

Abstract
Saccharomycotina comprises a diverse group of yeasts that includes numerous species of industrial or clinical relevance. Opportunistic pathogens within this clade are often assigned to the genus Candida but belong to phylogenetically distant lineages that also comprise non-pathogenic species. This indicates that the ability to infect humans has evolved independently several times among Saccharomycotina. Although the mechanisms of infection of the main groups of Candida pathogens are starting to be unveiled, we still lack sufficient understanding of the evolutionary paths that led to a virulent phenotype in each of the pathogenic lineages. Deciphering what genomic changes underlie the evolutionary emergence of a virulence trait will not only aid the discovery of novel virulence mechanisms but it will also provide valuable information to understand how new pathogens emerge, and what clades may pose a future danger. Here we review recent comparative genomics efforts that have revealed possible evolutionary paths to pathogenesis in different lineages, focusing on the main three agents of candidiasis worldwide: Candida albicans, C. parapsilosis and C. glabrata. We will discuss what genomic traits may facilitate the emergence of virulence, and focus on two different genome evolution mechanisms able to generate drastic phenotypic changes and which have been associated to the emergence of virulence: gene family expansion and interspecies hybridization.
Keywords: Candida, Saccharomycotina, pathogens, genomics, evolution
The growing availability of genomic information from the increasingly relevant Candida pathogens is helping us to unravel their recent evolution.
INTRODUCTION
Saccharomycotina is a subphylum of the Ascomycota. It contains a single class (Saccharomycetes), and is comprised mostly by yeasts that have naked asci, do not form fruiting bodies and can reproduce asexually by budding (Kurtzman, Fell and Boekhout 2011). Many budding yeast species are normal components of the human microbiota, inhabit our immediate environment, and some of them are traditionally used to ferment food or beverages (e.g. the baker's yeast Saccharomyces cerevisiae). In addition, some yeasts are considered opportunistic pathogens because they can cause disease under certain circumstances such as weakening of the immune system. Such opportunistic pathogenic yeasts constitute an important medical problem of increasing incidence. Indeed, yeast pathogens have become a major source of life-threatening nosocomial (i.e. hospital-acquired) infections since the 1980s, which is in part explained by recent medical progress. For instance, modern medical care has increased the survival of persons that are susceptible to yeasts opportunistic pathogens, such as premature neonates, elderly people, immunocompromised patients, as well as patients undergoing immunosuppressive chemotherapy. On the other hand, reliance on catheters, large-spectrum antibiotics and surgery are all factors that favor the spread and colonization of pathogenic yeasts (Turner and Butler 2014). Candidiasis is a medical term that refers to any superficial or invasive fungal infection caused by any type of Candida yeast. Despite recent advances, the mortality rates associated to invasive Candidiasis remain high at 30%–40%, and the treatment of such infections is complicated by the appearance of resistance to antifungals and the emergence of novel pathogenic species (Papon et al. 2013). The Candida genus itself is a very complex and heterogeneous taxon comprised of over 160 anamorphic (i.e. asexual reproductive stage) species, of which teleomorphs (i.e. sexual reproductive forms) belong to at least 16 different genera including Pichia, Debaryomyces or Saccharomyces (Guarro, Gené and Stchigel 1999). Thus, the Candida genus is poliphyletic and comprises species belonging to evolutionary distant clades. Given recent developments in fungal taxonomy, including increased reliance on genomic data and the ‘one fungus, one name’ rule (Hawksworth 2012), many Candida species are being renamed and the whole genus will likely be completely revamped in the coming years (Brandt and Lockhart 2012). Over 30 Candida (or formerly named Candida) species have been identified as etiological agents in candidiasis (Table 1). Although incidence may vary from region to region, four species generally account for over 95% of the cases: Candida albicans, C. glabrata, C. parapsilosis and C. tropicalis, generally in this order (Pfaller and Diekema 2007; Diekema et al.2012). After these four main players, other Candida species can be found as causative agents of candidiasis but with much lower incidence, followed by a growing list of species that are rarely found in infections, including single-case reports (Table 1).
Table 1.
List of 31 species reported as agents of candidasis. They are roughly classified in three groups according to their clinical incidence. Most incidence data come from a global study involving 141 hospitals from 41 countries over 10 years (1997–2007) (Pfaller et al.2010). Species marked with an asterisk were not listed in that study because they had not been discriminated as a different species, or because they have been reported from few cases elsewhere. Names between brackets indicate the currently accepted names for each Candida species.
| Incidence | Species |
|---|---|
| Common agents of candidiasis (5%–70%) | Candida albicans, C. glabrata, C. parapsilosis, C. tropicalis |
| Rare but may be locally common (0.1%–3%) | C. krusei (Pichia kudriavzevii), C. lusitanae (Clavispora lusitaniae), C. orthopsilosis*, C. metapsilosis*, C. guilliermondii (M. guilliermondii), C. kefyr (Kluyveromyces marxianus), C. inconspicua, C. famata (synonym: Debaryomyces hansenii), C. rugosa, C. dubliniensis, C. norvegensis (Pichia norvegensis), C. nivariensis*, C. bracarensis* |
| Rarely reported (<0.1% including single-case reports) | C. pelliculosa (Wickerhamomyces anomalus), C. subhashii*, C. lipolytica (Yarrowia lipolytica), C. sake, C. apicola, C. zeylanoides, C. valida (Pichia membranifaciens), C. intermedia, C. pulcherrima (Metschnikovia pulcherrima), C. haemulonii (C. heamulonis), C. stellatoidea, C. utilis (Cyberlindnera jardinii), C. humicola (Asterotremella humicola), C. lambica (Pichia fermentans), C. ciferrii (Trichomonascus ciferrii), C. colliculosa (Torulaspora delbrueckii), C. holmii (Kazachstania exigua), C. marina (Cryptococcus marinus), C. sphaerica (Kluyveromyces lactis) |
As mentioned above, despite their common genus name and their shared ability to infect humans, Candida pathogenic species can belong to phylogenetically distinct clades, which also contain non-pathogenic relatives. For instance, C. glabrata is a common pathogen that is closely related to the non-pathogenic species Saccharomyces cerevisiae, and C. parapsilosis is clearly a distinct lineage to that of C. albicans, and is more closely related to the non-pathogenic Lodderomyces elongisporus. Similarly, although C. tropicalis is not distantly related to C. albicans, it is sister to non-pathogenic species such as C. sojae. This indicates that the emergence of pathogenesis towards humans has occurred several times independently. Importantly, it is as yet poorly understood whether these different pathogenic clades may exploit common infection strategies or use lineage-specific mechanisms, or a combination of both. In addition, within each pathogenic clade, there is a large variability of virulence phenotypes across different strains (e.g. hyper and hypovirulent strains), and even some pathogenic strains have been described for generally non-pathogenic species such as S. cerevisiae (Anoop et al.2015). For many of the pathogenic species, however, it is unclear whether the human body constitutes their major ecological niche or whether they are adapted to a variety of environments. In fact, it is remarkable how little we know about the ecology of most yeast species. Ultimately, differences in virulence across species and strains are related to changes at the genomic level, and thus comparative genomics constitutes a promising research avenue.
Recent progress in genomics has facilitated the completion of the genomic sequences from numerous Candida pathogenic species and their non-virulent relatives, as well as of multiple isolates from the same species. Here we review recent advances in the understanding of how virulence traits in the different clades may have emerged. First, we provide an introduction to the computational and experimental approaches that can be used to trace what evolutionary mechanisms may have played a role in the evolution of a virulence trait. Then, we survey recent efforts focused on the three most important clades, according to their incidence: C. albicans, C. glabrata and C. parapsilosis. Finally, we discuss future avenues, including the application of high-throughput sequencing, meta-genomics and population genomics, to understand the recent evolution and epidemiology of fungal pathogens.
COMPARATIVE GENOMICS AND PHYLOGENOMICS TO DISCOVER AND UNRAVEL THE ORIGINS OF VIRULENCE TRAITS
The comparison of genomes under an evolutionary perspective (i.e. phylogenomics) constitutes a very powerful tool to understand the function, origin and evolution of biological processes of interest (Eisen 1998; Gabaldón 2008; Gabaldon and Marcet-Houben 2014). Given the widespread focus on pathogenesis, the genomes of virulent organisms have traditionally been the main target of whole genome sequencing projects. Comparison with non-pathogenic relatives constitutes a common strategy to identify and prioritize candidate virulence factors. In this respect the comparison of the pathogenic bacterium Listeria monocytogenes, with its non-pathogenic relative L. innocua, constitutes one of the first examples of the use of a comparative genomics approach to search for genes involved in virulence (Glaser et al.2001). In that study the researchers identified several species-specific genes, which were enriched in secreted or cell-wall proteins, including several known virulence factors that were found to be specific to L. monocytogenes. The basic rationale of this approach is simple. Related species are expected to share the genetic factors that determine their shared phenotypes, whereas they are expected to differ genetically in those genomic regions that determine their unique phenotypic characteristics. Differences in the presence and absence of genes is one obvious trait to focus on, and this approach has been shown to be extremely useful in pathogenic bacteria, where genomic islands, often transmitted across species through horizontal gene transfer, can determine the existence of pathogenicity to a given host, or of resistance to a certain spectrum of antibiotics (Gal-Mor and Finlay 2006). Two main problems underlie this approach. First of all, the two compared species are likely to differ in many more phenotypes than the one that constitutes the focus of our attention (i.e. virulence). In addition, most of the differing phenotypes or adaptations are likely to be unknown to us. Therefore, genetic differences will comprise those involved in all these many phenotypic differences. This problem scales up with the genetic and phenotypic divergence of the species compared. A second problem of the gene content comparison approach is that gene gain and loss is only one way in which changes in phenotype can be driven. Gene duplications, genomic re-arrangements and discrete point mutations in coding or non-coding regions may all determine key changes in phenotype, and would pass unnoticed in simple gene-content comparisons.
Taking into account these considerations, the phylogenomic approaches used to understand the evolution of pathogenesis have evolved in two main directions (see Fig. 1). On the one hand, the resolution of the comparison has increased by considering less divergent and a higher number of genomes. This includes genomes from more closely related species as well as several strains per species. On the other hand, the comparisons have developed into more sophisticated ones, including the study of large genomic re-arrangements as well as the detection of past evolutionary events through the use of a phylogenetic approach or the analyses of genomic variation. These methods include not only the detection of genes duplicated in a given lineage but also those under positive selection. This more detailed approach has been driven by parallel developments in genomics and computational methods.
Figure 1.

Schematic overview of comparative genomics methods to find virulence factors. In the center of the image a phylogenetic tree represents the evolutionary relationships between an idealized group of pathogenic and non-pathogenic species, where the ones drawn in red are pathogenic. Red stars indicate two points in the tree where virulence emerged. For species E, a list of strains is indicated on the right showing the presence of sequenced strains with different degrees of virulence. The boxes surrounding the tree indicate different kinds of analyses that can be performed to detect virulence traits. (A) Search for genomic re-arrangements. (B) Presence and absence of genes, shown as a table where the white squares represent missing genes and the gray and red square represent present genes. In addition, red squares indicate putative virulence factors. (C) Detection of differences between strains. Each arrow represents a gene. Red horizontal lines represent SNPs. (D) Detection of positive selection. Red horizontal lines represent non-synonymous SNPs and black horizontal lines represent synonymous SNPs. (E) Events that can be detected with the use of phylogenetic trees. Arrows represent genes. Gene duplication and loss is represented along with changes in gene order. (F) Gene expansions. Phylogenetic tree representing two independent expansion events in the pathogenic species. Expansion points are marked with a star.
Recent sequencing of several Candida pathogens and their close non-pathogenic relatives has enabled the use of comparative genomics approaches to discover virulence factors and trace their evolutionary emergence. Some such examples will be highlighted in the following sections. Before applying a comparative genomics approach, however, it is necessary to obtain an evolutionary scenario in which the relationships between pathogenic and non-pathogenic species are clearly delineated. In the following section we provide an overview of the evolutionary relationships among Candida species.
CANDIDA PATHOGENS ARE PHYLOGENETICALLY DIVERSE
To provide a glimpse of the diversity of currently sequenced Candida species, we used a phylogenomics approach, based on the analysis of 516 shared orthologous genes, to reconstruct the evolutionary relationships of 71 Saccharomycotina species (Fig. 2). We highlighted species currently named Candida and those whose anamorph name or synonym name are Candida, and marked in red those species that are well-established pathogens or those that are recognized as emerging fungal pathogens. As observed in the tree, Candida species are spread throughout the Saccharomycotina phylogeny, being present in most of the clades. In addition, Candida species are generally intermingled with species assigned to other genera, highlighting the polyphyly of the genus. Pathogenic species are mainly found in two different clades: the CTG group and the Nakaseomyces group (marked with blue dots on the tree). The only exception is Candida krusei, which branches close to the wine yeast Brettanomyces bruxellensis. Two other Candida species, C. boidinii and C. parapolymorpha (syn. Ogataea parapolymorpha), are found in the same clade as C. krusei, though both of them are considered non-pathogenic.
Figure 2.
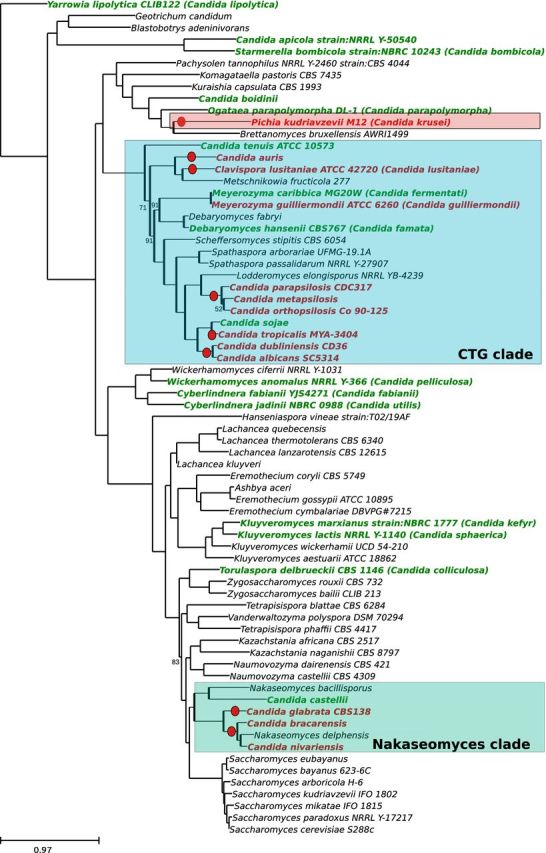
Phylogenetic relationships among Candida. A total of 516 genes detected as single copy genes in Saccharomyces species (Marcet-Houben and Gabaldón 2015) were used to perform a homology search against a proteome database formed by the 71 Saccharomycotina species included in the tree. A phylogenetic tree was reconstructed for each group of homologs using the phylome reconstruction pipeline (Huerta-Cepas et al.2014). Phylogenetic trees were examined with ETE (Huerta-Cepas, Dopazo and Gabaldón 2010), species-specific duplications were deleted and one sequence belonging to the duplicated clade was randomly chosen to represent the clade. Trees that, after filtering, contained one-to-one orthologs in at least three-fourth of the species were retained and their alignments were concatenated. The final alignment contained 190 110 amino acid positions. The phylogenetic tree was reconstructed using RAxML with the PROTGAMMALG model (Stamatakis, Ludwig and Meier 2005). Bootstraps were reconstructed using RAxML rapid bootstrap approach, supports below 100 are marked on the tree. For Candida species, red-colored leaves indicate pathogenic species while green-colored leaves indicate non-pathogenic species.
The CTG clade comprises several species that have the particularity of using an alternative genetic code in which the CUG codon, typically used for leucine, has been reassigned to serine and is ambiguously translated in the cytoplasm (Santos et al.2011). Interestingly, a reassignment of the same codon to alanine has apparently occurred in the lineage leading to Pachysolen tannophilus (Mühlhausen and Kollmar 2014), a distantly related yeast isolated from sulfite liquor. The CTG clade contains most of the pathogenic Candida species, including two of the three major candidiasis agents: C. albicans and C. parapsilosis. The group comprises many species and 19 have completely sequenced genomes. Out of the sequenced species nearly half (9) are from human pathogens. Rather than being monophyletic, pathogenic species appear interspersed within non-pathogenic ones, with at least six independent pathogenic lineages surrounded with non-pathogenic relatives. This includes (i) the clade containing C. albicans and C. dublinensis; (ii) a separate clade containing C. tropicalis, an emerging fungal pathogen which is naturally resistant to fluconazole and which is more closely related to the rarely pathogenic C. sojae than it is to C. albicans (Fig. 2); (iii) the C. parapsilosis species clade, comprising C. parapsilosis and the close relatives C. orthopsilosis and C. metapsilosis; (iv) a lineage comprising Meyerozyma guilliermondii (C. guilliermondii), and its close non-pathogenic relative M. caribbica (Candida fermentati); (v) a lineage containing Clavispora lusitaniae (Candida lusitaniae), grouped with the plant-associated yeast Metschnikowia fructicola; and (vi) a separate branch with the multidrug resistant C. auris.
The second main group of pathogenic Candida species is the Nakaseomyces, which is more closely related to Saccharomyces cerevisiae (Gabaldón et al.2013). The Nakaseomyces indeed is one of the several so-called post-WGD (post whole genome duplication) lineages, a group of species that diverged from an ancestor that duplicated its genome entirely (Wolfe and Shields 1997; Dujon 2010). Importantly, it has now become apparent that this WGD was triggered by an interspecies hybridization event (Marcet-Houben and Gabaldón 2015). The post-WGD clade is very diverse and contains many species of industrial interest, but is otherwise scarce in pathogenic species. Among the post-WGD clade, only the Nakaseomyces clade seems to be prone to present the ability to infect humans. It comprises three pathogenic species, with C. glabrata as the most well known and which ranks as the second-most common source of candidiasis worldwide. Candida bracarensis and C. nivarensis are two related emergent pathogens of growing incidence (Gabaldón and Carreté 2016). The three pathogens within the Nakaseomyces are not monophyletic, indicating that they may represent two, or even three parallel events of emergence of pathogenesis (Gabaldón et al.2013).
Altogether, from a phylogenetic perspective, Candida species do not have a single evolutionary origin, and the genus is destined to be completely redefined if we are to adhere to taxonomic principles. From a clinical perspective, we should look at Candida pathogens as a diverse set of infectious agents, despite their shared generic name and the common denomination of candidiasis for their infections. Indeed, the phylogenetic divergence between the four most prevalent Candida species (C. albicans, C. glabrata, C. parapsilosis and C. tropicalis) is reflected in their phenotypic diversity (Brunke and Hube 2013; Papon et al.2013).
Out of these four species, C. albicans is the one showing the highest morphological plasticity, being able to grow in the form of single cells (yeast form), pseudohyphae and true hyphae depending on the environment (Sudbery 2011; Thompson, Carlisle and Kadosh 2011; Modrzewska and Kurnatowski 2013; Priest and Lorenz 2015). In addition, C. albicans can switch to hyphal and pseudohyphal forms once phagocytosed and use filamentous growth to burst out and kill macrophages. From the other three species, only C. tropicalis is known to be able to switch to true hyphal form, a capability that is probably much more restricted than in the case of C. albicans (Priest and Lorenz 2015). The alternative genetic code of the CTG clade allows C. albicans to increase the diversity of its surface proteins, thus making it more difficult to detect by the immune system (Nather et al.2008; Miranda et al.2013). Little is known about the role in pathogenesis of this alternative codon in C. tropicalis and C. parapsilosis. Additionally, it is totally absent in the non-CTG clade species C. glabrata. Similarly, the response in face of the immune system of the different Candida species is highly variable. Candida glabrata shows adaptation to the conditions inside the phagosome and is able to live and even reproduce in this harsh environment during long periods of time (Fukuda et al.2013; Seider et al.2014). The other three species usually avoid phagocytosis in the first place, and switch to their hyphal or pseudohyphal stages in order to break free from phagocytic cells (Jiménez-López and Lorenz 2013). Candida glabrata is also the only of the four that is a haploid, although parasexual cycle in C. albicans can produce aneuploidies that can result in partial haploidy (Glöckner and Cornely 2015). Candida glabrata is also naturally resistant to azole compounds, and is the most common yeast to be able to acquire resistance to echinocandins (Pfaller and Diekema 2007). In contrast to other Candida species, C. glabrata is more prevalent in adults, where aging, use of antibiotics and the severity of other medical conditions are important risk factors (Pfaller and Diekema 2007; Guinea et al.2014). From the four, C. parapsilosis is the one that has lower lethality. It is also the only one that is not considered a human commensal and thus infections of C. parapsilosis are usually acquired from the environment and consistently avoided with proper preventive measures (Warnock 2007; Holland et al.2014). Candida parapsilosis is mostly related to the use of catheters, prosthetics and other similarly invasive medical devices. Lastly, C. tropicalis infections are usually restricted to patients with neutropenia and other blood malignancies and usually respond to azole antifungals (Pfaller and Diekema 2007; Guinea et al.2014). It is, however, more invasive than C. albicans and infections with this pathogen show higher mortality rates. Finally, beyond these generalities, the epidemiology of these fungal pathogens shows great variability among human populations and geographical regions (Pfaller and Diekema 2007; Warnock 2007; Guinea et al.2014).
In summary, the ability of Candida species to infect humans has emerged several times independently from each other from diverged non-pathogenic ancestors, and the extent to which they share virulence strategies is still an open question. From an evolutionary perspective, the parallel emergence of a common trait (i.e. the ability to infect humans) from different genomic backgrounds poses many intriguing questions: How many parallel adaptations emerged independently in more than one lineage? What set of genomic traits favored the emergence of virulence in each case? How many different paths may lead to the ability to infect humans? What evolutionary mechanisms have promoted the emergence of virulence? In the following sections, we provide an overview of recent studies that have addressed such and other questions in the main clades of pathogenic Candida.
GENOME VARIATION IN THE CTG CLADE AND EXPANSION OF CELL-WALL FAMILIES IN PATHOGENS
One of the first studies targeting genomic variation in Candida pathogens was conducted by Geraldine Butler and collaborators in 2009 (Butler et al.2009). That study expanded the set of genome sequences of Candida pathogens (two by then Candida albicans and C. glabrata), by adding five new species, C. tropicalis, C. parapsilosis, Lodderomyces elongisporus, C. guilliermondii and C. lusitaniae, and a new strain of C. albicans. One of the first findings of the genome comparison was the discovery of large variations in genome size within the CTG clade (up to 50% variation in size), despite a similar gene repertoire size, with gene numbers ranging from 5733 to 6318. Importantly, this study provided the first hint for an enriched cell-wall repertoire in Candida pathogens, as compared to non-pathogenic relatives. By examining the phylogenetic distribution of 9209 gene families present across the analyzed species, they identified 21 which were enriched in the more common pathogens. These included families encoding GPI-anchored cell-wall adhesins, secreted lipases, as well as oligopeptide transporters, and transcription factors.
Among the families associated to the cell wall that were found to be expanded in pathogenic species, there were some, such as Hyr/Iff and Als adhesins, that were found to be duplicated in tandem in some pathogenic species, with the paralogous gene clusters sometimes including five or six genes of the same family. Tandem duplications are an essential source of genetic novelty that have been found to commonly underlie novel phenotypic traits, and are generally indicative of recent directional selection (Conant and Wolfe 2008). In addition, these families were found to have high levels of sequence variation, including variations in number of intragenic tandem repeats. All these findings suggested that some virulence traits, such as an increased or more versatile adherence, may have emerged relatively fast by simple genome re-arrangements such as gene duplications followed by sequence divergence. An alternative approach identified 64 families likely subjected to positive selection in the lineages leading to highly pathogenic Candida species. Again, cell-wall proteins emerged in these analyses, together with families involved in hyphal and pseudohyphal filamentous growth, as well as in biofilm formation. Altogether, this study highlighted the importance of cell-wall components, probably mediating host–pathogen interactions, in shaping the ability to infect humans. Despite these important advances, the lack of close relatives for most of the pathogenic species included provided a limited resolution of the specificities of each pathogenic lineage.
The Butler et al. (2009) study also provided the first intraspecies genome comparison within a Candida pathogen, as it provided the genome sequence of a second strain belonging to a different Multilocus sequence type-based clade of C. albicans. The two strains were found to be highly colinear but they differed greatly in the extent of homozygous regions in their diploid genomes, indicating different events of loss of heterozygosity by break-induced recombination or recent passage through a parasexual cycle. More recent studies have increased our knowledge on the genetic variability at the species level in C. albicans, by analyzing the sequences of 21 independent clinical isolates (Hirakawa et al.2015) and 43 serial isolates from 11 distinct patients (Ford et al.2015). These analyses indicated that most genetic variation among C. albicans isolates corresponded to distinct patterns of loss of heterozygosis as well as total or partial chromosomal aneuploidies. These genomic changes underlaid important phenotypic variations such as up to 3-fold differences in growth rates, ability to filament or the resistance to antifungals. Importantly, these studies also showed that cell-wall proteins, including those mediating adhesion, were enriched among those gene families with accelerated evolutionary rates suggestive of positive selection. In the particular case of serial clinical isolates from patients undergoing azole treatments, loss of heterozygosity was found to be an important mechanism to mediate fast acquisition of reduced sensitivity to antifungals. In addition, point mutations affecting efflux pumps or drug targets appeared recurrently in isolates from several patients. However, it remains unclear whether this was the result of directional selection of standing variation in the patient's Candida population, or the result of the acquisition of novel mutations (Ford et al.2015).
THE CANDIDA PARAPSILOSIS COMPLEX AND A POTENTIAL ROLE OF HYBRIDIZATION
Candida parapsilosis was initially recognized as a single species and recently re-organized into three different species: C. parapsilosis sensu stricto, C. orthopsilosis and C. metapsilosis, respectively (Tavanti et al.2005). Notably, the three different species differ in their prevalence and their degree of virulence, C. parapsilosis being highly pathogenic and prevalent followed by C. orthopsilosis, and the least pathogenic and rarely isolated species C. metapsilosis. The first genome sequence of C. parapsilosis was provided by the above mentioned study (Butler et al.2009), and the main striking result was the low level of homozygosity of this genome, as compared to other diploid species in the same clade. In fact, C. parapsilosis presented only one heterozygous polymorphism per 15 553 bases, as compared to one every 200–600 in the other diploid species. Based on this finding and the fact that different C. parapsilosis strains are difficult to distinguish using standard molecular techniques (Tavanti et al.2010), it was later proposed that C. parapsilosis may have undergone a very recent population bottleneck (Sai et al.2011). In this context, it was also speculated that C. parapsilosis clinical isolates may represent a recent clonal expansion of a strain with increased virulence when compared with the closest relatives C. orthopsilosis and C. metapsilosis (Sai et al.2011). However, the sequencing of three additional isolates of clinical and environmental origins provided evidence against a recent clonal expansion in the clinical context (Pryszcz et al.2013). Indeed, a phylogenetic comparison of the clinical and environmental isolates showed that they were polyphyletic, suggesting multiple colonizations from the environment. In addition, all genomes analyzed, including those from clinical isolates, presented independent expansions (i.e. having different boundaries in the expanded segment) of a gene involved in arsenite resistance, which suggested recent directional selection in the environment rather than in the human body.
The first genome sequence of C. orthopsilosis was published in 2012 (Riccombeni et al.2012), and revealed a highly homozygous diploid genome. A comparison with the more pathogenic C. parapsilosis revealed that C. orthopsilosis had fewer copies of Als and Hyr/Iff families of cell-wall proteins, reinforcing the idea that the copy number of genes within these families may correlate with virulence. The sequencing of a second clinical C. orthopsilosis strain from Texas a few years later, however, was found to be highly heterozygous, with alternating blocks of homozygous and heterozygous genome regions (Pryszcz et al.2014). Importantly, heterozygous blocks were found to be composed of one haplotype virtually identical to the one of the previously sequenced strain, and another one showing a 4.5% divergence at the nucleotide level. Such level of divergence is high enough to correspond to a different species or subspecies, and it was concluded that this strain represented a hybrid. Even more striking was the finding that an independent clinical isolate from Singapore represented a virtually identical hybrid, suggesting a rapid global expansion of the hybrid lineage. In addition, as compared to the homozygous strain, the hybrid genome was found to have a higher number of copies of genes involved in virulence, suggesting that hybridization may have boosted its virulence.
The third and less virulent species of the complex, C. metapsilosis, was published in 2015 (Pryszcz et al.2015). Similar to the situation in the second published C. orthopsilosis genome, C. metapsilosis was found to be a hybrid between two lineages diverging 4.5% at the nucleotide level, and showing a chimeric genome with intercalated homozygous and heterozygous blocks. Here again, several globally distributed clinical isolates were found to derive from the same hybridization event, although in this case the 11 analyzed genomes were more divergent in terms of their patterns of homozygous and heterozygous blocks. The fact that the 11 analyzed C. metapsilosis isolates are hybrids descending from the same ancestor, and that none of the parental strains was present among the sequenced isolates strongly suggests that the parental lineages that formed this hybrid are not able to infect humans, and that the hybridization may have conferred this ability to the hybrids. Whether such hybrids can be found in nature or only associated to humans is still an open question, but the fact that the hybrids are somewhat divergent in their loss of heterozygosis patterns suggests they did not expand very recently. Altogether, these results argue for a new re-classification of the C. parapsilosis complex, in which at least five different lineages, or species, some of them with the ability to form hybrids exist. In addition, it raises the intriguing question of what role hybridization may play in the emergence of a virulent phenotype.
The impact of hybridization itself in pathogenicity is not fully understood, but there is evidence of hybridization in several other unrelated fungal human pathogens, such as Cryptococcus neoformans (Li et al.2012; Forche 2014), Coccidioides posadasi (Neafsey et al.2010) and Fusarium keratoplasticum (Short, O'Donnell and Geiser 2014). Among plant pathogens, hybridization is increasingly being recognized as an important factor in the evolution of virulence (Depotter et al.2016). Hybrid strains may combine dangerous phenotypes of two or more strains, such as virulence factors or antifungal resistance; and its role in the emergence of new species as prevalent pathogens should not be underestimated. Even more, the classical chromosomic instability typically associated to hybridization may itself be advantageous during the course of infection, as it may help the pathogen to adapt to the immune system or different drugs. The possibility that patient-specific hybridization arise during the course of infections, for instance between acquired microbes and commensals, is intriguing and should be taken into account when studying the evolution of the infectious process. Such events may be responsible of a fraction of infections by rarer organisms and the origin of novel pathogenic strains. However, whether there is a real high prevalence of hybrid strains in pathogenic fungi or the long list of examples is the consequence of a bias in our knowledge remains unclear.
CANDIDA GLABRATA AND THE NAKASEOMYCES CLADE
Candida glabrata was sequenced along with three other yeast species back in 2004 (Dujon et al.2004). Initial genome comparisons revealed that C. glabrata and Saccharomyces cerevisiae shared numerous blocks of duplicated genes implying that both species descended from the same tetraploid ancestor. However, as compared to S. cerevisiae, C. glabrata had fewer genes and a more compact genome, indicating a larger degree of genomic streamlining. Initial comparisons of both species with respect to their virulence potential identified the loss of some pathways such as that of the biosynthesis of nicotinic acid as a possible hallmark of adaptation to the human host (Roetzer, Gabaldón and Schüller 2011). More recently, the five other known species of the Nakaseomyces clade, including the two less-prevalent emerging pathogens C. bracarensis and C. nivarensis, were fully sequenced and compared under an evolutionary framework (Gabaldón et al.2013). This analysis established that many of the differences between C. glabrata and S. cerevisiae were shared by other members of the clade implying that they were ancient traits unrelated with a commensal or pathogenic lifestyle. In addition, contrary to what may have been expected, the three pathogenic species of the clade where closely related but not monophyletic, suggesting multiple independent acquisitions of the ability to infect humans. Remarkably, the genome comparisons unearthed a large correlation between the number of copies of a particular family of epithelial adhesins (EPA) and the pathogenecity of the species. This family of cell-wall proteins mediates adhesion of C. glabrata cells to different substrates including host tissues, and constitutes a well-known virulence factor in C. glabrata (de Groot et al.2013). Interestingly, a closer phylogenetic analysis of the evolution of this family revealed that the larger size of the EPA repertoire in the pathogenic species corresponded to two independent expansions, one specific to C. glabrata and another one preceding the divergence of C. bracarensis, C. nivarensis and the non-pathogenic relative Nakaseomyces delphensis, which subsequently reduced its EPA repertoire. Altogether, these findings suggest two independent acquisitions of the ability to infect humans by means of increasing the strength or versatility of adhesion. In a broader context, it underscores the importance of increased adhesion in pathogens of different clades, which may have originated by expansions of different families of cell-wall adhesins.
The availability of a single genome sequence for C. glabrata limits the assessment of intraspecific variation to studies focusing on chromosomal aneuploidies or on few genetic markers (Muller et al.2009; Ahmad et al.2014; Bolotin-Fukuhara and Fairhead 2014; Gabaldón and Carreté 2016). These analyses have revealed significant variation among C. glabrata isolates, including copy number variations in several gene clusters and changes in karyotypes. Some studies have shown the appearance of novel chromosomes that seem to have originated by segmental duplications or genomic translocations (Ahmad et al.2014). Even phylogenetically related isolates, as well as those evolved under normal laboratory conditions have been shown to display significant chromosomal rearrangements that can be linked to certain phenotypic changes (Bader et al.2012; Ahmad et al.2014), suggesting that C. glabrata may frequently remodel its genome as a response to certain selective pressures.
CONCLUDING REMARKS AND FUTURE PROSPECTS
The increasing availability of sequenced genomes from Candida pathogens and their close non-pathogenic relatives, and in some cases several genomes from each of the species, is having a clear impact on our understanding of how virulence appears and evolves across species. Phylogenetic analyses show that Candida pathogens are phylogenetically diverse, and that virulence has emerged multiple times among Saccharomycotina. It is anticipated that the whole taxonomy of the group will be reorganized to accommodate modern phylogenetic knowledge. In this regard most of the pathogenic species will have to be renamed, which will inevitably create confusion among clinicians and other researchers. At the same time, however, it may help to better understand their clearly different clinical and physiological traits. Genome comparisons are increasing our understanding of the similarities and specificities of each pathogen, which can often be correlated with different survival strategies within the host, and different behaviors in front of the host immune system or when faced with antibiotics. The closer analyses of the evolutionary path to virulence in the different Candida clades have revealed different evolutionary mechanisms, but strikingly similar patterns with respect to certain functional classes. In particular, the expansion of the cell-wall repertoire and more specifically of families involved in surface adhesion seems to be a common theme in most pathogenic clades analyzed. Yet, the specific families affected and the evolutionary mechanisms to achieve an expanded repertoire can differ broadly between clades. Thus different paths can lead to convergent properties of the cell surface such as a higher, or more versatile adherence.
Importantly, most of these adaptations seem to be driven by genomic re-arrangements involving duplications or hybridizations, which in one or a few steps can drive important phenotypic differences. This, in turn, suggests that the path to become an opportunistic pathogen is not a long one, but rather can be achieved by relatively few alterations from a non-pathogenic genetic background. Yet, some genetic backgrounds seem to be predisposed to cross that barrier. For instance, the presence of genes encoding adhesins in subtelomeric regions or other genomic locations prone to rearrangements may favor gene duplications, which would be selected in specific conditions. The role of hybridization deserves a special consideration, since human intervention, by altering the environment and promoting species invasions through global commerce and transport, may be favoring the appearance of new hybrids; some of which may have infective capabilities.
Despite recent progress, we are still far from understanding how virulence emerges and evolves across species. In this regard, population genomics approaches, as well as metagenomics hold the promise of increasing our knowledge on three fundamental questions that remain poorly addressed: (i) what are the recent selective pressures (at the species and population level) in human fungal pathogens? (ii) are opportunistic pathogens tightly adapted to humans, or do they colonize us from other niches in the environment? and (iii) what pre-adaptations in the environment favor a jump to an increase virulence in humans?
FUNDING
TG's group's research acknowledges support from grants, ‘ 2013–2017’ and cofounded by (ERDF); from the (AGAUR) []; from the under grant agreements and , and grant from the under the Marie Sklodowska-Curie grant agreement no .
Conflict of interest.None declared.
REFERENCES
- Ahmad KM, Kokošar J, Guo X, et al. Genome structure and dynamics of the yeast pathogen Candida glabrata. FEMS Yeast Res. 2014;14:529–35. doi: 10.1111/1567-1364.12145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anoop V, Rotaru S, Shwed PS, et al. Review of current methods for characterizing virulence and pathogenicity potential of industrial Saccharomyces cerevisiae strains towards humans. FEMS Yeast Res. 2015;15 doi: 10.1093/femsyr/fov057. [DOI] [PubMed] [Google Scholar]
- Bader O, Schwarz A, Kraneveld EA, et al. Gross karyotypic and phenotypic alterations among different progenies of the Candida Glabrata cbs138/atcc2001 reference strain. PLoS One. 2012;7:e52218. doi: 10.1371/journal.pone.0052218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotin-Fukuhara M, Fairhead C. Candida glabrata: a deadly companion? Yeast. 2014;31:279–88. doi: 10.1002/yea.3019. [DOI] [PubMed] [Google Scholar]
- Brandt ME, Lockhart SR. Recent taxonomic developments with Candida and other opportunistic yeasts. Curr Fungal Infect Rep. 2012;6:170–7. doi: 10.1007/s12281-012-0094-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunke S, Hube B. Two unlike cousins: Candida albicans and C. glabrata infection strategies. Cell Microbiol. 2013;15:701–8. doi: 10.1111/cmi.12091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler G, Rasmussen MD, Lin MF, et al. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature. 2009;459:657–62. doi: 10.1038/nature08064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant GC, Wolfe KH. Turning a hobby into a job: how duplicated genes find new functions. Nat Rev Genet. 2008;9:938–50. doi: 10.1038/nrg2482. [DOI] [PubMed] [Google Scholar]
- de Groot PWJ, Bader O, de Boer AD, et al. Adhesins in human fungal pathogens: glue with plenty of stick. Eukaryot Cell. 2013;12:470–81. doi: 10.1128/EC.00364-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depotter JR, Seidl MF, Wood TA, et al. Interspecific hybridization impacts host range and pathogenicity of filamentous microbes. Curr Opin Microbiol. 2016;32:7–13. doi: 10.1016/j.mib.2016.04.005. [DOI] [PubMed] [Google Scholar]
- Diekema D, Arbefeville S, Boyken L, et al. The changing epidemiology of healthcare-associated candidemia over three decades. Diagn Micr Infec Dis. 2012;73:45–8. doi: 10.1016/j.diagmicrobio.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Dujon B. Yeast evolutionary genomics. Nat Rev Genet. 2010;11:512–24. doi: 10.1038/nrg2811. [DOI] [PubMed] [Google Scholar]
- Dujon B, Sherman D, Fischer G, et al. Genome evolution in yeasts. Nature. 2004;430:35–44. doi: 10.1038/nature02579. [DOI] [PubMed] [Google Scholar]
- Eisen JA. Phylogenomics: improving functional predictions for uncharacterized genes by evolutionary analysis. Genome Res. 1998;8:163–7. doi: 10.1101/gr.8.3.163. [DOI] [PubMed] [Google Scholar]
- Forche A. Large-Scale chromosomal changes and associated fitness consequences in pathogenic fungi. Curr Fungal Infect Rep. 2014;8:163–70. doi: 10.1007/s12281-014-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CB, Funt JM, Abbey D, et al. The evolution of drug resistance in clinical isolates of Candida albicans. eLife. 2015;4:e00662. doi: 10.7554/eLife.00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda Y, Tsai H, Myers TG, et al. Transcriptional profiling of Candida glabrata during phagocytosis by neutrophils and in the infected mouse spleen. Infect Immun. 2013;81:1325–33. doi: 10.1128/IAI.00851-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabaldón T. Comparative genomics-based prediction of protein function. Method Mol Biol. 2008;439:387–401. doi: 10.1007/978-1-59745-188-8_26. [DOI] [PubMed] [Google Scholar]
- Gabaldón T, Carreté L. The birth of a deadly yeast: tracing the evolutionary emergence of virulence traits in Candida glabrata. FEMS Yeast Res. 2016;16 doi: 10.1093/femsyr/fov110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabaldon T, Marcet-Houben M. Phylogenomics for the study of fungal biology. In: Nowrousian M, editor. Fungal Genomics. Berlin: Springer Science & Business Media; 2014. pp. 61–79. [Google Scholar]
- Gabaldón T, Martin T, Marcet-Houben M, et al. Comparative genomics of emerging pathogens in the Candida glabrata clade. BMC Genomics. 2013;14:623. doi: 10.1186/1471-2164-14-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal-Mor O, Finlay BB. Pathogenicity islands: a molecular toolbox for bacterial virulence. Cell Microbiol. 2006;8:1707–19. doi: 10.1111/j.1462-5822.2006.00794.x. [DOI] [PubMed] [Google Scholar]
- Glaser P, Frangeul L, Buchrieser C, et al. Comparative genomics of Listeria species. Science. 2001;294:849–52. doi: 10.1126/science.1063447. [DOI] [PubMed] [Google Scholar]
- Glöckner A, Cornely OA. Candida glabrata - unique features and challenges in the clinical management of invasive infections. Mycoses. 2015;58:445–50. doi: 10.1111/myc.12348. [DOI] [PubMed] [Google Scholar]
- Guarro J, Gené J, Stchigel AM. Developments in fungal taxonomy. Clin Microbiol Rev. 1999;12:454–500. doi: 10.1128/cmr.12.3.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinea J, Martin G, Mannino D, et al. Global trends in the distribution of Candida species causing candidemia. Clin Microbiol Infec. 2014;20:5–10. doi: 10.1111/1469-0691.12539. [DOI] [PubMed] [Google Scholar]
- Hawksworth DL. Managing and coping with names of pleomorphic fungi in a period of transition. IMA Fungus. 2012;3:15–24. doi: 10.5598/imafungus.2012.03.01.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa MP, Martinez DA, Sakthikumar S, et al. Genetic and phenotypic intra-species variation in Candida albicans. Genome Res. 2015;25:413–25. doi: 10.1101/gr.174623.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland LM, Schröder MS, Turner SA, et al. Comparative phenotypic analysis of the major fungal pathogens Candida parapsilosis and Candida albicans. PLoS Pathog. 2014;10:e1004365. doi: 10.1371/journal.ppat.1004365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, Capella-Gutiérrez S, Pryszcz LP, et al. PhylomeDB v4: zooming into the plurality of evolutionary histories of a genome. Nucleic Acids Res. 2014;42:D897–902. doi: 10.1093/nar/gkt1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, Dopazo J, Gabaldón T. ETE: a python environment for tree exploration. BMC Bioinformatics. 2010;11:24. doi: 10.1186/1471-2105-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-López C, Lorenz MC. Fungal immune evasion in a model host-pathogen interaction: Candida albicans versus macrophages. PLoS Pathog. 2013;4:1–9. doi: 10.1371/journal.ppat.1003741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtzman C, Fell JW, Boekhout T. The Yeasts: A Taxonomic Study. Amsterdam: Elsevier; 2011. [Google Scholar]
- Li W, Averette AF, Desnos-Ollivier M, et al. Genetic diversity and genomic plasticity of Cryptococcus neoformans AD hybrid strains. G3 (Bethesda) 2012;2:83–97. doi: 10.1534/g3.111.001255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcet-Houben M, Gabaldón T. Beyond the whole-genome duplication: phylogenetic evidence for an ancient interspecies hybridization in the baker's yeast lineage. PLoS Biol. 2015;13:e1002220. doi: 10.1371/journal.pbio.1002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda I, Silva-Dias A, Rocha R, et al. Candida albicans CUG mistranslation is a mechanism to create cell surface variation. mBio. 2013;4:e00285–13. doi: 10.1128/mBio.00285-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrzewska B, Kurnatowski P. Selected pathogenic characteristics of fungi from the genus Candida. Ann Parasitol. 2013;59:57–66. [PubMed] [Google Scholar]
- Mühlhausen S, Kollmar M. Molecular phylogeny of sequenced Saccharomycetes reveals polyphyly of the alternative yeast codon usage. Genome Biol Evol. 2014;6:3222–37. doi: 10.1093/gbe/evu152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller H, Thierry A, Coppée J-Y, et al. Genomic polymorphism in the population of Candida glabrata: gene copy-number variation and chromosomal translocations. Fungal Genet Biol. 2009;46:264–76. doi: 10.1016/j.fgb.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Nather K, Munro CA, Albrecht A, et al. Generating cell surface diversity in Candida albicans and other fungal pathogens. FEMS Microbiol Lett. 2008;285:137–45. doi: 10.1111/j.1574-6968.2008.01263.x. [DOI] [PubMed] [Google Scholar]
- Neafsey DE, Barker BM, Sharpton TJ, et al. Population genomic sequencing of Coccidioides fungi reveals recent hybridization and transposon control. Genome Res. 2010;20:938–46. doi: 10.1101/gr.103911.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papon N, Courdavault V, Clastre M, et al. Emerging and emerged pathogenic Candida species: beyond the Candida albicans paradigm. PLoS Pathog. 2013;9:e1003550. doi: 10.1371/journal.ppat.1003550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller MA, Diekema DJ. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev. 2007;20:133–63. doi: 10.1128/CMR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller MA, Diekema DJ, Gibbs DL, et al. Results from the ARTEMIS DISK Global Antifungal Surveillance Study, 1997 to 2007: a 10.5-year analysis of susceptibilities of Candida species to fluconazole and voriconazole as determined by CLSI standardized disk diffusion. J Clin Microbiol. 2010;48:1366–77. doi: 10.1128/JCM.02117-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priest SJ, Lorenz MC. Characterization of virulence-related phenotypes in Candida species of the cug clade. Eukaryot Cell. 2015;14:931–40. doi: 10.1128/EC.00062-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryszcz LP, Németh T, Gácser A, et al. Unexpected genomic variability in clinical and environmental strains of the pathogenic yeast Candida parapsilosis. Genome Biol Evol. 2013 doi: 10.1093/gbe/evt185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryszcz LP, Németh T, Gácser A, et al. Genome comparison of Candida orthopsilosis clinical strains reveals the existence of hybrids between two distinct subspecies. Genome Biol Evol. 2014;6:1069–78. doi: 10.1093/gbe/evu082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryszcz LP, Németh T, Saus E, et al. The genomic aftermath of hybridization in the opportunistic pathogen Candida metapsilosis. PLoS Genet. 2015;11:e1005626. doi: 10.1371/journal.pgen.1005626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccombeni A, Vidanes G, Proux-Wéra E, et al. Sequence and analysis of the genome of the pathogenic yeast Candida orthopsilosis. PloS One. 2012;7:e35750. doi: 10.1371/journal.pone.0035750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roetzer A, Gabaldón T, Schüller C. From Saccharomyces cerevisiae to Candida glabrata in a few easy steps: important adaptations for an opportunistic pathogen. FEMS Microbiol Lett. 2011;314:1–9. doi: 10.1111/j.1574-6968.2010.02102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sai S, Holland LM, McGee CF, et al. Evolution of mating within the Candida parapsilosis species group. Eukaryot Cell. 2011;10:578–87. doi: 10.1128/EC.00276-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos MAS, Gomes AC, Santos MC, et al. The genetic code of the fungal CTG clade. C R Biol. 2011;334:607–11. doi: 10.1016/j.crvi.2011.05.008. [DOI] [PubMed] [Google Scholar]
- Seider K, Gerwien F, Kasper L, et al. Immune evasion, stress resistance, and efficient nutrient acquisition are crucial for intracellular survival of Candida glabrata within macrophages. Eukaryot Cell. 2014;13:170–83. doi: 10.1128/EC.00262-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short DPG, O'Donnell K, Geiser DM. Clonality, recombination, and hybridization in the plumbing-inhabiting human pathogen Fusarium keratoplasticum inferred from multilocus sequence typing. BMC Evol Biol. 2014;14:91. doi: 10.1186/1471-2148-14-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A, Ludwig T, Meier H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics. 2005;21:456–63. doi: 10.1093/bioinformatics/bti191. [DOI] [PubMed] [Google Scholar]
- Sudbery PE. Growth of Candida albicans hyphae. Nat Rev Microbiol. 2011;9:737–48. doi: 10.1038/nrmicro2636. [DOI] [PubMed] [Google Scholar]
- Tavanti A, Davidson AD, Gow NAR, et al. Candida orthopsilosis and Candida metapsilosis spp. nov. to replace Candida parapsilosis groups II and III. J Clin Microbiol. 2005;43:284–92. doi: 10.1128/JCM.43.1.284-292.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavanti A, Hensgens LAM, Mogavero S, et al. Genotypic and phenotypic properties of Candida parapsilosis sensu strictu strains isolated from different geographic regions and body sites. BMC Microbiol. 2010;10:203. doi: 10.1186/1471-2180-10-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DS, Carlisle PL, Kadosh D. Coevolution of morphology and virulence in Candida species. Eukaryot Cell. 2011;10:1173–82. doi: 10.1128/EC.05085-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner SA, Butler G. The Candida pathogenic species complex. Cold Spring Harb Perspect Med. 2014;4:a019778. doi: 10.1101/cshperspect.a019778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnock DW. Trends in the epidemiology of invasive fungal infections. Jpn J Med Mycol. 2007;48:1–12. doi: 10.3314/jjmm.48.1. [DOI] [PubMed] [Google Scholar]
- Wolfe KH, Shields DC. Molecular evidence for an ancient duplication of the entire yeast genome. Nature. 1997;387:708–13. doi: 10.1038/42711. [DOI] [PubMed] [Google Scholar]