

Abstract
Phenylpropanoids, such as flavonoids and stilbenoids, are of great commercial interest, and their production in Saccharomyces cerevisiae is a very promising strategy. However, to achieve commercially viable production, each step of the process must be optimised. We looked at carbon loss, known to occur in the heterologous flavonoid pathway in yeast, and identified an endogenous enzyme, the enoyl reductase Tsc13, which turned out to be responsible for the accumulation of phloretic acid via reduction of p-coumaroyl-CoA. Tsc13 is an essential enzyme involved in fatty acid synthesis and cannot be deleted. Hence, two approaches were adopted in an attempt to reduce the side activity without disrupting the natural function: site saturation mutagenesis identified a number of amino acid changes which slightly increased flavonoid production but without reducing the formation of the side product. Conversely, the complementation of TSC13 by a plant gene homologue essentially eliminated the unwanted side reaction, while retaining the productivity of phenylpropanoids in a simulated fed batch fermentation.
Keywords: Saccharomyces cerevisiae, flavonoids, phloretic acid, TSC13
Replacement of essential yeast reductase with corresponding gene from apple allows more efficient production of flavonoids by eliminating formation of side products.
INTRODUCTION
Phenylpropanoids and their flavonoid derivatives are natural constituents of the common human diet (Yao et al.2004). They are also commercially interesting and frequently marketed and used as dietary supplements (Chun, Chung and Song 2007; Vidak, Rozman and Komel. 2015), food colourants (He and Giusti 2010), and preservatives (Balasundram, Sundram and Samman 2006). Flavonoids are present in most plants, including berries, fruits and vegetables used for human consumption. In planta, these compounds serve several functions such as signalling, pigmentation, UV and pathogen protection, as well as providing plants with structural integrity (Ferrer et al.2008; Falcone Ferreyra, Rius and Casati 2012). In humans, numerous reports have linked them to health benefits due to various biological activities (recently reviewed by Yao et al.2004; Tapas, Sakarkar and Kakde 2008; Lu, Xiao and Zhang 2013).
Current commercial production of flavonoids relies almost exclusively on extraction from plants, which makes the process dependent on stable supplies of good quality raw materials. Furthermore, a number of interesting flavonoids are found only in rare plants, which may be difficult to access, or occur only in minute concentrations or in mixtures with similar compounds, making their isolation and purification difficult (Hatano et al.2000; Yu et al.2016). As an alternative, organic synthesis is also challenging since in many cases a mix of stereoisomers is produced and, hence, toxic solvents may be needed during the catalysis or purification processes (Fowler and Koffas 2009). As a response to these obstacles, the use of microorganisms as cell factories for industrial production is gaining popularity, and as such the yeast Saccharomyces cerevisiae has already been used, not only for production of flavonoids but also for a wide spectrum of products including e.g. fuels, pharmaceuticals, food ingredients and other chemicals (Abbott et al.2009; Keasling 2010; Hong and Nielsen 2012; Peralta-Yahya et al.2012; Nielsen et al.2013). Hence, the use of yeast for commercial production of flavonoids would seem to offer an excellent opportunity, being well suited for large-scale fermentation (Wang et al.2011; Baerends et al.2015; Katz et al.2015; Trantas et al.2015).
The flavonoid biosynthetic pathway in plants starts with the conversion of L-phenylalanine into trans-cinnamic acid through the non-oxidative deamination by phenylalanine ammonia lyase (PAL). Next, trans-cinnamic acid is hydroxylated at the para position to p-coumaric acid (4-hydroxycinnamic acid) by cinnamate-4-hydroxylase (C4H). C4H is a cytochrome P450 monooxygenase, needing regeneration by a cytochrome P450 reductase (CPR). Alternatively, the amino acid L-tyrosine can be converted into p-coumaric acid by tyrosine ammonia lyase (TAL). P-Coumaric acid is subsequently activated to p-coumaroyl-CoA by the 4-coumarate-CoA ligase (4CL). From here it branches out to give rise to flavonoids, stilbenoids, curcuminoids, lignins, etc. (Weisshaar and Jenkins 1998). In the case of most flavonoids, a chalcone synthase (CHS) and a chalcone isomerase (CHI) catalyse the condensation of p-coumaroyl-CoA with three molecules of malonyl-CoA, resulting in the formation of naringenin chalcone and finally naringenin (Fig. 1) (Weisshaar and Jenkins 1998; Ferrer et al.2008). Naringenin represents a key branch point, from which a large array of different flavonoids are synthesised.
Figure 1.

General phenylpropanoid pathway from phenylalanine to p-coumaroyl-CoA, where it branches out to flavonoids, stilbenoids, etc. The pathway to the key intermediate naringenin is shown. The DBR activity instead gives rise to DHCs. DBR, double-bound reductase; PAL, phenylalanine ammonia lyase; C4H, cinnamate-4-hydroxylase; CPR, cytochrome P450 reductase; TAL, tyrosine ammonia lyase; 4CL, 4-coumarate-CoA ligase; CHI, chalcone isomerase; CHS, chalcone synthase.
Several studies have reported the production of naringenin in Escherichia coli (Hwang et al.2003; Leonard et al.2007; Santos, Koffas and Stephanopoulos 2011; Wu et al.2014). However, since cytochrome P450 monooxygenases and their cognate reductases are not well expressed in bacteria (Hotze, Schröder and Schröder 1995), a bacterial host may not be optimal depending on the desired end product. In yeast, Ro and Douglas (2004) were the first to reconstruct the entry point of the plant phenylpropanoid pathway, by co-expressing PAL, C4H and a CPR. Building on these results, the naringenin biosynthetic pathway has subsequently been efficiently expressed in yeast, showing production titres close to those obtained in E. coli (Jiang, Wood and Morgan 2005; Yan, Kohli and Koffas 2005; Trantas, Panopoulos and Ververidis 2009; Koopman et al.2012), thus making S. cerevisiae a promising host for the production of phenylpropanoids and flavonoids. Another important consideration regarding the choice of production host is the ability to efficiently use cheap carbon sources, and the ability to funnel the carbon into the desired product. Thus, in addition to an optimised biosynthetic pathway, it is essential that the host metabolism can be adapted to the heterologous production during fermentation, and that undesired by-product formation, resulting in carbon loss, can be eliminated (Nevoigt 2008; Hong and Nielsen 2012). Previous reports using S. cerevisiae for expression of flavonoid pathways have described accumulation of such undesired by-products. In particular, several studies reported a significant accumulation of phloretic acid (dihydrocoumaric acid) (Beekwilder et al.2006; Koopman et al.2012; Luque et al.2014; Vos et al.2015) and in one case also phloretin (dihydrochalcone, DHC), a direct derivative of phloretic acid (Jiang, Wood and Morgan 2005). These authors speculated that phloretic acid is derived from p-coumaroyl-CoA in two steps: p-coumaroyl-CoA is first reduced to dihydro-coumaroyl-CoA by an unknown yeast reductase(s) and then dihydro-coumaroyl-CoA is hydrolysed, either spontaneously or by another unknown enzymatic reaction (Beekwilder et al.2006; Koopman et al.2012).
This hypothesis fits well with what is known from plants, in particular apples (Malus domestica), where phlorizin, the 2΄-glucoside of phloretin, is the major phenolic compound. The biosynthetic pathways of these compounds have been studied in some detail. Formation of phloretin depends on CoA-activated phloretic acid which in turn is the result of a p-coumaroyl-CoA double-bond reduction. Gosch et al. (2009) determined that incubation of p-coumaroyl-CoA with NADPH and an enzyme preparation from young apple leaves resulted in the formation of dihydro-coumaroyl-CoA. They proposed that dihydro-coumaroyl-CoA is synthesised from p-coumaroyl-CoA by an unknown NADPH-dependent double-bond reductase (DBR) (Fig. 1). Based on similarities to the enoyl reduction steps in fatty acid biosynthesis, they suggested a side activity of a fatty acid dehydrogenase to be involved. Based on these previous studies, in both yeast and plants, we decided to screen the Yeast Knock Out (YKO) collection for NAD(P)H-dependent reductases, with the aim of identifying the enzymatic activity responsible for phloretic acid formation in yeast. We identified Tsc13 as the main contributor to this activity, and then tested two different approaches aimed at reducing or eliminating the loss of carbon in this side reaction of the flavonoid pathway.
MATERIALS AND METHODS
Chemicals
Trans-cinnamic acid (CAS nr.: 140-10-3), p-coumaric acid (CAS nr.: 501-98-4), phloretic acid (CAS nr.: 501-97-3), naringenin (CAS nr.: 67604-48-2) and dihydro-cinnamic acid (CAS nr.: 501-52-0) were obtained from Sigma Aldrich, Copenhagen, Denmark. Phloretin (CAS nr.: 60-82-2) was obtained from Extrasynthese, Genay Cedex, France.
Yeast strains, transformation and cultivation
Strains constructed for the expression of flavonoids were derived from a Saccharomyces cerevisiae derivative of S288c (National Collection of Yeast Cultures, Norwich, UK, NCYC 3608). The YKO strains (Winzeler et al.1999) were selected from the YKO collections YSC1056 and YSC1057 obtained from Open Biosystems, (Huntsville, Madison, Alabama, USA) now part of Dharmacon (GE Health), http://dharmacon.gelifesciences.com/non-mammalian-cdna-and-orf/yeast-knockout-collection. From the YKO library, all NAD(P)H-dependent reductases and oxidoreductases with known substrate specificity were evaluated, and a total of 26 enzymes were selected based on structural similarity of their substrates to p-coumaroyl-CoA, e.g. the presence of enoyl double bonds. Most of the YKOs were homozygous diploids; however, for essential genes YKOs were obtained as heterozygous diploids (Table 1). Since an original wt is not provided with the YKO collection, the experiment was run without a genuine control strain, and strains were instead compared to each other based on relative production levels.
Table 1.
Selected NAD(P)H-dependent yeast oxidoreductases screened in this study (Winzeler et al.1999).
| Genotype | Systematic name |
|---|---|
| oye2/oye2 | YHR179W |
| ylr460c/ylr460c | YLR460C |
| adh7/adh7 | YCR105W |
| ydr541c/ydr541c | YDR541C |
| gre2/gre2 | YOL151W |
| adh6/adh6 | YMR318C |
| yml131w/yml131w | YML131W |
| ymr226c/ymr226c | YMR226C |
| oye3/oye3 | YPL171C |
| ypl088w/ypl088w | YPL088W |
| aad4/aad4 | YDL243C |
| aad6/aad6 | YFL056C |
| dfg10/dfg10 | YIL049W |
| zta1/zta1 | YBR046C |
| ari1/ari1 | YGL157W |
| aad3/aad3 | YCR107W |
| frd1/frd1 | YEL047C |
| shh3/shh3 | YMR118C |
| osm1/osm1 | YJR051W |
| lot6/lot6 | YLR011W |
| ayr1/ayr1 | YIL124W |
| yjr096w/yjr096w | YJR096W |
| ydl124w/ydl124w | YDL124W |
| TSC13/tsc13 | YDL015C |
| SDH3/sdh3 | YKL141W |
| ERG27/erg27 | YLR100W |
All yeast transformations were done using standard Li-acetate methods according to Gietz and Schiestl (2008). Transformed yeast cells were selected on synthetic complete (SC) medium lacking uracil (SC-Ura) (Illkirch Cedex, France, MP Biomedicals).
For each strain tested, six colonies were inoculated in 0.5 mL SC-Ura and incubated overnight at 30°C and 400 rpm in 96 well deep well plates (DWP). The following day, 50 μL of each culture was transferred into 0.5 mL of fresh SC medium or, for YKO library screening, into SC medium containing 5 μL of 100 mg mL−1p-coumaric acid in 96% ethanol. The transformants were then incubated for 96 h at 30°C and 400 rpm in 96 well DWP.
To test the production of phenylpropanoids in strains where TSC13 had been substituted by homologous genes, the cells were cultivated in synthetic feed-in-time (FIT) fed-batch medium (Baesweiler, Germany, m2p-labs) for 72 h at 30°C and 200 rpm in 24 well DWP. FIT medium was prepared according to the manufacturer's instructions and then supplemented with the supplied vitamin solution (final concentration 1% v/v), and the enzyme mix (final concentration 0.8% v/v) prior to use. Control strain used was BLY2 with an empty pRS416-GPD plasmid (described in Mumberg, Mailer and Funk 1995).
To test the growth of strains, they were cultivated in SC medium (2% glucose) for 72 h at 30°C and 200 rpm in 24 DWP. To confirm the results from the FIT batch experiment, a strain with MdECR and a control strain (BLY2 with pRS416-GPD) were cultivated in the synthetic fed-batch medium the (Baesweiler, Germany, m2p-labs) is already provided above for 72 h at 30°C and 180 rpm in 100 mL shake flasks (total volume 25 mL). For this experiment, FIT medium was prepared as described above but 0.4% v/v of the enzyme mix was added at three time points: immediately prior to use, at 22 h and at 46 h.
Plasmids and strains construction
Oligonucleotide primers used for cloning and verification of genomic integrations/deletions are listed in Tables S1–S3 (Supporting Information). All primers were synthesised by Integrated DNA Technologies, Leuven, Belgium. Constructed plasmids are listed in Tables S4–S6 (Supporting Information). Synthetic genes were codon optimised for S. cerevisiae and obtained from GeneArt (Naerum, Denmark, Thermo Fisher Scientific) (Table 2). Classic and USER cloning techniques (Nour-Eldin et al.2006) were used. Cloning was performed using E. coli DH5α competent cells, and transformants were selected on Luria-Bertani (LB) agar plates containing 100 mg mL1 of ampicillin.
Table 2.
Genes, accession numbers, EC numbers and source organism.
| Gene | Accession no | EC number | Organism |
|---|---|---|---|
| AtPAL2 | NP_190894 | 4.3.1.24. | Arabidopsis thaliana |
| AtC4H | NM_128601 | 1.14.13.11. | Arabidopsis thaliana |
| AtATR2 | NP_849472 | 1.6.2.4. | Arabidopsis thaliana |
| PhCHI | P11650 | 5.5.1.6. | Petunia hybrida |
| HaCHS | Q9FUB7 | 2.3.1.74. | Hypericum androsaemum |
| At4Cl | NP_188761 | 6.2.1.12. | Arabidopsis thaliana |
| MsCHI | P28012 | 5.5.1.6. | Medicago sativa |
| AtECR | NP_191096 | 1.3.1.93. | Arabidopsis thaliana |
| GhECR2 | ABV60089 | 1.3.1.38. | Gossypium hirsutum |
| MdECR | XP_008382818 | 1.3.1.93. | Malus domestica |
| AtPAL1 | L33677 | 4.3.1.24. | Arabidopsis thaliana |
| AnPAL1 | A2QIQ7 | 4.3.1.24. | Aspergillus niger |
| RtPAL | P11544 | 4.3.1.25. | Rhodosporidium toruloides |
Strains used for the reductase screening
For the YKO reductase screening, a plasmid coding for an incomplete naringenin pathway (At4CL, MsCHI and HaCHS) was assembled in vivo in the YKO strains using homologous recombination tags, essentially as described by Kuijpers et al. (2013). The fragments for the plasmid assembly were obtained by AscI digestion of the plasmids indicated in Table S4 (Supporting Information).
For the construction of BLY2 and BLY3 (Table 3), and strains with partial flavonoid pathways, the plasmids (Table S5, Supporting Information) were constructed as integrative Easy Clone vectors according to Jensen et al. (2014). Integration of the flavonoid pathway was done by homologous recombination as described by Shao, Zhao and Zhao (2009). Basic backbones are described by Mikkelsen et al. (2012), who also describes the techniques used for plasmid construction. Three assembler plasmids were used for each integration, each having homology either to each other or to a specific region of the yeast genome. Each of these plasmids already has terminator sequences, and promoters and gene coding sequences were pre-cloned into these plasmids by USER cloning (Nour-Eldin et al.2006). The KlURA3 marker on some of the integration vectors is flanked by direct repeats (DR). The KlURA3 marker was looped out after each transformation round, by recombination between the DRs and growing cells with 5-fluoroorotic acid (Lee and Da Silva 1997). Yeast genomic DNA was extracted according to Lõoke, Kristjuhan and Kristjuhan (2011), and genomic integration of all insertions/deletions was confirmed by PCR analysis.
Table 3.
Strains used in the present study, all based on S. cerevisiae S288C.
| BLY1 | MATalpha, hoΔ0, ura3Δ0::KanMX, pad1-fdc1Δ0::LoxP-NatMX-LoxP, aro10::DR |
|---|---|
| BLY2 | MATalpha, hoΔ0, ura3Δ0::KanMX, pad1-fdc1Δ0::LoxP-NatMX-LoxP, aro10::DR, X.2::DR pTDH3-AtPAL2-tPGI1/TEF2-C4H L5 ATR2-tCYC1/pPGK1-HaCHS-tENO2/pTEF1-PhCHI-tFBA1/pPDC1-At4Cl-tTDH2 |
| BLY3 | MATalpha, hoΔ0, ura3Δ0::KanMX, pad1-fdc1Δ0::LoxP-NatMX-LoxP, aro10::DR, X.2::DR/pTDH3-AtPAL2-tPGI1/TEF2-C4H L5 ATR2-tCYC1/pPGK1-HaCHS-tENO2/pTEF1-PhCHI-tFBA1/pPDC1-At4Cl-tTDH2, XI.2::DR pTDH3-AtPAL2-tPGI1/ pPGK1-HaCHS-tENO2, XVI-20::DR pTDH3-AtPAL2-tPGI1/TEF2-C4H L5 ATR2-tCYC1/ pPGK1-HaCHS-tENO2, X-4::DR pTEF1-HaCHS-tCYC1, X.3::DR/pTDH3-AtPAL2-tPGI1/pTEF2-AtPAL1-tCYC1/ pPGK1-HaCHS-tENO2/pTEF1-AnPAL1-tFBA1/pTPI1 AtPAL2 CO2-tADH1/pPDC1-RtPal -tTDH2, XI-5::DR/pTEF2-Ha CHS_co4-tCYC1/pPGK1-Ha CHS-tENO2/pTEF1-HaCHS_co1-tFBA1/pTPI1-HaCHS_co5-tADH1/pPDC1-HaCHS_co6-tTDH2, XII.5::DR pTEF1-Aro4 K229L-tCYC1 |
The reductases Tsc13 and Dfg10 were overexpressed on the multicopy plasmid pRS426-GPD (described in Mumberg, Mailer and Funk 1995) in strain BLY3. Strain BLY3, accumulating p-coumaric acid, was obtained by increasing the copy number of PAL genes and by boosting the supply of endogenous aromatic amino acids via the introduction of Aro4 K229L as described by Mckenna et al. (2014). Control strain used was BLY3 with an empty pRS426-GPD plasmid.
Strain for testing the TSC13 SSM library
Knockout of TSC13 results in non-viable cells due to its essential role as described by Kohlwein et al. (2001). In order to test tsc13 mutants, these were therefore introduced prior to deleting TSC13. The collection of tsc13 mutants (site saturation mutagenesis (SSM) library), plus a wild-type (wt) control, was initially introduced episomally on the pRS416-GPD plasmid (Mumberg, Mailer and Funk (1995)), before deleting the native TSC13. The TSC13 deletion cassette was constructed to contain the bleomycin resistance gene (ble), flanked by 500 bp regions both upstream and downstream of TSC13 (cloned in pROP671). A naringenin producer strain comprising either the wt TSC13 (control) or the SSM library plasmid mix was transformed with 300 ng of the TSC13 deletion cassette.
Tsc13 homology modelling
The homology model of Tsc13 was built based on the structure of a trans-2-enoyl reductase from Homo sapiens (Protein Data Bank ID 1YXM; Jansson et al. submitted). An initial sequence alignment was completed using EMBOSS Water Pairwise Sequence Alignment tool (Smith and Waterman 1981) using the standard alignment settings and the Pearson FASTA output. The homology model was constructed using the Homology Model tool in MOE (MOE 2013) using default settings, including the Protonate3D tool for placement of hydrogens and minimisation refinement of the final model with AMBER10:EHT force field. To mimic the natural cofactor NADP+, the crystal structure template 1YXM has an adenine molecule bound in the cofactor site. However, there is no substrate bound (apo state). Therefore, a hexanoyl-CoA substrate was modelled into the active site by superimposing the Tsc13 homology model on the active sites of 1YXM and 1W6U (Alphey et al.2005) using PyMol [PyMol]. Based on the resulting model, residues predicted to be within 4.5 Å from the putative active site were selected for mutagenesis.
SSM library screening
A total of 25 amino acids were predicted to be in the region of the Tsc13 active site: Arg8, Lys30, Phe62, Phe89, Cys90, Glu91, Tyr92, Leu93, Leu97, Val98, His99, Ser100, Leu101, Phe102, Leu105, Tyr138, Glu144, Gln150, Phe151, Thr155, Tyr182, Tyr219, Cys220, His221 and Ile222. The total TSC13 SSM library, produced by MAX randomisation (Nov 2012), was ordered from GeneArt (Thermo Fischer Scientific) and cloned into a pRS416-GPD plasmid. Approximately 30 000 primary transformants from the SSM library were collected and subjected to TSC13 deletion. Approximately 10 000 colonies, selected for deletion of wt TSC13, were obtained of which 3786 were finally tested. This number was derived from a calculation including a couple of assumptions and based on the work of Nov (2012). Briefly, we aimed to have a theoretical chance of 99.99% of finding one of the two best (K = 2) amino acids for each position that was mutated. Using the MAX randomisation, in which all amino acids are equally represented, the number of clones to screen was calculated according to Nov (2012) as 88 clones for each of the 25 mutations (http://stat.haifa.ac.il/∼yuval/toplib/). This number was further adjusted for an estimated knock-in efficiency of 70% (the ratio of clones with correct exchange of the TSC13 wt gene with a mutant) and a ratio of 83% correct mutant genes obtained from gene synthesis (as reported by the supplier). Hence the number of clones to screen was calculated as 25*88/(0.70*0.83) = 3786.
All clones were analysed by HPLC for production of p-coumaric acid, phloretic acid and naringenin. Two controls with wt TSC13 and two controls with only empty pRS416-GPD plasmids were included in each of the test units (96 well DWP). Mutants that displayed alterations in production patterns relative to the wt (average phloretic acid at least 20% lower or average naringenin at least 20% higher), were re-tested for production of phenylpropanoids, confirmed for TSC13 deletion, and finally the plasmid harbouring the mutated tsc13 was isolated and sequenced.
Introduction of TSC13 plant homologues
The native open reading frame (ORF) of TSC13 was replaced by gene homologues from Arabidospis thaliana (AtECR), Gossypium hirsutum (GhECR2) and Malus domestica (MdECR), according to the method described by Fairhead et al. (1996) using a split URA3 cassette. The homologues were under the control of the native TSC13 promoter. The split URA3 cassettes were amplified by PCR using the primers listed in Table S3 (Supporting Information). The PCR fragments used to create the split URA3 cassettes were stitched together by the USER enzyme (Kildegaard et al.2016), further ligated and finally PCR amplified. After transformation, correct insertion of the homologues was verified by PCR (using primers RES1056 TSC13 UP and RES1061 TSC13 DW) and further confirmed by sequencing of the PCR fragment.
Sample preparation and analytical methods
The optical density (OD) was measured at 600 nm in cuvettes using classical spectrophotometer or in MTP 96 microplates using an EnVision 2104 Plate Reader. For compound quantification, 100 μL from each culture was combined with 100 μL 96% ethanol, whirl mixed for 30 s at 1500 rpm and centrifuged for 10 min at 4000 × g. Supernatants were analysed by HPLC.
Phloretic acid, p-coumaric acid, cinnamic acid, dihydrocinnamic acid, naringenin and phloretin were measured at 225 nm using an Ultimate3000 HPLC with an Ultra C18 3μm Column (100 × 4.6 mm) operating at 40°C, coupled to a diode array detector. Solvent A was water and solvent B was acetonitrile, both containing 50 ppm trifluoroacetic acid. The flow rate was 1 mL/min with acetonitrile increasing from 20% at 0 min to 60% in 9 min.
RESULTS
Confirming the phenylpropanoid substrate of the endogenous reductase
The aim of this study was to identify the reductase responsible for the formation of phloretic acid in Saccharomyces cerevisiae and to obtain a strain capable of producing naringenin without accumulating phloretic acid. Hence, we first wanted to confirm p-coumaroyl-CoA as the intermediate being converted into phloretic acid. We constructed a small collection of yeast strains which expressed various enzymes of the phenylpropanoid pathway. We observed phloretic acid formation in a strain expressing AtPAL, AtC4H, AtCPR and At4CL, but not in strains expressing either AtPAL alone or AtPAL together with AtC4H and AtCPR (Fig. S1, Supporting Information). This indicated that p-coumaroyl-CoA, and not p-coumaric acid, is the actual substrate of the unknown yeast endogenous reductase, in agreement with what was suggested by Beekwilder et al. (2006) and Koopman et al. (2012). In a strain expressing only AtPAL and At4CL, we observed formation of dihydro-cinnamic acid, in addition to the presumed precursor cinnamic acid. In contrast, a strain expressing only AtPAL produced only cinnamic acid. This demonstrated that also cinnamoyl-CoA is a substrate for the endogenous reductase (Fig. S1).
Screening for an endogenous p-coumaroyl-CoA DBR
To narrow in on the endogenous activity responsible for phloretic acid formation, we looked at publicly available information about yeast enzymes. We assumed that the enzyme would be an NAD(P)H-dependent reductase, based on previous in vitro results with plant extracts (Gosch et al.2009), and we then further looked for enzymes of which the reported substrate showed some structural similarity to p-coumaroyl-CoA. From previous work in our labs, we also knew that overexpression of FAS1/FAS2 does not change the level of phloretic acid in strains expressing AtPAL and At4CL, so we excluded these enzymes. Hence, based on the above criteria, a collection of 26 genes were selected and the corresponding strains from the YKO collection were transformed with a plasmid carrying 4-coumarate-CoA ligase (At4CL) and cultivated for 96 h in medium supplemented with p-coumaric acid. We assumed that any loss of endogenous reductase activity on the p-coumaroyl-CoA would lead to lower production of phloretic acid. Therefore, instead of measuring p-coumaroyl-CoA product formation, which requires special analytical setup, we analysed the samples for consumption of p-coumaric acid and for production of phloretic acid. Out of the 26 strains tested, two YKO strains, TSC13/tsc13 and dfg10/dfg10, both consumed less p-coumaric acid and produced less phloretic acid (Fig. 2).
Figure 2.

Concentrations of p-coumaric acid remaining (orange) and phloretic acid produced (gray) by different YKO strains after 96 h of growth in SC media containing p-coumaric acid. Most knockouts were homozygous diploids with only TSC13/tsc13, SDH3/sdh3 and ERG27/erg27 tested as heterozygous diploids. Error bars represent standard deviation (n = 6).
Overexpression of TSC13 and DFG10 in a naringenin-producing strain
As a next step, we overexpressed TSC13 or DFG10 in a yeast strain which accumulates p-coumaroyl-CoA as an intermediate of the naringenin pathway. This strain, BLY3, had been engineered to improve the endogenous supply of aromatic amino acids, which had inadvertently resulted in an unbalanced naringenin pathway and, thus, in the accumulation of pathway intermediates. In this strain, TSC13 or DFG10 was overexpressed on a multicopy 2μ plasmid and analysed for any increase in the production of phloretic acid. The strain overexpressing DFG10 showed no change in the level of phloretic acid, p-coumaric acid or naringenin. In contrast, the strong overexpression of TSC13 resulted in a clear increase in the level of phloretic acid and the appearance of the derivative phloretin. Interestingly, this was mirrored by a sharp decrease in the naringenin concentration (Fig. 3). This was somewhat surprising since the level of p-coumaric acid, a direct precursor of naringenin chalcone/naringenin, remained unchanged. However, the observed compound levels might be explained by 4CL and/or CHS being rate limiting in the current strain. Thus, a constant level of p-coumaric acid may simply reflect a limitation in 4CL activity, with both Tsc13 and CHS being active only on the CoA activated compound. With these two enzymes competing for p-coumaroyl-CoA, the overexpression of Tsc13 would therefore leave less substrate for naringenin production. The increased pool of CoA activated dihydro-coumaric acid (phloretic acid) either dissociates to give rise to the observed increase in phloretic acid or is used by CHS to produce phloretin. With CHS being limiting that would further decrease the production of naringenin. In any case, the results clearly showed that Tsc13 is the main enzyme responsible for reduction of p-coumaroyl-CoA while Dfg10 plays only a minor role.
Figure 3.
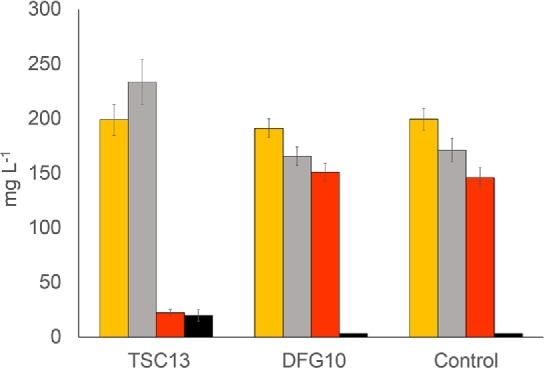
Phenylpropanoid production profile (p-coumaric acid (orange), phloretic acid (grey) naringenin (red) and phloretin (black)) in BLY3, overexpressing TSC13 or DFG10 on multicopy plasmid pRS426-GPD. Control strain is BLY3 with pRS426-GPD empty plasmid. Error bars represent standard deviation (n = 6).
Modelling the active site of Tsc13 for SSM screening
TSC13 is an essential gene, but we speculated whether it would be possible to engineer the corresponding enzyme, and thereby eliminate the non-specific activity on p-coumaroyl-CoA while maintaining its native activity. While a six-membrane-spanning topology, embedded in the ER membrane, has been proposed for yeast Tsc13 (Paul, Gable and Dunn 2007), there is, to our knowledge, no crystal structure or three-dimensional homology model of the enzyme available. Hence, we decided to create such a homology model in order to predict the active site of Tsc13. As Tsc13 contains a C-terminal 3-oxo-5α-steroid 4-dehydrogenase domain, the crystal structure of a similar protein, the 1YXM trans-2-enoyl CoA reductase, was selected as a basis for the model. For validation, the resulting Tsc13 homology model was submitted to the DALI server (http://ekhidna.biocenter.helsinki.fi/dali_server, Holm and Rosenström, Nucleic Acids Research, 2010, W545-W549) to search for known proteins with similar structures. The top 12 hits included chains A-D from 1YXM, but also 4FC6 and 4FC7 which have known trans-2-enoyl CoA reductase activity.
Based on the model, 25 residues predicted to be within 4.5 Å from the putative active site were selected for mutagenesis: Arg8*, Lys30*, Phe62*, Phe89, Cys90*, Glu91, Tyr92, Leu93, Leu97, Val98, His99, Ser100, Leu101, Phe102, Leu105, Tyr138*, Glu144, Gln150, Phe151*, Thr155*, Tyr182*, Tyr219, Cys220*, His221* and Ile222. Residues marked with an asterisk are predicted to be involved in binding of both the substrate and the co-factor NAD(P)H. These residues were then subjected to SSM and the library of mutants was cloned into a single copy yeast expression vector. The mutant library was introduced into the naringenin-producing strain, BLY2, before mass deletion of the native TSC13 gene.
To ensure a reasonable coverage of the SSM library, a total of 3786 clones were analysed for growth and metabolite production. This resulted in 18 clones which were confirmed both in terms of their altered phenylpropanoid production profile and for deletion of wt TSC13. Each specific tsc13 mutation was identified by sequencing. This narrowed down the number of specific mutations to nine different substitutions, involving five different positions. The level of phloretic acid was slightly lowered in case of L101 T, E 144C and H221C, whereas the level of naringenin was elevated in all mutants except L101 T and H221C (Fig. 4). We speculate that this increase might in fact be due to a reduced activity of the tsc13 mutants on p-coumaroyl-CoA, thus leaving more substrate available for naringenin formation.
Figure 4.
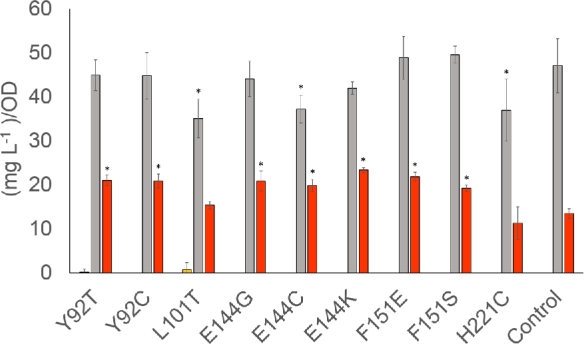
Phenylpropanoid production profile (p-coumaric acid (orange), phloretic acid (grey), naringenin (red)) in the naringenin-producing strain, where native TSC13 was replaced by different tsc13 mutants. Control strain is BLY2 with pRS416-GPD empty plasmid. Error bars represent standard deviation (n = 5). For statistical analysis, one-way ANOVA was used and the threshold of significance for the p value was 0.05. For all values significantly different than the control the bar are marked with asterisk.
Complementation of Tsc13 by plant homologues
An alternative way to circumvent unwanted phloretic acid formation would be the substitution of Tsc13 with an enzyme which is able to complement its normal function, but does not show activity on p-coumaroyl-CoA. Complementation of a non-functional Tsc13 with plant homologues from Arabidospis thaliana (AtECR) and Gossypium hirsutum (GhECR2) has been described previously (Gable et al.2004; Paul, Gable and Dunn 2007; Song et al.2009). In addition, a collection of TSC13 gene homologues from plants which were overexpressed in yeast did not exhibit any noticeable effect on phloretin production, suggesting a lack of DBR activity on p-coumaroyl-CoA (Eichenberger et al.2017). Based on these complementation and overexpression studies, we selected three homologues—A. thaliana AtECR, G. hirsutum GhECR2 and Malus domestica MdECR—in order to evaluate their ability to complement TSC13 in yeast, without interfering with the heterologous naringenin pathway. DNA sequences encoding each of the three homologues were inserted in place of the TSC13 ORF into the yeast genome by homologous recombination, thus placing these homologues under the control of the native regulatory sequences. The production of phloretic acid, p-coumaric acid and naringenin was evaluated in small-scale (DWP), fed-batch fermentation and, in addition, the growth of these strains was tested during batch growth on glucose.
Replacement of TSC13 with any of the three plant homologues AtECR, GhECR2 and MdECR resulted in viable cells, demonstrating the ability to, at least partially, complement the loss of Tsc13 cellular function. However, the growth in batch fermentation was clearly affected, and final OD600 after 72 h was substantially lower, reaching approximately 48%–55% compared to the wt control for the GhECR2 and MdECR, and just a bit more than 11% for the AtECR. The reduced biomass formation appeared to depend on whether glucose or ethanol was used as carbon source (Fig. S2, Supporting Information). Both GhECR and MdECR resulted in only slightly reduced growth rates on glucose (0.316 and 0.321 h−1, respectively, compared to 0.383 for the wt) whereas after the diauxic shift, the growth on ethanol was seriously impaired. In contrast, the AtECR strain grew poorly on both substrates (growth rate on glucose 0.096 h−1), and the formation of crystals could be observed in the medium (Fig. S3, Supporting Information). We did not investigate further the nature and composition of these crystals. The observed growth patterns are likely a result of suboptimal production, in the complemented strains, of the very long chain fatty acids (VLCFAs) which are important for structure and function of membranes (Tehlivets, Scheuringer and Kohlwein 2007). In the study of Song et al. (2009), the production of C26 fatty acids was shown to be reduced in a yeast strain complemented by the GhECR2, associated with a slight reduction in growth rate. A key point, demonstrated here by the batch fermentation results, was the poor complementation during growth on ethanol.
In order to evaluate phloretic acid formation, the complemented strains were regrown in DWP and, with the aim of preventing formation and growth on ethanol, using a simulated fed batch protocol based on the FIT enzymatic glucose release. Strains were grown for 72 h and finally cultures were analysed for production of flavonoid intermediates. In this fed batch setup, the final ODs of the complemented strains, as compared to the wt control, reached a relatively higher level than what was seen for the batch cultures (Fig. S4, Supporting Information). More interestingly, no phloretic acid was detected in the three strains complemented with plants homologues, whereas the control strain (BLY2) accumulated large amounts of this by-product (Fig. 5). Instead, the complemented strains accumulated p-coumaric acid, part of which was further metabolised to naringenin. Accumulation of p-coumaric acid, as opposed to naringenin, was expected due to the limited CHS activity in the background BLY2 strain. In addition, the supply of malonyl-CoA might have been negatively affected by an impaired synthesis of VLCFAs, for example, via the resulting accumulation of palmitoyl-CoA. This has previously been suggested to cause feedback inhibition of malonyl-CoA formation (Tehlivets, Scheuringer and Kohlwein 2007), which would then further reduce the production of naringenin. However, regarding the final level of p-coumaric acid and the observed differences between the three complemented strains, it is difficult to point to a single obvious explanation. Since it is only partially correlated to final OD, the reason(s) for this variation is unclear, but could be a result of various stress reactions of the cell.
Figure 5.
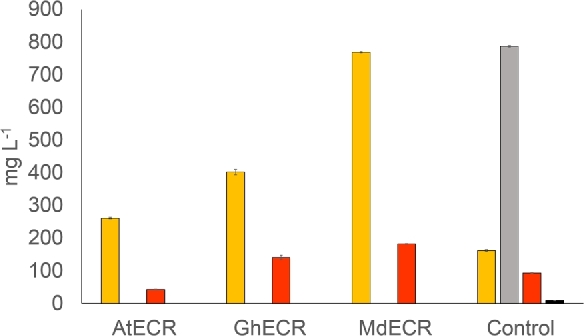
Production of flavonoid pathway intermediates (p-coumaric acid (orange), phloretic acid (gray), naringenin (red) and phloretin (black)) in BLY2 where TSC13 was replaced by homologous genes from three different organisms: A. thaliana, AtECR; G. hirsutum, GhECR2; and M. domestica, MdECR. Control strain is BLY2 with pRS416-GPD empty plasmid. Error bars represent standard deviation (n = 3).
As the most promising candidate, the MdECR strain was grown in 100 mL shake flask culture, in which the glucose release rate was further reduced, aiming to reach a final OD close to the wt control. This resulted in the gradual accumulation of p-coumaric acid and naringenin during 72 h (Fig. 6) in the MdECR-expressing strain, while growth was only marginally effected, as compared to the control (Fig. 7). In contrast to the wt control, which accumulated large amounts of phloretic acid, no by-product formation was seen in the MdECR strain, similar to our observation in the DWP experiment. Obviously, the rate of converting p-coumaric acid to naringenin is still limiting and far from optimal in this strain background. Nonetheless, the results demonstrate the feasibility of using plant homologues for complementing the Tsc13 in a controlled fermentation with slow glucose release, thereby eliminating the unwanted side activity of the native enzyme. The optimal growth rate for a commercial production would of course need to be determined for a strain with a fully optimised flavonoid pathway.
Figure 6.

Production of flavonoid pathway intermediates (p-coumaric acid (orange), phloretic acid (gray), naringenin (red) and phloretin (black)) in BLY2 (A) and in the strain where TSC13 was replaced by the homologous gene MdECR from M. domestica (B). The experiment was performed in shake flasks containing FIT media. Control strain is BLY2 with pRS416-GPD empty plasmid. Standard deviation varied between 1.4% and 8% of the total value for each point (n = 3).
Figure 7.
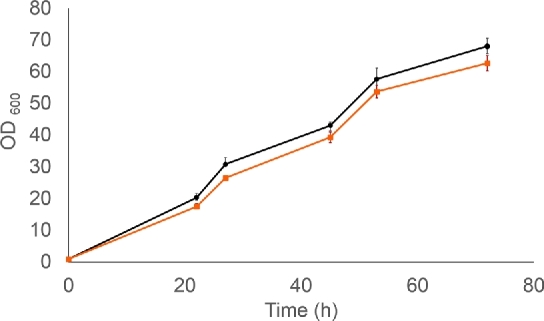
Growth of control strain BLY2 (black line with square marks) and the strain where TSC13 was replaced by the homologous gene MdECR from M. domestica (orange line with circle marks). The experiment was designed as described in Figure 6. Error bars represent standard deviation (n = 3).
DISCUSSION
Production of small biochemical molecules by fermentation, using genetically engineered host strains such as yeasts and bacteria, has emerged as one of the most promising trends in the recent quest for sustainable production methods. A major factor for achieving a commercially viable and successful fermentation process is the stable supply of cheap and renewable raw materials. An equally important factor is the efficient use of the raw material, which in terms of fermentation means a highly optimised production strain as well as a downstream process for recovery of the product. In the current work, we focused on optimising a yeast host strain for production of phenylpropanoid-derived compounds, with the specific goal of eliminating one of the major side products, phloretic acid, associated with this production.
Metabolic side reactions, such as the reduction of p-coumaroyl-CoA, drain carbon from the compound of interest. Hence, this enzymatic activity interferes with the industrial production of relevant flavonoids and stilbenoids. That p-coumaroyl-CoA is in fact the target for reduction had previously been suggested (Beekwilder et al.2006; Koopman et al.2012), and we confirmed this by showing that phloretic acid formation depends on the expression of 4CL, the CoA ligase.
An initial screen of host reductases identified the enzymes Tsc13 and Dfg10 as possible candidates, involved in the phloretic acid formation. TSC13 encodes an essential enzyme annotated as a trans-2-enoyl-CoA reductase. It catalyses the reduction of trans-2,3-enoyl-CoA intermediates in the elongation cycle of VLCFAs (Kohlwein et al.2001; Paul, Gable and Dunn 2007; Wakashima, Abe and Kihara 2014). DFG10 encodes a putative polyprenol reductase (Mösch and Fink 1997). In a naringenin-producing strain, overexpression of Tsc13, but not Dfg10, lead to increased phloretic acid production, and a concomitant decrease in naringenin, which strongly suggested that Tsc13 is the main enzyme responsible for the p-coumaroyl-CoA reductase activity in yeast, and we therefore decided to focus on this enzyme in an attempt to reduce by-product formation.
As a first approach, we used SSM (Chronopoulou and Labrou, 2011) to engineer the native Tsc13 by mutagenesis. Based on in silico modelling of the putative catalytic site, 25 amino acids were mutated. Unfortunately, the SSM library did not turn up any mutations which abolished phloretic acid production. This may be a natural consequence of the fact that TSC13 is an essential gene, and that major changes to the active site would be potentially lethal. Thus, mutations conferring smaller changes with less impact were selected by this approach.
Most interesting was the increase of naringenin production in several strains, while levels of phloretic and p-coumaric acids showed only minor changes. As shown in Fig. 8A, the aromatic ring of wt Y92 interacts with the carbonylic double bonds of CoA, stabilising the p-coumaroyl-CoA conformation in the active site, and this interaction is lost by mutation to C92 or T92. Also, in our model, wt F151 interacts with the conjugated electron system of p-coumaroyl-CoA, including the double bond to be reduced. Mutation of this residue to E151 or S151 would disrupt this stabilising interaction (Fig. 8B). These mutations may therefore reduce the affinity for p-coumaroyl-CoA, which in turn would make more substrate available for CHS, leading to the observed increase in naringenin. Regarding E144, it is not clear how mutations at this residue would affect the structural environment of the active site. Interestingly, all three residues are conserved between Tsc13 and the three plant homologues (Fig. S5, Supporting Information). We suspect that they are all essential for function at the active site, but that certain substitutions, which do not disrupt the native function in fatty acid synthesis, are tolerated. This corresponds well with the findings of Paul, Gable and Dunn (2007), who mutated several conserved residues to alanine, without Tsc13 loss of function.
Figure 8.
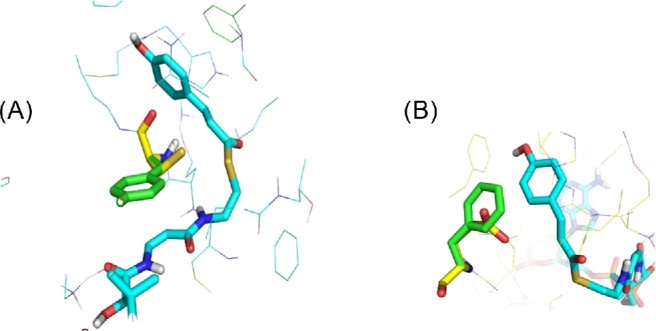
Overlay model of the T92C mutation, affecting the interaction of p-coumaroyl-CoA (cyan) with tyrosine (green) or cysteine (yellow) (A). Overlay model of the P151G mutation, affecting the interaction of p-coumaroyl-CoA (cyan) with phenylalanine (green) or glutamine (yellow) (B).
We believe the approach of using SSM to improve enzyme activity is viable, but in our case a better model or the crystal structure of the active site would be needed. Alternatively, a random mutagenesis approach could be combined with a more efficient screening assay. However, in the current study, we chose to not continue this line of work.
As an alternative to mutagenesis, we tested the feasibility of replacing the TSC13 gene with a plant homologue. One rationale for this approach was the fact that many plants are not known to produce DHCs despite having a continuous supply of p-coumaroyl-CoA. Thus, in these plants the enzymes involved in fatty acid synthesis may be less promiscuous. Alternatively, apples and other plant species known to produce DHCs might have specific enzymes dedicated to the reduction of p-coumaroyl-CoA, with other enoyl-reductases specific for fatty acid synthesis. This would of course not, a priori, exclude the possibility that the specificity for phenylpropanoids may have evolved from an enoyl-reductase of the fatty acid synthesis system. In the current study, we were interested in reductases that would not reduce p-coumaroyl-CoA while still being able to complement the natural activity of Tsc13 in the synthesis of VLCFAs. We therefore focused on reductases with functional similarity to Tsc13 (categorised as an EC 1.3.1.93), and selected three plant homologues from Arabidospis thaliana, Gossypium hirsutum and Malus domestica.
Complementation of Tsc13 has been demonstrated previously by two of those enzymes: Gable et al. (2004) reported that the AtECR homologue from A. thaliana (NP_191096) was able to complement a temperature-sensitive yeast Tsc13 mutant, and that it interacts with yeast Elo2 or Elo3, members of the VLCFA synthase complex. Paul, Gable and Dunn (2007) determined that AtECR has a membrane topology similar to Tsc13. We were able to confirm the ability of AtECR to complement Tsc13, although it did not seem to fully reconstitute function, as indicated by a decrease in specific growth rate in batch fermentation. Chemical analysis of a yeast strain expressing this homologue showed no phloretic acid production.
Another study has demonstrated complementation of Tsc13 in yeast by two homologues from cotton: GhECR1 (ABV60088) and GhECR2 (ABV60089) (Song et al.2009). This study showed that GhECR1/2 were both present in the ER fraction, indicating a cellular localisation similar to Tsc13. Both enzymes complemented the Tsc13 KO strain when expressed on plasmids, but whereas GhECR1 impaired growth substantially, GhECR2 did not affect growth to any larger extent. In the current study, we confirmed the ability of GhECR2 to complement Tsc13, although with more pronounced growth inhibition in batch fermentation than what was reported earlier (Song et al.2009). Also with this homologue we detected no production of phloretic acid.
Finally, the apple enzyme MdECR (XP_008382818) was tested in the current study. This enzyme was selected based on homology to AtECR and GhECR2 described above. Furthermore, it had very high similarity to the ENRL-1, which was previously found not to be involved in the production of DHCs (Dare et al.2013). In the current study, the strain expressing the MdECR homologue showed the best performance in terms of naringenin and p-coumaric acid production, and produced no phloretic acid. Growth of the complemented strain was somewhat inhibited when grown in batch cultivation. However, cultivating this strain in a fed-batch fermentation clearly improved the growth, most of all by preventing growth on ethanol.
In summary, all three plant homologues completely abolished phloretic acid formation, albeit with different abilities to complement native Tsc13. The growth inhibition we observed is likely linked to the synthesis of VLCFAs. In yeast, a large fraction of VLCFAs is used for the biosynthesis of sphingolipids and phosphatidylinositol, important components of the cell membrane, and disruption of Tsc13 is known to result in accumulation of fatty acids shorter than 26 carbons (Kohlwein et al.2001). Hence, incomplete complementation would lead to a deficiency in these long fatty acids. In a study on cotton ECRs (Song et al.2009), the strain complemented with GhECR2 showed a reduced level of VLCFAs compared to the wt strain, supporting the notion that this could be the reason for the growth inhibition. Preliminary results from our labs, where we overexpressed MdECR from a multicopy plasmid, indicated that further overexpression alone may not help. However, this could be due to the fact that Tsc13 normally functions as part of a larger complex, and that other components would also have to be overexpressed, or that interaction between these components, when including a non-native enzyme, is not optimal. Alternatively, the growth rate of a production strain might be adjusted to match the turnover rate of the heterologous reductase, e.g. by an appropriate fermentation process, which restores the balance of VLCFA synthesis and the overall fatty acid profile. The results from our shake flask fermentation (Fig. 7) appear to support this idea.
CONCLUSION
The native yeast Tsc13 VLCFA enoyl-reductase was identified as the main enzyme responsible for reducing heterologously produced p-coumaroyl-CoA. This side activity resulted in phloretic acid accumulation and thereby, serious carbon loss in the flavonoid biosynthetic pathway expressed in yeast.
Two different approaches to reduce this carbon loss were examined: mutation of native Tsc13 and replacement with a plant homologue. In the first approach, we obtained small improvements in the production of naringenin but were not able to eliminate the production of phloretic acid. In the second approach, we demonstrated that complementation of Tsc13 by plant homologues can completely eliminate the side product formation.
The possible effects of this Tsc13 complementation on the VLCFA synthesis machinery and the ensuing consequences of altering the composition of fatty acid derived were not investigated in this study, but would be an interesting subject for further studies. Here we achieved at least partial restoration of VLCFA acid synthesis, since loss of C26 fatty acids is lethal to yeast, and consequently we were able to do a fed-batch cultivation in which the complemented strain exhibited growth rates comparable to the wt Tsc13 strain. Obviously, for a final production strain, the optimal feed rate, and thus the maximum achievable growth rate, would have to be determined with an optimised heterologous pathway. The goal would be to avoid growth on ethanol, as is common in most fed-batch protocols, but also to prevent any accumulation of fatty acid intermediates that could disrupt the membrane structure and homeostasis, as well as preventing feedback inhibition of precursors like malonyl-CoA needed for flavonoid biosynthesis.
In short, we believe that, in particular, the latter approach is a viable route toward reducing carbon loss to undesired phloretic acid and, hence, for further optimisation of phenylpropanoid production in yeast. In combination with an otherwise balanced biosynthetic pathway, this should allow construction of flavonoid-producing cell factories with no side product formation and, consequently, a more efficient and economic industrial process.
Supplementary Material
Supplementary data are available at FEMSYR online.
Acknowledgments
The authors wish to thank Carlos Casado Vázquez and Richard J. S. Baerends for helpful discussions, Jørgen Hansen for reviewing this manuscript and Nicholas Milne for improving the language. Plasmids in Supplementary Table S5 and S6 were based on vectors kindly provided by Esben H. Hansen.
SUPPLEMENTARY DATA
Supplementary data are available at FEMSYR online.
FUNDING
The research of ME and MN is partially financed by the European Union Seventh Framework Programme under Grant agreements no. 613745, Promys and no. 613793, Bachberry.
Conflict of interest . Håvard Jenssen declares no competing interest. All other authors were supported by, or are directly employed by Evolva.
REFERENCES
- Abbott DA, Zelle RM, Pronk JT et al. Metabolic engineering of Saccharomyces cerevisiae for production of carboxylic acids: current status and challenges. FEMS Yeast Res 2009;9:1123–36 [DOI] [PubMed] [Google Scholar]
- Alphey MS, Yu W, Byres E et al. Structure and reactivity of human mitochondrial 2,4-dienoyl-CoA reductase. J Biol Chem 2005;280:3068–77 [DOI] [PubMed] [Google Scholar]
- Baerends RJS, Simon E, Meyer JP, et al. A Method For Producing Modified Resveratrol. 2015. WO 2015028324 A2, http://www.google.com/patents/WO2015028324A2?cl=tr (11 January 2016, date last accessed)
- Balasundram N, Sundram K, Samman S. Phenolic compounds in plants and agri-industrial by-products: antioxidant activity, occurrence, and potential uses. Food Chem 2006;99:191–203 [Google Scholar]
- Beekwilder J, Wolswinkel R, Jonker H et al. Production of resveratrol in recombinant microorganisms. Appl Environ Microb 2006;72:5670–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chronopoulou EG, Labrou NE. Site-saturation mutagenesis: a powerful tool for structure-based design of combinatorial mutation libraries. Curr Protoc Protein Sci 2011 [DOI] [PubMed] [Google Scholar]
- Chun OK, Chung SJ, Song WO. Estimated dietary flavonoid intake and major food sources of U.S. adults. J Nutr 2007;137:1244–52 [DOI] [PubMed] [Google Scholar]
- Dare AP, Tomes S, Cooney JM et al. The role of enoyl reductase genes in phloridzin biosynthesis in apple. Plant Physiol Biochem 2013;72:54–61 [DOI] [PubMed] [Google Scholar]
- Eichenberger M, Lehka B, Folly C et al. Metabolic engineering of Saccharomyces cerevisiae for de novo production of dihydrochalcones with known antioxidant, antidiabetic, and sweet tasting properties. Metabolic Engineering 2017;39:80–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairhead C, Llorente B, Denis F et al. New vectors for combinatorial deletions in yeast chromosomes and for gap-repair cloning using “split-marker” recombination. Yeast 1996;12:1439–57 [DOI] [PubMed] [Google Scholar]
- Falcone Ferreyra ML, Rius SP, Casati P. Flavonoids: biosynthesis, biological functions, and biotechnological applications. Front Plant Sci 2012;3:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer JL, Austin MB, Stewart C et al. Structure and function of enzymes involved in the biosynthesis of phenylpropanoids. Plant Physiol Biochem 2008;46:356–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler ZL, Koffas MAG. Biosynthesis and biotechnological production of flavanones: Current state and perspectives. Appl Microbiol Biot 2009;83:799–808 [DOI] [PubMed] [Google Scholar]
- Gable K, Garton S, Napier JA et al. Functional characterization of the Arabidopsis thaliana orthologue of Tsc13p, the enoyl reductase of the yeast microsomal fatty acid elongating system. J Exp Bot 2004;55:543–5 [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2008;2:31–4 [DOI] [PubMed] [Google Scholar]
- Gosch C, Halbwirth H, Kuhn J et al. Biosynthesis of phloridzin in apple (Malus domestica Borkh.). Plant Sci 2009;176:223–31 [Google Scholar]
- Hatano T, Aga Y, Shintani Y et al. Minor flavonoids from licorice. Phytochemistry 2000;55:9–13 [DOI] [PubMed] [Google Scholar]
- He J, Giusti MM. Anthocyanins: natural colorants with health-promoting properties. Annu Rev Food Sci Technol 2010;1:163–87 [DOI] [PubMed] [Google Scholar]
- Hong KK, Nielsen J. Metabolic engineering of Saccharomyces cerevisiae: a key cell factory platform for future biorefineries. Cell Mol Life Sci 2012;69:2671–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotze M, Schröder G, Schröder J. Cinnamate 4-hydroxylse from Catharanthus roseus, and a strategy for the functional expression of plant cytochrome P450 proteins as translational fusion with P450 reductase in Escherichia coli. FEBS Lett 1995;374:345–50 [DOI] [PubMed] [Google Scholar]
- Hwang E Il, Kaneko M, Ohnishi Y et al. Production of plant-specific flavanones by Escherichia coli containing an artificial gene cluster. Appl Environ Microb 2003;69:2699–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson A, Ng S, Arrowsmith C, et al. Crystal structure of perixomal trans 2-enoyl CoA reductase (PECRA) Submitted (MAY-2005) to the PDB data bank.
- Jensen NB, Strucko T, Kildegaard KR et al. EasyClone: method for iterative chromosomal integration of multiple genes in Saccharomyces cerevisiae. FEMS Yeast Res 2014;14:238–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Wood KV, Morgan JA. Metabolic engineering of the phenylpropanoid pathway in Saccharomyces cerevisiae metabolic engineering of the phenylpropanoid pathway in Saccharomyces cerevisiae. Appl Environ Microb 2005;71:2962–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz M, Smits HP, Förster J, et al. Metabolically Engineered Cells for the Production of Resveratrol or an Oligomeric or Glycosidically-Bound Derivative Thereof. 2015. US 9040269 B2. http://www.google.com/patents/US9040269 (11 January 2017, date last accessed)
- Keasling JD. Manufacturing molecules through metabolic engineering. Science (80-) 2010;330:1355–8 [DOI] [PubMed] [Google Scholar]
- Kildegaard KR, Jensen NB, Schneider K et al. Engineering and systems-level analysis of Saccharomyces cerevisiae for production of 3-hydroxypropionic acid via malonyl-CoA reductase-dependent pathway. Microb Cell Fact 2016;15:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlwein SD, Eder S, Oh CS et al. Tsc13p is required for fatty acid elongation and localizes to a novel structure at the nuclear-vacuolar interface in Saccharomyces cerevisiae. Mol Cell Biol 2001;21:109–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman F, Beekwilder J, Crimi B et al. De novo production of the flavonoid naringenin in engineered Saccharomyces cerevisiae. Microb Cell Fact 2012;11:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijpers NG, Solis-Escalante D, Bosman L et al. A versatile, efficient strategy for assembly of multi-fragment expression vectors in Saccharomyces cerevisiae using 60 bp synthetic recombination sequences. Microb Cell Fact 2013;12:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FW, Da Silva NA. Sequential delta-integration for the regulated insertion of cloned genes in Saccharomyces cerevisiae. Biotechnol Prog 1997;13:368–73 [DOI] [PubMed] [Google Scholar]
- Leonard E, Lim KH, Saw PN et al. Engineering central metabolic pathways for high-level flavonoid production in Escherichia coli. Appl Environ Microb 2007;73:3877–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lõoke M, Kristjuhan K, Kristjuhan A. Extraction of genomic DNA from yeasts for pcr- based applications. Biotechniques 2011;50:325–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu MF, Xiao ZT, Zhang HY. Where do health benefits of flavonoids come from? Insights from flavonoid targets and their evolutionary history. Biochem Bioph Res Co 2013;434:701–4 [DOI] [PubMed] [Google Scholar]
- Luque A, Sebai SC, Santiago-Schübel B et al. In vivo evolution of metabolic pathways by homeologous recombination in mitotic cells. Metab Eng 2014;23:123–35 [DOI] [PubMed] [Google Scholar]
- Mckenna R, Thompson B, Pugh S et al. Rational and combinatorial approaches to engineering styrene production by Saccharomyces cerevisiae. Microb Cell Fact 2014;13:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen MD, Buron LD, Salomonsen B et al. Microbial production of indolylglucosinolate through engineering of a multi-gene pathway in a versatile yeast expression platform. Metab Eng 2012;14:104–11 [DOI] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE) 2013. Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7. 08, 2016.
- Mösch HU, Fink GR. Dissection of filamentous growth by transposon mutagenesis in Saccharomyces cerevisiae. Genetics 1997;145:671–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Mailer R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds 1995;156:119–22 [DOI] [PubMed] [Google Scholar]
- Nevoigt E. Progress in metabolic engineering of Saccharomyces cerevisiae. Microbiol Mol Biol R 2008;72:379–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J, Larsson C, van Maris A et al. Metabolic engineering of yeast for production of fuels and chemicals. Curr Opin Biotechnol 2013;24:398–404 [DOI] [PubMed] [Google Scholar]
- Nour-Eldin HH, Hansen BG, Nørholm MHH et al. Advancing uracil-excision based cloning towards an ideal technique for cloning PCR fragments. Nucleic Acids Res 2006;34:e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nov Y. When second best is good enough: Another probabilistic look at saturation mutagenesis. Appl Environ Microb 2012;78:258–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Gable K, Dunn TM. A six-membrane-spanning topology for yeast and Arabidopsis Tsc13p, the enoyl reductases of the microsomal fatty acid elongating system. J Biol Chem 2007;282:19237–46 [DOI] [PubMed] [Google Scholar]
- Peralta-Yahya PP, Zhang F, del Cardayre SB et al. Microbial engineering for the production of advanced biofuels. Nature 2012;488:320–8 [DOI] [PubMed] [Google Scholar]
- Ro D, Douglas CJ. Reconstitution of the entry point of plant phenylpropanoid metabolism in yeast (Saccharomyces cerevisiae): implications for control of metabolic flux into the phenylpropanoid pathway. J Biol Chem 2004;279:2600–7 [DOI] [PubMed] [Google Scholar]
- Santos CNS, Koffas M, Stephanopoulos G. Optimization of a heterologous pathway for the production of flavonoids from glucose. Metab Eng 2011;13:392–400 [DOI] [PubMed] [Google Scholar]
- Shao Z, Zhao H, Zhao H. DNA assembler, an in vivo genetic method for rapid construction of biochemical pathways. Nucleic Acids Res 2009;37:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol 1981;147:195–7 [DOI] [PubMed] [Google Scholar]
- Song WQ, Qin YM, Saito M et al. Characterization of two cotton cDNAs encoding trans-2-enoyl-CoA reductase reveals a putative novel NADPH-binding motif. J Exp Bot 2009;60:1839–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapas A, Sakarkar D, Kakde R. Flavonoids as nutraceuticals: a review. Trop J Pharm Res 2008;7:1089–99 [Google Scholar]
- Tehlivets O, Scheuringer K, Kohlwein SD. Fatty acid synthesis and elongation in yeast. Biochim Biophys Acta 2007;1771:255–70 [DOI] [PubMed] [Google Scholar]
- Trantas EA, Koffas MA, Xu P et al. When plants produce not enough or at all: metabolic engineering of flavonoids in microbial hosts. Front Plant Sci 2015;6:1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trantas E, Panopoulos N, Ververidis F. Metabolic engineering of the complete pathway leading to heterologous biosynthesis of various flavonoids and stilbenoids in Saccharomyces cerevisiae. Metab Eng 2009;11:355–66 [DOI] [PubMed] [Google Scholar]
- Vidak M, Rozman D, Komel R. Effects of flavonoids from food and dietary supplements on glial and glioblastoma multiforme cells. Molecules 2015;20:19406–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos T, de la Torre Cortés P, van Gulik WM et al. Growth-rate dependency of de novo resveratrol production in chemostat cultures of an engineered Saccharomyces cerevisiae strain. Microb Cell Fact 2015;14:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakashima T, Abe K, Kihara A. Dual functions of the trans -2-enoyl-CoA reductase TER in the sphingosine 1-phosphate metabolic pathway and in fatty acid elongation. J Biol Chem 2014;289:24736–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Halls C, Zhang J et al. Stepwise increase of resveratrol biosynthesis in yeast Saccharomyces cerevisiae by metabolic engineering. Metab Eng 2011;13:455–63 [DOI] [PubMed] [Google Scholar]
- Weisshaar B, Jenkins G. Phenylpropanoid biosynthesis and its regulation. Curr Opin Plant Biol 1998;1:251–7 [DOI] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 1999;285:901–6 [DOI] [PubMed] [Google Scholar]
- Wu J, Yu O, Du G et al. Fine-tuning of the fatty acid pathway by synthetic antisense RNA for enhanced (2S)-naringenin production from L-tyrosine in Escherichia coli. Appl Environ Microb 2014;80:7283–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Kohli A, Koffas MAG. Biosynthesis of natural flavanones in Saccharomyces cerevisiae. Appl Environ Microb 2005;71:5610–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao LH, Jiang YM, Shi J et al. Flavonoids in food and their health benefits. Plant Foods Hum Nutr 2004;59:113–22 [DOI] [PubMed] [Google Scholar]
- Yu JS, Moon E, Choi SU et al. Asarotonide, a new phenylpropanoid with a rare natural acetonide group from the rhizomes of Acorus gramineus. Tetrahedron Lett 2016;57:1699–701 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data are available at FEMSYR online.