


Abstract
As molecular and cellular therapies advance in the clinic, the role of genetic regulation is becoming increasingly important for controlling therapeutic potency and safety. The emerging field of mammalian synthetic biology provides promising tools for the construction of regulatory platforms that can intervene with endogenous pathways and control cell behavior. Recent work has highlighted the development of synthetic biological systems that integrate sensing of molecular signals to regulated therapeutic function in various disease settings. However, the toxicity and limited dosing of currently available molecular inducers have largely inhibited translation to clinical settings. In this work, we developed synthetic microRNA-based genetic systems that are controlled by the pharmaceutical drug leucovorin, which is readily available and safe for prolonged administration in clinical settings. We designed microRNA switches to target endogenous cytokine receptor subunits (IL-2Rβ and γc) that mediate various signaling pathways in T cells. We demonstrate the function of these control systems by effectively regulating T cell proliferation with the drug input. Each control system produced unique functional responses, and combinatorial targeting of multiple receptor subunits exhibited greater repression of cell growth. This work highlights the potential use of drug-responsive genetic control systems to improve the management and safety of cellular therapeutics.
INTRODUCTION
The tools of synthetic biology are advancing our ability to design, modulate, and reprogram biological activity. Programmed cells can interface with complex biological systems and introduce novel functionality that is otherwise difficult to reproduce from nature. Recent advances in the field have led to growing interest in genetically engineering mammalian cells towards various applications in health and medicine (1,2). One area that has gained significant interest is in cell-based therapy, where cells are used as therapeutic agents to treat diseases. Unlike small-molecule drugs, cells have inherent therapeutic capabilities that enable them to sense signals, localize to specific tissue environments, and execute complex tasks (3–5). These features may potentially be harnessed to treat a range of disorders, and indeed, revolutionary clinical trials have highlighted the promise of using engineered cells as therapy (6–13).
One example that has recently gained significant attention is the use of engineered T cells as therapeutic agents. T cells offer an attractive platform because of their innate ability to survey the body for specific molecular signatures and exhibit targeted cytotoxicity. They can be readily isolated from the blood and genetically manipulated and expanded ex vivo to generate a personalized cellular therapy. Researchers have genetically modified T cells to redirect their killing specificity towards cancer cells via the expression of engineered T cell receptors (14–16) and chimeric antigen receptors (CARs) (17–19); these synthetic receptors can significantly boost the immune response from antigen-stimulated T cells. In particular, clinical trials with CAR T cells have demonstrated remarkable success in treating B cell hematological malignancies (7,8,10,12,20). T cells have also been engineered to express therapeutic payloads (i.e. IL-12) to enhance T cell function (21,22). The localized delivery of cytokines, chemokines and other immune effectors may aid in boosting the immune response to overcome the immunosuppressive environment that is characteristic of solid tumors.
Despite the promise of engineered cells as therapy, one of the primary concerns is the lack of control over cell behavior and function when the cells are inside a patient. Engineered cells can exhibit potent effector functions, and the challenge in predicting their efficacy and response in vivo stresses the need for strategies that can effectively intervene with and control cell behavior. CAR T cells have shown incredible efficacy but also severe (and in some cases fatal) toxicities that were difficult to anticipate (14,15,23–27). Therefore, numerous efforts have been directed towards improving the safety profile of genetically modified T cells, such as controlling cell death with suicide switches (28,29) and engineering more sophisticated CARs (30–34).
As an alternative strategy, we explored the use of RNA-based, conditional gene expression systems for modulating T cell behavior. Synthetic RNA switches that link the detection of molecular input signals to regulated gene expression events have been constructed using a variety of regulatory mechanisms on the levels of transcription, translation, RNA splicing, mRNA stability, and post-translational processes (35,36). These RNA-based controllers integrate sensing (encoded by an RNA aptamer) and gene-regulatory functions (encoded by an RNA regulatory element) into a compact framework. RNA control systems avoid the immunogenicity of protein components, and their small genetic footprint facilitates translation to therapeutic applications. Since RNA aptamers can be generated de novo to diverse molecular ligands (37), these RNA platforms offer the potential to develop genetic control systems that are tailored to sense application-specific molecular inputs. By implementing small-molecule control systems in T cells, clinicians may administer a drug input to precisely control timing and release of therapeutic payload. In contrast to using suicide switches, this strategy will be advantageous in tailoring treatment to cases of varying severities, while maintaining T cell therapeutic activity. A recent study demonstrated the use of small molecules to control CAR reconstitution and subsequent signaling (31). However, the rapamycin analog used as the trigger molecule has a short half-life that may limit its clinical applicability, and ligand-responsive dimerization domains are difficult to reengineer and be adapted to other input molecules.
In this work, we developed drug-responsive, microRNA (miRNA)-based gene regulatory systems that are capable of modulating cell proliferation in T cells. These miRNA switches are responsive to the biologically inactive ingredient of leucovorin ((6R)-folinic acid), a pharmaceutical drug that has ideal pharmacokinetic properties for clinical use, thus overcoming the barriers inherent to common small molecule-regulated genetic control systems. We constructed various drug-responsive miRNA switch systems and applied them to regulate multiple components in the cytokine signaling pathway. We show that these systems can effectively modulate T cell proliferation in response to drug input by exerting tight control over critical upstream signaling molecules. The miRNA switches described in this work may also be adapted to target other endogenous genes and tailored for therapeutic applications that require genetic control.
MATERIALS AND METHODS
Plasmids and reagents
All plasmids were constructed using standard molecular biology cloning techniques (see Supplementary Materials for details). DNA synthesis was performed by Integrated DNA Technologies (Coralville, IA, USA) or the Protein and Nucleic Acid Facility (Stanford, CA, USA). Enzymes for cloning, including restriction enzymes, polymerases, and ligases, were obtained from New England Biolabs (Ipswich, MA, USA). All cloned constructs were sequence-verified by Elim Biopharmaceuticals (Hayward, CA, USA). DNA assembly was performed using Gibson cloning (NEB) and the MultiSite Gateway Cloning System (Life Technologies) following manufacturer's instructions. (6R)-, (6S)- and (6R,S)-folinic acid were obtained from Schircks Laboratories (Jona, Switzerland).
T cell culture maintenance
The CTLL-2 murine T cell line was obtained from ATCC and maintained in RPMI-1640 medium (Lonza) supplemented with 10% heat-inactivated FBS (Hyclone), 2 mM sodium pyruvate (Gibco), and 4.5 g/l d-(+)-glucose (Sigma-Aldrich). Cells were fed 100 U/ml IL-2 every 72 hours and maintained at densities between 0.02 and 0.5 × 106 cells/ml. In addition, CTLL-2 platform lines were cultured in 0.25 mg/ml hygromycin (Life Technologies), and stably integrated cell lines were cultured in 0.1 mg/ml zeocin (Life Technologies).
microRNA design
The web-based BLOCK-iT RNAi Designer tool (Thermo Fisher Scientific) was used to identify candidate miRNA sequences. All miRNAs were designed to bind in the ORF or 3’ UTR with 100% homology to their target mRNA per the algorithm.
Plasmid DNA transfection in CTLL-2 cells
All transfections in CTLL-2 cells were performed with an Amaxa Nucleofector II Device and the Mouse T Cell Nucleofector Kit (Lonza) following manufacturer's protocols. The electroporations were performed using 1 × 106 cells with 4 μg of total plasmid DNA (encoding miRNA switch). For stable integrations, the destination vector and integrase vector were combined in a 1:1 mass ratio. Each transfected sample was diluted 2-fold in supplemented media with IL-2, split into the desired number of conditions, and treated with the indicated concentration of drug input. For transient assay, the cells were harvested for antibody staining and fluorescence quantification after 24 h. For stable integrations, the cells were selected in 0.1 mg/ml zeocin (Life Technologies).
Cell surface staining for cytokine receptor expression
Cells were harvested for cytokine receptor staining by washing each sample once with 500 μl HBSS, incubating with either 0.75 μl PE-conjugated mouse IL-2Rβ (BioLegend, clone 5H4) or 2.5 μl PE-conjugated mouse γc (BioLegend, clone TUGm2) antibody in HBSS in a total volume of 50 μl for 15 min at 4°C in the dark, washing twice with 500 μl HBSS, and analyzing on a MACSQuant VYB flow cytometer (Miltenyi Biotec). Fluorescence intensities were quantified as the geometric mean intensity observed in the gated population. Reported expression levels represent the mean ± SD of three replicate samples.
Generation of a clonal CTLL-2 platform line
To generate a clonal CTLL-2 platform cell line for site-specific integration of transgenes, we used modified vectors obtained from the Jump-In TI Gateway System (Life Technologies) and followed manufacturer's protocols. Briefly, the process involved: (i) creating the platform cell line, (ii) sorting the transformants into single clones and (iii) screening the individual clonal cell lines. A transient transfection was performed with 2 × 106 CTLL-2 cells with 10 μg of plasmid DNA (1:1 ratio of pJTI Bxb1 tTA2-Zeo and pJTI PhiC31 Int). Twenty four hours following transfection, the cells were placed in fresh media with IL-2 and diluted to a total volume of 40 ml in a T75 flask, and hygromycin (Life Technologies) was added 24 h later to a final concentration of 0.25 mg/ml to select for stable transformants. The media was replenished every week thereafter, and the cells were split as necessary to avoid overcrowding. After 3 weeks of selection and growth, the cells were stained with a 7-AAD viability dye (Life Technologies) and sorted into single, viable clones on a BD Influx (BD Biosciences) cell sorter. Individual clones were sorted into 200 μl of media supplemented with IL-2 and hygromycin. The clonal populations were individually selected after >2 weeks of growth by clonal expansion and viability, and the successfully expanded clones were individually screened by stably integrating a GFP reporter (pJTI pTet-GFP) using the Bxb1 integrase (pCMV-Bx). Screening was necessary to determine the level of gene expression at the integration loci. We selected a clone that displayed stable, robust and inducible expression of GFP over time.
Growth assays
Cells were harvested, washed twice with HBSS, and seeded at 0.025 × 106 cells/ml in the absence or presence of 100 U/ml IL-2 (depending on whether or not the cells stably expressed the IL-15 proliferative cytokine). (6R)-FA was added to the indicated concentrations, and the cultures were sequentially expanded over time into larger mediums as necessary. Cell count was obtained by assaying 50 μl of cell culture on a Quanta Cell Lab flow cytometer (Beckman Coulter). Total cell count was calculated by the formula:
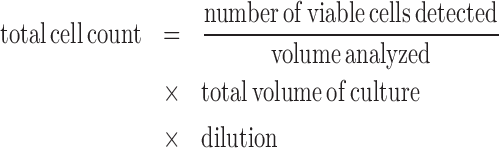 |
where dilution indicates the split ratio when expanding into a larger medium. Reported total cell counts represent the mean ± SD of three replicate samples. For the 24-day growth experiment assaying response to drug input availability, cells were washed twice with HBSS on day 12, split into two equal volumes, and reseeded at 0.05 × 106 cells/ml. (6R)-FA was added to one of the two volumes, and water was added to the other.
Cell surface staining for CD19 expression
CD19 staining was performed by washing 0.5 × 106 cells once with 500 μl HBSS (Gibco), incubating with 5 μl PE-conjugated CD19 antibody (Beckman Coulter) in HBSS in a total volume of 25 μl for 15 min at 4°C in the dark, washing twice with 500 μl HBSS, and analyzing on a MACSQuant VYB flow cytometer (Miltenyi Biotec).
ELISA analysis of IL-15 levels
Cells were seeded at 0.01 × 106 cells/ml in T25 flasks. After 5 days, the cell culture supernatant was collected for IL-15 measurement using the Human IL-15 Quantikine ELISA Kit (R&D Systems) following manufacturer's protocols. The absorbance was measured at 450 nm (reference set at 540 nm) using a Tecan Safire Microplate Reader (Tecan). A standard curve (constructed from standards provided in the kit) was used to determine the concentration of IL-15. Reported concentrations represent the mean ± SD of three replicate samples.
Statistical analysis
Reported gene-regulatory activities represent the mean ± SD of at least two replicate samples (n = 2 or n = 3). Cell count measurements represent mean ± SD of three replicate samples (unless otherwise indicated). Statistical significance was calculated using Student's t-test and indicated by asterisks (n = 3; **P < 0.01).
RESULTS
Identification of a clinically applicable, small-molecule drug for regulating RNA switches
A major barrier to the translation of RNA-based controllers into therapeutic applications is the lack of synthetic RNA switches that sense clinically suitable, small-molecule ligands. The theophylline aptamer has been the model aptamer of choice for mammalian cell applications that involve small-molecule regulation (38,39), but the toxicity and narrow therapeutic index of theophylline limit its practical use in physiological settings. We therefore focused our attention on designing RNA switches that respond to a clinically relevant input molecule, such as the pharmaceutical drug folinic acid (FA), a commonly used adjuvant in cancer chemotherapy (40,41). FA has been previously approved for clinical administration by the U.S. FDA as a racemic form ((6R,S)-FA; known as leucovorin) or as the single diastereomer ((6S)-FA; known as levoleucovorin). (6S)-FA is naturally occurring and biologically active, but the (6R)-FA diastereomer is heterologous and not biologically active (Figure 1A). Pharmacokinetic studies have shown that (6R)-FA is metabolized slower and has a significantly longer plasma half-life than that of (6S)-FA (42). We performed a toxicity study of (6R)-FA to verify that it is nontoxic at concentrations used for regulating RNA switch activity (Supplementary Figure S1A). This combination of low toxicity, biological stability, and absence in natural physiological environments make (6R)-FA an ideal input signal for small molecule-regulated control systems.
Figure 1.

Rational design of a (6R)-FA-responsive miRNA switch to regulate endogenous genes. (A) Chemical structures of (6S)-FA (biologically active) and (6R)-FA (biologically inactive). (B) Predicted secondary structure of the FAt8-3 aptamer. The equilibrium dissociation constants (Kd) to (6R)-FA and (6S)-FA were measured using surface plasmon resonance and represent the mean and standard deviation of three independent experiments. (C) Schematic representation of miRNA switch design. An IL-2Rβ-targeting miRNA switch was constructed by integrating the FAt8-3 aptamer into the basal segment domain of an IL-2Rβ-targeting miRNA. In the upright orientation, stems I and III of FAt8-3 are aligned towards the terminal loop and basal segment of the miRNA, respectively. In the rotated orientation, stems I and III of FAt8-3 are aligned towards the basal segment and terminal loop of the miRNA, respectively. Blue, aptamer; black, miRNA. (D) Panel of miRNA switch designs. For each aptamer orientation (upright or rotated), a panel of switch designs were constructed with subtle structural modifications in the bulge loop size and nucleotide positioning. Switch designs with a rotated aptamer orientation contain two bulge loops, and the indicated size represents the top bulge loop. In selected designs, specific base pairs above the bulge loop were mutated to create an extra bulge. See Supplementary Figure S2 for the predicted secondary structures. (E) Gene-regulatory activities of the miRNA switch panel analyzed by transient transfection in CTLL-2 cells. IL-2Rβ expression levels were examined by cell surface staining in the absence and presence of (6R)-FA, and the mean fluorescence intensity was normalized to those of cells transfected with a construct that lacks a miRNA switch. β-miR represents an IL-2Rβ-targeting miRNA that lacks an aptamer. (F) Predicted secondary structure of β-FAmiR12. IL-2Rβ-targeting sequence is highlighted in red. The nucleotide highlighted in cyan represents a modification to the original FAt-8-3 aptamer bulge loop. (G) Multiple-copy miRNA expression improves gene-regulatory activity of β-FAmiR12. β-FAmiR12 incorporates a modified FAt8-3 aptamer in the basal segment domain of β-miR. IL-2Rβ expression was examined as described in (E). Reported IL-2Rβ expression represents mean ± SD from two replicate samples.
Several RNA aptamers to (6R)-FA have been previously generated using in vitro selection strategies (43). These aptamers have dissociation constants <400 nM at physiologically relevant Mg2+ concentrations (0.5 mM) and ligand specificities up to three orders of magnitude higher than those of structurally similar molecules. Based on the individual structure and sequence flexibility of these aptamers, we chose the FAt8-3 aptamer as the sensor for designing an RNA switch (Figure 1B and Supplementary Figure S1B).
Design of a (6R)-FA-responsive microRNA switch to target endogenous genes
MicroRNAs (miRNAs) are a type of small noncoding RNA that mediates sequence-specific, post-transcriptional gene silencing through the RNAi pathway. The ability of miRNAs to silence endogenous genes offers a potential regulatory mechanism for the modulation of key signaling molecules that affect T cell growth. Previous work has demonstrated the construction of ligand-responsive miRNA switches by integrating an RNA aptamer into the basal segment of a miRNA hairpin to confer ligand-responsive activity (44). Ligand binding to the aptamer inhibits proper maturation of the miRNA, thereby leading to a reduction in RNAi-mediated gene silencing and a subsequent increase in target gene expression. However, the previous miRNA switches were designed using the theophylline aptamer, which contains a relatively compact 15 nucleotide (nt) core binding site (45). In contrast, the FAt8-3 aptamer contains a single 13 nt bulge loop enclosed by 3 stem loops. Given the dissimilar structures of the two aptamers, we created new miRNA switch designs to support integration of the FAt8-3 aptamer.
We rationally designed a (6R)-FA-responsive miRNA switch by integrating the FAt8-3 aptamer into the basal segment domain of an IL-2Rβ-targeting miRNA (Figure 1C). IL-2Rβ is a cytokine receptor that is displayed on the surface of T cells, and we used IL-2Rβ expression as the transient readout to assay miRNA activity in CTLL-2 cells (an IL-2-dependent murine T cell line). We used stems I and III of FAt8-3 as points for integration into a miRNA hairpin that encodes an IL-2Rβ-targeting miRNA sequence (identified through screening). The upright orientation involves aligning stem I towards the terminal loop and stem III towards the basal segment of the miRNA, and the rotated orientation exhibits the reverse alignment. Previous studies have shown that gene-silencing activity is highly sensitive to the structure of the miRNA, and a sufficient bulge size in the miRNA basal segment region is required for proper processing by ribonucleases (46). Therefore, we made a panel of switch designs for each orientation containing various structural alterations in the basal segments of the miRNA switch (Figure 1D and Supplementary Figure S2). To examine gene-regulatory activity, we performed transient transfection assays in CTLL-2 cells and measured IL-2Rβ expression via surface antibody staining in the presence and absence of (6R)-FA (Figure 1E).
Characterization data of initial switch designs containing the unmodified FAt8-3 aptamer confirms the importance of a sufficient bulge loop size. In particular, β-FAmiR3 exhibited a loss of knockdown activity when FAt8-3 was integrated in the basal segment region of β-miR (an IL-2Rβ-targeting miRNA), hence suggesting that the 13 nt bulge loop in FAt8-3 was insufficient to render proper maturation of the miRNA in vivo. Therefore, we made subtle structural modifications by truncating the aptamer domain and increasing the bulge loop size to facilitate Drosha processing of the miRNA (44). We examined 14 and 15 nt bulge loop sizes by adding nucleotides to various positions within the bulge loop. We also mutated specific base pairs above the bulge loop to create a small bulge to decrease the structural rigidity of the miRNA hairpin. However, extensive modifications of FAt8-3 abolished sensitivity of the aptamer to (6R)-FA, so we introduced subtle changes to determine the design space that afforded a balance between gene-silencing activity and ligand responsiveness.
The majority of switch designs in the panel either lost gene-silencing activity or exhibited minimal responsiveness to (6R)-FA. In the rotated orientation, three of five switch designs retained gene-silencing activity, but only β-FAmiR18 exhibited a response to (6R)-FA (18% increase in gene expression). In the upright orientation, most of the switch designs lost gene-silencing activity, likely due to an insufficient bulge loop size. However, β-FAmiR12 exhibited an optimal combination of gene-silencing activity (20% knockdown) and responsiveness to input (17% increase in gene expression). β-FAmiR12 contains a truncated and modified FAt8-3 aptamer, where an extra C is placed in the region between stems II and III of FAt8-3 (Figure 1F). Multiple-copy miRNA expression led to increased gene silencing (57% knockdown) and improved dynamic range (35% difference in gene expression between the presence and absence of drug input) (Figure 1G). Binding assays via surface plasmon resonance confirmed that β-FAmiR12 exhibits a similar binding affinity for (6R)-FA compared to that of the unmodified FAt8-3 aptamer (Supplementary Figure S3). Based on the gene-silencing and regulatory activities, we selected β-FAmiR12 as the optimal miRNA switch design for regulating endogenous gene expression in response to (6R)-FA.
(6R)-FA-responsive miRNA switches exhibit regulation of cytokine receptor expression
Cytokines often share receptor subunits and can mediate functionally overlapping immune responses. For example, IL-2 and IL-15 both utilize IL-2Rβ (CD122) and the common γ chain (γc; also known as CD132) as signaling subunits in their respective heterotrimeric receptors (47). IL-2 and IL-15 can thus mediate common intracellular signaling pathways leading to T cell activation and proliferation (48). In addition, γc also confers responsiveness to other cytokines such as IL-4, IL-7, IL-9 and IL-21. The promiscuous roles played by IL-2Rβ and γc in T cell signaling indicate that they may be effective targets for regulation. Moreover, since receptor subunits control upstream signaling events, their regulatory effects may be further amplified by downstream signaling cascades.
Using our optimized switch design, we also constructed miRNA switches to target the γc cytokine receptor subunit, which is critical for mediating T cell proliferation. Overall, we identified IL-2Rβ- and γc-targeting miRNA sequences that show effective silencing of their intended target receptor with high specificity (Supplementary Figure S4). To assess the regulatory effects on cytokine receptor expression, we generated T cell lines stably expressing various (6R)-FA-responsive miRNA switches. Methods for generating stable cell lines typically rely on random integration of the gene of interest or transduction with viruses. However, random integration of genetic constructs into the genome is inefficient, nonspecific, and often yields inconsistent levels of gene expression due to genomic position effects (49). In order to accurately compare the regulatory activities of various miRNA switches, it was imperative that the genetic constructs are stably expressed at similar levels to minimize any biological discrepancies. Therefore, we generated a clonal CTLL-2 platform line for site-specific integration (Supplementary Figure S5A). We also encoded a tetracycline transactivator (tTA2) in the genome of the CTLL-2 platform line to enable conditional expression of the integrated genetic construct. We verified that gene expression at the integrated loci was robust, consistent, durable, and controllable by doxycycline (Supplementary Figure S5B and S5C). This method of targeted integration obviates the complications from transgene copy variation and genomic position, thus enabling more accurate comparison of the activities of different regulatory elements such as miRNA switches.
We generated clonal CTLL-2 cell lines that harbored various miRNA switches encoded in the 3’ UTR of a GFP reporter (Figure 2A). The constructs were site-specifically integrated into the genome to ensure comparable levels of miRNA expression. We designed constructs with single and multiple copies of IL-2Rβ- or γc-targeting miRNA switches (β(nx) or γ(nx), where ‘n’ represents switch copy number). We also assembled co-targeting switch systems (β(nx)-γ(nx)) to examine potential synergistic effects of silencing both receptor subunits.
Figure 2.

IL-2Rβ- and γc-targeting miRNA switches exhibit drug-mediated control of gene silencing. (A) Schematic representation of the miRNA switch system stably integrated in CTLL-2 cells. The miRNA switches were encoded in the 3’ UTR of a GFP reporter and target endogenously expressed IL-2Rβ and/or γc, which form part of the heterotrimeric IL-2 receptor. miRNA maturation results in formation of mature miRNA and subsequent degradation of the mRNA transcript. Exogenous IL-2 was fed to the cells. Blue, aptamer; black, miRNA; ‘x’, miRNA switch copy number; IL-2R, IL-2 receptor. (B) (6R)-FA-mediated regulation of IL-2Rβ and γc cytokine receptor expression. Clonal cell lines harboring the indicated miRNA switch systems were treated with the specified concentrations of (6R)-FA. IL-2Rβ and γc expression were examined by cell surface staining, and the mean fluorescence intensity was normalized to those of cells harboring a non-targeting miRNA switch. See Supplementary Figure S6 for representative flow cytometry histograms. Reported IL-2Rβ and γc expression represent mean ± SD from three replicate samples.
Examination of cytokine receptor expression by cell surface staining revealed that both β(nx) and γ(nx) effectively silenced their respective targets (>90% for IL-2Rβ and >60% for γc), and that receptor levels could be partially rescued by (6R)-FA (Figure 2B and Supplementary Figure S6). Combinatorial expression of β(nx) and γ(nx) also silenced both receptors, demonstrating that co-targeting switch systems can simultaneously regulate the expression of multiple endogenous targets. However, multiple-copy miRNA expression only further reduced cytokine receptor levels by a small percentage, suggesting that gene silencing via RNAi may have reached a maximum threshold. We also assayed for GFP expression as a gauge to estimate the extent of miRNA maturation in the presence and absence of drug input. Processing of the primary miRNA by Drosha results in transcript cleavage and subsequent mRNA degradation, resulting in reduced translation of the gene (GFP) encoded in the miRNA construct. We observed a proportional decrease in GFP expression with increasing miRNA switch copies, and GFP expression was elevated in the presence of (6R)-FA, thus correlating well with the downstream gene-silencing activities (Supplementary Figure S7).
IL-2Rβ- and γc-targeting miRNA switches exhibit tunable inhibition of T cell proliferation
To examine effects on cell phenotype, we treated clonal cells harboring various miRNA switch systems with (6R)-FA and monitored growth in the presence of IL-2 over a 12-day period. We selected 12 days for the time course of the experiment because CTLL-2 cells double every ∼30 h, so 12 days approximately equal 10 cell doublings. Phenotypic responses, such as cell growth, are often delayed compared to changes in gene expression and thus require multiple cell doublings to exhibit a notable response. We incrementally titrated GFP-miRNA expression via a tetracycline-repressible promoter to determine the complete phenotypic response of the cells at different miRNA expression levels (Figure 3A). When expression was suppressed, all cell lines exhibited normal growth. However, when miRNA expression was gradually increased, we observed unique phenotypes of growth inhibition for each cell line, and systems with higher miRNA switch copies rendered greater repression of cell growth. In contrast, a structurally identical, non-targeting miRNA switch (nt miR) did not produce specific growth-inhibitory effects, but cell proliferation was slightly reduced by ∼20% at high GFP-miRNA expression possibly due to increased energetic demands from constitutive protein expression.
Figure 3.

IL-2Rβ- and γc-targeting miRNA switches exhibit tunable inhibition of T cell proliferation. (A) Complete regulatory response of cell growth at various miRNA expression levels. Clonal cell lines harboring miRNA switch systems targeting IL-2Rβ and/or γc were treated with the specified concentrations of (6R)-FA for five days, and GFP-miRNA expression was titrated via a tet-repressible promoter. ‘nt miR’, non-targeting miRNA switch. (B) T cell proliferation response mediated by (6R)-FA-responsive miRNA switch systems. Clonal cell lines in (A) at full miRNA expression were treated with the specified concentrations of (6R)-FA for 12 days. See Supplementary Figure S8A for individual growth curves. Italicized numbers (red) indicate fold-change in cell count between 0 and 250 μM (6R)-FA. (C) Effects of (6S)-FA and (6R)-FA on T cell proliferation and GFP expression. A clonal cell line harboring β(2x) was treated with the specified concentrations of (6S)-FA and (6R)-FA for 12 days. Cell count and GFP mean fluorescence intensity (MFI) were measured via flow cytometry, and GFP MFI was normalized to those of cells harboring nt miR. Reported measurements represent mean ± SD from three replicate samples. n = 3; **P < 0.01. Two-tailed P values were calculated with Student's t-test.
Following the same experimental setup as described above, we performed time course studies at full miRNA expression (no doxycycline) and observed effective inhibition of cell proliferation by both β(nx) and γ(nx), but cell growth was rescued by (6R)-FA (Figure 3B and Supplementary Figure S8A). Systems harboring a single miRNA switch exhibited up to a 7-fold increase in growth in the presence of (6R)-FA. However, multiple-copy switch systems rendered stronger repression of cell growth, resulting in up to 100-fold regulatory dynamic range (fold-change in cell count between the presence and absence of (6R)-FA). Surprisingly, γ(3x) was not more potent than γ(2x) at repressing cell growth, suggesting a point of diminishing return with >2 copies of this particular γc-targeting miRNA sequence. Combinatorial expression of β(1x)-γ(1x) exhibited a 5-fold higher regulatory dynamic range than either miRNA switch alone, indicating that the gene-silencing activities of each miRNA were maintained and non-redundant. Quantification of the growth response indicates that combinatorial targeting of multiple cytokine receptors rendered greater changes in cell doubling time (DT), with up to a 5-fold change in DT between untreated and (6R)-FA-treated cell populations (Supplementary Figure S8B). Although multiple-copy switch systems only marginally reduced IL-2Rβ and γc receptor expression, the phenotypic results indicate that small changes in cytokine receptor expression may exhibit potent downstream regulatory responses.
To examine the specificity of the regulatory response, we treated clonal cells harboring β(2x) with either (6S)- or (6R)-FA and monitored growth over time (Figure 3C). We selected β(2x) as a representative switch system to illustrate the contrast in response—cells treated with (6R)-FA displayed a >100-fold increase in cell count after 12 days of growth compared to the untreated population, whereas cells treated with (6S)-FA did not exhibit a significant growth response. Assessment of GFP expression levels further supported the observed downstream phenotypic responses—GFP expression was elevated only in the presence of (6R)-FA.
miRNA switch systems exhibit dynamic control over autonomous T cell growth
Therapeutic applications will require genetic control systems that are dynamic and capable of responding in near real time to input signals. To investigate whether the miRNA switch systems react dynamically to changes in drug input levels, we monitored the growth of cells in response to the addition and withdrawal of (6R)-FA. At the beginning of the experiment (day 0), miRNA expression was induced and each cell line was split into two volumes, to which (6R)-FA was added to one. The cell lines were maintained in one condition (with or without (6R)-FA) for 12 days followed by another 12 days in the reverse condition (without or with (6R)-FA). Since CTLL-2 cells are dependent on exogenous cytokine, cell growth over long periods may be sensitive to the timing and levels of IL-2 supplemented to the culture medium. Therefore, we stably integrated a proliferative cytokine (IL-15) along with the individual miRNA switch systems into the CTLL-2 platform line to support autonomous cell division without exogenous cytokine input (Figure 4A and Supplementary Figure S9). This strategy ensured that the observed phenotypic responses were entirely due to (6R)-FA-mediated regulation of cytokine receptor expression.
Figure 4.

Multi-targeted miRNA switch systems enable dynamic control of autonomous T cell growth. (A) Schematic representation of the miRNA switch system stably integrated in autonomously dividing CTLL-2 cells. The miRNA switches target endogenously expressed IL-2Rβ and/or γc, which form part of the heterotrimeric IL-15 receptor. Clonal cell lines also expressed a transgene encoding a cell surface marker (CD19) and proliferative cytokine (IL-15). Stably integrated cells were selected by gating for GFP+CD19+ cells (see Supplementary Figure S9). Blue, aptamer; black, miRNA; ‘x’, miRNA switch copy number; IL-15R, IL-15 receptor. (B) Dynamic control of autonomous T cell growth. Clonal cell lines harboring the indicated miRNA switch systems were treated with or without (6R)-FA for 12 days. On day 12, the cell lines were reseeded and split into two equal volumes, to which (6R)-FA was added to one. Growth was monitored for an additional 12 days. The cell lines were grown in the absence of exogenously fed cytokine. Cell count measurements represent mean ± SD from three replicate samples. Italicized numbers (red) indicate fold-change in cell count on day 24. n = 3; **P < 0.01. Two-tailed P values were calculated with Student's t-test.
Results indicate that T cell proliferation was dynamically regulated, displaying up to 4-fold higher growth after (6R)-FA addition and up to 26-fold diminished growth following (6R)-FA removal (Figure 4B). Cells harboring the β(2x)-γ(2x) system showed stronger repression of cell growth and a greater response to changes in drug availability compared to those expressing the β(1x)-γ(1x) system. However, the stronger inhibition in the absence of drug input also resulted in a relatively longer recovery phase, indicating that the cells may require a longer exposure to drug in order to generate an effective growth response. Assessment of GFP expression shows that transcript levels also correlated dynamically with (6R)-FA treatment (Supplementary Figure S10). In contrast, cells expressing a non-targeting miRNA switch (nt miR) did not exhibit significant changes in growth.
DISCUSSION
Synthetic biological systems have vast potential in health and medical applications. In particular, cell-based therapies, which are quickly emerging as the next-generation therapeutic, represent prime indications for cellular reprogramming. Cells have inherent biological advantages and unique functionalities that may be harnessed to treat diseases that have thus far eluded effective treatment. However, the complexity and therapeutic potency of engineered cells also present challenges and safety concerns that must be addressed. Recent efforts in the field of mammalian synthetic biology have focused on tackling these issues by encoding novel regulatory systems that interface with endogenous signaling pathways to modulate biological function or cell behavior (50–52). However, in general there are limited gene-regulatory systems that are suitable for broad application in clinical settings. Synthetic inducible promoter systems are used extensively in research, but they rely on transcriptional regulatory proteins that are heterologous in nature and unsuitable for stable expression in physiological conditions. Most of the chemical inducers are also too toxic to administer at sufficiently high concentrations in human subjects.
In this work, we developed a synthetic, drug-responsive miRNA-based regulatory system that has broad utility in biological systems that require genetic control. Through rational design and structural engineering, we constructed miRNA switches that are controllable by (6R)-FA, the biologically inactive ingredient of the pharmaceutical drug leucovorin. Unlike other commonly used small-molecule drugs (e.g., theophylline), (6R)-FA exhibits no observable toxicity to the cell line at concentrations used for regulating RNA switch activity, thus permitting safe and prolonged administration in translational applications. Importantly, the miRNA switches exhibit high specificity to (6R)-FA and avoid potential nonspecific activation by structurally similar molecules. We pursued a design strategy to target critical upstream signaling components (IL-2Rβ and γc) in the T cell proliferation pathway, thereby using natural amplification cascades to generate enhanced downstream functional responses.
The miRNA control systems, each encoding a different combination and/or switch copy, produced unique functional responses and regulatory dynamic ranges. Combinatorial targeting of IL-2Rβ and γc exhibited a greater repression of cell growth than targeting either receptor alone, highlighting the synergistic potential of silencing multiple endogenous genes. The co-targeting switch systems also functioned dynamically in response to changes in (6R)-FA availability, exerting both positive and negative regulation of autonomous cell growth following the addition and withdrawal of (6R)-FA, respectively. The growth response to (6R)-FA addition was weaker than that for drug withdrawal, likely due to the delay in miRNA degradation. Studies have shown that mature miRNAs in mammalian cells can exhibit half-lives of greater than 24 h (53), thus prolonging their gene-silencing effects even after their maturation is halted. Therefore, we would expect the addition of (6R)-FA to gradually (rather than immediately) reverse the inhibitory effects on cell growth. Nevertheless, the cells were able to expand from a dormant state given sufficient time. In contrast to suicide switches where cells are irreversibly eliminated in an all-or-none approach, the miRNA control systems enable tunable regulation of T cell growth, which may be advantageous in preserving T cell effector function.
The miRNA switches described in this work are also versatile and may be applied to control the expression of other endogenous genes. For example, checkpoint inhibitory receptors (e.g. CTLA-4) play major roles in modulating the activation of physiological immune responses from T cells and thus represent prime targets for regulation (54). The targeting sequence in the miRNA hairpin can be readily altered to change the target of interest without affecting the switch's responsiveness to (6R)-FA. To create more sophisticated control systems, synthetic miRNA clusters may be assembled to simultaneously regulate multiple signaling pathways. The development of RNA aptamers to other small molecules may also facilitate the construction of more advanced RNA-based control systems, where additional inputs are used to implement layers of regulation. However, the field is limited by the lack of available aptamers that sense clinically suitable drugs. Advancements in aptamer selection, such as the incorporation of high-throughput technologies (i.e. next-generation sequencing), will facilitate the generation of new aptamers to clinically-relevant targets (55,56).
Based on the measured binding affinity of the aptamer, the effective doses of (6R)-FA required to trigger a full response from the switch was higher than anticipated. This discrepancy is likely due to a lowered intracellular concentration of the drug. Since (6R)-FA is an unnatural isomer of folinic acid, it may not be as readily transported into cells as compared to the natural 6S isomer (57). Therefore, the actual concentration of (6R)-FA that is available for binding to the miRNA switch is likely lower than the concentration dissolved in the cell culture medium. The concentrations of (6R)-FA used in this study (50–500 μM) were also generally higher than the reported steady-state serum concentrations of (6R)-FA (∼20–100 μM) (42,57). However, the miRNA switch systems exhibited responses at concentrations as low as 50 μM, and combinatorial targeting of multiple cytokine receptors enhanced the regulatory dynamic range of cell growth. To improve the robustness of the system, miRNA switches may be applied to regulate multiple key targets within a signaling pathway. Alternatively, the individual miRNA switch may be improved by screening a library of mutations in the miRNA basal segments to identify new switch designs that exhibit greater silencing of target gene. miRNA switch designs that are better substrates for Drosha processing and that maintain high affinity for (6R)-FA may achieve enhanced gene-regulatory activities at lower concentrations of (6R)-FA.
Although still in its infancy, mammalian synthetic biology is already inspiring the design and development of next-generation therapeutics (22,31–34). Along with new technologies in genome editing and biomarker discovery (58,59), synthetic regulatory systems and genetic circuitries support the design of engineered cells that have higher specificity, increased potency, and diversified functional outputs. Furthermore, the evolving tool kit of cell engineering will enable researchers to build more advanced cell therapy platforms by incorporating novel modes of regulation, such as exogenous control of therapeutic output and dosage, to generate versatile and safer therapeutics. The recent progress in the field highlights the potential of using synthetically engineered biological systems in advancing medical innovations.
Supplementary Material
ACKNOWLEDGEMENTS
We thank K. Wei for assistance with cloning, A. Chang for identifying aptamer sequences, and M. McKeague and J. Xiang for assistance with the Biacore. The Bxb1 attB and attP sequences and the Bxb1 integrase were generously provided by Dr Michele Calos at Stanford University. The cytokines for cell culture were generously provided by Dr Michael Jensen at Seattle Children's Hospital. Cell sorting for this project was done on instruments in the Stanford Shared FACS Facility.
Author Contributions: R.S.W. and C.D.S. conceived of the project, designed the experiments, analyzed the results and wrote the manuscript. R.S.W. and Y.Y.C. performed the experiments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Defense Advanced Research Projects Agency [HR0011-11-2-0002 to C.D.S.]; Stanford Bio-X Institute (fellowship to R.S.W.). C.D.S. is a Chan Zuckerberg Biohub investigator. Funding for open access charge: Discretionary funds.
Conflict of interest statement. None declared.
REFERENCES
- 1. Chen Y.Y., Smolke C.D.. From DNA to targeted therapeutics: bringing synthetic biology to the clinic. Sci. Transl. Med. 2011; 3:106ps142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lienert F., Lohmueller J.J., Garg A., Silver P.A.. Synthetic biology in mammalian cells: next generation research tools and therapeutics. Nat. Rev. Mol. Cell Biol. 2014; 15:95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mason C., Brindley D.A., Culme-Seymour E.J., Davie N.L.. Cell therapy industry: billion dollar global business with unlimited potential. Regen. Med. 2011; 6:265–272. [DOI] [PubMed] [Google Scholar]
- 4. Fischbach M.A., Bluestone J.A., Lim W.A.. Cell-based therapeutics: the next pillar of medicine. Sci. Transl. Med. 2013; 5:179ps177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buzhor E., Leshansky L., Blumenthal J., Barash H., Warshawsky D., Mazor Y., Shtrichman R.. Cell-based therapy approaches: the hope for incurable diseases. Regen. Med. 2014; 9:649–672. [DOI] [PubMed] [Google Scholar]
- 6. Culme-Seymour E.J., Davie N.L., Brindley D.A., Edwards-Parton S., Mason C.. A decade of cell therapy clinical trials (2000-2010). Regen. Med. 2012; 7:455–462. [DOI] [PubMed] [Google Scholar]
- 7. Kalos M., Levine B.L., Porter D.L., Katz S., Grupp S.A., Bagg A., June C.H.. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011; 3:95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Porter D.L., Levine B.L., Kalos M., Bagg A., June C.H.. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011; 365:725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brentjens R.J., Davila M.L., Riviere I., Park J., Wang X., Cowell L.G., Bartido S., Stefanski J., Taylor C., Olszewska M. et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013; 5:177ra138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grupp S.A., Kalos M., Barrett D., Aplenc R., Porter D.L., Rheingold S.R., Teachey D.T., Chew A., Hauck B., Wright J.F. et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013; 368:1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van Nood E., Vrieze A., Nieuwdorp M., Fuentes S., Zoetendal E.G., de Vos W.M., Visser C.E., Kuijper E.J., Bartelsman J.F., Tijssen J.G. et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N. Engl. J. Med. 2013; 368:407–415. [DOI] [PubMed] [Google Scholar]
- 12. Davila M.L., Riviere I., Wang X., Bartido S., Park J., Curran K., Chung S.S., Stefanski J., Borquez-Ojeda O., Olszewska M. et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014; 6:224ra225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kochenderfer J.N., Dudley M.E., Kassim S.H., Somerville R.P., Carpenter R.O., Stetler-Stevenson M., Yang J.C., Phan G.Q., Hughes M.S., Sherry R.M. et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015; 33:540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Parkhurst M.R., Yang J.C., Langan R.C., Dudley M.E., Nathan D.A., Feldman S.A., Davis J.L., Morgan R.A., Merino M.J., Sherry R.M. et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011; 19:620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morgan R.A., Chinnasamy N., Abate-Daga D., Gros A., Robbins P.F., Zheng Z., Dudley M.E., Feldman S.A., Yang J.C., Sherry R.M. et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013; 36:133–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Robbins P.F., Morgan R.A., Feldman S.A., Yang J.C., Sherry R.M., Dudley M.E., Wunderlich J.R., Nahvi A.V., Helman L.J., Mackall C.L. et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011; 29:917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sadelain M., Brentjens R., Riviere I.. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013; 3:388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maus M.V., Grupp S.A., Porter D.L., June C.H.. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood. 2014; 123:2625–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kalos M., June C.H.. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. 2013; 39:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F. et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014; 371:1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pegram H.J., Lee J.C., Hayman E.G., Imperato G.H., Tedder T.F., Sadelain M., Brentjens R.J.. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012; 119:4133–4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roybal K.T., Williams J.Z., Morsut L., Rupp L.J., Kolinko I., Choe J.H., Walker W.J., McNally K.A., Lim W.A.. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell. 2016; 167:419–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brentjens R., Yeh R., Bernal Y., Riviere I., Sadelain M.. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol. Ther. 2010; 18:666–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cameron B.J., Gerry A.B., Dukes J., Harper J.V., Kannan V., Bianchi F.C., Grand F., Brewer J.E., Gupta M., Plesa G. et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 2013; 5:197ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Linette G.P., Stadtmauer E.A., Maus M.V., Rapoport A.P., Levine B.L., Emery L., Litzky L., Bagg A., Carreno B.M., Cimino P.J. et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013; 122:863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maus M.V., Haas A.R., Beatty G.L., Albelda S.M., Levine B.L., Liu X., Zhao Y., Kalos M., June C.H.. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013; 1:26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morgan R.A., Yang J.C., Kitano M., Dudley M.E., Laurencot C.M., Rosenberg S.A.. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010; 18:843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Di Stasi A., Tey S.K., Dotti G., Fujita Y., Kennedy-Nasser A., Martinez C., Straathof K., Liu E., Durett A.G., Grilley B. et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011; 365:1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou X., Di Stasi A., Tey S.K., Krance R.A., Martinez C., Leung K.S., Durett A.G., Wu M.F., Liu H., Leen A.M. et al. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood. 2014; 123:3895–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kloss C.C., Condomines M., Cartellieri M., Bachmann M., Sadelain M.. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 2013; 31:71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu C.Y., Roybal K.T., Puchner E.M., Onuffer J., Lim W.A.. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015; 350:aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Roybal K.T., Rupp L.J., Morsut L., Walker W.J., McNally K.A., Park J.S., Lim W.A.. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell. 2016; 164:770–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ma J.S., Kim J.Y., Kazane S.A., Choi S.H., Yun H.Y., Kim M.S., Rodgers D.T., Pugh H.M., Singer O., Sun S.B. et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E450–E458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rodgers D.T., Mazagova M., Hampton E.N., Cao Y., Ramadoss N.S., Hardy I.R., Schulman A., Du J., Wang F., Singer O. et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E459–E468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liang J.C., Bloom R.J., Smolke C.D.. Engineering biological systems with synthetic RNA molecules. Mol. Cell. 2011; 43:915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chang A.L., Wolf J.J., Smolke C.D.. Synthetic RNA switches as a tool for temporal and spatial control over gene expression. Curr. Opin. Biotechnol. 2012; 23:679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ellington A.D., Szostak J.W.. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990; 346:818–822. [DOI] [PubMed] [Google Scholar]
- 38. Chen Y.Y., Jensen M.C., Smolke C.D.. Genetic control of mammalian T-cell proliferation with synthetic RNA regulatory systems. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:8531–8536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ketzer P., Kaufmann J.K., Engelhardt S., Bossow S., von Kalle C., Hartig J.S., Ungerechts G., Nettelbeck D.M.. Artificial riboswitches for gene expression and replication control of DNA and RNA viruses. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:E554–E562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Flombaum C.D., Meyers P.A.. High-dose leucovorin as sole therapy for methotrexate toxicity. J. Clin. Oncol. 1999; 17:1589–1594. [DOI] [PubMed] [Google Scholar]
- 41. Mini E., Trave F., Rustum Y.M., Bertino J.R.. Enhancement of the antitumor effects of 5-fluorouracil by folinic acid. Pharmacol. Ther. 1990; 47:1–19. [DOI] [PubMed] [Google Scholar]
- 42. Newman E.M., Straw J.A., Doroshow J.H.. Pharmacokinetics of diastereoisomers of (6R,S)-folinic acid (leucovorin) in humans during constant high-dose intravenous infusion. Cancer Res. 1989; 49:5755–5760. [PubMed] [Google Scholar]
- 43. McKeague M., Wang Y.H., Smolke C.D.. In vitro screening and in silico modeling of RNA-based gene expression control. ACS Chem. Biol. 2015; 10:2463–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Beisel C.L., Chen Y.Y., Culler S.J., Hoff K.G., Smolke C.D.. Design of small molecule-responsive microRNAs based on structural requirements for Drosha processing. Nucleic Acids Res. 2011; 39:2981–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zimmermann G.R., Wick C.L., Shields T.P., Jenison R.D., Pardi A.. Molecular interactions and metal binding in the theophylline-binding core of an RNA aptamer. RNA. 2000; 6:659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Beisel C.L., Bayer T.S., Hoff K.G., Smolke C.D.. Model-guided design of ligand-regulated RNAi for programmable control of gene expression. Mol. Syst. Biol. 2008; 4:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ring A.M., Lin J.X., Feng D., Mitra S., Rickert M., Bowman G.R., Pande V.S., Li P., Moraga I., Spolski R. et al. Mechanistic and structural insight into the functional dichotomy between IL-2 and IL-15. Nat. Immunol. 2012; 13:1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fehniger T.A., Caligiuri M.A.. Interleukin 15: biology and relevance to human disease. Blood. 2001; 97:14–32. [DOI] [PubMed] [Google Scholar]
- 49. Liang Z., Breman A.M., Grimes B.R., Rosen E.D.. Identifying and genotyping transgene integration loci. Transgenic Res. 2008; 17:979–983. [DOI] [PubMed] [Google Scholar]
- 50. Morsut L., Roybal K.T., Xiong X., Gordley R.M., Coyle S.M., Thomson M., Lim W.A.. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell. 2016; 164:780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xie M., Ye H., Wang H., Charpin-El Hamri G., Lormeau C., Saxena P., Stelling J., Fussenegger M.. beta-cell-mimetic designer cells provide closed-loop glycemic control. Science. 2016; 354:1296–1301. [DOI] [PubMed] [Google Scholar]
- 52. Ye H., Xie M., Xue S., Charpin-El Hamri G., Yin J., Zulewski H., Fussenegger M.. Self-adjusting synthetic gene circuit for correcting insulin resistance. Nat. Biomed. Eng. 2017; 1:0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Marzi M.J., Ghini F., Cerruti B., de Pretis S., Bonetti P., Giacomelli C., Gorski M.M., Kress T., Pelizzola M., Muller H. et al. Degradation dynamics of microRNAs revealed by a novel pulse-chase approach. Genome Res. 2016; 26:554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pardoll D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012; 12:252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Riley K.R., Gagliano J., Xiao J., Libby K., Saito S., Yu G., Cubicciotti R., Macosko J., Colyer C.L., Guthold M. et al. Combining capillary electrophoresis and next-generation sequencing for aptamer selection. Anal. Bioanal. Chem. 2015; 407:1527–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Spiga F.M., Maietta P., Guiducci C.. More DNA-aptamers for small drugs: a capture-SELEX coupled with surface plasmon resonance and high-throughput sequencing. ACS Comb. Sci. 2015; 17:326–333. [DOI] [PubMed] [Google Scholar]
- 57. Bertrand R., Jolivet J.. Lack of interference by the unnatural isomer of 5-formyltetrahydrofolate with the effects of the natural isomer in leucovorin preparations. J. Natl. Cancer Inst. 1989; 81:1175–1178. [DOI] [PubMed] [Google Scholar]
- 58. Nissim L., Perli S.D., Fridkin A., Perez-Pinera P., Lu T.K.. Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells. Mol. Cell. 2014; 54:698–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Brunger J.M., Zutshi A., Willard V.P., Gersbach C.A., Guilak F.. CRISPR/Cas9 editing of murine induced pluripotent stem cells for engineering inflammation-resistant tissues. Arthritis Rheumatol. 2017; 69:1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.