

Abstract
Objective
FX006 is a novel, microsphere‐based, extended‐release formulation of triamcinolone acetonide for intraarticular (IA) injection designed to maintain treatment concentration in the joint and provide prolonged analgesic benefits in patients with osteoarthritis (OA) of the knee. This study was undertaken to compare the analgesic benefits of 2 FX006 doses with saline placebo injection.
Methods
In this phase IIb study, participants with knee OA (Kellgren/Lawrence grade 2–3) and average daily pain (ADP) intensity ≥5 to ≤9 (on a 0–10 Numerical Rating Scale) were randomized (1:1:1) to receive single IA injections of FX006 32 mg (n = 104) or 16 mg (n = 102) or saline placebo (n = 100). The primary end point was the least squares mean (LSM) change from baseline to week 12 in weekly mean ADP intensity scores for FX006 32 mg versus saline placebo.
Results
The primary end point was not met (LSM change at week 12 −3.1 with FX006 32 mg versus −2.5 with saline placebo; LSM difference [95% confidence interval] −0.58 [−1.22, 0.07]) (P = 0.08). However, improvements in ADP intensity were significantly greater with FX006 32 mg than saline placebo at weeks 1–11 and week 13. Improvements in ADP intensity were significantly greater with FX006 16 mg versus saline placebo at weeks 1–9. A dose‐response effect in duration of maximal analgesic effect was evident (13 weeks with 32 mg versus 9 weeks with 16 mg), with FX006 32 mg providing increased therapeutic benefit relative to FX006 16 mg. All treatments were well tolerated.
Conclusion
Although the primary end point was not met, our findings indicate a prolonged reduction in symptoms with FX006 with an evident dose response and a safety profile similar to saline placebo.
Osteoarthritis (OA) of the knee is characterized by pain, progressive cartilage destruction, subchondral bone changes, and joint inflammation 1. Treatment guidelines recommend intraarticular (IA) corticosteroids 2. Standard IA corticosteroids provide moderate improvements in pain, but the magnitude of benefit rapidly wanes after injection 3. FX006, an extended‐release formulation of triamcinolone acetonide in 75:25 poly(lactic‐co‐glycolic acid) microspheres, was designed to maintain prolonged concentrations of triamcinolone acetonide in the joint, with the intent to improve analgesic effect and reduce systemic exposure versus commercially available triamcinolone acetonide crystalline suspension. In a pharmacokinetic study (ClinicalTrials.gov identifier: NCT02637323), FX006 demonstrated prolonged residency in synovial fluid and reduced systemic exposure following IA injection versus triamcinolone acetonide crystalline suspension in people with knee OA 4.
In the first clinical study of efficacy following a single IA injection of FX006 (target low, mid, and high doses of 10 mg, 40 mg, and 60 mg, respectively) in patients with knee OA (ClinicalTrials.gov identifier: NCT01487161) 5, the mid dose of FX006 yielded pain relief superior to triamcinolone acetonide crystalline suspension 40 mg with treatment differences achieving statistical significance at weeks 5–10 (all P < 0.05) and numerical improvement at each of weeks 2–12. Pain relief with the high dose of FX006 compared with triamcinolone acetonide crystalline suspension was similar to that for the mid dose through week 6, but diminished from weeks 7 to 12. Pain relief was numerically improved with the low dose of FX006 compared with triamcinolone acetonide crystalline suspension at each of weeks 2–12, but the effect did not achieve statistical significance. Hence, the mid dose of FX006 was concluded to be the most efficacious tested in that trial 5 and further evaluation was deemed appropriate.
The present study (ClinicalTrials.gov identifier: NCT02116972) was conducted to confirm the appropriate target dose of FX006 and further assess FX006 efficacy and safety. Two notable differences exist between the previous dose‐ranging study 5 and the present one with regard to administration and dosing of FX006. First, the injection volume increased from 3 ml in the previous study to 5 ml in the present study following adjustment of the diluent volume to enhance microsphere dispersion and reduce aggregation. Second, the FX006 doses reported here (16 mg and 32 mg) reflect the amount of drug received by patients following an ~20% reduction during reconstitution, as determined by dose‐delivery studies.
Patients and Methods
In this phase IIb, double‐blind, parallel‐group, dose‐ranging, single‐injection study, participants were randomized (1:1:1, block of 6; by a centralized interactive web randomization system) to receive a single 5‐ml IA injection of FX006 16 mg, FX006 32 mg, or saline placebo. Participants and assessors were blinded with regard to treatment administered by a nonblinded injector. Details related to randomization, blinding, and the injection procedure are provided in the Supplementary text, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40364/abstract. After screening, participants were seen on day 1 (baseline) and at weeks 4, 8, 12, 16, 20, and 24.
Eligible participants (ages ≥40 years, body mass index ≤40 kg/m2) had knee OA according to the American College of Rheumatology clinical and radiologic criteria 6 for ≥6 months before screening, a Kellgren/Lawrence grade of 2 or 3 on a centrally read screening radiograph 7, pain in the index knee (defined as the most painful knee in participants with bilateral disease) on >15 days of the previous month, and a mean average daily pain (ADP) intensity score ≥5 and ≤9 (on an 11‐point Numerical Rating Scale [NRS]). Patients were excluded if they had ipsilateral hip OA, other arthritic/immune‐mediated inflammatory disorders, or unstable knee joints (e.g., torn anterior cruciate ligament) within 12 months of screening or received prior IA corticosteroids within 3 months of screening, prior IA hyaluronic acid injections in the index knee within 6 months of screening, prior FX006 at any time, or intramuscular, oral, inhaled, intranasal, or topical corticosteroids within 2 weeks of screening.
From screening through week 24, participants logged daily ADP intensity scores via an interactive voice response system, using a 0–10 NRS (where 0 = “no pain” and 10 = “pain as bad as you can imagine”). Participants completed the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) 8 on day 1 prior to randomization and at weeks 4, 8, 12, 16, 20, and 24, and the Patient Global Impression of Change (PGIC) 9 at weeks 4, 8, 12, 16, 20, and 24.
Safety was evaluated via adverse events (AEs) spontaneously reported or discovered by the investigator from information obtained via patient electronic diaries, routine physical/laboratory evaluations, and assessments of the index knee by an investigator who was blinded with regard to treatment group. After informed consent was obtained, and for ≥7 days pretreatment, analgesic medications for index knee pain were not to be taken or used during the study with the exception of study‐issued acetaminophen (≤3,000 mg/day in sponsor‐provided 500‐mg tablets) as rescue pain treatment.
The primary efficacy end point, change from baseline to week 12 in weekly mean ADP intensity scores for FX006 32 mg versus saline placebo in the full analysis set (all randomized and treated participants), was analyzed using longitudinal mixed model for repeated measures (MMRM) methodology on observed data with no imputation for missing data (see the Supplementary text, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40364/abstract). A sample size of ~300 participants (100 per treatment arm) was estimated to provide 80% power (2‐sided alpha level of 0.05) if the true underlying primary end point treatment effect was 1.0 on the 0–10 NRS.
All secondary efficacy end points were first compared between FX006 32 mg and saline placebo, followed by FX006 16 mg versus saline placebo. Predefined key secondary end points (in order of step‐down testing) were change from baseline to week 12 in WOMAC physical function and PGIC scores, and changes in weekly mean ADP intensity scores from baseline to weeks 16, 20, and 24. Additional secondary outcomes included percent of responders according to Outcome Measures in Rheumatology (OMERACT)–Osteoarthritis Research Society International (OARSI) “strict” criteria (defined as ≥50% improvement and an absolute improvement of ≥20 points from baseline in either ADP intensity or the WOMAC physical function subscale) 10 at weeks 4, 8, 12, 16, 20, and 24; change from baseline to each week (except weeks 12, 16, 20, and 24 as outlined above, which are specified as the primary and secondary end points) in weekly mean ADP intensity score; change from baseline to each of weeks 4, 8, 12, 16, 20, and 24 in the WOMAC pain subscale; change from baseline to each of weeks 4, 8, 16, 20, and 24 in the WOMAC physical function subscale and PGIC; and time to onset of pain relief (time to first ADP intensity assessment showing >30% improvement from baseline).
To quantify the magnitude of difference between FX006 16 mg and 32 mg and saline placebo, standardized effect sizes were determined post hoc using methods described previously 11 (see the Supplementary text, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40364/abstract). Safety summaries included treated patients.
This trial was conducted according to Good Clinical Practice guidelines. Each site's governing ethics body approved the protocol, and participants provided written informed consent.
Results
Patient disposition and baseline characteristics. The trial was conducted from April 29, 2014 to August 8, 2015. Participants were screened at 48 sites (43 in the US and 5 in Canada) and enrolled at 44 sites (40 in the US and 4 in Canada). Among 310 randomized participants, 306 were treated (4 randomized to saline placebo were not treated). Approximately 8% and 18% of participants prematurely discontinued participation through week 12 (primary end point) and week 24 (study completion), respectively (Figure 1). Ultrasound guidance during IA injection was used in 4 patients.
Figure 1.
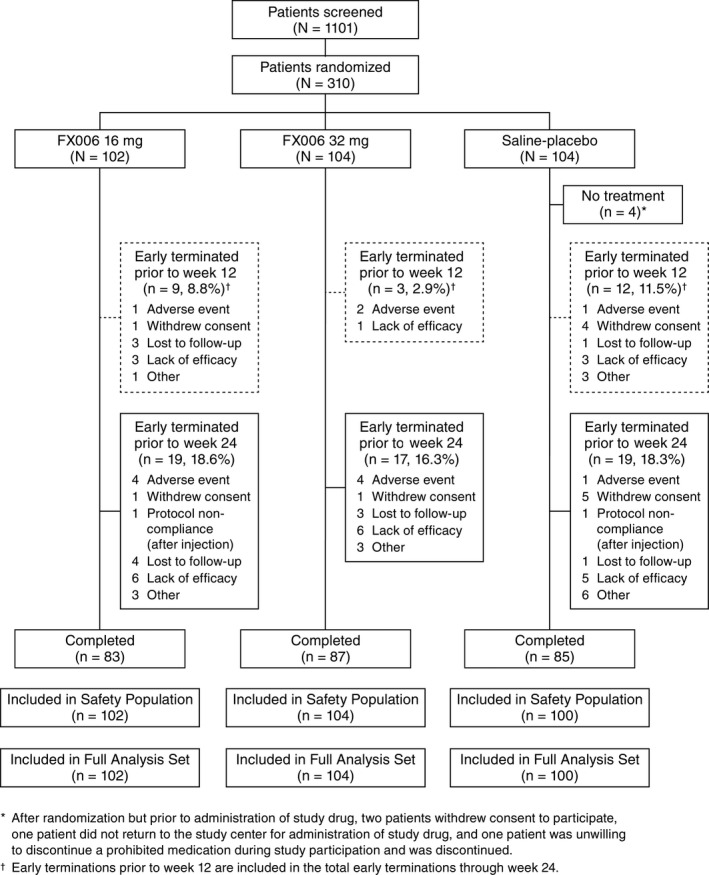
Disposition of the patients.
All baseline characteristics, except sex, were generally well‐balanced across arms. A higher proportion of men comprised the FX006 32 mg group (51%) compared with the 16 mg group (39%) or saline placebo group (39%). Baseline ADP intensity scores (mean 6.6) indicated substantial daily pain at a moderate level of intensity (Table 1).
Table 1.
Baseline demographic and disease characteristics of the patients with OA (full analysis set)a
| FX006 16 mg (n = 102) | FX006 32 mg (n = 104) | Saline placebo (n = 100) | Total (n = 306) | |
|---|---|---|---|---|
| Male, no. (%) | 40 (39.2) | 53 (51.0) | 39 (39.0) | 132 (43.1) |
| White, no. (%) | 81 (79.4) | 85 (81.7) | 82 (82.0) | 248 (81.0) |
| Age at consent, mean ± SD years | 58.2 ± 8.34 | 58.7 ± 8.06 | 59.7 ± 8.23 | 58.8 ± 8.20 |
| BMI, mean ± SD kg/m2 | 30.6 ± 4.86 | 31.0 ± 4.55 | 31.2 ± 5.11 | 30.9 ± 4.84 |
| Weight category, no. (%) | ||||
| Underweight (BMI <18.5) | 2 (2.0) | 0 (0.0) | 0 (0.0) | 2 (0.7) |
| Normal weight (BMI 18.5–24.9) | 12 (11.8) | 10 (9.6) | 10 (10.0) | 32 (10.5) |
| Overweight (BMI 25.0–29.9) | 32 (31.4) | 35 (33.7) | 33 (33.0) | 100 (32.7) |
| Obesity class I (BMI 30.0–34.9) | 37 (36.3) | 39 (37.5) | 29 (29.0) | 105 (34.3) |
| Obesity class II (BMI 35.0–39.9) | 19 (18.6) | 18 (17.3) | 27 (27.0) | 64 (20.9) |
| Morbid obesity (BMI ≥40.0) | 0 (0.0) | 2 (1.9) | 1 (1.0) | 3 (1.0) |
| Type of knee OA, no. (%) | ||||
| Unilateral | 45 (44.1) | 46 (44.2) | 38 (38.0) | 129 (42.2) |
| Bilateral | 57 (55.9) | 58 (55.8) | 62 (62.0) | 177 (57.8) |
| Years since diagnosis, mean ± SD | 6.7 ± 6.66 | 7.2 ± 7.27 | 6.4 ± 5.79 | 6.8 ± 6.60 |
| Kellgren/Lawrence grade, no. (%) | ||||
| 2 | 33 (32.4) | 28 (26.9) | 37 (37.0) | 98 (32.0) |
| 3 | 69 (67.6) | 76 (73.1) | 63 (63.0) | 208 (68.0) |
| Prior index knee surgeries/interventions, no. (%) | ||||
| Surgery or procedure | 20 (19.6) | 17 (16.3) | 15 (15.0) | 52 (17.0) |
| IA steroid injection | 32 (31.4) | 35 (33.7) | 24 (24.0) | 91 (29.7) |
| IA hyaluronic acid injection | 6 (5.9) | 11 (10.6) | 19 (19.0) | 36 (11.8) |
|
ADP intensity, mean ± SD (0–10 scale) |
6.6 ± 0.97 | 6.5 ± 1.01 | 6.7 ± 1.08 | 6.6 ± 1.02 |
|
WOMAC pain subscale, mean ± SD (0–4 scale) |
2.3 ± 0.62 | 2.1 ± 0.58 | 2.3 ± 0.65 | 2.2 ± 0.62 |
|
WOMAC stiffness subscale, mean ± SD (0–4 scale) |
2.5 ± 0.74 | 2.4 ± 0.71 | 2.4 ± 0.68 | 2.4 ± 0.71 |
|
WOMAC physical function subscale, mean ± SD (0–4 scale) |
2.3 ± 0.68 | 2.1 ± 0.57 | 2.3 ± 0.65 | 2.2 ± 0.63 |
The Patient Global Impression of Change (PGIC) is the patient's report of change since baseline; thus, there is no PGIC assessment at baseline. OA = osteoarthritis; BMI = body mass index; IA = intraarticular; ADP = average daily pain; WOMAC = Western Ontario and McMaster Universities Osteoarthritis Index.
Efficacy. The primary end point was not met. The least squares mean (LSM) change in ADP intensity at week 12 was −3.1 with FX006 32 mg versus −2.5 with saline placebo. The LSM difference in ADP intensity (95% confidence interval [95% CI]) for FX006 32 mg versus saline placebo at week 12 was −0.58 (−1.22, 0.07) (P = 0.08). However, FX006 32 mg resulted in significant improvements in ADP intensity versus saline placebo at each of weeks 1–11 (P ≤ 0.036) and week 13 (P ≤ 0.039) and numerical improvements at week 12 and each of weeks 14–24 (Figure 2A). Patients receiving FX006 16 mg had significant improvements in ADP at weeks 1–9; thereafter, the difference in pain scores between FX006 16 mg and saline placebo was small and not statistically significant (Figure 2A). As such, a dose‐response effect was evident in the duration of maximal effect. FX006 16 mg and 32 mg had similar median time to onset of analgesic effect (day 4), which was more rapid than saline placebo (day 8). The maximum magnitude of analgesic effect was similar for FX006 16 mg and 32 mg (achieved at weeks 4–5; LSM difference −1.22 for FX006 16 mg at week 4, −1.23 for FX006 16 mg at week 5, −1.36 for FX006 32 mg at week 4, and −1.34 for FX006 32 mg at week 5) (Figure 2A).
Figure 2.
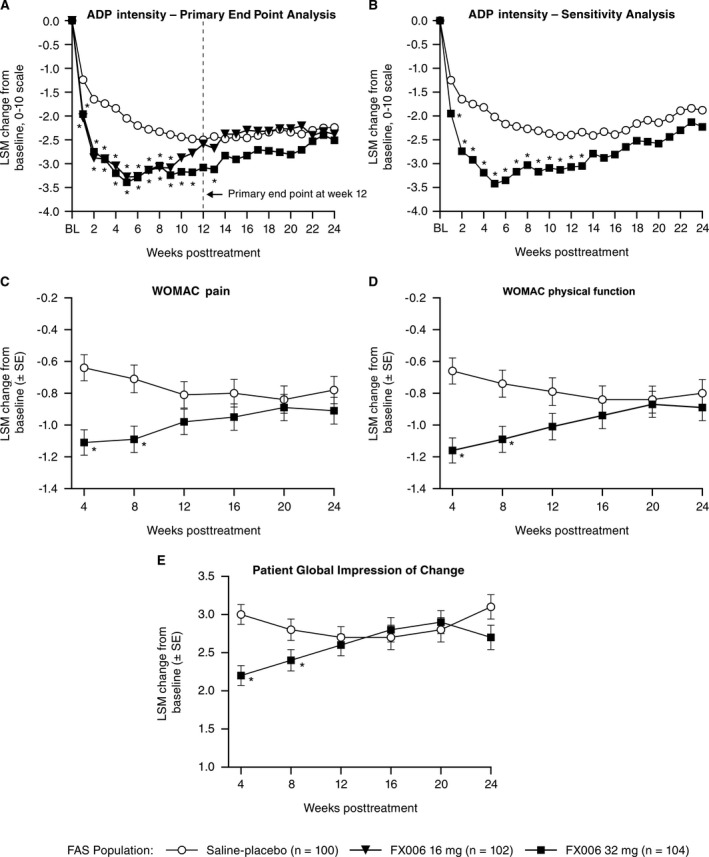
A, Least squares mean (LSM) change from baseline (BL) in weekly mean average daily pain (ADP) intensity scores at the primary end point at week 12 and key secondary end points at weeks 16, 20, and 24 (observed data; mixed model for repeated measures). B, Sensitivity analysis of ADP intensity (imputed data; last observation carried forward/baseline observation carried forward). C, Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain subscale. D, WOMAC physical function subscale (key secondary end point at week 12). E, Patient Global Impression of Change score (key secondary end point at week 12) through week 24. FAS = full analysis set. Values are the LSM ± SEM. * = P < 0.05.
Sensitivity analyses of the primary end point (last observation carried forward [LOCF]/baseline observation carried forward [BLOCF]) addressing patient discontinuations prior to week 12 showed that ADP intensity was significantly improved with FX006 32 mg versus saline placebo at each visit from weeks 1 to 13 (all P < 0.05), including week 12 (LSM difference [95% CI] −0.67 [−1.32, −0.02]; P = 0.042) (Figure 2B). Results of a multiple imputation sensitivity analysis of the primary end point demonstrated consistent differences between FX006 32 mg and saline placebo in ADP intensity changes from baseline to week 12 (LSM difference [95% CI] −0.65 [−1.30, 0.01]; P = 0.053) (data not shown). Consistent results were observed from a post hoc exploratory primary end point analysis performed with site added to the MMRM as a covariate (week 12 LSM difference [95% CI] −0.72 [−1.39, −0.05]; P = 0.034) (data not shown). Inclusion of the 4 patients who were randomized and not treated (i.e., the intent‐to‐treat population) yielded results identical to those of the analysis based on the full analysis set population, because none of these 4 patients had efficacy data after their screening visit.
Results from secondary analyses also favored FX006 32 mg compared with saline placebo. FX006 32 mg was associated with significantly improved WOMAC pain (Figure 2C), WOMAC physical function (Figure 2D), and PGIC (Figure 2E) scores, and resulted in a higher proportion of patients who achieved OMERACT‐OARSI “strict” responder criteria versus saline placebo at week 4 and week 8 (Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40364/abstract). Numerical advantage was maintained at week 12 for all of these end points except OMERACT‐OARSI “strict” responders. Secondary end point findings for FX006 16 mg versus saline placebo showed a similar pattern, with a reduced duration of effect, no trend favoring FX006 16 mg over saline placebo for improvements in ADP intensity at week 12, and lower OMERACT‐OARSI “strict” response rates at week 8 (Figure 2A and Supplementary Table 1).
Results from post hoc standardized effect size determinations indicated that the effect sizes based on ADP were consistently lower than those for the WOMAC pain subscale. The ADP effect sizes for FX006 32 mg at weeks 4, 8, and 12 were 0.27, 0.13, and 0.12, respectively, and the effect sizes for the WOMAC pain subscale were 0.72, 0.54, and 0.27, respectively. For each instrument, the effect sizes for FX006 16 mg were similar to those of FX006 32 mg at weeks 4 and 8. Consistent with prespecified secondary end points, effect sizes for both ADP and the WOMAC pain subscale were markedly lower for FX006 16 mg versus FX006 32 mg at week 12 (Supplementary Table 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40364/abstract).
Safety. Similar proportions of participants reported AEs through week 24 across treatment arms (Table 2). Most AEs were grade 1 or 2, nonserious, and considered unrelated to the study drug by investigators who were blinded with regard to treatment group. No deaths occurred. Serious AEs occurred in 4 (1.9%) of the FX006‐treated patients (left distal femur fracture in the 16 mg group and worsening left ankle OA, myocardial infarction, and rheumatoid arthritis in the 32 mg group). All were unrelated to study drug. No AE was consistent with postinjection flare. AEs causing study discontinuation occurred in 8 (3.9%) of the FX006‐treated patients and 1 (1.0%) of the saline placebo–treated patients (Table 2). The onset of such events was not temporally associated with study drug administration. All of these events were considered unrelated to study drug except for 1 grade 2 AE (in the FX006 16 mg group) and 1 grade 3 AE (in the saline placebo group). The incidence of index knee–related AEs was relatively low given the study population. AEs related to the injection procedure were observed in 2.0%, 0%, and 6.0% of FX006 16 mg–, FX006 32 mg–, and saline placebo–treated patients, respectively.
Table 2.
Summary of AEs among all treated patientsa
| FX006 16 mg (n = 102) | FX006 32 mg (n = 104) | Saline placebo (n = 100) | |
|---|---|---|---|
| Patients with ≥1 AE | 43 (42.2) | 46 (44.2) | 43 (43.0) |
| AEs occurring in >2.0% of patients in any treatment groupb | |||
| Arthralgia (any joint) | 10 (9.8) | 8 (7.7) | 16 (16.0) |
| Back pain | 3 (2.9) | 2 (1.9) | 2 (2.0) |
| Bronchitis | 3 (2.9) | 2 (1.9) | 2 (2.0) |
| Headache | 3 (2.9) | 4 (3.8) | 2 (2.0) |
| Joint swelling | 4 (3.9) | 5 (4.8) | 5 (5.0) |
| Ligament sprain | 4 (3.9) | 4 (3.8) | 2 (2.0) |
| Nasopharyngitis | 2 (2.0) | 2 (1.9) | 4 (4.0) |
| Neck pain | 0 (0.0) | 3 (2.9) | 0 (0.0) |
| Sinusitis | 3 (2.9) | 2 (1.9) | 1 (1.0) |
| Toothache | 0 (0.0) | 3 (2.9) | 1 (1.0) |
| Patients with ≥1 serious AE | 1 (1.0) | 3 (2.9) | 0 (0.0) |
| Patients with ≥1 AE leading to study discontinuation | 4 (3.9)c | 4 (3.8)d | 1 (1.0)e |
| Drug‐related | 1 (1.0)f | 0 (0.0) | 1 (1.0)f |
| Due to serious AE | 0 (0.0) | 1 (1.0)g | 0 (0.0) |
| Patients with ≥1 index knee–related AE | 15 (14.7) | 14 (13.5) | 17 (17.0) |
| Index knee–related AEs occurring in >2.0% of patients in any treatment groupb | |||
| Arthralgia | 8 (7.8) | 7 (6.7) | 14 (14.0) |
| Joint swelling | 4 (3.9) | 4 (3.8) | 3 (3.0) |
| Ligament sprain | 3 (2.9) | 1 (1.0) | 0 (0.0) |
| Patients with ≥1 index knee–related serious AE | 1 (1.0) | 1 (1.0) | 0 (0.0) |
| Patients with ≥1 index knee–related AE leading to study discontinuation | 4 (3.9)c | 3 (2.9)d | 1 (1.0)e |
| Drug‐related | 1 (1.0)f | 0 (0.0) | 1 (1.0)f |
| Due to serious AE | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Values are the number (%).
Listed in alphabetical order.
Joint effusion (day 131, grade 1, not related to study drug), arthralgia (day 57, grade 2, definitely related to study drug), and joint swelling (day 57, grade 3, not related to study drug) in 1 patient each, and arthralgia, arthritis, joint swelling, and synovial cyst all in 1 patient (day 88, all grade 2 and either not related or unlikely related to study drug). All adverse events (AEs) were index knee related.
Joint swelling (day 84, grade 1, not related to study drug), worsening ankle osteoarthritis (OA) (day 37, grade 2, not related to study drug, serious), positive Klebsiella test (day 21, subsequently considered to be a false‐positive finding by the investigator, grade 3, not related to study drug), and synovitis (day 112, grade 3, unlikely related to study drug). All AEs were index knee related except ankle OA.
Arthralgia (day 41, grade 3, probably related to study drug); index knee related.
Arthralgia.
Ankle OA.
Discussion
The primary end point of this study, significant improvement in ADP intensity at week 12 with FX006 32 mg versus placebo, was not achieved (P = 0.08); however, significant improvement with FX006 32 mg was seen at all time points from week 1 to week 11 (P ≤ 0.036) and at week 13 (P = 0.039). The placebo response at week 12 (LSM reduction of −2.5) was the largest reported over the 24‐week study. Studies show that placebo effects may confound the interpretation of clinical data 12 and are more pronounced with IA versus other routes of administration 13. Differences in saline placebo response across study sites were noted; a post hoc exploratory analysis with a site covariate included in the MMRM accounted for the variability in site responses, and the model demonstrated statistical significance for the primary end point at week 12 (P = 0.034). Sensitivity analyses (LOCF/BLOCF) that addressed patient discontinuations prior to week 12 indicated that FX006 32 mg was significantly superior to saline placebo at weeks 1–13 (P ≤ 0.042). Results of a multiple imputation analysis demonstrated consistent differences between FX006 32 mg and saline placebo in ADP intensity changes from baseline to week 12.
Although FX006 16 mg and 32 mg had comparable median times to onset of analgesia (day 4) and provided similar and significant maximal analgesic effects (beginning at approximately week 5) versus saline placebo, maximal effect persisted longer with FX006 32 mg (to approximately week 13) than 16 mg (to approximately week 9). Numerically larger pain relief, as measured by ADP intensity, was maintained with FX006 32 mg versus saline placebo at all time points through week 24. Other measures of OA signs/symptoms (WOMAC physical function subscale, PGIC, WOMAC pain subscale, and OMERACT‐OARSI “strict” response) were significantly improved with FX006 32 mg through week 8 with strong trends remaining at week 12; results for FX006 16 mg versus saline placebo showed a similar pattern but were not as robust or long lasting. A previous clinical study of FX006 in patients with OA demonstrated a dose‐dependent increase in synovial fluid concentrations of triamcinolone acetonide at week 6 following IA injection 14. It is postulated that there is a critical synovial fluid concentration required to maintain an analgesic effect and that the loss of analgesic effect after week 9 with FX006 16 mg is attributable to synovial fluid triamcinolone acetonide levels dropping below that critical concentration.
The clinical relevance of these findings was assessed with post hoc analysis of standardized effect size 11, a measure used to quantify the magnitude of difference between 2 treatment groups. An effect size >0.3 is considered an important change for a patient‐reported outcome 15. For FX006 32 mg compared with saline placebo, effect size for the WOMAC pain instrument exceeded 0.30 at weeks 4 and 8 and approached 0.30 at week 12. Effect size with the 16‐mg dose was comparable to that of the 32‐mg dose at week 4 but was notably lower at week 8, and no active treatment effect was seen at week 12. Effect sizes assessed with ADP were consistently lower than those for the WOMAC pain subscale for both FX006 doses at each of these time points. Across a large number of trials, the multi‐item, knee OA–specific WOMAC pain instrument has proved to be a more sensitive measure of treatment effect than the single‐item, general purpose ADP intensity 0–10 NRS 11. Overall, both FX006 doses demonstrated systemic and local safety profiles similar to saline placebo. No FX006 dose relationship in AEs was apparent.
In conclusion, although the study's primary end point was not met, the dose effect on duration of analgesic efficacy observed in this phase IIb study of people with knee OA confirms that FX006 32 mg confers increased therapeutic benefit relative to FX006 16 mg with similar safety, and that improvements in pain afforded by FX006 32 mg were of a magnitude that would be important to patients.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Conaghan had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Conaghan, Lufkin, Johnson, Bodick.
Acquisition of data
Lufkin, Bodick.
Analysis and interpretation of data
Conaghan, Cohen, Berenbaum, Lufkin, Johnson, Bodick.
Role of the Study Sponsor
Flexion Therapeutics sponsored and funded this study. Together with guidance from regulatory authorities, a subset of the authors (including the corresponding author) worked with the sponsor to develop the study design, study protocol, and statistical analysis plan, and subsequently to interpret study data. Site management and monitoring were provided by contract research organizations (CROs). Data collection and database maintenance were performed by a CRO. A full audit trail was maintained from the time of data entry to database lock. The database was then securely transferred to a separate CRO for the conduct of statistical analyses (by Karen Ozer and Teresa Curto, Cytel, Waltham, MA). All authors participated in data interpretation and manuscript development in collaboration with a professional medical writer/editor (Michelle L. Perate, MS) funded by the sponsor. All authors had full access to the study data, and the corresponding author takes final responsibility for the decision to submit for publication. No author external to the sponsor received financial compensation to write the manuscript. Publication of this article was not contingent upon approval by Flexion Therapeutics.
Supporting information
Supplementary text
Supplementary Table 1
Supplementary Table 2
Acknowledgments
The authors thank the FX006‐2014‐006 study participants and their enrolling investigators.
ClinicalTrials.gov identifier: NCT02116972.
The views expressed herein are those of the authors and do not necessarily reflect those of the NHS, the NIHR, or the Department of Health.
Supported by Flexion Therapeutics. Dr. Conaghan's work is supported in part by the NIHR Leeds Biomedical Research Centre.
Dr. Conaghan has received consulting fees from AbbVie, Flexion Therapeutics, Infirst, Medivir, Merck Serono, Novartis, and Ono Pharmaceutical (less than $10,000 each). Dr. Cohen has received consulting fees from Flexion Therapeutics (less than $10,000). Dr. Berenbaum has received consulting fees, speaking fees, and/or honoraria from AbbVie, Biogaran, Biogen, Expanscience, Flexion Therapeutics, IBSA, Janssen, Merck Serono, Novartis, Pfizer, Sanofi, Servier, TRB Chemedica, and UCB (less than $10,000 each). Ms Lufkin owns stock or stock options in Flexion Therapeutics. Dr. Johnson has received consulting fees from Acura Pharmaceuticals, Flexion Therapeutics, Iroko Pharmaceuticals, iX Biopharma, and Tolmar (more than $10,000 each). Dr. Bodick owns stock or stock options in Flexion Therapeutics, holds a patent for corticosteroids for the treatment of joint pain, and has patent applications pending related to corticosteroid formulations for maintaining corticosteroid synovial fluid formulations, corticosteroid formulations and methods for the treatment of joint pain in patients with type 2 diabetes mellitus, and corticosteroid formulations and methods for the treatment of joint pain in patients with diabetes.
References
- 1. Goldring SR, Goldring MB. Clinical aspects, pathology and pathophysiology of osteoarthritis. J Musculoskelet Neuronal Interact 2006;6:376–8. [PubMed] [Google Scholar]
- 2. McAlindon TE, Bannuru RR, Sullivan MC, Arden NK, Berenbaum F, Bierma‐Zeinstra SM, et al. OARSI guidelines for the non‐surgical management of knee osteoarthritis. Osteoarthritis Cartilage 2014;22:363–88. [DOI] [PubMed] [Google Scholar]
- 3. Da Costa BR, Hari R, Juni P. Intra‐articular corticosteroids for osteoarthritis of the knee. JAMA 2016;316:2671–2. [DOI] [PubMed] [Google Scholar]
- 4. Kraus VB, Conaghan PG, Aazami HA, Mehra P, Kivitz AJ, Lufkin J, et al. Synovial and systemic pharmacokinetics of triamcinolone acetonide following intra‐articular injection of an extended‐release microsphere‐based formulation (FX006) or standard crystalline suspension in patients with knee osteoarthritis. Osteoarthritis Cartilage 2017. E‐pub ahead of print. [DOI] [PubMed] [Google Scholar]
- 5. Bodick N, Lufkin J, Willwerth C, Kumar A, Bolognese J, Schoonmaker C, et al. An intra‐articular, extended‐release formulation of triamcinolone acetonide prolongs and amplifies analgesic effect in patients with osteoarthritis of the knee: a randomized clinical trial. J Bone Joint Surg Am 2015;97:877–88. [DOI] [PubMed] [Google Scholar]
- 6. Altman R, Asch E, Bloch D, Bole G, Borenstein D, Brandt K, et al. Development of criteria for the classification and reporting of osteoarthritis: classification of osteoarthritis of the knee. Arthritis Rheum 1986;29:1039–49. [DOI] [PubMed] [Google Scholar]
- 7. Kellgren JH, Lawrence JS. Radiological assessment of osteo‐arthrosis. Ann Rheum Dis 1957;16:494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. WOMAC Osteoarthritis Index . WOMAC 3.1 Index: knee and hip osteoarthritis index. URL: http://www.womac.org/womac/index.htm.
- 9. Dworkin RH, Turk DC, Farrar JT, Haythornthwaite JA, Jensen MP, Katz NP, et al. Core outcome measures for chronic pain clinical trials: IMMPACT recommendations. Pain 2005;113:9–19. [DOI] [PubMed] [Google Scholar]
- 10. Pham T, van der Heijde D, Altman RD, Anderson JJ, Bellamy N, Hochberg M, et al. OMERACT–OARSI initiative: Osteoarthritis Research Society International set of responder criteria for osteoarthritis clinical trials revisited. Osteoarthritis Cartilage 2004;12:389–99. [DOI] [PubMed] [Google Scholar]
- 11. Dworkin RH, Peirce‐Sandner S, Turk DC, McDermott MP, Gibofsky A, Simon L, et al. Outcome measures in placebo‐controlled trials of osteoarthritis: responsiveness to treatment effects in the REPORT database. Osteoarthritis Cartilage 2011;19:483–92. [DOI] [PubMed] [Google Scholar]
- 12. Abhishek A, Doherty M. Mechanisms of the placebo response in pain in osteoarthritis. Osteoarthritis Cartilage 2013;21:1229–35. [DOI] [PubMed] [Google Scholar]
- 13. Bannuru RR, McAlindon TE, Sullivan MC, Wong JB, Kent DM, Schmid CH. Effectiveness and implications of alternative placebo treatments: a systematic review and network meta‐analysis of osteoarthritis trials. Ann Intern Med 2015;163:365–72. [DOI] [PubMed] [Google Scholar]
- 14. Bodick N, Lufkin J, Willwerth C, Hauben J, Kumar A, Boen P, et al. Prolonged joint residency of triamcinolone acetonide after an intra‐articular injection of FX006, a sustained release formulation for the treatment of osteoarthritis [abstract]. Osteoarthritis Cartilage 2015;23 Suppl:A360–1. [Google Scholar]
- 15. Zhang W, Nuki G, Moskowitz RW, Abramson S, Altman RD, Arden NK, et al. OARSI recommendations for the management of hip and knee osteoarthritis. Part III. Changes in evidence following systematic cumulative update of research published through January 2009. Osteoarthritis Cartilage 2010;18:476–99. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary text
Supplementary Table 1
Supplementary Table 2