

Abstract
The Gaucher Outcome Survey (GOS) is an international Gaucher disease (GD) registry established in 2010 for patients with a confirmed GD diagnosis, regardless of GD type or treatment status, designed to evaluate the safety and long‐term effectiveness of velaglucerase alfa and other GD‐related treatments. As of February 25, 2017, 1209 patients had enrolled, the majority from Israel (44.3%) and the US (31.4%). Median age at GOS entry was 40.4 years, 44.1% were male, and 13.3% had undergone a total splenectomy. Most patients had type 1 GD (91.5%) and were of Ashkenazi Jewish ethnicity (55.8%). N370S/N370S was the most prevalent genotype, accounting for 44.2% of genotype‐confirmed individuals (n = 847); however, there was considerable variation between countries. A total of 887 (73.4%) patients had received ≥1 GD‐specific treatment at any time, most commonly imiglucerase (n = 587), velaglucerase alfa (n = 507), and alglucerase (n = 102). Hematological and visceral findings at the time of GOS entry were close to normal for most patients, probably a result of previous treatment; however, spleen volume of patients in Israel was almost double that of patients elsewhere (7.2 multiples of normal [MN] vs. 2.7, 2.9 and 4.9 MN in the US, UK and rest of world), which may be explained by a greater disease severity in this cohort. This analysis aimed to provide an overview of GOS and present baseline demographic and disease characteristics of participating patients to help improve the understanding of the natural history of GD and inform the overall management of patients with the disease.
1. INTRODUCTION
Gaucher disease (GD) is an autosomal recessive disease caused by pathogenic variations in the GBA1 gene, which encodes the lysosomal enzyme β‐glucocerebrosidase, resulting in deficient activity of this enzyme.1 GD affects approximately 1 in 40 000 individuals worldwide1 and is characterized by glucocerebroside accumulation in lysosomes, resulting in disease manifestations primarily in the organs of the monocyte‐macrophage system.
GD may be regarded as a phenotypic continuum2 owing to its highly variable clinical presentation; however, patients continue to be classified into the traditionally recognized clinical variants of GD, types 1, 2, or 3, to facilitate patient management. Type 1, the non‐neuronopathic form, is typically characterized by splenomegaly, hepatomegaly, thrombocytopenia, and anemia.1 It is the most common form, estimated to affect 90% of diagnosed patients in Europe, North America, and Australia.1 According to a recent literature analysis of individuals who manifest the disease, the prevalence ranges from 0.70 to 1.75 in 100 000.3 There is a higher prevalence of type 1 GD among people of Ashkenazi Jewish ethnicity, with a birth incidence of about 1 in 850.1
As with other rare diseases, the development of effective management strategies for GD may be hampered by an incomplete understanding of the natural history of the disease, as well as the challenge of conducting large, long‐term, placebo‐controlled clinical trials. Since 1991, the standard of care for GD has been enzyme replacement therapy (ERT), initially with placenta‐derived alglucerase (now discontinued).4 Although regulatory status and approved indications may differ between countries, ERTs available for GD include imiglucerase (Cerezyme®, Sanofi/Genzyme Corporation, USA), produced in Chinese hamster ovary cells and differing from the native human enzyme by a single amino acid substitution;5, 6 taliglucerase alfa (Elelyso®, Pfizer, USA/Protalix, Israel), produced in a carrot‐cell system and having the same core amino acid sequence as the native human enzyme, differing by 2 and 7 amino acids at the N‐terminus and C‐terminus, respectively;7, 8, 9 and velaglucerase alfa (VPRIV®, Shire, USA), produced in a human cell line and having the same amino acid sequence as the human enzyme.10 Substrate reduction therapy has also been available since 2002, and includes miglustat (Zavesca®, Actelion, Switzerland)11 and eliglustat (Cerdelga®, Sanofi/Genzyme, USA).12 The pharmacological chaperone ambroxol is also under investigation for the treatment of patients with both neuronopathic and non‐neuronopathic forms of GD.13, 14
Observational studies, including prospective long‐term patient registries, can help to expand the knowledge base for rare diseases, allowing clinical information to be collected from larger, more heterogeneous populations than otherwise possible by clinical trials. This can help to better define the population distribution of the disease and its variability in presentation or severity, and assess response to treatment to inform management strategies. The Gaucher Outcome Survey (GOS) is an international GD‐specific registry established in 2010 for patients with a confirmed GD diagnosis, regardless of GD type or treatment status. The primary objectives of GOS are to evaluate the safety and long‐term effectiveness of velaglucerase alfa and other GD‐related treatments, to gain a better understanding of the natural history of GD, and to serve as a database for evidence‐based management of GD.
Here we provide an overview of GOS and present the baseline demographic and general disease characteristics of participating patients in the registry. This analysis also provides a baseline for additional analyses and makes comparisons between specific subpopulations within GOS.
2. MATERIALS AND METHODS
GOS is an ongoing registry designed to document the clinical outcome of patients with GD over time, with 31 active sites in 11 countries as of February 25, 2017 (Argentina, Brazil, France, Israel, Italy, Paraguay, Poland, Russia, Spain, the UK, and the US). The registry was initiated in September 2010 by Shire Human Genetic Therapies, Inc, in collaboration with leading international experts on GD, as part of post‐marketing commitments to regulatory authorities. The management, administration, and infrastructure of GOS is funded by Shire. The specific aims of GOS are to better characterize the global GD population, to monitor the natural history of treated and untreated patients with GD of any type, and to enhance understanding of the effectiveness and safety of current GD treatments through observing the evolving nature of the disease among patients treated to varying degrees and at different stages. GOS is directed by a steering committee that includes international experts (investigators) and medical personnel from Shire. The steering committee meets on an annual basis to review the progress in the operation, database management, and analysis of GOS.
2.1. Patients
Patients with a diagnosis of GD of any type (based on enzymatic assay and/or GBA1 genotyping) are enrolled into GOS on a voluntary basis under the direction of their physician in accordance with routine clinical practice. Written informed consent is obtained from patients, or the patient's parent or legal guardian for those <18 years of age worldwide or <16 years of age in the UK, and assent is obtained where appropriate. Patients currently participating in ongoing clinical trials are excluded from enrollment. Patients can withdraw from the registry at any time for any reason without prejudice to their future care.
2.2. Data collection
All patients’ data are entered in an electronic case report form (eCRF), completed by authorized registry personnel at each site after having obtained the consent of each patient. The development of the eCRF involved 2 different vendors to optimize the final version for use in this registry. A secure web‐based application is used for entering data, allowing for remote data entry at GOS sites. Upon first data entry, an identification code is generated by the system for patient identification to ensure patient anonymity, with only the sex and month/year of birth accessible to the sponsor. Once a patient has been enrolled, data are entered into GOS at baseline (defined as time of GOS entry) and at the usual routine clinical evaluations performed by their physician in their ongoing management and care. In addition, historical data from their medical records can be entered retrospectively. The core variables captured via the eCRF include baseline characteristics (eg, month and year of birth, gender, height, and weight); GD type and genotype; splenectomy status; laboratory findings (eg, hemoglobin concentration, platelet counts, and plasma chitotriosidase activity); abdominal imaging of liver and spleen volume (volumetric magnetic resonance imaging, computed tomography, or ultrasound); orthopedic imaging; GD treatment information; and adverse event data. In the current analysis, patient demographic and baseline characteristics; liver and spleen volume; hematological parameters; and treatment status are summarized for patients at the time of GOS entry.
The quality of the data entered into GOS by the sites is automatically checked using predefined, established limits set within the registry database program. Additional data checks are performed by the data managers and site monitors to detect inconsistencies. If abnormal, confounding, or missing data (due to a parameter not being measured or recorded) are detected for a database entry, a request for clarification is electronically sent to the investigator. The interface of the database was designed to reflect routine clinical practice, while remaining “user‐friendly” following input from GOS investigators and study coordinators.
All patient data are handled in accordance with relevant global and local regulations and best practice and in accordance with Good Pharmacoepidemiological Practice (GPP), Good Research for Comparative Effectiveness principles, and the principles of the International Conference on Harmonization Good Clinical Practice (GCP) guidelines. GOS is monitored by an independent clinical research organization.
2.3. Analysis
Analyses were conducted on patient baseline data (defined as at time of GOS entry) extracted from the database for all patients enrolled into GOS on February 25, 2017. All‐patient analyses included all patients with available data for the chosen variable. For analyses of treatment status, treated patients were defined as those with one or more records of a GD‐specific treatment and a treatment start date specified. Untreated patients were defined as individuals for whom there was a recorded indication of not having received GD‐specific therapy and no record of GD‐specific treatment at any time. Patients not meeting the criteria for classification as “treated” or “untreated”, that is, with no information on treatment status or treatment start date, were classified as “missing treatment status” and excluded from treatment status analyses. All analyses were regarded as exploratory. Countries with fewer than 100 patients enrolled in GOS are grouped in the “rest of world” (ROW) category, which includes Argentina, Brazil, Spain, France, Italy, Paraguay, Poland, and Russia.
Spleen and liver volumes were measured predominantly by 3D ultrasound in Israel, as well as a few sites elsewhere in the world, and were recalculated using an algorithm of conversion to be comparable to computed tomography (CT) measurements as previously described.15 Briefly, estimated spleen CT = 539.5 + 0.344 (spleen ultrasound volume) and estimated liver CT = 320.86 + 0.317 (liver ultrasound volume), where ultrasound organ volume was calculated as the product of the 3 largest organ dimensions: longitudinal, transverse at maximal plane, and true anteroposterior.
3. RESULTS
3.1. Population demographics
A total of 1209 patients at 31 active sites in 11 countries had participated in the study as of February 25, 2017. The majority of patients were from Israel (44.3%), the US (31.4%), and the UK (9.8%), with the remaining 14.5% from Poland, Brazil, Russia, Paraguay, France, Spain, Argentina, and Italy (Supporting information, Figure S1). Table 1 outlines the baseline characteristics of these patients overall. The median (interquartile range [IQR]) patient age at GOS entry was 40.4 (26.5–56.8) years. Most (1106/1209; 91.5%) patients enrolled in GOS were classified as type 1 GD, based on assessment by the treating physician, with 4 (0.3%) and 37 (3.1%) classified as type 2 and type 3 GD, respectively (missing GD information for 62 [5.1%] patients). All 4 patients classified as type 2 GD were in the US. The majority of patients (675/1209; 55.8%) were of Ashkenazi Jewish ethnicity and were located mainly in Israel (498/675; 73.8%) and the US (172/675; 25.5%). Overall, 238 (19.7%) patients with GD had undergone a splenectomy (recorded as 21 [8.8%] partial and 161 [67.6%] total splenectomies, with a further 56 [23.5%] “other” or “unspecified”); these subgroups will be evaluated in a separate analysis.
Table 1.
Baseline characteristics of patients enrolled in GOS as of February 25, 2017 (N = 1209)
|
UK (n = 119) |
Israel (n = 536) |
US (n = 380) |
ROWa
(n = 174) |
Overall patients (N = 1209) |
|
|---|---|---|---|---|---|
| Sex | |||||
| Male | 53 (44.5) | 247 (46.1) | 165 (43.4) | 68 (39.1) | 533 (44.1) |
| Female | 66 (55.5) | 289 (53.9) | 215 (56.6) | 106 (60.9) | 676 (55.9) |
| Ethnicity | |||||
| Ashkenazi Jewish | 3 (2.5) | 498 (92.9) | 172 (45.3) | 2 (1.1) | 675 (55.8) |
| Other | 52 (43.7) | 37 (6.9) | 190 (50.0) | 116 (66.7) | 395 (32.7) |
| Missing information | 64 (53.8) | 1 (0.2) | 18 (4.7) | 56 (32.2) | 139 (11.5) |
| Median (IQR) age at GOS entry, years |
50.7 (35.5–60.9) |
39.3 (27.8–54.5) |
40.6 (23.8–59.0) |
36.3 (24.1–48.9) |
40.4 (26.5–56.8) |
| Type of GD | |||||
| Type 1 | 106 (89.1) | 518 (96.6) | 351 (92.4) | 131 (75.3) | 1106 (91.5) |
| Type 2 | 0 | 0 | 4 (1.1) | 0 | 4 (0.3) |
| Type 3 | 10 (8.4) | 5 (0.9) | 17 (4.5) | 5 (2.9) | 37 (3.1) |
| Missing information | 3 (2.5) | 13 (2.4) | 8 (2.1) | 38 (21.8) | 62 (5.1) |
| Diagnosis confirmationb | |||||
| Biochemical analysis (enzyme assay) | 25 (23.1) | 196 (28.7) | 137 (29.7) | 111 (63.8) | 469 (32.0) |
| GBA1 genotype testing | 73 (67.6) | 444 (65.1) | 267 (57.8) | 81 (46.6) | 865 (59.0) |
| Family history of GD | |||||
| Yes | 21 (17.6) | 212 (39.6) | 118 (31.1) | 52 (29.9) | 403 (33.3) |
| No | 45 (37.8) | 305 (56.9) | 216 (56.8) | 73 (42.0) | 639 (52.9) |
| Missing information | 53 (44.5) | 19 (3.5) | 46 (12.1) | 49 (28.2) | 167 (13.8) |
| Treatment status | |||||
| Treated | 111 (93.3) | 341 (63.6) | 314 (82.6) | 121 (69.5) | 887 (73.4) |
| Untreated | 4 (3.4) | 152 (28.4) | 13 (3.4) | 5 (2.9) | 174 (14.4) |
| Missing information | 4 (3.4) | 43 (8.0) | 53 (13.9) | 48 (27.6) | 148 (12.2) |
| Splenectomy status | |||||
| Intact spleen | 76 (63.9) | 452 (84.3) | 305 (80.3) | 138 (79.3) | 971 (80.3) |
| Total splenectomy | 33 (27.7) | 66 (12.3) | 39 (10.3) | 23 (13.2) | 161 (13.3) |
| Partial splenectomy | 1 (0.8) | 11 (2.1) | 8 (2.1) | 1 (0.6) | 21 (1.7) |
| Not specified/other | 9 (7.6) | 7 (1.3) | 28 (7.4) | 12 (6.9) | 56 (4.6) |
Abbreviations: GD, Gaucher disease; GOS, Gaucher Outcome Survey; IQR, interquartile range; ROW, rest of world.
Values are n (%) unless otherwise stated.
Argentina, Brazil, France, Italy, Paraguay, Poland, Spain, and Russia.
Excluding patients with prenatal diagnosis. Not mutually exclusive categories. Data available for n = 82 (UK), n = 473 (Israel), n = 320 (US), n = 133 (ROW), and n = 1008 (overall) patients.
In the overall population, N370S/N370S was the single most prevalent genotype, accounting for 374 (44.2%) of genotype‐confirmed individuals (n = 847), compared with 21.8% with N370S/other or N370S/unknown, 9.9% with N370S/84GG, 11.0% with N370S/L444P, 2.2% with L444P/L444P, 0.5% with L444P/other, and 10.4% with other genotypes (Supporting information, Figure S2). However, there was considerable variation between countries, with the N370S/N370S genotype occurring in 259 (58.9%) patients in Israel (n = 440) compared with 98 (37.4%) in the US (n = 262), 9 (12.0%) in ROW (n = 75), and 8 (11.4%) in the UK (n = 70). In the UK, mutations other than N370S or L444P comprised 31.4% of known genotypes, compared with 17.3% in ROW, 15.6% in the US, and 2.7% in Israel (Supporting information, Figure S2).
3.2. Treatment status
A total of 887 (73.4%) of 1209 patients enrolled in GOS had received GD‐specific treatments at any time, while 174 (14.4%) patients were untreated; treatment information was not available for 148 (12.2%) patients (Table 1; Figure 1). Of the treated patients, 843 (95.0%) had type 1 GD, 3 (0.3%) had type 2 GD, and 35 (3.9%) had type 3 GD (GD information missing for 6 patients) (Figure 1). The most widely used GD‐specific treatments were imiglucerase (n = 587), velaglucerase alfa (n = 507), alglucerase (n = 102), taliglucerase alfa (n = 93), and miglustat (n = 91). Patients from the UK were more likely to be receiving GD‐specific treatment at GOS entry (93.3% vs. 73.4% overall), while almost one‐third (28.4%) of patients from Israel were untreated at GOS entry, compared with <5% of patients in the UK, US, and ROW (Table 1). Further analyses relating to treatment patterns in GD patients enrolled in GOS have been reported separately.16
3.3. Hematological and visceral findings at GOS entry
Overall, the majority of patients (73.5%; 889/1209; missing n = 227) had normal hemoglobin concentrations of ≥11.0 g/dL (females and patients ≤12 years of age17) or ≥12.0 g/dL (males17) at GOS entry and 58.9% (712/1209; missing n = 224) had platelet counts ≥120 × 109/L (Table 2). All patients with available data at GOS entry (n = 501) had a liver volume ≤2.5 multiples of normal (MN) at GOS entry (median [IQR] 1.0 [0.9–1.2] MN) and most patients with an intact spleen and available data at GOS entry (393/405; 97.0%) had a spleen volume ≤15 MN17 (median [IQR] 6.1 [4.2–8.2] MN; Table 3). There were small differences in liver volume (MN) between patients aged <18 or ≥18 years and between different geographical locations (Table 3); however, the median [IQR] spleen volume of patients in Israel (7.2 [5.9‐9.3] MN) was almost double that of patients in the US, UK, and ROW (2.9 [2.0–4.6] MN, 2.7 [2.2–3.6] MN, and 4.9 [3.6‐5.4] MN, respectively; Table 3). Overall, 62.2% of patients had moderate splenomegaly (>5 and ≤15 MN), rising to 84.5% in the Israel cohort. The proportion of patients with severe splenomegaly (>15 MN) was consistent across geographical locations, between 0 and 4%.
Table 2.
Hemoglobin and platelet ranges in patients enrolled in the Gaucher Outcome Survey as of February 25, 2017 (N = 1209)
|
UK (n = 119) |
Israel (n = 536) |
US (n = 380) |
ROWa
(n = 174) |
Overall (n = 1209) |
|
|---|---|---|---|---|---|
| Hemoglobin concentrationb, n (%) | |||||
| Below | 4 (3.4) | 49 (9.1) | 23 (6.1) | 17 (9.8) | 93 (7.7) |
| Normal | 102 (85.7) | 421 (78.5) | 278 (73.2) | 88 (50.6) | 889 (73.5) |
| Missing | 13 (10.9) | 66 (12.3) | 79 (20.8) | 69 (39.7) | 227 (18.8) |
| Platelet count, n (%) | |||||
| <60 × 109/L | 3 (2.5) | 29 (5.4) | 11 (2.9) | 5 (2.9) | 48 (4.0) |
| ≥60–120 × 109/L | 12 (10.1) | 149 (27.8) | 41 (10.8) | 23 (13.2) | 225 (18.6) |
| ≥120 × 109/L | 91 (76.5) | 292 (54.5) | 252 (66.3) | 77 (44.3) | 712 (58.9) |
| Missing | 13 (10.9) | 66 (12.3) | 76 (20.0) | 69 (39.7) | 224 (18.5) |
Abbreviation: ROW, rest of world.
Argentina, Brazil, France, Italy, Paraguay, Poland, Spain, and Russia.
Hemoglobin ranges for women and children (≤12 years of age): below: <11.0 g/dL; normal: ≥11.0 g/dL. Hemoglobin ranges for men: below: <12.0 g/dL; normal: ≥12.0 g/dL.17
Table 3.
Liver and spleen volume in patients enrolled in the Gaucher Outcome Survey as of February 25, 2017 (N = 1209)
| All patientsn = 1209 | <18 years of agen = 153 | ≥18 years of agen = 1056 | ||||
|---|---|---|---|---|---|---|
| n | Median (IQR) | n | Median (IQR) | n | Median (IQR) | |
| Liver volume, MN | 501 | 1.0 (0.9–1.2) | 32 | 1.1 (1.0–1.4) | 469 | 1.0 (0.9–1.2) |
| UK | 42 | 0.8 (0.7–0.9) | 0 | – | 42 | 0.8 (0.7–0.9) |
| Israel | 324 | 1.1 (1.0–1.3) | 15 | 1.2 (1.0–1.4) | 309 | 1.1 (1.0–1.2) |
| US | 122 | 1.0 (0.8–1.1) | 17 | 1.1 (0.9–1.4) | 105 | 1.0 (0.8–1.1) |
| ROWa | 13 | 0.8 (0.5–0.9) | 0 | – | 13 | 0.8 (0.5–0.9) |
| Spleen volume, MNb | 405 | 6.1 (4.2–8.2) | 31 | 7.8 (4.6–10.7) | 374 | 6.1 (4.1–8.1) |
| UK | 27 | 2.7 (2.2–3.6) | 0 | – | 27 | 2.7 (2.2–3.6) |
| Israel | 267 | 7.2 (5.9–9.3) | 15 | 8.6 (8.0–13.8) | 252 | 7.1 (5.9–9.2) |
| US | 97 | 2.9 (2.0–4.6) | 15 | 4.6 (3.0–7.3) | 82 | 2.6 (1.9–4.3) |
| ROWa | 14 | 4.9 (3.6–5.4) | 1 | 8.6 (8.6–8.6) | 13 | 4.9 (3.6–5.4) |
Abbreviations: IQR, interquartile range; MN, multiples of normal.
Argentina, Brazil, France, Italy, Paraguay, Poland, Spain, and Russia.
Patients with an intact spleen only.
3.4. Skeletal findings at GOS entry
At GOS entry, 413 (34.2%) of the enrolled patients (n = 1209) had at least one skeletal abnormality on orthopedic imaging: 96 (11.8%) abnormalities were avascular necrosis, 176 (21.5%) were bone marrow infiltration, and 123 (15.1%) were Erlenmeyer flask deformities, while fractures accounted for 30 (3.7%) abnormalities. Among 350 patients with available data, bone pain was reported in 111 (31.7%) patients and was classified (as interpreted by the treating physician) as severe for 20 (5.7%) patients. Analysis of orthopedic imaging data will be reported in more detail in a separate report.
4. DISCUSSION
GOS is an ongoing, global, GD‐specific disease registry with >1200 patients included from 11 countries, with a preponderance from 3 countries. The registry was initiated in 2010 by Shire in collaboration with expert investigators from around the globe. The analysis outlined here is an overview of the initial description of GOS‐enrolled patients and focuses on baseline demographic and disease characteristics at the time of enrollment into GOS. These findings provide context for clinical outcomes analyses from GOS, which will be reported separately.
The countries enrolling the greatest numbers of patients with GD are Israel and the US, consistent with the high proportion of patients with Ashkenazi Jewish ethnicity in GOS (55.8%). Similarly, albeit 16 years ago, the majority of patients enrolled in the international Collaborative Gaucher Group (ICGG) registry were from the US, Israel, and Europe,18 all of which are regions where type 1 GD is the predominant type. Countries with greater frequencies of types 2 and 3 GD, including non‐Israeli Middle East, the Indian subcontinent, China, Japan, and Korea,19 are under‐represented in both the GOS and ICGG registries, and thus findings cannot be assumed to be representative of the wider GD population.
The distribution of GD genotypes was found to vary across the major enrolling countries, reflective of the high proportion of patients from Israel in this analysis compared with the ICGG registry (44.3% vs 17%18) and the prevalence of the N370S allele among the Ashkenazi Jewish population.20 The majority (58.9%) of patients in Israel were N370S homozygous, consistent with a milder phenotype,21 but there was a more even distribution of the 3 major genotypes in the US (N370S/N370S [37.4%], N370S/other [19.5%], and N370S/L444P [13.4%]). A broader range of mutations was apparent in the UK, with only 8 (11.4%) patients having an N370S homozygous genotype and 22 (31.4%) having mutations other than N370S or L444P.
In contrast to findings from the ICGG registry, in which most patients had low hemoglobin and platelet levels and enlarged liver and spleen volumes,18 hematological and visceral findings at the time of GOS entry were close to normal for the majority of patients, probably as a result of previous treatment. Nevertheless, some patients entered GOS with more severe disease characteristics likely to benefit from ERT. It will be informative to monitor the response of the GOS population to long‐term ERT use, particularly for patients with clinical signs of disease at GOS entry who receive velaglucerase alfa. Previous studies have shown that velaglucerase alfa normalized hematological, visceral, and skeletal disease parameters in treatment‐naïve patients with type 1 GD in the clinical trial setting,22 and it would be interesting to see if similar results are obtained in a “real‐world” setting.
Although most clinical parameters were consistent across the countries included in this study, some geographical differences in spleen size were observed. Abdominal imaging results showed that the mean spleen volume of patients in Israel (7.2 MN) was higher than that of patients in other countries (2.7, 2.9, and 4.9 MN in the UK, US, and ROW, respectively). A larger proportion of patients from Israel had moderate splenomegaly (>5 and ≤15 MN) compared with elsewhere (84.6% vs. 62.2% patients overall), although there was no increase in the proportion of patients with severe splenomegaly (>15 MN), and minimum and maximum values were consistent across all countries.
An important consideration is the abdominal imaging methodologies utilized in GOS. Abdominal imaging was performed using magnetic resonance imaging (MRI)/CT in most participating countries, whereas 3D ultrasound was used in Israel (and a few other sites), with results converted to estimated MRI/CT values using the calculation published by Elstein et al.15 Although this calculation has been used previously with ICGG registry data,18, 23, 24, 25 spleen sizes stratified by country have not previously been reported. Other methodologies for conversion of 3D ultrasound to MRI/CT values are available; for example, a calculation published by Yetter et al.26 When applied to these data, the latter calculation yields smaller spleen sizes for the Israeli population than was obtained using the Elstein et al. calculation, although still higher than the values obtained by MRI and CT elsewhere (data not shown). These findings highlight a need for further evaluation of this calculation, applied to larger numbers of patients with both MRI/CT and US results from Israel.
The relatively larger spleen sizes in Israel compared with other countries in this analysis may also be explained by a potentially greater disease severity in this cohort. Considering hematological parameters, one‐third of patients from Israel had platelet counts <120 × 109/L, indicative of thrombocytopenia, compared with 12%‐16% elsewhere, while hemoglobin levels were similar. Liver volumes, also measured using 3D ultrasound in Israel and subject to similar conversion calculations, were only marginally elevated in Israel (1.1 MN vs. 0.8‐1.0 MN elsewhere), with no differences in the proportion of patients with hepatomegaly. Compared with the overall population, more untreated patients were enrolled in GOS from Israel compared with other countries (28.4% vs. <5% in the UK, US, and ROW); there were, however, no substantial differences in median spleen size between the untreated patient cohort in Israel and the overall untreated group (6.7 vs. 6.5 MN), indicating treatment status is unlikely to explain the differential spleen sizes observed. Median spleen sizes were slightly elevated for the Israeli treated cohort compared with the overall treated group (7.5 vs. 6.5 MN), which might be in part related to the lower dose of ERT used in this country.16 These findings suggest that the enlarged spleen sizes in Israeli patients may reflect genuine differences from other countries, rather than being a result of differing imaging modalities, and warrants further investigation.
Registries can provide a valuable source of real‐world data for rare conditions like GD, where data collection from clinical trials would be challenging. However, evaluation of registry data has several limitations. Owing to the nature of participation in GOS (at the discretion of patients and their treating physicians), data input is reliant on the choice and subsequent declaration of the participating physician; data may therefore be incomplete, missing, based on broad criteria, or subject to selection bias, particularly in the reporting of treated patients over untreated patients. A few sites initially enrolled only treated patients or those who had been treated solely with velaglucerase alfa. While physicians are encouraged to enter information for all their GD patients, regardless of treatment status or the treatment they receive, there are other disease‐ and treatment‐specific registries for GD, and some sites may not be able to enroll all patients into all registries for which they are eligible.
As an ongoing registry, the structure and design of GOS is evolving in order to reflect the changing demographic characteristics of the GD population and to present data on relevant clinical outcomes. Previous analyses of GOS data have evaluated trends in treatment patterns16 and the impact of treatment during pregnancy,27 while ongoing analyses will examine patient comorbidities, skeletal abnormalities, patients with type 3 GD, and outcomes with long‐term treatment.
In conclusion, the findings from this evaluation of baseline data provide insights into the key demographic and disease characteristics of patients with GD who are enrolled in GOS and, together with future evaluations, will help to improve understanding of the natural history of the disease and the overall management of patients with GD.
CONFLICT OF INTEREST
Nothing to report.
DISCLOSURE STATEMENT
AZ reports receiving consulting fees or other remuneration, including speaker fees, from Pfizer and Shire; receiving research support from Pfizer and Shire; assisting in the design of and/or participating in clinical studies using products manufactured by Shire; and current or recent participation in a clinical trial sponsored by Shire. AZ's institution receives support from Genzyme, Pfizer, and Shire for participation in their respective registries (ICGG, TALIAS, and GOS, respectively). NB reports receiving consulting fees or other remuneration, including speaker fees, from Sanofi‐Genzyme and Shire. NB's institution (Beaujon University Hospital) has received grants from Sanofi‐Genzyme and Shire. BB reports receiving consulting fees or other remuneration, including speaker fees, from Actelion, Genzyme, and Shire; and research support from Actelion, Genzyme, and Shire. PD reports receiving consulting fees or other remuneration, including speaker fees, from Shire. DE was employed by Shaare Zedek Medical Center, Jerusalem, during the planning of this analysis and received travel expenses and honoraria for presentations at professional meetings from Shire; as of January 2016, she is employed as a contractor for Shire. DF‐S reports receiving consulting fees or other remuneration, including speaker fees, from Shire. PG reports receiving research support from Actelion, Genzyme, and Shire; assisting in the design of and/or participating in clinical studies using products manufactured by Shire; and current or recent participation in a clinical trial sponsored by Shire. OG‐A reports membership on company advisory boards or similar committees for Genzyme, Protalix, and Shire; receiving consulting fees or other remuneration, including speaker fees, from Actelion, Genzyme, Pfizer, and Shire; receiving research support from Alexion, Amicus, Genzyme, Pfizer, Protalix, and Shire; assisting in the design of and/or participating in clinical studies using products manufactured by Genzyme, Protalix, and Shire; current or recent participation in clinical trials sponsored by Genzyme, Protalix, and Shire. HL reports membership on company advisory boards or similar committees for Shire; receiving consulting fees or other remuneration, including speaker fees, from BioMarin and Shire; receiving research support from Amicus, BioMarin, GlaxoSmithKline, Pfizer, Sanofi‐Genzyme, Shire, and Ultragenyx. EL reports membership on company advisory boards or similar committees; receiving consulting fees or other remuneration, including speaker fees; and receiving research support from Genzyme and Shire. ZP is an employee of Shire. IVDS reports membership on company advisory boards or similar committees; receiving consulting fees or other remuneration, including speaker fees; and research support from Shire.
Figure 1.
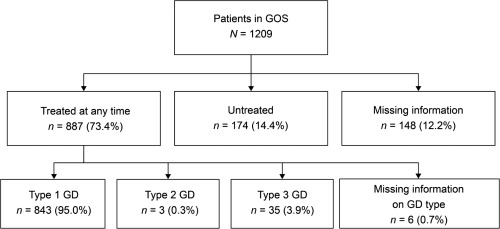
Treatment status of patients enrolled in GOS as of February 25, 2017 (N = 1209). Treated at any time: patients who had one or more records of a GD‐specific treatment and a treatment start date specified. Untreated: patients for whom there was a recorded indication of not having received GD‐specific treatment and no indication of GD‐specific treatment at any time. Missing information: patients who were missing information on treatment status or treatment start date. GD, Gaucher disease; GOS, Gaucher Outcome Survey
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article.
Supplementary Information
ACKNOWLEDGMENTS
This research was funded by the sponsor, Shire Human Genetic Therapies Inc. Under the direction of the authors, Julia Cope, Sally Hassan, and Lindsay Napier, of Excel Scientific Solutions, provided writing assistance for this publication. Editorial assistance in formatting, proofreading, copyediting, and fact checking was also provided by Excel Scientific Solutions. Svetlana Bizjajeva and Jaco Botha, from Shire International GmbH, performed statistical analyses for this study and reviewed and edited the manuscript for scientific accuracy. Shire International GmbH provided funding to Excel Scientific Solutions for support in writing and editing this manuscript.
Zimran A, Belmatoug N, Bembi B, et al. Demographics and patient characteristics of 1209 patients with Gaucher disease: Descriptive analysis from the Gaucher Outcome Survey (GOS). Am J Hematol. 2018;93:205–212. https://doi.org/10.1002/ajh.24957
Funding information Shire Human Genetic Therapies Inc.
REFERENCES
- 1. Zimran A, Elstein D, Gaucher disease and related lysosomal storage diseases In: Kaushansky K, Lichtman M, Prchal J, et al., eds. Williams Hematology. 9th ed New York, NY: McGraw‐Hill; 2016. [Google Scholar]
- 2. Sidransky E. Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab. 2004;83(1–2):6–15. [DOI] [PubMed] [Google Scholar]
- 3. Nalysnyk L, Rotella P, Simeone JC, et al. Gaucher disease epidemiology and natural history: a comprehensive review of the literature. Hematology. 2017;22(2):65–73. [DOI] [PubMed] [Google Scholar]
- 4. Barton NW, Brady RO, Dambrosia JM, et al. Replacement therapy for inherited enzyme deficiency–macrophage‐targeted glucocerebrosidase for Gaucher's disease. N Engl J Med. 1991;324(21):1464–1470. [DOI] [PubMed] [Google Scholar]
- 5. Grabowski GA, Barton NW, Pastores G, et al. Enzyme therapy in type 1 Gaucher disease: comparative efficacy of mannose‐terminated glucocerebrosidase from natural and recombinant sources. Ann Intern Med. 1995;122(1):33–39. [DOI] [PubMed] [Google Scholar]
- 6. Zimran A, Elstein D, Levy‐Lahad E, et al. Replacement therapy with imiglucerase for type 1 Gaucher's disease. Lancet. 1995;345(8963):1479–1480. [DOI] [PubMed] [Google Scholar]
- 7. Zimran A, Durán G, Giraldo P, et al. Long‐term efficacy and safety results of taliglucerase alfa through 5years in adult treatment‐naive patients with Gaucher disease. Blood Cells Mol Dis. 2016;S1079-9796(16):30087. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 8. Zimran A, Brill‐Almon E, Chertkoff R, et al. Pivotal trial with plant cell‐expressed recombinant glucocerebrosidase, taliglucerase alfa, a novel enzyme replacement therapy for Gaucher disease. Blood. 2011;118(22):5767–5773. [DOI] [PubMed] [Google Scholar]
- 9. Pfizer Inc. ELELYSO® (taliglucerase alfa) injection : Highlights of prescribing information. Dec 2016. Available at: http://labeling.pfizer.com/ShowLabeling.aspx?id=798. Accessed on: Feb 2, 2017.
- 10. Zimran A, Altarescu G, Philips M, et al. Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48‐month experience. Blood. 2010;115(23):4651–4656. [DOI] [PubMed] [Google Scholar]
- 11. Cox T, Lachmann R, Hollak C, et al. Novel oral treatment of Gaucher's disease with N‐butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet. 2000;355(9214):1481–1485. [DOI] [PubMed] [Google Scholar]
- 12. Lukina E, Watman N, Arreguin EA, et al. Improvement in hematological, visceral, and skeletal manifestations of Gaucher disease type 1 with oral eliglustat tartrate (Genz‐112638) treatment: 2‐year results of a phase 2 study. Blood. 2010;116(20):4095–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Narita A, Shirai K, Itamura S, et al. Ambroxol chaperone therapy for neuronopathic Gaucher disease: A pilot study. Ann Clin Transl Neurol. 2016;3(3):200–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zimran A, Altarescu G, Elstein D. Pilot study using ambroxol as a pharmacological chaperone in type 1 Gaucher disease. Blood Cells Mol Dis. 2013;50(2):134–137. [DOI] [PubMed] [Google Scholar]
- 15. Elstein D, Hadas‐Halpern I, Azuri Y, et al. Accuracy of ultrasonography in assessing spleen and liver size in patients with Gaucher disease: comparison to computed tomographic measurements. J Ultrasound Med. 1997;16(3):209–211. [DOI] [PubMed] [Google Scholar]
- 16. Deegan P, Fernandez‐Sasso D, Giraldo P, et al. Treatment patterns from 647 patients with Gaucher disease: an analysis from the Gaucher Outcome Survey. Blood Cells Mol Dis. 2016;S1079-9796(16):30213. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 17. Pastores GM, Weinreb NJ, Aerts H, et al. Therapeutic goals in the treatment of Gaucher disease. Semin Hematol. 2004;41(4 Suppl 5):4–14. [DOI] [PubMed] [Google Scholar]
- 18. Charrow J, Andersson HC, Kaplan P, et al. The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med. 2000;160(18):2835–2843. [DOI] [PubMed] [Google Scholar]
- 19. Burrow TA, Barnes S, Grabowski GA. Prevalence and management of Gaucher disease. Pediatr Health Med Ther. 2011;2:59–73. [Google Scholar]
- 20. Zimran A, Gelbart T, Westwood B, et al. High frequency of the Gaucher disease mutation at nucleotide 1226 among Ashkenazi Jews. Am J Hum Genet. 1991;49(4):855–859. [PMC free article] [PubMed] [Google Scholar]
- 21. Horowitz M, Pasmanik‐Chor M, Borochowitz Z, et al. Prevalence of glucocerebrosidase mutations in the Israeli Ashkenazi Jewish population. Hum Mutat. 1998;12(4):240–244. [DOI] [PubMed] [Google Scholar]
- 22. Zimran A, Elstein D, Gonzalez DE, et al. Treatment‐naive Gaucher disease patients achieve therapeutic goals and normalization with velaglucerase alfa by 4years in phase 3 trials. Blood Cells Mol Dis. 2016;[Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 23. El‐Beshlawy A, Tylki‐Szymanska A, Vellodi A, et al. Long‐term hematological, visceral, and growth outcomes in children with Gaucher disease type 3 treated with imiglucerase in the International Collaborative Gaucher Group Gaucher Registry. Mol Genet Metab. 2017;120(1–2):47–56. [DOI] [PubMed] [Google Scholar]
- 24. Weinreb NJ, Goldblatt J, Villalobos J, et al. Long‐term clinical outcomes in type 1 Gaucher disease following 10 years of imiglucerase treatment. J Inherit Metab Dis. 2013;36(3):543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mistry PK, Weinreb NJ, Kaplan P, et al. Osteopenia in Gaucher disease develops early in life: response to imiglucerase enzyme therapy in children, adolescents and adults. Blood Cells Mol Dis. 2011;46(1):66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yetter EM, Acosta KB, Olson MC, et al. Estimating splenic volume: sonographic measurements correlated with helical CT determination. AJR Am J Roentgenol. 2003;181(6):1615–1620. [DOI] [PubMed] [Google Scholar]
- 27. Lau H, Belmatoug N, Deegan P, et al. Reported outcomes of 453 pregnancies in patients with Gaucher disease: an analysis from the Gaucher outcome survey. Blood Cells Mol Dis. 2016;S1079-9796(16):30225. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found online in the supporting information tab for this article.
Supplementary Information