


Abstract
Gene regulatory network perturbations contribute to the development and progression of cancer, however, molecular determinants that mediate transcriptional perturbations remain a fundamental challenge for cancer biology. We show that transcriptional perturbations are widely mediated by long noncoding RNAs (lncRNAs) via integration of genome-wide transcriptional regulation with paired lncRNA and gene expression profiles. Systematic construction of an LncRNA Modulator Atlas in Pan-cancer (LncMAP) reveals distinct types of lncRNA regulatory molecules, which are expressed in multiple tissues, exhibit higher conservation. Strikingly, cancers with similar tissue origin share lncRNA modulators which perturb the regulation of cell cycle and immune response-related functions. Furthermore, we identified a large number of pan-cancer lncRNA modulators with potential clinical significance, which are differentially expressed in cancer or are strongly correlated with drug sensitivity across cell lines. Further stratification of cancer patients based on lncRNA-mediated transcriptional perturbations identifies subtypes with distinct survival rates. Finally, we made a user-friendly web interface available for exploring lncRNA-mediated transcriptional perturbations across cancer types. Our study provides a systems-level dissection of lncRNA-mediated regulatory perturbations in cancer, and also presents a valuable tool and resource for investigating the function of lncRNAs in cancer.
INTRODUCTION
Human cancers are complex diseases involving multiple genetic and epigenetic changes in the genome (1,2). With the development of high-throughput sequencing, a number of studies have provided an ever-expanding survey of genetic aberrations in cancer (3,4). Multiple new cancer-related genes have been identified, and these genes formed gene regulatory network to play critical roles in cancer. Gene regulatory network perturbations have been demonstrated to contribute to the development and progression of cancer, however, the molecular determinants of the gene regulatory network perturbations remains a fundamental challenge in cancer.
In addition, emerging evidence has indicated that long noncoding RNAs (lncRNAs) play key roles in a wide range of biological processes (5), and their expressions are strikingly cell-type and tissue-specific (6). Given that lncRNAs are key regulators of gene expression, recent studies suggested that lncRNAs are involved in the tumor initiation and progression through diverse mechanisms (7–9). Although a considerable portion of human genome is transcribed as lncRNAs, the vast majority are functionally uncharacterized. Therefore, it is another challenge for identification of lncRNAs involved in carcinogenesis and characterization of their functions in specific cancer context.
Gene transcription is often regulated by transcription factors (TFs) that bind to cis-regulatory elements in a sequence-specific fashion (10,11). While TFs are proved to be the primary engines, emerging evidence have demonstrated that the regulation of TF to its targets are also modulated by lncRNAs. For instance, lncRNA GAS5 has been found to bind to the DNA binding domain of the TFs by acting as a decoy, thus competes with DNA for binding to the TFs (12). In addition, Yolanda et al. have demonstrated that two lncRNA targets of P53 can affect the binding of this TF to other targets, and thus modulate the P53 transcriptional network in cancers (13). Another study has shown that lncRNA CASC11 can interact with hnRNP-K and activates the WNT/β-catenin pathway to promote growth and metastasis in colorectal cancer (14). A recent study has suggested that MALAT1 can regulate E2F1 activity (15), which is a crucial determinant of cell cycle progression. Moreover, cancer associated lncRNA-p21 is associated with hnRNP-K and represses P53-dependent transcriptional responses (16). The lncRNA CCAT1-L also plays key roles in regulating the activity of MYC (17). These findings suggest that lncRNAs are emerging as important regulators of TF functions in cancer. However, systematical identification of lncRNA modulators in cancer are urgently needed.
Thus, we recently developed a computational approach LncMod to comprehensively identify perturbed lncRNA–TF–gene triplets by combining both lncRNA and gene expression profiles with TF–target relationships (18). Applying this method to glioblastoma, we identify cancer-related lncRNA–TF–gene triplets, such as HOTAIR-MXI1-CD58/PRKCE and HOTAIR-ATF5-NCAM1. The integration of lncRNA modulators into transcriptional regulatory networks is expected to lay the foundation for development of novel lncRNA biomarkers and therapeutic agents. However, many questions about the common or specific lncRNA-mediated TF regulatory perturbation mechanism in different types of cancer have not been fully addressed, such as which lncRNAs mediate transcriptional dysregulation across tumor types (pan-cancer modulators) or in a specific cancer (cancer-specific modulators)? What about the potential functions, regulatory roles and biological insights of the pan-cancer lncRNA modulators? Thus, systematic analysis of the lncRNA-mediated transcription perturbation and their clinical applications is urgent and necessary.
Recent RNA-Seq datasets over large cancer patient cohorts, such as The Cancer Genome Atlas (TCGA) provide us an unprecedented opportunity for identifying lncRNA-medicated gene regulatory network perturbations across cancer types in a systematic way (Figure 1A). Here, we performed an integrative analysis of paired lncRNA and mRNA expression profiles, genome-wide transcriptional regulatory networks as well as functional datasets to investigate the functional regulation of lncRNAs. Based on the proposed computational method LncMod (Figure 1B), we identified the widespread lncRNA mediated transcriptional regulatory perturbations in 20 types of cancer. Pan-cancer modulators were with distinct features, including tissue specificity and conservation. Moreover, we also identified several lncRNA modulators were associated with drug activity and survival time of patients. Overall, our genome-wide analyses identified a comprehensive set of candidate noncoding biomarkers with pan-cancer potential, and provided novel insights into the functions of lncRNA in cancer.
Figure 1.

An integrative framework identifies widespread lncRNA-mediated transcriptional network perturbations in pan-cancer. (A) Global transcriptional network perturbations were observed across cancer types. Global lncRNA modulators that mediated the network perturbations were analyzed based on proposed LncMod method. The identified modulators were analyzed for different functional characteristics, including cancer specificity, differential expression, cancer hallmark, drug activity and clinical association. (B) The framework to identify lncRNA modulators across cancer types. Firstly, TF–gene regulation were identified based on ChIP-Seq datasets. Second, regression analysis was used to identify context-specific regulation based on gene expression. Next, lncRNA mediated transcriptional network perturbations in each cancer type were discovered by the modified LncMod method and further classified as six regulatory patterns.
MATERIALS AND METHODS
The lncRNA and mRNA transcriptome landscape across tumor types
The genome-wide lncRNA expression across 20 types of cancer were directly obtained from ‘The Atlas of Noncoding RNAs in Cancer’ (TANRIC) (19). In total, 12 727 lncRNAs were quantified for the expression levels as reads per kilo base per million mapped reads (RPKM). To ensure detection reliability and reduce noise, we applied two filters used in one previous study in each cancer type to identify the expressed lncRNAs (20). First, lncRNAs whose 10th-percentile RPKM value is equal to 0 were eliminated and the second, we selected only lncRNAs whose 90th-percentile RPKM value is greater than 0.1 for further analysis. The expression value of each lncRNA was log2(RPKM + 0.05)-transformed.
We obtained the corresponding gene expression profiles of these cancer samples from TCGA. Only samples with paired lncRNA and mRNA expression profiles were used in this study (Supplementary Table S1), that is the samples were with both lncRNA and mRNA expression profiles. In total, the expression of 18 999 protein coding genes were measured. The expression values were also log2(RPKM + 0.05) transformed.
Identifying the TF–gene regulatory interactions across cancers
To identify the TF–gene regulation in each cancer type, we firstly downloaded the TF–gene regulation from ChIPBase (21), which is an integrated resource for decoding TF binding maps from ChIP-Seq data. In total, we obtained 504 522 regulatory interactions among 107 TFs and 16 417 genes. As the TF–gene regulation in each cancer is context-dependent, we used linear regression to evaluate the pairwise TF–gene expression association. In this model, mRNA 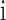 expression (log2),
expression (log2), 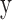 , changes as a linear function of TF
, changes as a linear function of TF  expression (log2),
expression (log2),  , in the
, in the 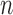 tumor samples of a given cancer type:
tumor samples of a given cancer type:
 |
in this model, 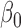 is the intercept,
is the intercept, 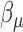 is the regression coefficients for TF expression variable. We used the ordinary least squares method to obtain an estimate for the TF coefficient and test the null hypothesis that the expression level of TF is not associated with change in expression of gene. The TF–gene regulation with Bonferroni adjusted P-value <0.01 were selected for further analysis.
is the regression coefficients for TF expression variable. We used the ordinary least squares method to obtain an estimate for the TF coefficient and test the null hypothesis that the expression level of TF is not associated with change in expression of gene. The TF–gene regulation with Bonferroni adjusted P-value <0.01 were selected for further analysis.
Identification of lncRNA modulators across cancers
Here, we proposed a framework to identify the lncRNA modulators. First, lncRNAs, TFs, and target genes were filtered based on the expression variation across samples (‘range constraint’). Individual TFs (gTF) and target genes (gt) were selected for further analysis based on their variation across samples (log2 IQR>0.58). For each lncRNA modulator gm, we sorted the cancer samples based on the expression of gm. Then the top and bottom 25% of samples in terms of lncRNA expression were contrasted, which were defined as H-group and L-group. Downstream analysis was only performed on TFs that were not differentially expressed between H-group and L-group. Target genes are required to differentially express between two conditions (P-adjusted < 0.01 and fold change > 1.5). Next, each possible lncRNA–TF–gene triplet was tested to determine whether lncRNA altered the TF–gene regulation. The Spearman correlation coefficient between TF and gene was used to measure the TF–gene regulation in H-group (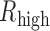 ) and L-group (
) and L-group (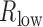 ) separately. To guarantee that TF regulated gene in at least one condition, we required that the absolute value of either
) separately. To guarantee that TF regulated gene in at least one condition, we required that the absolute value of either 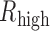 or
or 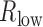 was >0.4. In addition, only TF–gene pairs with the absolute value of difference between
was >0.4. In addition, only TF–gene pairs with the absolute value of difference between 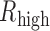 and
and 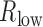 >0.45 were further analyzed. Then, we used Fisher's test of difference between two correlation coefficients as previous studies (22). Firstly, these correlations between TFs and genes were transformed by Fisher transformation as follow:
>0.45 were further analyzed. Then, we used Fisher's test of difference between two correlation coefficients as previous studies (22). Firstly, these correlations between TFs and genes were transformed by Fisher transformation as follow:
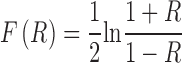 |
Next, we defined the rewiring score,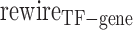 , which ranges between 0 and 1, with larger value indicating more rewiring effect between TF and gene.
, which ranges between 0 and 1, with larger value indicating more rewiring effect between TF and gene.
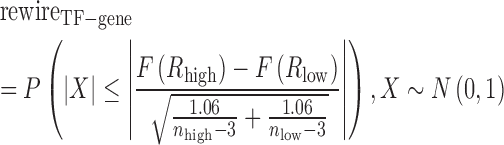 |
where 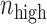 and
and  are the number of samples in H-group and L-group, separately. To determine whether the difference between H-group and L-group is significantly high. The samples were randomly sorted and we recalculated the rewire score, this process was repeated 100 times. The P value is the fraction of rewire score in random conditions that was larger than that in the real conditions P-values were Bonferroni-corrected for the total number of candidate lncRNA–TF–gene triplets. All lncRNA–TF–gene triplets with P-adjusted <0.01 were regarded as significant. In addition, all the triplets were classified as six patterns based on the correlation coefficient and the changes of rewiring scores (Supplementary Table S2) (18).
are the number of samples in H-group and L-group, separately. To determine whether the difference between H-group and L-group is significantly high. The samples were randomly sorted and we recalculated the rewire score, this process was repeated 100 times. The P value is the fraction of rewire score in random conditions that was larger than that in the real conditions P-values were Bonferroni-corrected for the total number of candidate lncRNA–TF–gene triplets. All lncRNA–TF–gene triplets with P-adjusted <0.01 were regarded as significant. In addition, all the triplets were classified as six patterns based on the correlation coefficient and the changes of rewiring scores (Supplementary Table S2) (18).
Tissue specificity and conservation analysis of lncRNA modulators
To evaluate the tissue specificity of an lncRNA modulator, we relied on Cabili et al. (23) and used an entropy-based measure. Here, we assembled a consensus lncRNA transcriptome by curating hundreds of RNA-Seq datasets across normal human tissues from 16 independent studies (24). The tissue specificity score of cancer specific lncRNA modulators, moderate lncRNA modulators and pan-cancer modulators were compared with Wilcox rank sum test. PhastCons scores were used for measure the conservation of lncRNA modulators. The PhastCons scores for 100 genomes were downloaded from UCSC Table Browser (25). The average PhastCons score for each nt position of lncRNA was computed as conservation.
Identification of differentially expressed lncRNAs in cancer
To identify differentially expressed lncRNAs in each cancer type, we used two methods. Firstly, the lncRNAs with expression level 0 in <30% samples in both normal and tumor samples were subjected to t-test. LncRNAs with fold change greater than twice and FDR <0.01 were identified as differentially expressed. If the expression levels of the lncRNAs were 0 in >30% tumor or normal samples, we used on/off analysis. For each lncRNA, we determined its expression in binary fashion: On (expressed, RPKM > 0), OFF (not detected, RPKM = 0). We next calculated the frequency of expression in normal and cancer samples. LncRNAs expressed twice more frequently in cancer than that in normal samples were selected as ‘On in cancer’, whereas lncRNAs not expressed twice more often were identified as ‘OFF in cancer’. The significance of the contingency between ON/OFF and cancer/normal status was tested by Fisher's exact test. The threshold of FDR <0.01 was used.
Drug–lncRNA associations across cancer cell lines
To evaluate the effects of drug on lncRNA expression, we downloaded the lncRNA expression profiles in cancer cell lines from TANRIC. These cancer cell lines were obtained from Cancer Cell Line Encyclopedia (CCLE) (26). In addition, we also downloaded the drug screening data from CCLE, and calculated the correlations between lncRNA expression levels and IC50 values of 24 drugs across cell lines. We used the Spearman rank correlations to detect significant drug–lncRNA correlations with a coefficient (absolute value) cutoff of 0.3 and FDR <0.01.
Functional analysis of lncRNA modulators
Although a number of methods had been proposed to predict the functions of lncRNAs, such as co-expression, co-localization. Here, we proposed that investigating the functions of these target genes in lncRNA–TF–gene triplets might provide new insights of the functions of these lncRNAs. Thus, the function enrichment analysis was carried out via the targets of triplets to determine the functions of lncRNAs by hypergeometric test. Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways with adjusted P values <0.01 and including at least two interesting genes were considered. Moreover, we used Gene Set Enrichment Analysis (GSEA) to identify the functions of genes regulated by multiple lncRNA modulators. Firstly, genes were ranked by the number of lncRNA modulators that regulated them in each cancer type. Next, we computed the average rank for each gene in 20 types of cancer. The ranked gene list was subject to GSEA analysis and functions with P-value <0.05 were identified.
Identification of survival-related lncRNA–TF–gene triplets
To identify the lncRNA–TF–gene triplets that are related with cancer patient survival, we randomly divided the tumor samples into discovery and validation datasets without age and sex difference. Two subsets had the similar number of patients. We then used univariate Cox regression analysis to evaluate the association between survival time and the expression level of each lncRNA, TF, or gene. The regression coefficients with a plus sign indicated that increased expression of this element was associated with decreased survival (risky factors), and, conversely, a minus sign indicated that increased expression of the element was associated with increased survival (protective factors). We then constructed a mathematical formula for survival prediction, taking into account both the strength and direction for each factor in the triplet with respect to survival. As in one of our previous studies (18), the risk score for each patient i was calculated as follows:
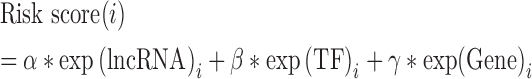 |
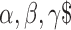 were the regression coefficient of lncRNA, TF and gene, respectively. All tumor samples in the discovery dataset were thus assigned to high-risk and low-risk groups using the median risk score as the cut-off. The coefficient and cut-off derived from the discovery dataset were directly applied to expression data of the corresponding validation dataset to divide the tumor samples in the validation dataset into high-risk and low-risk groups. The Kaplan–Meier method was used to estimate the overall survival time for the two subgroups, and differences in survival time were analyzed using the log rank test. Moreover, we linked the triplets that share at least two elements and constructed the triplets’ network. Network cliques were identified and then these cliques were also subjected to the survival analysis. Samples were divided into two groups based on the expression of elements in cliques, and log-rank test was used to test the survival difference. For the independent dataset validation, the expression of lncRNA, TF and gene were firstly Z-score transformed. Next, we clustered the tumor samples based on the expression profile of lncRNA, TF and gene. Tumor samples were clustered into two groups, and log-rank test was used to estimate the difference in survival time.
were the regression coefficient of lncRNA, TF and gene, respectively. All tumor samples in the discovery dataset were thus assigned to high-risk and low-risk groups using the median risk score as the cut-off. The coefficient and cut-off derived from the discovery dataset were directly applied to expression data of the corresponding validation dataset to divide the tumor samples in the validation dataset into high-risk and low-risk groups. The Kaplan–Meier method was used to estimate the overall survival time for the two subgroups, and differences in survival time were analyzed using the log rank test. Moreover, we linked the triplets that share at least two elements and constructed the triplets’ network. Network cliques were identified and then these cliques were also subjected to the survival analysis. Samples were divided into two groups based on the expression of elements in cliques, and log-rank test was used to test the survival difference. For the independent dataset validation, the expression of lncRNA, TF and gene were firstly Z-score transformed. Next, we clustered the tumor samples based on the expression profile of lncRNA, TF and gene. Tumor samples were clustered into two groups, and log-rank test was used to estimate the difference in survival time.
RESULTS
Widespread lncRNA-mediated transcriptional perturbations across 20 cancer types
Integration of genome-wide TF–gene regulation and paired lncRNA–mRNA expression profiles across 20 types of cancer (Supplementary Table S1), we investigated the landscape of lncRNA mediated transcriptional perturbations in cancer. This process mainly involved five steps (Figure 1B). Firstly, based on high-through ChIP-Seq datasets, TF–gene bindings were obtained. As transcriptional regulation had been demonstrated to be context specific (27–29), we thus incorporated gene expression to identify the active TF–gene regulation and constructed transcriptional network in each cancer type. Next, the modified LncMod method was used to identify lncRNA modulators in each cancer type (see details in methods). Finally, the identified lncRNA–TF–gene triplets were classified into six patterns based on their activity changes. In total, about 6234 samples across 20 types of cancer were analyzed in our study (Figure 2A). These cancer types were primarily involved in eight classes, including brain (brain lower grade glioma, LGG; glioblastoma multiforme, GBM), genitourinary (kidney chromophobe, KICH; kidney renal clear cell carcinoma, KIRC; kidney renal papillary cell carcinoma, KIRP; bladder urothelial carcinoma, BLCA; prostate adenocarcinoma, PRAD), Thoracic (lung squamous cell carcinoma, LUSC; lung adenocarcinoma, LUAD), gastrointestinal (rectum adenocarcinoma, READ; colon adenocarcinoma, COAD; stomach adenocarcinoma, STAD; liver hepatocellular carcinoma, LIHC), gynecologic system (cervical squamous cell carcinoma and endocervical adenocarcinoma, CESC; ovarian serous cystadeno carcinoma, OV; uterine corpus endometrial carcinoma, UCEC), breast cancer (BRCA), skin cancer (skin cutaneous melanoma, SKCM), and head and neck cancer types (head and neck squamous carcinoma, HNSC; thyroid carcinoma, THCA).
Figure 2.

Widespread lncRNA-mediated transcriptional regulation perturbations in 20 types of cancer. (A) The paired gene and lncRNA expression profiles and number of samples for each cancer type. The numbers 1–20 indicate different types of cancer and cancer types are ordered by their tissue origin. (B) The proportion of TF–gene regulatory interactions in each cancer type. (C) The pie chart shows the proportion of TF–gene regulatory interactions occurring in different number of cancer types. (D) The proportion of lncRNA–TF–gene triplets in each cancer type. (E) The proportion of lncRNA–TF–gene triplets of different regulatory patterns in each cancer type. Different color lines indicate distinct regulatory patterns, including inverts inhibition, inverts activation, attenuates activation, attenuates inhibition, enhances activation and enhances inhibition.
Based on genome-wide expression correlation analysis, we identified cancer context-specific TF–gene regulatory interaction. As a result, we observed that only a small proportion (range from 1.14% to 35.15%) of transcriptional regulation were active in a specific cancer context (Figure 2B). In addition, ∼30.51% (101 117/331 399) of these TF–gene regulation were observed only in one cancer type; just 0.66% (2219/331 399) regulation were active in >16 cancer types (Figure 2C). These results highlight the transcriptional regulation specificity across cancer types. Next, lncRNA modulators were identified in each cancer type. However, we found that the majority of TF–gene regulation remain stable and approximate 0.026% to 0.46% of all possible lncRNA–TF–gene triplets were identified in these cancer types (Supplementary Figure S1). Moreover, we observed that although the proportion of TF–gene regulation in SKCM is limited (Figure 2D), the proportion of detected triplets in this cancer was higher than other cancers. This observation suggest that the widespread transcriptional perturbations in this cancer type, which might be caused by environmental stimulation. Next, we classified the detected triplets in each cancer into six classes as one of our previous studies (18). As shown in Figure 2E, the majority of lncRNAs regulated the TF activity in a fine manner but not to alter the direction of TF regulation. Together, the identified lncRNA–TF–gene triplet landscape provides a valuable resource to investigate the functions of lncRNAs across cancer types.
Conserved lncRNA modulators elucidate critical functions across cancer types
The landscape of lncRNA-mediated transcriptional perturbation allows us to investigate the distinct roles of lncRNA across cancer types. We next investigated the extent to which lncRNA mediated perturbation events contributed to cancer specificity. First, we computed the number of cancer types that lncRNA modulators occurred. We found that the distribution of this number follows a bimodal distribution, indicating that there are distinct types of lncRNA modulators (Figure 3A). LncRNA modulators tended to be highly cancer type-specific, as 17% of the lncRNA modulators were detected in only one cancer type (Figure 3A). In addition, we found that the majority of these cancer specific modulators were observed in LGG (24.26%) and STAD (15.15%) (Supplementary Figure S2A). In contrast, our analysis also revealed a small subset of lncRNA modulators (n = 1580) that were detected across multiple cancer types (>15 cancer types). Thus, we divided the modulators into three groups based on the number of cancer types detected: specific modulators, moderate modulators and pan-cancer modulators (Figure 3A).
Figure 3.

Different types of lncRNA modulators and regulatory similarity of cancer types with similar tissue origin. (A)The number of lncRNA modulators that occur in different number of cancer types. LncRNA modulators are classified into three types: cancer specific modulators (occurring in only one cancer type), moderate modulators (occurring in 2–15 cancer types) and pan-cancer modulators (occurring in more than 16 cancer types). (B) The degree distribution of pan-cancer modulators and other modulators across 20 types of cancer. Each cancer type is represented by two boxes, while the below box is the degree distribution for pan-cancer modulators and the above one is the distribution for other modulators. ***P < 0.001 and **P < 0.05, Wilcox rank sum test. (C) Tissue-specific score distribution for different types of lncRNA modulators. (D) Conservation score distribution for different types of lncRNA modulators. (E and F) The biological processes enriched by genes in the common triplets identified in cancer types with similar tissue origin: LGG and GBM in (E), and COAD and READ in (F).
To determine their roles in cancer, we first compared degree of these three types of modulators, which was defined as the number of transcriptional regulation perturbations they mediated. We found that pan-cancer modulators were with significantly higher degree than other modulators (Figure 3B, P-value <0.05 for 18/20 cancer types, Wilcox rank sum test), indicating that pan-cancer modulators were likely to play pivot roles in cancer. Tissue specific expression might contribute to the roles of lncRNA modulators in cancer (30), we thus computed the tissue specific index for lncRNA modulators based on expression in 20 tissues (see details in methods). We found that pan-cancer modulators exhibited significantly lower tissue specific index (Figure 3C, P-values <0.05, Wilcox rank sum tests), suggesting they were widely expressed across tissues. Moreover, we found that pan-cancer modulators were with higher conservation than moderate and specific modulators (Figure 3D, P-values <0.05, Wilcox rank sum tests). Taken together, these specific features of pan-cancer modulators further highlight their critical roles in cancer.
Cancer types with similar tissue origins share lncRNA-mediated transcriptional perturbation patterns
Lines of evidence have indicated that cancer types with similar tissue origins share multiple molecular features, such as protein coding gene expression, miRNA expression and lncRNA expression (31–33). However, whether cancer types with similar tissue origins exhibit similar lncRNA regulatory patterns is unknown. To address this question, we computed a paired similarity score based on lncRNA mediated transcriptional dysregulation events in each cancer. This analysis indicated that cancer types with similar tissue origins showed similar lncRNA mediated transcriptional dysregulation patterns, such as LGG and GBM, COAD and READ (Figure 3E and F, P < 1.0e–32, hypergeometric test). These observations suggest that related mechanisms might operate in cancer types with similar tissue origins.
To explore the conserved function of lncRNA modulators in similar cancer types, we performed the functional enrichment analysis based on the target genes of lncRNA modulators mediated that were detected commonly. In total, 441 and 771 lncRNA–TF–gene triplets were identified as common in these two types of cancer (Figure 3E and F, up-panel). Functional analysis indicated that these lncRNA modulators primarily play roles in cell cycle and immune response pathways (Figure 3E–F and Supplementary Figure S2). These results indicate that lncRNA-mediated transcriptional perturbation in cancer provides a predictor of cytotoxic immune cell infiltration and cell cycle. Furthermore, lncRNA mediated transcriptional perturbation profiling may help identify cancer patients most likely to respond to immunotherapy.
LncRNA modulators with potential biomedical significance across cancer types
Based on the genome-wide lncRNA expression profiles across cancer types, we performed a comprehensive survey to assess the potential biomedical significance of lncRNAs. For 14 TCGA cancer types with available normal samples, we found that large numbers of lncRNAs exhibited significant differential expression between tumor and normal samples (Figure 4A, FDR < 0.01, Student's t-test). Moreover, ∼7515 lncRNAs switched their expression profiles between tumor and normal samples were observed (FDR < 0.01, Fisher's exact test), extending the candidate list for functional validation. In addition, we also randomly perturbed the labels of the samples and recalculated the Fisher's exact test P-values, we found that all of those differential expressed lncRNAs we identified were all with P-values <0.05. However, we found that the majority (86.7–98.3%) of the differentially expressed lncRNA modulators were identified based on t-test. LncRNAs had been demonstrated to have strong tissue specific expression patterns, we next investigated the distribution of the differentially expressed lncRNAs across cancer types. This analysis revealed that either the switched lncRNAs or differentially expressed lncRNAs were cancer specific, 199 lncRNAs showed recurrent differentially expression in more than seven types of cancer (Figure 4B).
Figure 4.

Landscape of differentially expressed lncRNAs across cancer types. (A)The proportion of differentially expressed lncRNAs in each type of cancer. There are three bars for each cancer type, the first one shows the proportion of lncRNAs with expression shift, the second one shows the proportion of lncRNAs with expression switch on/off, and the third one shows the union of the first and second ones. Purple, upregulated or switch on; blue, down-regulated or switch off. (B) Pie charts show the proportion of differentially expressed lncRNAs identified in different numbers of cancer types. LncRNAs identified in more than 7 cancer types are recurrent lncRNAs. (C) Overlap of pan-cancer modulators and the recurrent differentially expressed lncRNAs. P < 2.2e–16, hypergeometric test. (D) The cancer hallmarks enriched by recurrent differentially expressed pan-cancer lncRNA modulators. Each row represents an lncRNA and each column represents a Gene Ontology term. The size of the circles correspond to different P-values (hypergeometric test). GO terms belong to the same hallmarks are indicated by the green bars. (E) LncRNA PVT1 mediated transcriptional regulation perturbations in cancer. White nodes represent TFs, genes in different hallmarks are marked with different colors. (F) The expression of PVT1 in normal and cancer samples across cancer types. (G) LncRNA modulator CDKN2B-AS1 mediated transcriptional regulation perturbations. (H) The expression of CDKN2B-AS1 in normal and cancer samples. ***P < 0.001 and **P < 0.05, t-test.
Furthermore, the recurrent differentially expressed lncRNAs were significantly enriched in pan-cancer modulators (Figure 4C, P < 1.0e–32, hypergeometric test). These 88 differentially expressed pan-cancer modulators were mainly identified based on the t-test method. To further explore the roles these pan-cancer lncRNA modulators, we performed functional enrichment analysis of their mediated genes for cancer hallmarks (Figure 4D). This analysis revealed one or more hallmarks were enriched across different lncRNA modulators. One example is PVT1, a long non-coding RNA encoded by the human PVT1 gene, which is located in the well-known cancer-related region 8q24 (34). Multiple molecular mechanisms of action of this lncRNA were revealed (35–37), including participating in DNA rearrangements, encoding miRNAs, and interacting with MYC. We found that this lncRNA might mediate the transcriptional regulatory perturbation of MYC to its target genes (Figure 4E). Expression analysis also revealed that this lncRNA was differentially expressed in the majority (85.7% or 12/14) of cancer types (Figure 4F, FDR < 0.05). Another example is the antisense transcript CDKN2B-AS1, which is located within the CDKN2A/CDKN2B tumor suppressor locus. A number of studies had demonstrated the expression dysregulation of this lncRNA (38–40). Here, we found that this lncRNA mediated transcriptional perturbation of many tumor associated genes, such as WNT5A, ERBB3 and PRKCB, thus involved in the tissue invasion and metastasis, sustained angiogenesis and self-sufficiency in growth signal hallmarks (Figure 4G). This lncRNA was differentially expressed in 85.7% (12/14) of cancer types (Figure 4H, FDR < 0.05). Moreover, we also found some other cancer-related lncRNAs that were annotated in LncRNADisease (41) and Lnc2Cancer (42) can mediate transcriptional perturbations (Supplementary Table S3), such as AP000330.8, DLEU2 (43) and VPS9D1-AS1 (44) (Supplementary Figure S3). These results indicate that identification of lncRNA mediated transcriptional perturbation increased our understanding of the role of lncRNAs in tumorigenesis.
Potential drug targeted lncRNA modulators across cancer types
Previous studies and our above results have indicated that lncRNAs may affect normal gene expression and disease progression, making lncRNAs as a new class of targets for drug discovery (45,46). However, their mechanisms of actions are unknown. To identify drug-related lncRNAs in cancer, we thus explored the association of lncRNA expression and drug activity in hundreds of cancer cell lines. We downloaded the drug screening data from Cancer Cell Line Encyclopedia (CCLE) (https://portals.broadinstitute.org/ccle/home) and calculated the Spearman correlation coefficient between lncRNA expression levels and the IC50 values of 24 drugs. At the cutoff (absolute value) of 0.3, we obtained the drug–lncRNA associations (Figure 5A). Analysis of these drug–lncRNA associations, we observed that most (58.47% or 466/797) of the lncRNAs were associated with one drug, while the expression of RBPMS-AS1 was associated with eight drugs (Figure 5B). In contrast, five drugs’ activities were only associated with the expression of one lncRNA while six drugs’ activities were associated with more than 50 lncRNAs (Figure 5C), suggesting their wide therapeutic effects.
Figure 5.

Drug activity related lncRNAs across cancer types. (A) The association of drug activity and lncRNA expression in cell lines. Each dot represents a pair of drug–lncRNA, and two lines indicate the correlation coefficients as 0.3 and –0.3. (B) The number of lncRNAs associated with different numbers of drugs. (C) The number of drugs associated with different numbers of lncRNAs. (D–F) Examples of lncRNA–TF–gene triplets with distinct regulatory patterns. The cartoons show the regulatory pattern of each triplet. The drugs that target the genes were marked. The below boxplots shows the expression of TFs and genes in lncRNA high expression group and low expression group. The dot-lines show the correlation between TF–gene in two subgroups. Green, lncRNA low expression group; purple, lncRNA high expression group.
Moreover, we also discovered some lncRNA modulators could regulate the expression of known drug targets. Histone deacetylases (HDAC) have been identified as therapeutic targets due to their functions in regulating DNA structure and organization (49). Panobinostat (also known as LBH589) which is a novel inhibitor of class I and II HDACs, and can also target HDAC7 (50,51). We observed that the pan-cancer lncRNA modulator RP3–402G11.28 can mediated the regulation between NFKB1 and HDAC7, and lncRNA high expression can regulate the expression of HDAC7 (Figure 5D). Another example is the target of Irinotecan-TOP1, which controls and alters the topologic states of DNA during transcription (52). We found that pan-cancer lncRNA modulator TMEM191A could also regulate the expression of the drug target (Figure 5E), these results suggest that combining the lncRNA with the Irinotecan might contribute to the therapy of multiple types of cancer. Moreover, alterations of cell cycle regulators have been implicated in human cancer, including CDK6 (53). Here, we found that pan-cancer lncRNA modulator RP11–490M8.1 could regulate the expression of CDK6 (Figure 5F). PD0332991 had been demonstrated to be an orally active, highly selective inhibitor of CDK6 by blocking retinoblastoma (Rb) phosphorylation (54). These results indicated that PD0332991 might regulate the expression of CDK6 synergistically with lncRNA RP11–490M8.1. Some other representative examples were also illustrated in Supplementary Figure S4. Taken together, these results suggest that investigating the lncRNA mediated transcriptional dysregulation might reveal some lncRNA molecular synergistically with small molecular drugs to regulate the expression of target genes, these lncRNA modulators might be candidate noncoding drugs for therapy of cancer.
Clinical relevance of tumor subtypes revealed by lncRNA modulators
Our above analysis indicated that we could infer the functions of lncRNA modulators based on their regulated genes. Evidence in regulatory network biology analysis had suggested that genes regulated by multiple regulators were with critical roles in cancer (47). Thus, we investigated the functions of genes regulated by multiple lncRNA modulators in various types of cancer. Genes were ranked by the number of lncRNA modulators that regulated them and then we ranked the genes by the mean rank in 20 types of cancer. Gene set enrichment analysis (GSEA) analysis (48) indicated that genes regulated by multiple modulators were enriched in chromosome organization (Figure 6A, FDR = 0.003), regulation of immune response (Figure 6B, FDR = 0.004) and DNA repair (Figure 6C, FDR = 0.010), regulation of angiogenesis and cell migration (Supplementary Figure S5A–C). These results suggest widespread transcriptional dysregulation of immune genes and DNA repair genes.
Figure 6.

Clinically associated lncRNA–TF–gene triplets across cancer types. (A–C) Functions enriched by the genes regulated by multiple number of lncRNAs in cancer. (A) Chromosome organization; (B) regulation of immune response and (C) DNA repair. (D) The framework for identification of survival-related triplets in each cancer type. (E) The survival landscape of lncRNA–TF–gene triplets. X-axis, –log(P-value) in discovery set and Y-axis, –log(P) in the validation set. Each dot represents a triplet in each cancer type. Dots are marked by the same color as the cancer types. Two lines indicated P-value less than 0.05. (F) The lncRNA–TF–gene clique identified in GBM. (G) Kaplan–Meier plot of survival for GBM samples with different risk scores. (left) Discovery set; (middle) validation set; (right) independent dataset. The survival difference among clusters is calculated by log-rank test. Red line, high-risk group; blue, low-risk group.
Previous studies and our current results demonstrated that lncRNA modulators are expressed in a tissue-specific fashion and undergo expression changes during cancer development or progression. These specific properties render them valid candidates as prognostic biomarkers. We thus integrated the clinical data to explore the prognostic associated lncRNA–TF–gene triplets (Figure 6D). Tumor samples in each cancer type were randomly divided into discovery and validation sets without age and sex difference. Next, we trained a model in the discovery set and tumor samples in the discovery set were classified as low-risk group and high-risk group based on a risk score (see details in Materials and Nethods). lncRNA–TF–gene triplets with log-rank P-values <0.01 in both discovery and validation sets were identified as clinical associated triplets. For all 20 cancer types, we identified 7.68% clinical-associated triplets (Figure 6E) and the majority of triplets were discovered in LGG (Supplementary Figure S5D, 75.8%) and KIRC (21.1%).
Interestingly, we identified TPTEP1-JUN-DDR2 triplet was associated with patients’ survival in discovery (Supplementary Figure S5E, P = 9.97e–5, log-rank test) and validation sets (P = 5.34e–4, log-rank test). TPTEP1 was identified as a pan-cancer modulator here, which is a pseudogene and had been demonstrated to be differentially expressed in cancer (49). In addition, DNA repair gene DDR2 had been found to regulate several cellular functions including cell adhesion and migration, and proliferation (50,51). Next, we validated the prognostic effect of this triplet in another independent Chinese LGG dataset (52). We found that the expression of this triplet could also distinguish tumor samples with different survival time (Supplementary Figure S5E, P = 2.98e–12, log-rank test). Next, we constructed a network by linking the survival-related triplets that share at least two elements. Several triplet-cliques were identified in GBM. For instance, one clique mainly mediated by LEF1-AS1 (Figure 6F) was identified to be associated with survival in discovery (Figure 6G, P = 0.004, log-rank test) and validation set (Figure 6G, P = 0.005, log-rank test). It was also validated in another Chinese dataset (52), which including 100 GBM tumor samples (Figure 6G, P = 0.001, log-rank test). These transcriptional regulation were mainly involved the transcription factor CEBPA, which had been found to control genes of the mesenchymal signature of gliomas and FBP1 physically interacts with p53, functions as a regulator of p53-regulatory proteins (53). We also identified another prognostic related triplet-clique which was mediated by C20orf166-AS1 (Supplementary Figure S5F and G) in GBM. Specifically, we found that the log-rank P-values for the cliques were less than those of individual elements in both discovery, testing and independent datasets. These results suggest the critical roles of these triplet-cliques in cancer and provide potential clinical usage of these biomarkers.
A user-friendly web interface for exploring the lncRNA-mediated transcriptional perturbations across cancer types
In order to facilitate the researchers to use the lncRNA mediated transcriptional perturbation resource in 20 types of cancer, we constructed a user-friendly web interface LncMAP (LncRNA Modulator Atlas in Pan-cancer, http://www.bio-bigdata.com/LncMAP), which allows users to flexibly obtain information about their lncRNAs of interest. This resource is valuable for both experimental and computational researchers. This platform provides a web interface for users to search and download data sets in the database (Supplementary Figure S6). Specifically, we provided experimental users several ways to query the lncRNA mediated transcription perturbations as well as their association with the drug activity. First, users can query the platform for exploring the triplets that involved the interesting lncRNA, TF or target gene in specific cancer type. When input an interesting lncRNA, TF or gene, this platform will return a list of matched entries. The results included all the parameters used for identification of the lncRNA–TF–gene triplets, including the Z-scores in two groups and the regulatory pattern of the triplets. Moreover, we stored the drug–lncRNA associations in this platform. Then, the users can generate or download the detailed annotations for listed entries to explore the association between lncRNAs and drug activities. In the search result page, the correlation coefficients and P-values were also listed.
In order to facilitate the users to explore the clinically associated lncRNA–TF–gene triplets in specific cancer, we provided the search interface for users to find the survival information of the interesting triplets. The log-rank P-values in discovery and validation sets for each triplets were included in the searching result page. Moreover, we also provided the tool for users to obtain the transcriptional regulation among TFs and genes in specific cancer context. Specifically, we also provided several ways for visualization of the expression of interesting lncRNAs, TFs and genes, the TF–gene correlation changes mediated by lncRNAs, network of TF–gene regulation, and network visualization of drug–lncRNA associations (Supplementary Figure S6). These visualization provided flexible ways for global view of the lncRNA functions in cancer. All data in the resource can be freely downloaded from the ‘download’ page for further computational analyses. A detailed tutorial for the usage of the resource is available in the ‘Help’ page.
DISCUSSION
Transcription regulatory network perturbations were widely observed in various types of cancer. Identification of determinants that mediated such perturbations is one of the major challenges in cancer biology (54). In this study, we have introduced a computational framework for identification of lncRNA-mediated transcription perturbations in a diverse set of cancer types by integrating genome wide transcriptional regulation with gene expression datasets. The diversity of the perturbed transcriptional regulation, and their association with specific lncRNA modulators highlight the broad heterogeneity underlying various cancer types. The majority of the active transcriptional regulation and the lncRNA modulators were cancer specific. Applied the proposed method to 20 types of cancer, we identified widespread transcriptional regulation mediated by lncRNAs and identify three types of lncRNA modulator, in which pan-cancer lncRNA modulators were with critical functional features, including tissue-specific expression and high conservation. Moreover, we also identify the drug-related and survival related lncRNA-TF-triplets in various types of cancer. All these results were provided in a user friendly resource for both computational and biological researchers, which will deepen our understanding of the roles of lncRNAs in cancer.
Despite this heterogeneity, our analysis also identified a type of lncRNA modulators that mediated transcription perturbations in multiple types of cancer (pan-cancer modulators). These lncRNA modulators mediated widespread transcription perturbations and widely expressed in multiple tissues, were much more conserved. All these features highlighted their critical roles in cancer. Moreover, our pan-cancer analysis revealed that cancer types with similar tissue origin significantly shared lncRNA-mediated transcription perturbations. These lncRNAs were mainly involved in cell cycle and immune response pathways. These discoveries related to lncRNAs would provide a comprehensive understanding of immune regulation and provide novel insights into the lncRNA-based immune therapy. Moreover, integrated the genome wide lncRNA expression we found that these pan-cancer lncRNA modulators show perturbed expression in multiple cancer types. Our analyses also predict the involvement of many known lncRNA regulators (such as PVT1 and CDKN2B-AS1) in cancer-associated pathways, thus revealing putative onco-lncRNAs and tumor suppressors, and yielding potential drug target candidates.
Based on the drug activity screen data, we also revealed the associations among lncRNA modulators and cancer drugs. The ability of lncRNAs to fine-tune gene expression makes them as potential targets for drug development. However, the process for drug discovering is never easy. Uncertainty about how lncRNAs function makes the identification and development of lncRNA based drug target even more challenging (55). Our analyses not only revealed the associations between specific lncRNA and drug, but also provided the potential functional pattern of this lncRNA. As our understanding of lncRNAs mediated transcriptional perturbation and their regulatory mechanisms improve, design of effective lncRNA based target will gain a firmer foundation and the likelihood of clinical success to cure cancer will increase.
Moreover, we found the majority of the lncRNA regulated genes were enriched in DNA repair and immune response. These observations indicated the widespread perturbations of DNA repair and immune functions in cancer. The rapidly advancing field of cancer immunology has produced several new methods for treating cancer, called immunotherapies, which increase the strength of immune responses against tumors (56). The identified lncRNA modulators as important regulators of immune genes have shed light on our understanding of the link between lncRNA world and immune regulation. To facilitate users to analyze the immune-related lncRNA modulators, we obtained the immune-related genes from ImmPort project (57) and provided these triples as an independent resource (Supplementary Table S4). Finally, further stratification of cancer patients based on their lncRNA–TF–gene triplets identifies different subtypes with distinct clinical features, including survival rates.
In summary, we have systematically characterized widespread lncRNA-mediated transcription perturbations across cancer types and presented an integrative computational method to identify lncRNA modulators that mediated such perturbations. Our method and results presented here will be useful for investigators who explore the functions of lncRNAs through rapidly emerging next-generation sequencing applications in cancer. We hope that the method and resource that we provide here will serve as an inspiration for future investigating lncRNA functions in diverse cancer types.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
FUNDING
National Program on Key Basic Research Project (973 Program) [2014CB910504]; Creative Research Groups of the National Natural Science Foundation of China [81421063]; National High Technology Research and Development Program of China (863 Program) [2014AA021102]; National Natural Science Foundation of China [91439117, 61473106, 31571331, 61502126, 61603116]; NCI Transition Career Development Award [1K22CA214765-01]; China Postdoctoral Science Foundation [2016T90309, 2015M571436, 2016M591544, LBH-Z14134, LBH-Z16142]; Natural Science Foundation of Heilongjiang Province [QC2015020]; Weihan Yu Youth Science Fund Project of Harbin Medical University, University Nursing Program for Young Scholars with Creative Talents in Heilongjiang Province [UNPYSCT-2016189, UNPYSCT-2017060]; Harbin Special Funds of Innovative Talents on Science and Technology Research Project [2015RAQXJ091, 2017RAQXJ164]; Cancer Prevention and Research Institute of Texas (CPRIT) New Investigator Grant [RP160021 to N.S.]; AASLD Foundation Pinnacle Research Award (to N.S.). Funding for open access charge: Research grant.
Conflict of interest statement. None declared.
REFERENCES
- 1. You J.S., Jones P.A.. Cancer genetics and epigenetics: two sides of the same coin?. Cancer Cell. 2012; 22:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shen H., Laird P.W.. Interplay between the cancer genome and epigenome. Cell. 2013; 153:38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xu J., Wang Z., Li S., Chen J., Zhang J., Jiang C., Zhao Z., Li J., Li Y., Li X.. Combinatorial epigenetic regulation of non-coding RNAs has profound effects on oncogenic pathways in breast cancer subtypes. Brief. Bioinform. 2016; doi:10.1093/bib/bbw099. [DOI] [PubMed] [Google Scholar]
- 4. Zhang J., Baran J., Cros A., Guberman J.M., Haider S., Hsu J., Liang Y., Rivkin E., Wang J., Whitty B. et al. International Cancer Genome Consortium Data Portal–a one-stop shop for cancer genomics data. Database. 2011; 2011:bar026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012; 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huarte M. The emerging role of lncRNAs in cancer. Nat. Med. 2015; 21:1253–1261. [DOI] [PubMed] [Google Scholar]
- 7. Yang G., Lu X., Yuan L.. LncRNA: a link between RNA and cancer. Biochim. Biophys. Acta. 2014; 1839:1097–1109. [DOI] [PubMed] [Google Scholar]
- 8. Schmitt A.M., Chang H.Y.. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016; 29:452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. White N.M., Cabanski C.R., Silva-Fisher J.M., Dang H.X., Govindan R., Maher C.A.. Transcriptome sequencing reveals altered long intergenic non-coding RNAs in lung cancer. Genome Biol. 2014; 15:429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Whitfield T.W., Wang J., Collins P.J., Partridge E.C., Aldred S.F., Trinklein N.D., Myers R.M., Weng Z.. Functional analysis of transcription factor binding sites in human promoters. Genome Biol. 2012; 13:R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Won K.J., Ren B., Wang W.. Genome-wide prediction of transcription factor binding sites using an integrated model. Genome Biol. 2010; 11:R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu Y., Zhao J., Zhang W., Gan J., Hu C., Huang G., Zhang Y.. lncRNA GAS5 enhances G1 cell cycle arrest via binding to YBX1 to regulate p21 expression in stomach cancer. Scientific Rep. 2015; 5:10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sanchez Y., Segura V., Marin-Bejar O., Athie A., Marchese F.P., Gonzalez J., Bujanda L., Guo S., Matheu A., Huarte M.. Genome-wide analysis of the human p53 transcriptional network unveils a lncRNA tumour suppressor signature. Nat. Commun. 2014; 5:5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang Z., Zhou C., Chang Y., Zhang Z., Hu Y., Zhang F., Lu Y., Zheng L., Zhang W., Li X. et al. Long non-coding RNA CASC11 interacts with hnRNP-K and activates the WNT/beta-catenin pathway to promote growth and metastasis in colorectal cancer. Cancer Lett. 2016; 376:62–73. [DOI] [PubMed] [Google Scholar]
- 15. Yang L., Lin C., Liu W., Zhang J., Ohgi K.A., Grinstein J.D., Dorrestein P.C., Rosenfeld M.G.. ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell. 2011; 147:773–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huarte M., Guttman M., Feldser D., Garber M., Koziol M.J., Kenzelmann-Broz D., Khalil A.M., Zuk O., Amit I., Rabani M. et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010; 142:409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xiang J.F., Yin Q.F., Chen T., Zhang Y., Zhang X.O., Wu Z., Zhang S., Wang H.B., Ge J., Lu X. et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014; 24:513–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li Y., Wang Z., Wang Y., Zhao Z., Zhang J., Lu J., Xu J., Li X.. Identification and characterization of lncRNA mediated transcriptional dysregulation dictates lncRNA roles in glioblastoma. Oncotarget. 2016; 7:45027–45041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li J., Han L., Roebuck P., Diao L., Liu L., Yuan Y., Weinstein J.N., Liang H.. TANRIC: an interactive open platform to explore the function of lncRNAs in cancer. Cancer Res. 2015; 75:3728–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yan X., Hu Z., Feng Y., Hu X., Yuan J., Zhao S.D., Zhang Y., Yang L., Shan W., He Q. et al. Comprehensive genomic characterization of long non-coding RNAs across human cancers. Cancer Cell. 2015; 28:529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang J.H., Li J.H., Jiang S., Zhou H., Qu L.H.. ChIPBase: a database for decoding the transcriptional regulation of long non-coding RNA and microRNA genes from ChIP-Seq data. Nucleic Acids Res. 2013; 41:D177–D187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiang W., Mitra R., Lin C.C., Wang Q., Cheng F., Zhao Z.. Systematic dissection of dysregulated transcription factor-miRNA feed-forward loops across tumor types. Brief. Bioinform. 2016; 17:996–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cabili M.N., Trapnell C., Goff L., Koziol M., Tazon-Vega B., Regev A., Rinn J.L.. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011; 25:1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang C., Li Y., Zhao Z., Lu J., Chen H., Ding N., Wang G., Xu J., Li X.. Identifying and functionally characterizing tissue-specific and ubiquitously expressed human lncRNAs. Oncotarget. 2016; 7:7120–7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rosenbloom K.R., Armstrong J., Barber G.P., Casper J., Clawson H., Diekhans M., Dreszer T.R., Fujita P.A., Guruvadoo L., Haeussler M. et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 2015; 43:D670–D681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehar J., Kryukov G.V., Sonkin D. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012; 483:603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang S., Tian D., Tran N.H., Choi K.P., Zhang L.. Profiling the transcription factor regulatory networks of human cell types. Nucleic Acids Res. 2014; 42:12380–12387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goode D.K., Obier N., Vijayabaskar M.S., Lie A.L.M., Lilly A.J., Hannah R., Lichtinger M., Batta K., Florkowska M., Patel R. et al. Dynamic gene regulatory networks drive hematopoietic specification and differentiation. Dev. Cell. 2016; 36:572–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li Y., Shao T., Jiang C., Bai J., Wang Z., Zhang J., Zhang L., Zhao Z., Xu J., Li X.. Construction and analysis of dynamic transcription factor regulatory networks in the progression of glioma. Scientific Rep. 2015; 5:15953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Iyer M.K., Niknafs Y.S., Malik R., Singhal U., Sahu A., Hosono Y., Barrette T.R., Prensner J.R., Evans J.R., Zhao S. et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015; 47:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hoadley K.A., Yau C., Wolf D.M., Cherniack A.D., Tamborero D., Ng S., Leiserson M.D., Niu B., McLellan M.D., Uzunangelov V. et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014; 158:929–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rosenfeld N., Aharonov R., Meiri E., Rosenwald S., Spector Y., Zepeniuk M., Benjamin H., Shabes N., Tabak S., Levy A. et al. MicroRNAs accurately identify cancer tissue origin. Nat. Biotechnol. 2008; 26:462–469. [DOI] [PubMed] [Google Scholar]
- 33. Xu Q., Chen J., Ni S., Tan C., Xu M., Dong L., Yuan L., Wang Q., Du X.. Pan-cancer transcriptome analysis reveals a gene expression signature for the identification of tumor tissue origin. Modern Pathol. 2016; 29:546–556. [DOI] [PubMed] [Google Scholar]
- 34. Cui M., You L., Ren X., Zhao W., Liao Q., Zhao Y.. Long non-coding RNA PVT1 and cancer. Biochem. Biophys. Res. Commun. 2016; 471:10–14. [DOI] [PubMed] [Google Scholar]
- 35. Tseng Y.Y., Moriarity B.S., Gong W., Akiyama R., Tiwari A., Kawakami H., Ronning P., Reuland B., Guenther K., Beadnell T.C. et al. PVT1 dependence in cancer with MYC copy-number increase. Nature. 2014; 512:82–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tseng Y.Y., Bagchi A.. The PVT1-MYC duet in cancer. Mol. Cell. Oncol. 2015; 2:e974467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim H.P., Cho G.A., Han S.W., Shin J.Y., Jeong E.G., Song S.H., Lee W.C., Lee K.H., Bang D., Seo J.S. et al. Novel fusion transcripts in human gastric cancer revealed by transcriptome analysis. Oncogene. 2014; 33:5434–5441. [DOI] [PubMed] [Google Scholar]
- 38. Shi X., Sun M., Liu H., Yao Y., Song Y.. Long non-coding RNAs: a new frontier in the study of human diseases. Cancer Lett. 2013; 339:159–166. [DOI] [PubMed] [Google Scholar]
- 39. Martinez-Fernandez M., Feber A., Duenas M., Segovia C., Rubio C., Fernandez M., Villacampa F., Duarte J., Lopez-Calderon F.F., Gomez-Rodriguez M.J. et al. Analysis of the Polycomb-related lncRNAs HOTAIR and ANRIL in bladder cancer. Clin. Epigenet. 2015; 7:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nie F.Q., Sun M., Yang J.S., Xie M., Xu T.P., Xia R., Liu Y.W., Liu X.H., Zhang E.B., Lu K.H. et al. Long noncoding RNA ANRIL promotes non-small cell lung cancer cell proliferation and inhibits apoptosis by silencing KLF2 and P21 expression. Mol. Cancer Ther. 2015; 14:268–277. [DOI] [PubMed] [Google Scholar]
- 41. Chen G., Wang Z., Wang D., Qiu C., Liu M., Chen X., Zhang Q., Yan G., Cui Q.. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013; 41:D983–D986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ning S., Zhang J., Wang P., Zhi H., Wang J., Liu Y., Gao Y., Guo M., Yue M., Wang L. et al. Lnc2Cancer: a manually curated database of experimentally supported lncRNAs associated with various human cancers. Nucleic Acids Res. 2016; 44:D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morenos L., Chatterton Z., Ng J.L., Halemba M.S., Parkinson-Bates M., Mechinaud F., Elwood N., Saffery R., Wong N.C.. Hypermethylation and down-regulation of DLEU2 in paediatric acute myeloid leukaemia independent of embedded tumour suppressor miR-15a/16-1. Mol. Cancer. 2014; 13:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang L., Xu L., Wang Q., Wang M., An G.. Dysregulation of long non-coding RNA profiles in human colorectal cancer and its association with overall survival. Oncol. Lett. 2016; 12:4068–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wahlestedt C. Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat. Rev. Drug Discov. 2013; 12:433–446. [DOI] [PubMed] [Google Scholar]
- 46. Ling H., Fabbri M., Calin G.A.. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013; 12:847–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Knaack S.A., Siahpirani A.F., Roy S.. A pan-cancer modular regulatory network analysis to identify common and cancer-specific network components. Cancer Inform. 2014; 13:69–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. PNAS. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liang Q., Ding J., Xu R., Xu Z., Zheng S.. The novel human endogenous retrovirus-related gene, psiTPTE22-HERV, is silenced by DNA methylation in cancers. Int. J. Cancer. 2010; 127:1833–1843. [DOI] [PubMed] [Google Scholar]
- 50. Xie B., Lin W., Ye J., Wang X., Zhang B., Xiong S., Li H., Tan G.. DDR2 facilitates hepatocellular carcinoma invasion and metastasis via activating ERK signaling and stabilizing SNAIL1. J. Exp. Clin. Cancer Res.: CR. 2015; 34:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Toy K.A., Valiathan R.R., Nunez F., Kidwell K.M., Gonzalez M.E., Fridman R., Kleer C.G.. Tyrosine kinase discoidin domain receptors DDR1 and DDR2 are coordinately deregulated in triple-negative breast cancer. Breast Cancer Res. Treat. 2015; 150:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bao Z.S., Chen H.M., Yang M.Y., Zhang C.B., Yu K., Ye W.L., Hu B.Q., Yan W., Zhang W., Akers J. et al. RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res. 2014; 24:1765–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dixit U., Liu Z., Pandey A.K., Kothari R., Pandey V.N.. Fuse binding protein antagonizes the transcription activity of tumor suppressor protein p53. BMC cancer. 2014; 14:925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Goodarzi H., Elemento O., Tavazoie S.. Revealing global regulatory perturbations across human cancers. Mol. Cell. 2009; 36:900–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Matsui M., Corey D.R.. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 2017; 16:167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Antonioli L., Blandizzi C., Pacher P., Hasko G.. Immunity, inflammation and cancer: a leading role for adenosine. Nat. Rev. Cancer. 2013; 13:842–857. [DOI] [PubMed] [Google Scholar]
- 57. Bhattacharya S., Andorf S., Gomes L., Dunn P., Schaefer H., Pontius J., Berger P., Desborough V., Smith T., Campbell J. et al. ImmPort: disseminating data to the public for the future of immunology. Immunol. Res. 2014; 58:234–239. [DOI] [PubMed] [Google Scholar]
- 58. Falkenberg K.J., Johnstone R.W.. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014; 13:673–691. [DOI] [PubMed] [Google Scholar]
- 59. Geng L., Cuneo K.C., Fu A., Tu T., Atadja P.W., Hallahan D.E.. Histone deacetylase (HDAC) inhibitor LBH589 increases duration of gamma-H2AX foci and confines HDAC4 to the cytoplasm in irradiated non-small cell lung cancer. Cancer Res. 2006; 66:11298–11304. [DOI] [PubMed] [Google Scholar]
- 60. Beckers T., Burkhardt C., Wieland H., Gimmnich P., Ciossek T., Maier T., Sanders K.. Distinct pharmacological properties of second generation HDAC inhibitors with the benzamide or hydroxamate head group. Int. J. Cancer. 2007; 121:1138–1148. [DOI] [PubMed] [Google Scholar]
- 61. Garcia-Carbonero R., Supko J.G.. Current perspectives on the clinical experience, pharmacology, and continued development of the camptothecins. Clin. Cancer Res. 2002; 8:641–661. [PubMed] [Google Scholar]
- 62. Tadesse S., Yu M., Kumarasiri M., Le B.T., Wang S.. Targeting CDK6 in cancer: state of the art and new insights. Cell Cycle. 2015; 14:3220–3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Finn R.S., Dering J., Conklin D., Kalous O., Cohen D.J., Desai A.J., Ginther C., Atefi M., Chen I., Fowst C. et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res.: BCR. 2009; 11:R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.