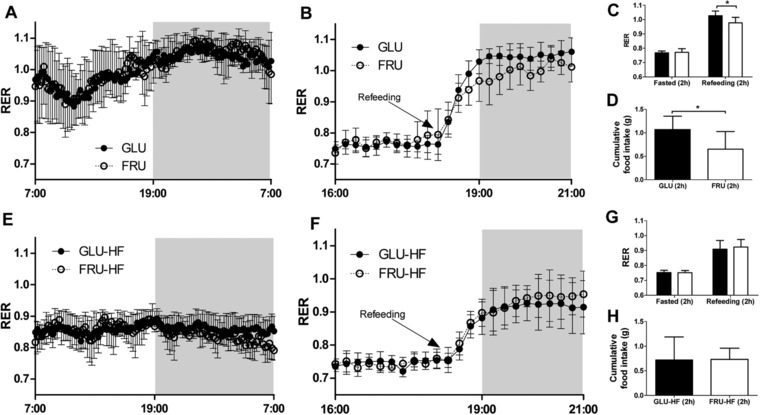
Respiratory exchange ratio (RER) during basal conditions and fasting‐refeeding challenge in males during the intervention (week 5) and during the HF diet (week 14). A) RER for males on glucose or fructose post‐weaning diet (week 5) during 24 hour basal measurement and B) during fasting‐refeeding challenge. C) Average RER for fasting‐refeeding challenge, values represent the average of two hours before and two hours after refeeding for males on GLU or FRU diet (week 5). D) Cumulative food intake for the first two hours after refeeding in the fasting‐refeeding challenge for GLU and FRU males (week 5). E) RER for GLU‐HF and FRU‐HF males (week 14) during 24 hour basal measurement and F) during fasting‐refeeding challenge. G) Average RER for fasting‐refeeding challenge, values represent the average of two hours before and two hours after refeeding for GLU‐HF and FRU‐HF males (week 14). H) Cumulative food intake for the first two hours after refeeding in the fasting‐refeeding challenge for GLU‐HF and FRU‐HF males (week 14). Values represent mean ± SD, n = 6–9. * p < 0.05.
Figure 3.
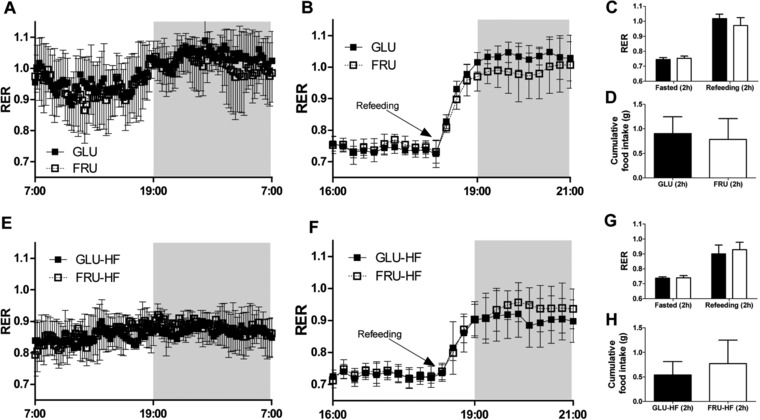
Respiratory exchange ratio (RER) during normal conditions and fasting‐refeeding challenge in females during the intervention (week 5) and during the HF diet (week 14). A) RER for females on glucose or fructose post‐weaning diet (week 5) during 24 hour basal measurements and B) during fasting‐refeeding challenge. C) Average RER for fasting‐refeeding challenge, values represent the average of two hours before and two hours after refeeding for females on GLU or FRU diet (week 5). D) Cumulative food intake for the first two hours after the refeeding in the fasting‐refeeding challenge for GLU and FRU females (week 5). E) RER for GLU‐HF and FRU‐HF females (week 14) during 24 h basal measurement and F) during fasting‐refeeding challenge. G) Average RER for fasting‐refeeding challenge, values represent the average of two hours before and two hours after refeeding for GLU‐HF and FRU‐HF (week 14). H) Cumulative food intake for the first two hours after the refeeding in the fasting‐refeeding challenge for GLU‐HF and FRU‐HF females (week 14). Values represent mean ± SD, n = 8–9.
Basal blood glucose levels were similar, and the response to the OGTT in week 11 was not significantly different between GLU‐HF and FRU‐HF in both males and females (Figure 4).
Figure 4.
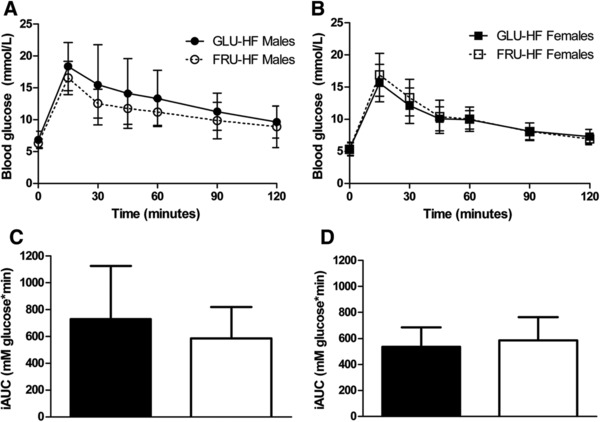
Oral glucose tolerance test (OGTT) in week 11. A) Blood glucose levels for males and B) females. C) Incremental area under the curve (iAUC) data for males and D) females. Black bars represent GLU‐HF groups, white bars FRU‐HF groups. Values represent mean ± SD, n = 12 for males, n = 14 for females.
At sacrifice, liver, pancreas, mesenteric white adipose tissue (WAT), and gonadal WAT weights were not different between GLU‐HF and FRU‐HF (Table 3), reflecting whole body FM observations. There was a trend for higher circulating leptin levels in GLU‐HF females compared to FRU‐HF females (p = 0.058), while adiponectin levels were not different (Table 3). In males, no difference in leptin or adiponectin levels was found (Table 3). Liver TG content in week 15 was not significantly altered by post‐weaning diet for males or females (Table 3). Insulin levels in week 15 were significantly higher for GLU‐HF females than FRU‐HF females, while blood glucose levels were not significantly affected (Figure 5B and D), even though these mice received the same HF diet for 9 weeks. Consequently, HOMA‐IR was higher for GLU‐HF compared to FRU‐HF females (Figure 5F), suggesting higher insulin resistance in GLU‐HF females than in FRU‐HF females. In males, blood glucose, insulin, or HOMA‐IR indexes were not significantly different in week 15 (Figure 5A, C and E).
Table 3.
Organ weights, serum parameters and hepatic TG levels at section (week 15)
| Organ weights | Serum Parameters | ||||||
|---|---|---|---|---|---|---|---|
| Liver weight (g) | Pancreas weight (g) | Mesenteric WAT weight (g) | Gonadal WAT weight (g) | Leptin (ng ml–1) | Adiponectin (μg mL–1) | Liver TG (μg mg–1 protein) | |
| GLU‐HF Males | 1.19 ± 0.35 | 0.50 ± 0.15 | 0.63 ± 0.27 | 0.66 ± 0.29 | 29.55 ± 16.99 | 14.23 ± 2.96 | 341 ± 226 |
| FRU‐HF Males | 1.10 ± 0.38 | 0.44 ± 0.12 | 0.51 ± 0.25 | 0.59 ± 0.34 | 20.63 ± 12.70 | 14.92 ± 3.88 | 254 ± 136 |
| GLU‐HF Females | 0.90 ± 0.20 | 0.35 ± 0.09 | 0.43 ± 0.26 | 0.41 ± 0.25 | 14.46 ± 9.70 | 17.55 ± 4.13 | 194 ± 94 |
| FRU‐HF Females | 0.83 ± 0.17 | 0.33 ± 0.07 | 0.30 ± 0.10 | 0.30 ± 0.13 | 8.66 ± 4.57 | 17.74 ± 4.47 | 178 ± 64 |
Data represent mean ± SD; n = 11–12 for males, n = 14 for females. No significant differences were observed between dietary groups.
Figure 5.
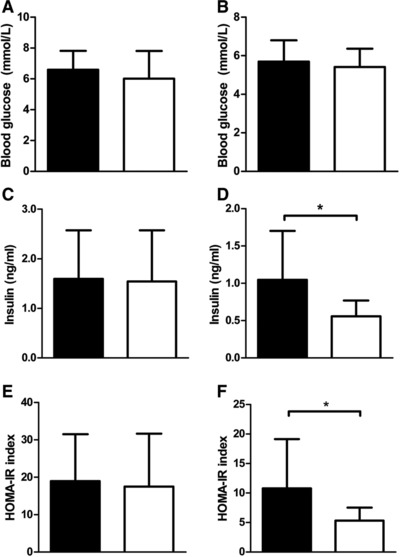
Blood glucose, insulin levels, and HOMA‐IR index week 15. A) Blood glucose levels (two hours fasted) for males and B) females. C) Serum insulin levels (ng/mL) males and D) females. E) HOMA‐IR index for males and F) females. Black bars represent GLU‐HF groups, white bars for the FRU‐HF groups. Values represent mean ± SD, n = 12 for males, n = 13–14 for females. *p < 0.05.
Pancreata of females were analyzed histologically (Supporting Information Figure S2) to see whether the effect on serum insulin was reflected in β‐cell density or mass. The percentage of β‐cell area (0.67% ± 0.17% and 0.75% ± 0.23% of total pancreas area) was not different between the GLU‐HF and FRU‐HF females, nor was β‐cell mass (Figure 6).
Figure 6.
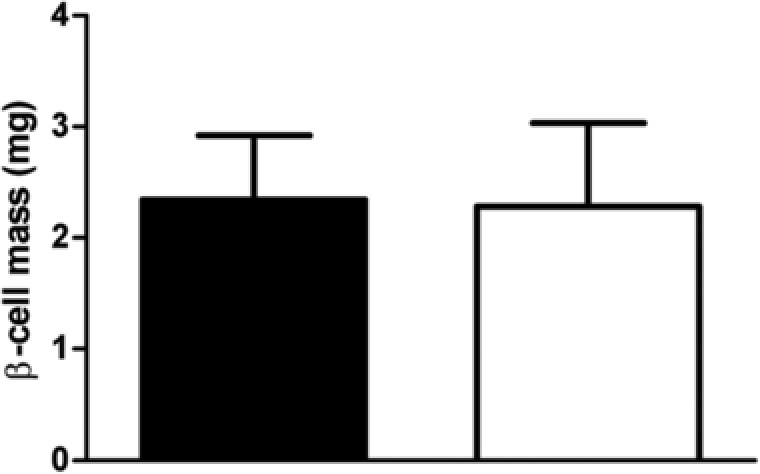
Pancreatic β‐cell mass in females in week 15. Black bars represent GLU‐HF females, white bars represent FRU‐HF females. Values represent mean ± SD, n = 6.
4. Discussion
Overall, the post‐weaning 3 week intervention with fructose versus glucose as part of pelletized diets neither significantly affected BW or BC (adiposity) directly, nor did it differently affect BW or adiposity in later life when the animals were switched to a fructose‐free HF diet for 9 weeks. Remarkably, serum insulin levels at the end of the study appeared to be lower in females fed fructose post‐weaning compared to females fed glucose post‐weaning.
In this study, two challenge tests were included to assess metabolic health: the fasting–refeeding challenge in InCa, and the classical OGTT. It is nowadays recognized that health is not a static state, but includes adaptation to changing environmental conditions and the flexibility to cope with these: the better the ability to adapt the healthier.30 The ability to adapt, or phenotypic flexibility, may be especially useful in nutrition research, where effects are usually small.30 Metabolic flexibility, or “the ability of a system to adjust fuel oxidation to fuel availability”,31 was tested with the fasting–refeeding challenge both during the post‐weaning intervention and during the HF period. Fasting–refeeding challenge did not show differences in the males on HF or in the females at both time points, indicating the metabolic flexibility is not affected nor programmed. Unfortunately, male GLU mice had a higher feed intake upon refeeding than FRU male mice, thereby hampering the ability to draw conclusions regarding the metabolic flexibility of these mice, as higher food intake can cause a higher RER.
The OGTT is a classically used challenge test. In this study, the OGTT in week 11 indicated that glucose tolerance was not affected by the type of monosaccharide in the post‐weaning diet.
However, basal insulin levels in the females in week 15 (thus after 9 weeks HF diet) suggest that insulin sensitivity was lower in the GLU‐HF females than in the FRU‐HF females. Possibly, insulin sensitivity was altered, yet this was not reflected by the OGTT in week 11, likely because impaired insulin sensitivity precedes altered glucose tolerance.32 Thus, insulin levels may have risen higher in GLU‐HF females than in FRU‐HF females during the OGTT in week 11, yet were still adequate for normal glucose homeostasis. Further studies are needed to elucidate whether extending the 40 en% HF diet period will ultimately lead to altered glucose tolerance by metabolic programming in early life.
The difference in fasting serum insulin levels between GLU‐HF and FRU‐HF females could not be explained by a difference in pancreatic β‐cell mass (Figure 6). Our intervention started at the post‐weaning state, thus excluding the transition phase from lactation to solid foods during weaning, which was shown to trigger a discrete maturation step of β‐cells, elevating the mitogenic and secretory responses to glucose in mice.33 Nonetheless, it might be that effects of the post‐weaning intervention on β‐cell mass were overruled by effects of the HF diet given in later life, as an HF diet can also lead to increased β‐cell mass and enlarged islets.34 Alternatively, peripheral insulin resistance in skeletal muscle, liver, or adipose tissue might underlie increased insulin production without altered β‐cell mass. Measurements of circulating insulin levels during an OGTT challenge, an euglycemic insulin clamp study, or an insulin inhibition test will be best for future in‐depth studies focused on insulin sensitivity in females. Overall, the evidence is not strong enough to conclude fructose versus glucose post‐weaning on later life insulin sensitivity is beneficial, but with confidence we can conclude that fructose compared to glucose does not show adverse effects on later life health for all parameters analyzed.
To investigate the metabolic programming of the post‐weaning diet on later life health, the fructose and glucose diets were replaced by an HF diet after 3 weeks. It was recently shown that continuing on a fructose rich diet (compared to a glucose rich diet) affects BW gain adversely: young male mice on a fructose diet (18 en%) gained more BW than on a glucose diet (18 en%), although this difference only appeared after 4 weeks.35 Here, FM in females had a tendency to be higher in the FRU group at the end of the intervention, suggesting FM could be negatively affected if the diet had been continued. Interestingly however, the trend in FM did not persevere when the females were fed HF diet, and serum leptin levels, a marker for adiposity, in week 15 even showed a reverse trend. In addition, the two‐way ANOVA analysis showed a significant interaction between post‐weaning diet and time. This may suggest that the females fed the (more adverse) fructose diet are slightly better able to cope with the adverse later life HF diet environment. These results in the females could be in line with the mismatch hypothesis in the developmental origins of health and disease,36 which postulates that a mismatch between the early life and the mature environment increases the risk of metabolic disease.
A limitation in this study is the quantification of FI during the 3 week intervention period with glucose and fructose diets, which was far less reliable for the fructose diet due to its crumbliness. The lower hardness and the wet appearance of the fructose intervention diet can be explained by the fact that fructose is more hygroscopic (water attracting) than glucose.37 That FI was not higher when measured in the 24 h basal measurement in the InCa (Table 1) seemed to suggest that intake of fructose and glucose diets were not different. Also the HF diet was crumbly due to its increased lipid content, but both groups received the same diet likely affecting FI measurements similarly.
Glucose and fructose are considered to have differential effects on brain appetite and reward pathways, as it is thought that fructose stimulates food intake via hypothalamic signaling, while glucose inhibits food intake (reviewed in 38). The difference in appetite effects is likely hormone mediated, as insulin, leptin, and glucagon‐like peptide‐1 release are stimulated by glucose, yet not by fructose, while the reduction in ghrelin is more pronounced with glucose intake.
Yet, even if the animals on the fructose intervention diet had a higher intake, this did not result in negative effects on the parameters studied here. This suggests that fructose indeed does not program adversely compared to glucose. In fact, insulin levels and insulin resistance as indicated by HOMA‐IR index appeared more adverse in GLU‐HF females compared to FRU‐HF females, even after 9 weeks HF feeding.
In conclusion, this study showed that, at least when incorporated in solid food, fructose and glucose are comparable given their direct physiological effect. Moreover, no adverse programming effects of dietary fructose in the post‐weaning diet in comparison to glucose on BW, adiposity, glucose tolerance, and metabolic flexibility were observed. If anything, for females, fructose rather than glucose in post‐weaning diet possibly enhanced insulin sensitivity in adulthood. Our results underscore and warrant future nutritional studies that are carefully designed to exclude effects of caloric load and focus on understanding mechanisms of effect of individual monosaccharides.
Abbreviations
- BC
body composition
- BW
body weight
- EE
energy expenditure
- FI
food intake
- FM
fat mass
- FRU
group fed fructose diet in early life
- FRU‐HF
group fed fructose diet in early life and high fat diet in later life
- GLU
group fed glucose diet in early life
- GLU‐HF
group fed glucose diet in early life and high fat diet in later life
- HF
high fat
- Inca
indirect calorimetry
- LM
lean mass
- RER
respiratory exchange ratio
- WAT
white adipose tissue
Conflict of Interest
AO is employee of Nutricia Research B.V., Utrecht, The Netherlands. The other authors declare no conflict of interest.
Supporting information
Supporting Information
Supporting Information
Acknowledgments
LB and JFC contributed equally to this work. AO, EvS, JFC, JK, HS and LB designed animal studies; JFC, HS and LB executed the animal studies, JFC and HS executed Inca experiments; EvS, JFC and HS analyzed Inca data, IvdS and LB performed laboratory analysis, which was analyzed by EvS, IvdS, JK and LB; AO, EvS, IvdS, JFC, JK, HS and LB wrote or critically revised the manuscript. This project was funded by the Dutch Technology Foundation STW (13509), which is part of the Netherlands Organization for Scientific Research (NWO), and which is partly funded by the Ministry of Economic Affairs. The authors like to thank Auke Vellinga and the staff of the animal facilities for their help in the execution of the experiment.
Bouwman L. M. S., Fernández‐Calleja J. M. S., Swarts H. J. M., van der Stelt I., Oosting A., Keijer J., van Schothorst E. M., Mol. Nutr. Food Res. 2018, 62, 1700315 https://doi.org/10.1002/mnfr.201700315
References
- 1. Fernandez‐Twinn D. S., Ozanne S. E., Ann. N. Y. Acad. Sci. 2010, 1212, 78. [DOI] [PubMed] [Google Scholar]
- 2. Langley‐Evans S. C., J. Hum. Nutr. Diet. 2015, 28, 1. [DOI] [PubMed] [Google Scholar]
- 3. Pico C., Palou M., Priego T., Sanchez J., Palou A., Front. Physiol. 2012, 3, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lucas A., Am. J. Clin. Nutr. 2000, 71, 602. [DOI] [PubMed] [Google Scholar]
- 5. D'Alessandro M. E., Oliva M. E., Ferreira M. R., Selenscig D., Lombardo Y. B., Chicco A., Clin. Exp. Pharmacol. Physiol. 2012, 39, 623. [DOI] [PubMed] [Google Scholar]
- 6. D'Alessandro M. E., Oliva M. E., Fortino M. A., Chicco A., Food Funct. 2014, 5, 446. [DOI] [PubMed] [Google Scholar]
- 7. Samuelsson A. M., Matthews P. A., Jansen E., Taylor P. D., Poston L., Front. Physiol. 2013, 4, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rawana S., Clark K., Zhong S., Buison A., Chackunkal S., Jen K. L., J. Nutr. 1993, 123, 2158. [DOI] [PubMed] [Google Scholar]
- 9. Srinivasan M., Dodds C., Ghanim H., Gao T., Ross P. J., Browne R. W., Dandona P., Patel M. S., Am. J. Physiol. Endocrinol. Metab. 2008, 295, E895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vadlamudi S., Hiremagalur B. K., Tao L., Kalhan S. C., Kalaria R. N., Kaung H. L., Patel M. S., Am. J. Physiol. 1993, 265, E565. [DOI] [PubMed] [Google Scholar]
- 11. Wielinga P. Y., Harthoorn L. F., Verschuren L., Schoemaker M. H., Jouni Z. E., van Tol E. A., Kleemann R., Kooistra T., Mol. Nutr. Food Res. 2012, 56, 1081. [DOI] [PubMed] [Google Scholar]
- 12. Oosting A., Kegler D., Wopereis H. J., Teller I. C., van de Heijning B. J., Verkade H. J., van der Beek E. M., Pediatr. Res. 2012, 72, 362. [DOI] [PubMed] [Google Scholar]
- 13. Moser P. B., Berdanier C. D., J. Nutr. 1974, 104, 687. [DOI] [PubMed] [Google Scholar]
- 14. Newens K. J., Walton J., J. Hum. Nutr. Diet. 2016, 29, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moore J. B., Fielding B. A., Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 303. [DOI] [PubMed] [Google Scholar]
- 16. Bray G. A., Popkin B.M., Diabetes care 2014, 37, 950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lustig R. H., Adv. Nutr. 2013, 4, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sievenpiper J. L., de Souza R. J., Cozma A. I., Chiavaroli L., Ha V., Mirrahimi A., Curr. Opin. Lipidol. 2014, 25, 8. [DOI] [PubMed] [Google Scholar]
- 19. Tappy L., BMC Biol. 2012, 10, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. White J. S., Adv. Nutr. 2013, 4, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tappy L., Le K. A., Physiol. Rev. 2010, 90, 23. [DOI] [PubMed] [Google Scholar]
- 22. Sun S. Z., Empie M. W., Nutr. Metab. (Lond.) 2012, 9, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hoevenaars F. P., van Schothorst E. M., Horakova O., Voigt A., Rossmeisl M., Pico C., Caimari A., Kopecky J., Klaus S., Keijer J., Genes Nutr. 2012, 7, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reeves P. G., Nielsen F. H., Fahey G. C., J. Nutr. 1993, 123, 1939. [DOI] [PubMed] [Google Scholar]
- 25. Hoevenaars F. P. M., Keijer J., Swarts H. J., Snaas‐Alders S., Bekkenkamp‐Grovenstein M., van Schothorst E. M., Exp. Physiolog. 2013, 98, 1053. [DOI] [PubMed] [Google Scholar]
- 26. Duivenvoorde L. P., van Schothorst E. M., Swarts H. M., Kuda O., Steenbergh E., Termeulen S., Kopecky J., Keijer J., PLoS One 2015, 10, e0128515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Constantinides C., Mean R., Janssen B. J., ILAR J. 2011, 52, e21. [PMC free article] [PubMed] [Google Scholar]
- 28. van Dijk T. H., Laskewitz A. J., Grefhorst A., Boer T. S., Bloks V. W., Kuipers F., Groen A. K., Reijngoud D. J., Lab. Anim. 2013, 47, 79. [DOI] [PubMed] [Google Scholar]
- 29. Kruit J. K., Wijesekara N., Westwell‐Roper C., Vanmierlo T., de Haan W., Bhattacharjee A., Tang R., Wellington C. L., LutJohann D., Johnson J. D., Brunham L. R., Verchere C. B., Hayden M. R., Diabetes 2012, 61, 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stroeve J. H., van Wietmarschen H., Kremer B. H., van Ommen B., Wopereis S., Genes Nutr. 2015, 10, 459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Galgani J. E., Moro C., Ravussin E., Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DeFronzo R. A., Tripathy D., Diabetes Care 2009, 32, S157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stolovich‐Rain M., Enk J., Vikesa J., Nielsen F. C., Saada A., Glaser B., Dor Y., Dev. Cell 2015, 33, 238. [DOI] [PubMed] [Google Scholar]
- 34. Roat R., Rao V., Doliba N. M., Matschinsky F. M., Tobias J. W., Garcia E., Ahima R. S., Imai Y., PloS One 2014, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rendeiro C., Masnik A. M., Mun J. G., Du K., Clark D., Dilger R. N., Dilger A. C., Rhodes J. S., Sci. Rep. 2015, 5, 9589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gluckman P. D., Hanson M. A., Beedle A. S., Am. J. Hum. Biol. 2007, 19, 1. [DOI] [PubMed] [Google Scholar]
- 37. Donnelly B. J., Fruin J. C., Scallet B. L., Cereal Chem. 1973, 50, 512. [Google Scholar]
- 38. Page K. A., Melrose A. J., Curr. Opin. Behav. Sci. 2016, 9, 7.26858972 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information