

Abstract
Lymph node expansion during immune responses is accompanied by rapid vascular expansion. The reestablishment of quiescence and stabilization of the newly expanded vasculature and the regulatory mechanisms involved have not been well studied. We show that while initiation of vascular expansion in immune-stimulated nodes is associated with upregulated endothelial cell proliferation, increased high endothelial venule trafficking efficiency and VCAM-1 expression, and disrupted perivascular fibroblastic reticular cell organization, the reestablishment of vascular quiescence and stabilization after expansion is characterized by reversal of these phenomena. While CD11cmed cells are associated with the initiation of vascular expansion, CD11chiMHCIImed dendritic cells accumulate later and their short-term depletion in mice abrogates the reestablishment of vascular quiescence and stabilization. CD11chiMHCIImed cells promote endothelial cell quiescence in vitro and, in vivo, mediate quiescence at least in part by mediating reduced lymph node VEGF. Disrupted vascular quiescence and stabilization in expanded nodes is associated with attenuated T cell-dependent B cell responses. These results describe a novel mechanism whereby CD11chiMHCIImed dendritic cells regulate the reestablishment of vascular quiescence and stabilization after lymph node vascular expansion and suggest that these dendritic cells function in part to orchestrate the microenvironmental alterations required for successful immunity.
Keywords: Spleen and lymph nodes, Endothelial cells, Dendritic cells, Stromal cells, Rodent
Introduction
Lymph node blood vessels are the portals of entry for circulating immune cells and play critical roles in the orchestration and regulation of immune responses. Specialized postcapillary venules with cuboidal endothelial cell morphology are termed high endothelial venules (HEV) and mediate the constitutive trafficking of cells from the blood by displaying adhesion molecules and chemokines that allow for the rolling, adhesion, and transmigration of cells across the endothelium (1). During immune responses, lymph node blood vessels demonstrate functional plasticity, with transient increases in HEV trafficking capabilities and endothelial cell proliferation along with vascular expansion. These vascular changes likely serve at least in part to increase delivery of antigen-specific and other cells to optimize the developing immune response (2–7). During angiogenesis, vascular expansion is followed by vascular maturation whereupon the newly expanded vasculature is stabilized and endothelial cell activity is downregulated (8–10). Studies have begun to delineate the events and regulation of early vascular activation and expansion after immune stimulation (2, 4–6, 11), but the reestablishment of vascular quiescence and stabilization after expansion has not been well studied.
Vascular expansion occurs within a few days after lymph node stimulation. Endothelial cell proliferation is upregulated by day 2 and vascular expansion is detectable as soon as day 3 (7, 12). We have identified a role for dendritic cells (DCs) in the initiation of vascular expansion, as depletion of CD11c+ cells abrogates the upregulation of endothelial cell proliferation, and DC injection can stimulate upregulation of endothelial cell proliferation even in mice lacking mature T and B cells. DCs stimulate increased endothelial cell proliferation at least in part by upregulating lymph node vascular endothelial growth factor (VEGF) levels, as VEGF levels are upregulated within a day of immune stimulation and the increased endothelial cell proliferation and subsequent vascular expansion are sensitive to VEGF blockade (6, 13). B cells also contribute to lymph node VEGF levels, and lymphatic expansion, which occurs in conjunction with the blood vessel expansion, is dependent on B cells (14). The lymph node immune cells, then, regulate the early process of vascular expansion, presumably to prepare the microenvironment for the ensuing immune response. We have observed that upon immunization with ovalbumin in complete Freund adjuvant (OVA/CFA), the proliferative vascular expansion is followed by a period of downregulated endothelial cell proliferation and maintenance of the expanded endothelial cell numbers (6). The role of immune cells in mediating the early vascular expansion raises the possibility that immune cells also mediate the subsequent reestablishment of endothelial cell quiescence and vascular stabilization.
In addition to proliferation and expansion, the lymph node vasculature also undergoes phenotypic alterations during immune responses. HEV endothelial cells in stimulated lymph nodes appear metabolically activated, with increased polyribosomes and prominent rough endoplasmic reticulum (7). Within a day after DC injection, CXCL9 on HEV is upregulated and may mediate increased entry of activated T cells and NK cells (2, 3). Similarly, the adhesion molecule CD62P is upregulated and mediates effector memory T cell entry (4). Less well characterized in terms of HEV phenotypic alterations is the general increase in trafficking of naïve lymphocytes that occurs in the same time frame (3, 5), although this process may be dependent on DCs as lymph nodes remain small when CD11c+ cells are depleted (6). Peripheral node addressin (PNAd) is a group of fucosylated and sialylated proteins that mark peripheral node HEV (15), and downregulated expression of PNAd protein cores and synthesizing enzymes within days of lymph node stimulation is accompanied by upregulated expression of MADCAM-1, a cell adhesion molecule normally expressed on mucosal HEV (1, 11). These phenotypic alterations are transient with acute stimulation and correspond to the period of increased endothelial cell proliferation and vascular expansion. Whether there are also other phenotypic alterations and whether HEV phenotype is regulated in conjunction with downregulation of endothelial cell proliferation are open questions.
Lymph node blood vessels are suspended in a reticular network composed of collagen-rich fibrils ensheathed by fibroblastic reticular cells (FRCs). FRCs synthesize the extracellular matrix components of the fibrils and provide a surface upon which lymphocytes migrate (16). FRCs also function to express the chemokines that promote T and B cell compartmentalization and IL-7 that maintains T cell numbers (17, 18). The FRC-covered fibrils form a continuous network with the vessel walls. At HEV, FRCs resemble pericytes of other microvascular beds in that they form a sheath several layers thick around the vessels (19). There, FRCs regulate parenchymal entry of bloodborne cells after they have crossed the endothelium (16) and can potentially regulate vasopermeability. The compartment between the FRCs and the fibrillar core in the reticular fibrils are conduits for small molecules. These conduits continue to the FRCHEV interface and small molecules can reach the endothelial cells through the conduit system, providing a potential means of vascular regulation (19–21). FRCs associated with blood vessel walls and also with nearby fibrils are the main expressers of VEGF mRNA in homeostatic and immune-stimulated lymph nodes, suggesting a role for perivascular and nearby FRCs in regulating lymph node endothelial cell proliferation (13). Because of the physical and functional association between the endothelial cells and FRCs, regulation of vascular activity is likely to involve regulation of both endothelial cells and nearby FRCs.
In this study, we characterize the reestablishment of vascular quiescence and stabilization, identify CD11chiMHCIImed DCs as critical mediators of the process, and show that disruption of this process is associated with attenuated T cell-dependent humoral responses. We show that, after vascular expansion, endothelial cell quiescence is reestablished with downregulation of the previously upregulated endothelial cell proliferation, HEV trafficking efficiency, and VCAM-1 expression. The newly expanded vasculature is also stabilized, with the reassembly of FRC around and near by vessels. While CD11cmed cells are associated with initiation of vascular expansion, CD11chiMHCIImed DCs accumulate in large numbers near vessels at the onset of vascular quiescence and stabilization. Depletion of CD11chiMHCIImed DCs disrupts this process and these DCs can promote endothelial cell quiescence in vitro. CD11chiMHCIImed DCs mediate the downregulation of endothelial cell proliferation at least in part by regulating lymph node VEGF levels. Disruption of the reestablishment of vascular quiescence and stabilization is associated with attenuated development of T cell-dependent B cell responses. Our results provide new insights into the regulation of the lymph node vasculature and describe a novel function for CD11chiMHCIImed DCs in regulating the lymph node microenvironment.
Materials and Methods
Mice
B6 CD11c-DTR mice originally from Jackson Laboratory and C57BL/6 mice from Jackson Laboratory, Taconic Farms or National Cancer Institute were used at 6–10 weeks of age. All animal procedures were performed in accordance with the regulations of the Institutional Animal Use and Care Committee at the Hospital for Special Surgery.
BMDC preparation
BMDCs were generated as described (6). Briefly, bone marrow cells were cultured with 8–10% supernatant from GM-CSF-expressing J558L cells, matured with LPS on day 7, harvested on day 8, washed, and injected at 1×106 cells per hind footpad.
Flow cytometry
Flow cytometry analysis was as described (6). Briefly, lymph nodes were digested with type II collagenase (Worthington, Lakewood, NJ) and cells were stained for flow cytometry. Unless otherwise specified, all antibodies were from BD Biosciences (San Jose, CA). For analysis of endothelial cell proliferation, mice received 2mg of BrdU (Sigma, St. Louis, MO) 18 hours and 1 hour before sacrifice and 0.8mg/ml BrdU in drinking water in between. Cells were stained with indicated endothelial cell markers and with anti-BrdU-AlexaFluor647 (Invitrogen). For VCAM-1 expression measurement, cells were stained for endothelial cell markers and with biotinylated anti-VCAM-1 (clone 429(MVCAM.A1)) followed by streptavidin-allophycocyanin (Invitrogen, Carlsbad, CA) or streptavidin-pacific blue (Invitrogen). The level of VCAM-1 was analyzed by flow cytometry.
Other antibodies that were used for flow cytometry were: anti-CD45 (30-F11), anti-CD31 (MEC13.3), anti-PNAd (MECA-79), anti-CD11c (HL3), anti-IAb (anti-MHCII, clone AF6), anti-rat IgM (as a secondary antibody for anti-PNAd staining, Jackson Immunoresearch, West Grove, PA).
Depletion of CD11chiMHCIImed cells
CD11c-DTR mice are transgenic for a simian DTR-GFP fusion protein that is driven by the CD11c promoter and that allows for the depletion of CD11c+ cells upon DT injection(22). Unless otherwise specified, mice received intraperitoneal injections of 100ng DT (Calbiochem, San Diego, CA) or inactive mutant diphtheria toxin cross reactive material (CRM 197, Calbiochem). For CD11c-DTR bone marrow chimeras, 200ng DT was injected intraperitoneally. The increased dose of DT for the chimeras was found by us to be necessary for consistent depletion of CD11chiMHCIImed cells (unpublished observations) and is in line with the higher dosing of DT used in CD11c-DTR chimeric mice in other studies (23, 24). For day 7 endothelial cell proliferation studies, BrdU was first injected 8 hours after DT injection on day 6.
Aortic ring culture
The aortic ring culture was based on that described by Berger et al (25). One millimeter aortic rings were placed 1 ring per well of 12-well tissue culture plates coated with 0.0025U/ul thrombin solution (Sigma). Fibrinogen (0.1%)(Sigma) and CD11c+ cell subsets were added slowly and incubated at 37°C for 10 minutes to promote gel formation. M199 (Invitrogen) with 1% fungizone (Invitrogen), 0.04mg/ml gentamicin (Mediatech), 0.5% ε-amino-caproic acid (Sigma) and 2% B6 mouse serum were added and samples were incubated at 37°C and 5% CO2. On day 3, BrdU was added at 0.025μg/ml. On day 4, type II collagenase (564 U/ml) with RPMI, 0.5% BSA, and 40 μg/ml DNase I were added and wells were shaken at 50rpm for 30 minutes at 37°C. Three rings were pooled as one sample and digested for another 30 minutes. The suspension was triturated 40 times and EDTA was added to 10mM. Cells were incubated 5 more minutes, passed through a 70-μm filter, and prepared for flow cytometry.
CD11c+ cell subset sorting
Mice were immunized with OVA/CFA at multiple sites. On day 8, draining cervical, axillary, brachial, inguinal, popliteal and lumbar nodes were pooled and collagenase digested. T and B cells were depleted using biotinylated CD3 and B220 antibodies and magnetic separation (MACS, Miltenyi Biotec, Auburn, CA). Live gated CD11c+ subsets were then sorted using a FACS Vantage.
Immunohistochemistry
Immunohistochemical staining of fresh frozen sections was performed as described(6). Antibodies used included CD31, CD11c, desmin (Labvision/Thermo Scientific, Fremont, CA), and gp38 (clone 8.1.1, Developmental Studies Hybridoma Bank, Iowa City, IA). Horseradish peroxidase or alkaline phosphatase-conjugated secondary antibodies were from Jackson Immunoresearch (West Grove, PA).
Quantitation of vessels with disrupted FRC organization
Sections stained for desmin and CD31 were assessed for percent of T zone HEV with disorganized FRCs per high power (40x objective) field. HEV were scored as having disorganized FRCs if the FRC sheath was loosely organized, with splaying of FRCs at angles away from vessel wall or across endothelium.
Evans blue assays
For detection of Evans blue permeability in frozen sections, mice received tail vein injections of 200μl of 3.75 mg/ml Evans blue (Sigma) and were perfused with PBS after 10 minutes to wash out intravascular dye. Nodes were then harvested and frozen for cryosectioning.
For measuring the Evans blue content in tissues, 250μl of 2% Evans blue was intravenously injected 4 hours before sacrificing mice. Bilateral lymph nodes were harvested, incubated in 150ul formamide for 24 hours at 37 °C and supernatant were measured in a microplate reader at 620nm. A serial dilution of 2% Evans blue was used to generate a standard curve for each experiment, and the tissue Evans blue content was expressed relative to the content in 2% Evans Blue.
Generation of bone marrow chimeras
Wild-type B6 mice were lethally irradiated with 875 rads using an Xray source, injected with wild-type or CD11c-DTR bone marrow, and then allowed to reconstitute for a minimum of 6 weeks as described in (13).
Lymph node VEGF measurements
Lymph nodes were solubilized in lysis buffer and subject to VEGF measurement usingDuoSet-mouse VEGF (R&D Systems, Minneapolis, MN) as previously described(6).
Anti-FBS antibody detection
ELISA plates were coated with 1% FBS overnight at 4°C and blocked with 1 mg/ml OVA for 2 hours. Serum diluted 1000-fold in PBT buffer (0.5% BSA, 0.4% Tween-20, PBS) was added for 2 hours. Bound antibody was detected with anti-mouse IgG-HRP (Southern Biotech, Birmingham, Alabama) and 3,3′,5,5′-tetramethylbenzidine substrate (Sigma). Results were normalized to results obtained from a standard batch of reactive serum.
Results
Vascular quiescence and stabilization after lymph node vascular expansion
We have previously shown that subcutaneous injection of bone marrow-derived DCs (BMDCs) stimulates increased endothelial cell proliferation in draining lymph nodes by day 2 and expansion of endothelial cell numbers by day 5 (6). We further characterized the alterations in endothelial cell activity over time in this system. Upregulated endothelial cell proliferation and vascular expansion was followed after day 5 by a phase of downregulated endothelial cell proliferation and maintenance of endothelial cell numbers (Fig. 1, A–C). During this second phase, lymph node cellularity was also reduced (Fig. 1D). We measured the efficiency of HEV cell trafficking by determining the number of injected splenocytes entered per PNAd+ endothelial cell. HEV trafficking efficiency was upregulated at day 1 but was downregulated to below baseline by day 6 (Fig. 1E). VCAM-1 expression on PNAd+ endothelial cells was analyzed by flow cytometry (supplemental Fig. 1) and showed a similar pattern of regulation (Fig. 1F), suggesting that alterations in HEV endothelial cell phenotype contributed to alterations in HEV trafficking efficiency. Together, these results suggested that while the initiation of vascular expansion is characterized by upregulated endothelial cell proliferation, HEV trafficking efficiency, and VCAM-1 expression, endothelial cell quiescence is reestablished after vascular expansion, with downregulated endothelial cell proliferation, HEV trafficking efficiency, and VCAM-1 expression.
Figure 1. Reestablishment of vascular quiescence and stabilization after expansion.

Mice received footpad injection of BMDCs on day 0 and draining popliteal lymph nodes were harvested at denoted times. For (A–F), cells were analyzed by flow cytometry. (A) Endothelial cell proliferation as determined by the percentage of CD45−CD31+ endothelial cells that are BrdU+. (B) Number of endothelial cells per lymph node. (C) Number of PNAd+ (and CD45−CD31+) endothelial cells per lymph node. (D) Number of total cells per lymph node. (E) HEV trafficking efficiency. About 5×107 CFSE-labeled splenocytes were injected intravenously and the number of CFSE+ cells that entered the lymph node at 30 minutes was divided by the number of PNAd+ endothelial cells. For (A–E), each symbol represents one mouse, and results are representative of experiments performed at least three times. (F) VCAM-1 expression on PNAd+ endothelial cells as determined by mean fluorescence intensity of VCAM-1 staining. n≥3 each time point; error bars represent SD. *=p<.05 with the Student’s t test when compared to day 0. (G–H) FRC organization around vessels in the T zone over time using (G) desmin or (H) gp38 to mark FRCs. Arrows point to examples of FRC that are protruding from the vessel wall at day 2. (I) Percent of T zone HEV that have disrupted FRC organization over time. At least 12 fields over a minimum of 2 lymph nodes were assessed for each time point. ***=p<.001 with the Student’s t test.
As pericytes contribute to the stabilization of vessels after vascular expansion (8, 10), we examined the organization of the perivascular FRCs. FRCs throughout the lymph node can be identified by their expression of desmin (16, 18, 26). At homeostasis, the majority of T zone HEV were ensheathed by desmin+ cells arranged in a tightly packed laminar structure (Fig. 1, G (left panel) and I). However, by day 2 after BMDC injection, FRC organization around vessels appeared disrupted. FRCs around most HEV were more loosely organized and individual FRCs were visible, with some protruding or splayed out at an angle perpendicular to the vessel wall (Fig. 1, G (middle panel) and I). By day 8, FRCs were reassembling around HEV in a more tightly organized structure (Fig. 1, G (right panel) and I). Staining for gp38, another marker of FRCs (18), showed the same alterations in FRC organization over time (Fig. 1H). In addition to altered perivascular FRC organization, FRCs associated with fibrils near vessels also appeared to undergo organizational alterations, with the fibrillar desmin appearing less continuous and more ragged at day 2 and recovering by day 8 (Fig. 1G). These data suggested that the reestablishment of endothelial cell quiescence after day 5 is accompanied by FRC reassembly that likely contributes to the reestablishment of vascular stabilization.
CD11cmed and CD11chiMHCIImed cells accumulate with different kinetics
We have previously shown that DCs are involved in the initiation of vascular expansion (6) and wanted to ask whether they could also regulate the reestablishment of vascular quiescence and stabilization. We examined whether the presence of different CD11c+ cell populations correlated with the initiation of vascular expansion versus the subsequent reestablishment of vascular quiescence and stabilization. CD11c+MHCII+ cells were composed mostly of CD11cmedMHCIImed and CD11cmedMHCIIhi cells at day 0 (Fig. 2,A,B, and E) and these two populations accumulated rapidly after BMDC injection, reaching peak or near-peak numbers by day 2 (Fig. 2B). The CD11cmed cells that accumulated included the injected BMDCs, which had a CD11cmedMHCIImed-hi phenotype (Fig. 2D). In contrast, the CD11chiMHCIImed cells accumulated more slowly, with numbers rising more steeply after day 2 and peaking at day 5 (Fig. 2, A and B). The CD11chiMHCIImed cells were endogenous cells, as BMDCs remained CD11cmed in phenotype through their peak accumulation at day 5 and subsequent reduction in numbers (data not shown)(6). The small population of CD11chiMHCIIhi cells showed relatively slow and low levels of accumulation (Fig. 2B). The initiation of vascular expansion, then, was characterized by a relative predominance of CD11cmed cells, resulting in a low ratio of CD11chiMHCIImed cells to CD11cmed cells (Fig. 2C). In contrast, the onset of the reestablishment of vascular quiescence and stabilization at day 5 was characterized by increased accumulation of CD11chiMHCIImed cells and an increase in the CD11chiMHCIImed cell to CD11cmed cell ratio (Fig. 2C). These results raised the possibility that CD11cmed cells mediated the initiation of vascular expansion while CD11chiMHCIImed cells mediated the reestablishment of vascular quiescence and stabilization.
Figure 2. CD11cmed cells are associated with initiation of vascular expansion while CD11chi MHCIImed cells accumulate later.

(A–C) Mice received BMDCs on day 0 and nodes were harvested at denoted times. (A) Flow cytometry density plots showing CD11c+ subsets at days 0 and 5. (B) Number of cells in each CD11c+ subset over time. (C) Ratio of CD11chiMHCIImed (R1) cell numbers to CD11cmed (R2+R3) cell numbers over time. n=6–8 mice per time point. For (B–C), *=p<.05 when compared with day 0 values using the Student’s t test. For (B), asteriks refer to the comparison of a particular CD11c+ subset with the same subset at day 0. (D) Flow cytometry dot plot showing the level of CD11c and MHCII expression by injected BMDCs in the draining nodes at day 2. BMDCs are shown in green while endogenous lymph node cells are shown in red. Representative of at least 10 mice. (E–I) Mice received footpad injection of 20μl of 1mg/ml OVA/CFA on day 0 and draining popliteal nodes were examined on day 2. (E) Number of cells in each CD11c+ subset (as denoted in (A)) at day 0 and day 2 after OVA/CFA. n=3–4 mice for each time point. (F–G) CD11c-DTR mice were injected with 100ng DT or control inactive mutant toxin (CRM) in the right footpad at 8 hours before OVA/CFA injection in the same footpad. (F) Number of cells in each CD11c+ subset in the draining nodes at day 2. (G) Lymph node endothelial cell proliferation. n=4 mice per condition. (H–I) CD11c-DTR mice received intraperitoneal injection of 100ng DT or CRM at 8 hours before OVA/CFA injection of the right footpad. (H) Number of cells in each CD11c+ subset. (I) Endothelial cell proliferation. n=6 mice per condition. For (E–I), *=p<.05 and ***=p<.001. For (F and H), asteriks refer to the comparison of a particular CD11c+ subset with the same subset in CRM-treated mice.
We further assessed the role of CD11cmed cells in the initiation of vascular expansion. OVA/CFA injection also led to rapid accumulation of predominantly CD11cmed cells and slower accumulation of CD11chiMHCIImed cells at day 2 (Fig. 2E), further supporting a role for CD11cmed cells in initiating vascular expansion. Preliminary experiments had indicated that subcutaneous and intraperitoneal DT injection in CD11c-DTR mice had different effects on the early accumulation of CD11cmed cells and we used these differences to test the role of CD11cmed cells in the initial upregulation of endothelial cell proliferation. Subcutaneous and intraperitoneal DT at 8 hours prior to OVA/CFA at day 0 resulted in a similar fold reduction in CD11chiMHCIImed cell numbers at day 2 when compared to the respective CRM-treated controls (7.4-fold +/− standard deviation 4.4 for subcutaneous DT and 4.6-fold +/− 1.8 for intraperitoneal DT; p=.2). In contrast, CD11cmed populations were less well reduced with intraperitoneal DT injection than with subcutaenous DT (Fig. 2, F and H)(for CD11cmed cells as a whole, 10.8-fold +/−7.2 for subcutaneous DT and 3.6-fold +/− 1.8 for intraperitoneal DT; p<.05). Endothelial cell proliferation is upregulated by OVA/CFA (Fig. 2G) from the basal proliferation rate of about 1% (see Fig. 1A) and subcutaneous DT dramatically reduced the upregulation of endothelial cell proliferation (Fig. 2G). Intraperitoneal DT, however, led to a less dramatic reduction of the upregulation (Fig. 2I). These results indicated a correlation between the extent of reduction in CD11cmed cell numbers and the extent of reduction in the upregulation of endothelial cell proliferation and were consistent with the idea that CD11cmed cells mediated the initiation of vascular expansion.
CD11chiMHCIImed cell depletion disrupts endothelial cell quiescence
We asked whether CD11chiMHCIImed cells mediated the reestablishment of endothelial cell quiescence that was observed after day 5. Intraperitoneal injection of DT in CD11c-DTR mice at day 6 depleted CD11chiMHCIImed cells but not CD11cmed cells or the small population of CD11chiMHCIIhi cells (Fig. 3, A and B). CD11chiMHCIImed cells at day 7 consist mainly of CD8−CD11b+ cells (70%) and CD8+CD11b− cells (16.5%), with 64% of CD8−CD11b+ subset expressing F4/80, and all subsets were equally depleted after DT treatment (supplemental Fig. 2). The depletion reduced the ratio of CD11chiMHCIImed cells to CD11cmed cells (Fig. 3C) and led to upregulation of endothelial cell proliferation on day 7, thereby abrogating the downregulation of endothelial cell proliferation normally observed at this time point (Fig. 3D). Both PNAd+ and PNAd− endothelial cells showed increased proliferation, although the effect was more pronounced in the PNAd+ population (Fig. 3, E and F). Depletion of CD11chiMHCIImed cells also upregulated HEV trafficking efficiency (Fig. 3G) and VCAM-1 expression (Fig. 3H). Total lymph node cellularity was modestly increased (Fig. 3I), potentially at least in part reflecting the increased trafficking. These results suggested that CD11chiMHCIImed DCs that accumulate upon vascular expansion are important for the reestablishment of vascular quiescence.
Figure 3. Depletion of CD11chiMHCIImed cells disrupts endothelial cell quiescence.

(A–I) Wild-type (WT) or CD11c-DTR (DTR) mice were injected with BMDCs on day 0 and received intraperitoneal injection of 100ng DT or CRM on day 6. Popliteal nodes were harvested at day 7. (A) Flow cytometry plot showing effect on CD11c+ subsets with DT treatment of DTR mice. (B) Number of cells in each CD11c+ subset. (C)Ratio of CD11chiMHCIImed (R1) cell numbers to CD11cmed (R2+R3) cell numbers. (D) Proliferation of total CD45−CD31+ endothelial cell population. (E) Proliferation of CD45−CD31+PNAd+ HEV endothelial cells. (F) Proliferation of CD45−CD31+PNAd− endothelial cells. (G) HEV trafficking efficiency. (H) VCAM-1 expression level on PNAd+ endothelial cells. (I) Total cell number per lymph node. For (B–I), n ≥ 6 mice for each condition. *=p<0.05; **= p < 0.01; ***= p < 0.001 with the Student’s t test. (J–K) Unstimulated CD11c-DTR mice received DT or CRM at day 0 and were examined at day 2. (J) Number of cells in each CD11c+ subset. (K) Proliferation of CD45−CD31+ endothelial cells. For (J–K), n ≥ 4 mice for each condition. *=p<0.05. For (J), asterik refers to comparison with the same CD11c+ subset in CRM-treated mice.
CD11chiMHCIImed cells were also present at homeostasis (Fig. 2, B and E; Fig. 3J), albeit in smaller numbers than in stimulated nodes (Fig. 2B). They were specifically depleted in unstimulated mice by DT treatment, and this depletion resulted in an increase of the basal endothelial cell proliferation (Fig. 3, J and K). These results suggested that CD11chiMHCIImed cells also mediate lymph node vascular quiescence during homeostasis.
CD11chiMHCIImed cells are localized near vessels and promote endothelial cell quiescence in vitro
We examined the localization of CD11chiMHCIImed cells relative to the vasculature. CD11chiMHCIImed cells comprised the vast majority of CD11chi cells from day 5 onward (Fig. 2B), suggesting that the localization of CD11cbright cells in frozen sections reflected the localization of CD11chiMHCIImed cells. Consistent with this notion, CD11cbright cells appeared numerous at day 7 but appeared relatively sparse in DT-treated CD11c-DTR mice depleted of CD11chiMHCIImed cells (supplemental Fig. 3). In the T zone, CD11cbright cells were enriched in regions that were rich in blood vessels and lymphatic sinuses. These regions were under and between follicles and in vessel-rich cords that coursed toward the medulla (Fig. 4A). In the medullary cords, CD11cbright cells aggregated around HEV and small blood vessels, and, to a lesser extent, at lymphatic sinus walls (Fig. 4B). Consistent with other studies showing DC-FRC associations (16, 26, 27), CD11cbright DCs were associated with FRCs at both reticular fibrils and vessel walls (Fig. 4C). The localization of CD11chiMHCIImed cells around and near vessels suggested that these cells could potentially act directly on local vessels to modulate vascular quiescence and stabilization.
Figure 4. CD11chiMHCIImed cells localize near vessels.

Immunohistochemical staining of day 8 lymph nodes. The CD11c stain was developed lightly so that CD11cbright cells were most apparent. Blood vessels are CD31bright and lymphatic sinuses are CD31med. (A–B) Localization of CD11chi cells relative to vasculature in the (A) T zone and (B) medullary cords. “F” denotes B cell follicles. (C) Localization of CD11chi cells relative to desmin+ FRCs. For (A–C), figures are representative of at least 3 lymph nodes over 3 experiments.
We asked whether CD11chiMHCIImed cells could promote endothelial cell quiescence in vitro. Because FRCs may regulate endothelial cell activity (13), we cultured aortic rings that provided a source of both endothelial and fibroblast-type cells. Culturing aortic rings in fibrin gels stimulates proliferative outgrowths of endothelial cells with a microvascular phenotype (28). CD11chiMHCIImed cells were added to these cultures and specifically reduced endothelial cell proliferation (Fig. 5, A and B). The area covered by the outgrowths and total endothelial cell numbers remained unchanged (data not shown). These results further establish that CD11chiMHCIImed cells can mediate endothelial cell quiescence and suggest that they are able to act directly on vessels.
Figure 5. CD11chiMHCIImed cells mediate endothelial cell quiescence in vitro.
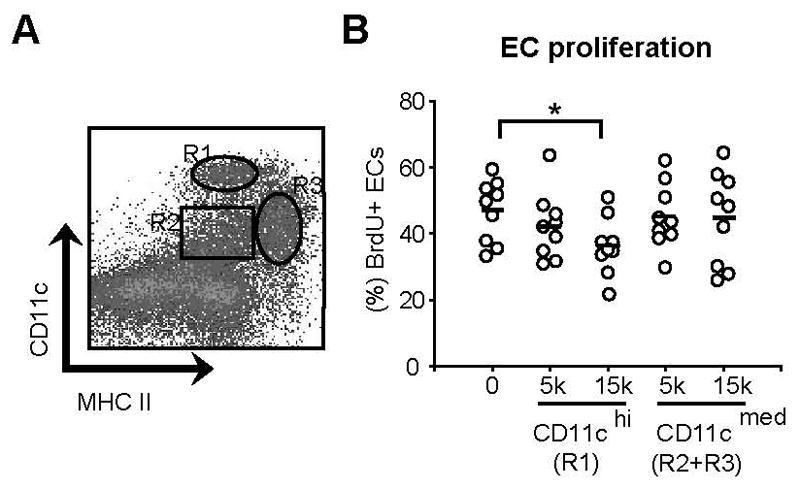
Aortic rings were cultured with sorted CD11c+ cell subsets and were analyzed by flow cytometry on day 4. (A) Flow cytometry plot showing the gating for sorting of CD11chiMHCIImed (R1) or CD11cmed (R2+R3) cells. (B) Proliferation of CD45−CD31+ endothelial cells from the aortic ring culture. Each symbol represents 1 pooled sample (see Material and Methods). *=p < 0.05 with the Student’s t test. p=0.7 for comparison between control culture and 15K CD11cmed cell coculture.
CD11chiMHCIImed cell depletion disrupts vascular stabilization
Depletion of CD11chiMHCIImed cells at day 6 also disrupted FRC reassembly. Similar to the appearance of vessels at day 2 after lymph node stimulation (Fig. 1, G and H), the FRCs around the majority of T zone HEV were more loosely organized, with many cells oriented away from the vessel wall (Fig. 6A–D; supplemental Fig. 4A). Some FRCs also appeared displaced toward the lumen and were associated with the bodies of endothelial cells (Fig. 6, A and B; supplemental Fig. 4A). The vessels were more severely affected than the vessels at day 2, as HEV tended to look more angular and less rounded (Fig. 6, A–C; supplemental Fig. 4A). FRCs associated with small non-HEV blood vessels, lymphatic sinuses, and reticular fibrils near T zone HEV also appeared more disorganized (Fig. 6A; supplemental Fig. 4A), suggesting a generalized effect on the FRCs of vessel-rich regions in the T zone. FRCs associated with HEV of the medullary cords were also disrupted (supplemental Fig. 4B).
Figure 6. Depletion of CD11chiMHCIImed cells disrupts FRC reassembly and vascular barrier function.

Mice were injected with BMDCs on day 0, received intraperitoneal DT or CRM on day 6, and were analyzed on day 7. (A–B) Desmin+ FRC organization around and near T zone HEV at (A) 160x and (B) 240x. Arrows highlight examples of perivascular FRCs that are more loosely associated or protruding out from the vessel wall. (C) gp38+ FRC organization around HEV. Arrows highlight examples of perivascular FRCs that are protruding out from the vessel wall. For (A–C), photos are representative of at least 5 mice per condition. (D) Percentage of T zone HEV with disrupted FRC organization. At least 19 fields over 5 lymph nodes were assessed for each condition in CD11c-DTR mice. **=p<.001. (E) Relative vessel permeability as assessed by Evans blue extravasation. Evans blue has red fluorescence and vascular permeability is reflected by the relative degree of red fluorescence (47). Photos are representative of at least 3 mice per condition.
We injected Evans blue dye intravenously to test blood vessel permeability. More Evans blue accumulated in the CD11chiMHCIImed cell-depleted lymph nodes, suggesting that the disrupted blood vessels were more permeable to small solutes (Fig. 6E). Together, these results suggested that, in addition to promoting endothelial quiescence, CD11chiMHCIImed cells regulate the reestablishment of vascular stabilization.
Vascular quiescence and stabilization is disrupted in CD11c-DTR-->wild type bone marrow chimeras
To exclude the possibility that the disrupted vascular quiescence and stabilization upon DT treatment of CD11c-DTR mice reflected direct effects of DT on non-hematopoietic cells such as endothelial cells and FRCs, we made bone marrow chimeras using CD11c-DTR donors and lethally irradiated wild-type recipients. Depletion of CD11chiMHCIImed cells on day 6 after BMDC injection led to an increased proliferation of total endothelial cells with a more pronounced effect on PNAd+ endothelial cell proliferation, increased HEV trafficking efficiency, and increased HEV VCAM-1 expression (Fig. 7, A–F). Vascular permeability was increased and FRC reassembly around vessels was disrupted (Fig. 7, G and H). These results further supported a role for CD11chiMHCIImed cells in reestablishing vascular quiescence and stabilization after lymph node vascular expansion.
Figure 7. Depletion of CD11chiMHCIImed cells in CD11c-DTR bone marrow chimeras disrupts vascular quiescence and stabilization.

Wild-type mice reconstituted with CD11c-DTR bone marrow received footpad injection of BMDCs on day 0 and 200ng DT or CRM intraperitoneally on day 6. Popliteal nodes were harvested at day 7. CD11c+ cell subsets were gated as in Figs. 2A and 3A. (A) CD11chiMHCIImed cell numbers. (B) Ratio of CD11chiMHCIImed (R1) cells to CD11cmed (R2+R3) cells. (C) Proliferation of total CD45−CD31+ endothelial cell population. (D) Proliferation of PNAd+ endothelial cells. (E) Relative HEV trafficking efficiency. Values in DT-treated mice were normalized to that of the CRM-treated controls in each experiment. (F) VCAM-1 expression level on PNAd+ endothelial cells. For (A–F), n = 4–8 mice per group. (G) Vascular permeability as indicated by Evans blue content in lymph nodes. n=4 mice per group. (H) Percent of T zone HEV that have disrupted FRC organization. Results represent at least 24 fields over 5 lymph nodes for each condition. For (A–H), *=p<0.05; **= p < 0.01 with the Student’s t test.
CD11chiMHCIImed cells also mediate vascular quiescence and stabilization in OVA/CFA-stimulated nodes
We asked whether CD11chiMHCIImed cells could also mediate the reestablishment of vascular quiescence and stabilization in lymph nodes stimulated with OVA/CFA. The initial rapid accumulation of CD11cmed cells (Fig. 2E and supplemental Fig. 5A) and reduction of the ratio of CD11chiMHCIImed cells to CD11cmed cells (supplemental Fig. 5B) was followed by accelerated CD11chiMHCIImed cell accumulation and progressive upregulation of the ratio from day 5 on (supplemental Fig. 5, A and B). Consistent with our previous findings (6), endothelial cell proliferation peaked at days 5–8, corresponding with the plateau of vascular expansion, and was subsequently downregulated by day 10 (supplemental Fig. 5, C and D). Trafficking efficiency was upregulated early and then, similar to BMDC-stimulated nodes, was downregulated past baseline by day 10 (supplemental Fig. 5E). VCAM-1 was also downregulated past baseline at day 10, but, in contrast to stimulation with BMDCs, VCAM-1 was also at lower than baseline at day 1 (supplemental Fig. 5F). Similar to lymph nodes stimulated with BMDC, early disorganization of FRC was followed by reassembly (supplemental Fig. 5G). CD11chiMHCIImed cell accumulation and an increased ratio of CD11chiMHCIImed cells to CD11cmed cells correlated, then, with downregulation of endothelial cell proliferation, reduction of trafficking efficiency and VCAM-1 below baseline, and FRC reassembly.
OVA/CFA-stimulated CD11c-DTR mice were injected intraperitoneally with DT to deplete CD11chiMHCIImed cells at day 9 or 10, a time point just after the peak of endothelial cell proliferation. Twenty-four hours after DT injection, endothelial cell proliferation, trafficking efficiency, and VCAM-1 expression were increased and FRC reassembly was disrupted (supplemental Fig. 5, H–M). These results suggested that CD11chiMHCIImed cells also mediate vascular quiescence and stabilization upon immunization with OVA/CFA and support a general role for these cells in regulating the reestablishment of vascular quiescence and stabilization upon lymph node stimulation.
CD11chiMHCIImed cells mediate downregulated VEGF levels
We have previously shown that VEGF is upregulated by day 1 after lymph node stimulation and is an important mediator of the initial upregulation of endothelial cell proliferation (6). In comparison to the elevated VEGF level at day 2 after BMDC injection (Fig. 8A), VEGF was downregulated at day 8, raising the possibility that the downregulation of endothelial cell proliferation during the reestablishment of quiescence was in part due to VEGF downregulation. Depletion of CD11chiMHCIImed cells at day 6 led to increased VEGF levels at day 7 (Fig. 8B), suggesting that CD11chiMHCIImed cells mediated the relative downregulation of VEGF levels during the period of endothelial cell quiescence. Anti-VEGF abrogated the increase in endothelial cell proliferation that occurred with depletion of CD11chiMHCIImed cells (Fig. 8C), suggesting that the increase in VEGF induced by CD11chiMHCIImed cell depletion was an important factor in disrupting endothelial cell quiescence. Together, these results suggested that CD11chiMHCIImed cells mediate downregulation of endothelial cell proliferation after vascular expansion at least in part by mediating the downregulation of lymph node VEGF levels.
Figure 8. CD11chiMHCIImed cells mediate downregulated lymph node VEGF levels.

(A) Relative VEGF levels in draining lymph nodes with time after BMDC injection. VEGF levels at each time point were normalized to that of unstimulated lymph nodes taken from control mice at the same time. n = 4 mice for each time point. *=p < 0.05 and **=p<0.01 with Student’s t test when compared to day 0. (B–C) Mice received BMDCs on day 0, intraperitoneal DT or CRM on day 6, and were examined on day 7. (B) Lymph node VEGF levels upon CD11chiMHCIImed cell depletion. n= 6–9 mice per group; *=p < 0.05. (C) Anti-VEGF blocks the increase in endothelial cell proliferation induced by CD11chiMHCIImed cell depletion. Mice received 200μg of anti-VEGF (R&D Systems) or control goat IgG on day 6. Each symbol represents 1 mouse. Results representative of 2 similar experiments.
Disrupted vascular quiescence does not lead to vascular expansion
We asked whether the increased endothelial cell proliferation induced by CD11chiMHCIImed cell depletion resulted in expanded endothelial cell numbers. Endothelial cell numbers remained constant up to 11 days after continuous depletion of CD11chiMHCIImed cells (supplemental Fig. 6, A, C, and E), despite increased endothelial cell proliferation at all time points (supplemental Fig. 6, B, D, and F). This is in contrast to the first few days after immunization when increased endothelial cell proliferation led to expanded endothelial cell numbers (6) (Fig. 1, A and B). These results suggested that endothelial cell survival was compromised upon CD11chiMHCIImed cell depletion at day 6, potentially because of the severity of vessel destabilization, because endothelial cells require CD11chiMHCIImed cells for survival after proliferation, or because other microenvironmental elements present early after immunization are lacking at day 6.
Disrupted reestablishment of vascular quiescence and stabilization is associated with attenuated T cell-dependent B cell responses
Increased lymphocyte entry upon disruption of vascular quiescence and stabilization could potentially increase competition for lymphocyte survival factors and disrupted FRC organization could potentially interfere with lymphocyte migration and expression of stromal-derived lymphocyte survival factors. We thus asked whether disrupting the reestablishment of vascular quiescence and stabilization was associated with disruption of the developing immune response. The injected BMDCs are cultured with fetal bovine serum (FBS) and they induced the development of germinal centers and anti-FBS IgG titers by day 8. CD11chiMHCIImed cell depletion at day 6 led to reduced germinal center B cell numbers and anti-FBS IgG at day 8 (Fig. 9, A and B), suggesting attenuation of the T cell-dependent B cell response. IgM-secreting plasma cells induced during splenic T cell-independent responses express DTR-GFP fusion protein in CD11c-DTR mice and are directly depleted by DT injection (29), raising the possibility that the reduced anti-FBS IgG reflected direct plasma cell depletion by DT rather than a downstream effect of CD11chiMHCIImed cell depletion. Anti-FBS IgM titers were not reduced by DT injection (Fig. 9C), arguing against a significant contribution of direct plasma cell depletion. We observed that about 25% of IgG+B220low plasma cells expressed GFP (supplemental Fig. 7). However, DT injection led to reduction of both GFP+ and GFP- plasma cell numbers (supplemental Fig. 7), suggesting that although a percentage of plasma cells may be directly depleted by DT, the reduction in anti-FBS IgG likely also reflects attenuated plasma cell development. Since it was possible that antigen was transferred from BMDCs to endogenous CD11chiMHCIImed cells (30), CD11chiMHCIImed cell depletion may have led to attenuated B cell responses by interfering with antigen presentation and consequent T cell activation. However, the numbers of CD25+, CD69+ and CD44+ CD4+ T cells were unchanged (supplemental Fig. 8), suggesting that a lack of antigen-presenting function at day 6 did not contribute to the attenuated B cell response at day 8. Together, our results suggested that CD11chiMHCIImed cell depletion and disrupted reestablishment of vascular quiescence and stabilization were associated with attenuated development of T cell-dependent B cell responses.
Figure 9. Depletion of CD11chiMHCIImed cells leads to attenuated B cell responses.

Mice received BMDCs on day 0 and intraperitoneal DT or CRM on day 6. Serum or lymph nodes were harvested on day 8. (A) Relative numbers of B220+peanut agglutinin+ germinal center B cells. The number of germinal center B cells in DT-treated mice was normalized to that of CRM-treated mice from the same experiment. n = 4–8 mice per group; *=p < 0.05 with the Student’s t test. (B) Serum anti-FBS IgG titers. (C) Serum anti-FBS IgM titers from same mice as in (B). For (B–C), each symbol represents 1 mouse; *=p < 0.05 with the Student’s t test.
CD11cmed cell accumulation is regulated by CD11cmedMHChi cells
We showed that intraperitoneal DT treatment of CD11c-DTR mice at homeostasis depleted CD11chiMHCmed cells but not CD11cmed cells (Fig. 3J) but that the same DT treatment followed by OVA/CFA immunization resulted in reduced accumulation of CD11cmedMHChi cells at day 2 (Fig. 2H). CD11cmedMHChi cells represent DCs that migrate from the skin (31) and, upon immunization, large numbers of these cells accumulate in the first 2 days (supplemental Fig. 5A). We hypothesized that the reduced CD11cmedMHChi cells with OVA/CFA reflected an indirect effect of CD11chiMHCIImed cell depletion on CD11cmedMHChi cell migration or survival rather than direct CD11cmedMHChi cell depletion. We thus tested the effects of intraperitoneal DT on the accumulation of injected wild-type BMDCs in CD11c-DTR mice. BMDC accumulation in the draining lymph nodes on day 2 was lower in DT-treated mice than in control CRM-treated mice (supplemental Fig. 9A) and there was a corresponding reduction in the upregulation of endothelial cell proliferation (supplemental Fig. 9B). Together, our data suggest that while intraperitoneal DT injection directly depletes CD11chiMHCIImed cells, in the setting of a large influx of CD11cmed cells from the skin (such as during the first two days of an immune response), intraperitoneal DT can also indirectly reduce the accumulation of CD11cmed cells in lymph nodes.
Discussion
We delineate a novel role for CD11chiMHCIImed DCs in mediating lymph node vascular quiescence and stabilization. We show that these cells accumulate in large numbers near vessels upon vascular expansion in immune-stimulated nodes and mediate the subsequent quiescence and stabilization of the newly expanded vasculature. CD11chiMHCIImed DCs are considered to be classical or conventional DCs that function to stimulate T cells (32–34). Our results suggest the existence of a DC-vasculature axis whereby CD11chiMHCIImed DCs, in addition to their T cell-stimulating functions, facilitate lymph node function by mediating vascular quiescence and stabilization.
Our studies were mostly conducted in BMDC-stimulated mice, but examination of OVA/CFA-stimulated mice supported a general role for CD11chiMHCIImed DCs in reestablishing vascular quiescence and stabilization in immune-stimulated nodes. In both BMDC- and OVA/CFA-stimulated nodes, early CD11cmed cell accumulation was followed by increased CD11chiMHCIImed cell accumulation and upregulation of the CD11chiMHCIImed cell: CD11cmed cell ratio. OVA/CFA-stimulated nodes demonstrated a broader peak of endothelial cell proliferation, delayed plateauing of endothelial cell expansion, and a more progressive rise in the CD11chiMHCIImed cell: CD11cmed cell ratio, perhaps reflecting the prolonged stimulation by the deposited OVA/CFA. Generally, however, the kinetics of CD11cmed cell and CD11chiMHCIImed cell accumulation and the regulation of vascular quiescence and stabilization were similar to that of BMDC-stimulated lymph nodes. The main difference between the two systems was that, although VCAM-1 was at below baseline after vascular expansion in both systems, VCAM-1 was upregulated early in BMDC-stimulated nodes but was downregulated early in OVA/CFA-stimulated nodes. This suggests that the exact pattern of cell adhesion molecule regulation in the first 1–2 days after lymph node stimulation is stimulus-specific. Consistent with this thought, we have found that PNAd+ is upregulated at day 1 in OVA/CFA-stimulated nodes (unpublished observations) and subcutaneous OVA/LPS, similar to injection of LPS-stimulated BMDCs, induces early upregulation of VCAM-1 (unpublished observations). Together, our data indicate that, although some of the parameters associated with early vascular activation may be stimulus-specific, the events associated with the reestablishment of vascular quiescence and stabilization seem consistent between systems and CD11chiMHCIImed cells have a general role in mediating quiescence and stabilization.
We observed that FRC organization adjacent to and near blood vessels was modulated over time and by CD11chiMHCIImed cells. Disrupted FRC organization upon CD11chiMHCIImed cell depletion was associated with increased vascular permeability and disrupted FRC organization at day 2 correlates with the early increase in vascular permeability that has been described (7). These results are consistent with the idea that FRCs function at least in part to regulate lymph node vascular permeability. FRCs are also key sources of chemokines (17), IL-7 (18), and VEGF (13), and our results indicating the modulation of VEGF over time and with CD11chiMHCIImed cell depletion raise the possibility that multiple FRC functions may be linked to the state of FRC organization.
Our results suggested that, after vascular expansion, CD11chiMHCIImed cells mediate downregulation of endothelial cell proliferation at least in part by mediating downregulation of lymph node VEGF levels. VEGF-expressing FRCs (13) and CD11chiMHCIImed cells are both located around vessels and on nearby fibrils, raising the possibility that CD11chiMHCIImed cells downregulate endothelial cell proliferation by acting directly on FRCs to downregulate FRC expression of VEGF. This model does not preclude alternate scenarios whereby DC-derived soluble mediators flow through the fibril-associated conduit system (19–21) to modulate endothelial cell activity directly. Indeed, Jung and colleagues have recently shown that uterine CD11chi DCs mediate implantation site angiogenesis and vascular stabilization and that they express TGFβ1 (35) that can potentially downregulate endothelial cell activity (36, 37).
The CD11chiMHCIImed DCs at day 7 consisted of mainly CD8+ CD11b− and CD8−CD11b+ subsets and all subsets were equally depleted in CD11c-DTR mice. Either subset could potentially regulate vascular function. Interestingly, recovery of altered HEV PNAd-associated molecules and MADCAM-1 is dependent on B cells and lymphotoxin β receptor (LTβR) signals (11). B cells express LTβR signals and LTβR signals can regulate CD8-CD11chiMHCIImed DC numbers (38), raising the possibility that B cell-DC interactions regulate the reestablishment of vascular quiescence and stabilization and that CD8–CD11chiMHCIImed DCs are the relevant subset.
Our results suggested a role for CD11cmed cells in mediating the upregulation of endothelial cell proliferation at the onset of vascular expansion. The injected BMDC that triggered the upregulation had a CD11cmedMHCIImed-hi phenotype and stimulated the rapid accumulation of endogenous CD11cmedMHCIIhi and CD11cmedMHCIImed cells. OVA/CFA also stimulated the rapid accumulation of predominantly CD11cmed cells, and, in DT-treated mice, the extent of upregulation of endothelial cell proliferation correlated with the extent of CD11cmed cell accumulation. CD11cmedMHCIIhi cells include skin-derived DCs and CD11cmedMHCIImed cells include skin-derived DCs, bloodborne monocyte-derived DCs, plasmacytoid DCs (31, 32, 39). Further studies will be needed to identify the exact CD11cmed subpopulation(s) involved in initiating vascular expansion and also whether they could be related in lineage to the late-accumulating CD11chiMHCIImed cells.
Our studies mainly focused on the role of CD11chiMHCIImed DCs in mediating the reestablishment of vascular quiescence and stabilization after vascular expansion. However, our experiments also suggested that they regulate vascular activity at homeostasis, as their specific depletion in homeostatic mice by intraperitoneal DT led to increased basal endothelial cell proliferation. The maintenance of homeostatic vascular activity may be important for the accumulation of skin-derived DCs and the robust upregulation of endothelial cell proliferation that occurs at day 2 after inflammatory stimulation, as intraperitoneal DT at homeostasis attenuated the accumulation of CD11cmedMHCIIhi cells after OVA/CFA and of injected BMDCs and also attenuated upregulation of endothelial cell proliferation. We observed that CD11chiMHCIImed cell depletion disrupted FRC organization around sinuses, raising the possibility that the reduced accumulation of skin-derived DCs reflects disrupted lymphatic function. CD11chiMHCIImed cells at homeostasis, then, may regulate vascular quiescence and stabilization that is important for the accumulation of antigen-bearing DCs and for full vascular expansion in the early phase of an immune response.
Our results suggest that the reestablishment of vascular quiescence and stabilization may contribute to the optimal development of T cell-dependent B cell responses. While it is possible that antigen was transferred from the injected BMDCs to endogenous CD11chiMHCIImed DCs and the reduced B cell response upon CD11chiMHCIImed cell depletion at day 6 was related to the lack of antigen presentation to T cells, activation of T cells that participate in the B cell response between days 6 and 8 is likely to have occurred at an earlier time point before CD11chiMHCIImed cell depletion (40, 41) and we also observed that activated T cell numbers were unchanged. Instead, we hypothesize that the attenuated B cell response is related to the following factors: 1) Increased B cell trafficking induced by CD11chiMHCIImed cell depletion may increase competition for limited survival factors that are needed for optimal germinal center development. 2) FRC are a source of survival factors (18, 42), and disrupted FRC organization may be associated with attenuated expression of these survival factors. 3) Antigen-specific T cells accumulate under and between B cell follicles and migrate into follicles during germinal center development (41, 43). These areas of accumulation are HEV and CD11chiMHCIImed cell-rich areas. Potentially, disrupted FRC organization around vessels and nearby fibrils in these areas leads to disrupted T cell migration and follicular entry, thereby limiting the amount of T cell help. Further studies will be needed to determine the mechanism(s) by which vascular quiescence and stabilization may contribute to the developing immune response.
Our findings describe a novel role for DCs in regulating lymph node vascular quiescence and stabilization and have potential implications for treating autoimmune and lymphoproliferative diseases. Autoimmune diseases such as systemic lupus erythematosus characterized by pathogenic autoantibodies are associated with enlarged lymph nodes with abundant vasculature (44) and the survival and aggressiveness of lymphomas may be related to the state of the vasculature (45, 46). Targeting lymph node CD11chiMHCIImed DCs for their effects on the vasculature may be a means to disrupt autoimmune and lymphoproliferative processes.
Supplementary Material
Acknowledgments
Funding support:
This work was funded by NIAID R01-AI069800, Arthritis Foundation Investigator Award and Lupus Research Institute award (TL), CRI Tumor Immunology Predoctoral Fellowship (TT), and T32-AR007517 (AC).
We thank Drs. Lionel Ivashkiv and Eric Pamer and Lu lab members for helpful discussions.
Abbreviations
- DC
dendritic cell
- FRC
fibroblastic reticular cell
- HEV
high endothelial venule
- VEGF
vascular endothelial growth factor
References
- 1.von Andrian UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nat Rev Immunol. 2003;3:867–878. doi: 10.1038/nri1222. [DOI] [PubMed] [Google Scholar]
- 2.Guarda G, Hons M, Soriano SF, Huang AY, Polley R, Martin-Fontecha A, Stein JV, Germain RN, Lanzavecchia A, Sallusto F. L-selectin-negative CCR7− effector and memory CD8+ T cells enter reactive lymph nodes and kill dendritic cells. Nat Immunol. 2007;8:743–752. doi: 10.1038/ni1469. [DOI] [PubMed] [Google Scholar]
- 3.Martin-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A, Sallusto F. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol. 2004;5:1260–1265. doi: 10.1038/ni1138. [DOI] [PubMed] [Google Scholar]
- 4.Martin-Fontecha A, Baumjohann D, Guarda G, Reboldi A, Hons M, Lanzavecchia A, Sallusto F. CD40L+ CD4+ memory T cells migrate in a CD62P-dependent fashion into reactive lymph nodes and license dendritic cells for T cell priming. J Exp Med. 2008;205:2561–2574. doi: 10.1084/jem.20081212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soderberg KA, Payne GW, Sato A, Medzhitov R, Segal SS, Iwasaki A. Innate control of adaptive immunity via remodeling of lymph node feed arteriole. Proc Natl Acad Sci U S A. 2005;102:16315–16320. doi: 10.1073/pnas.0506190102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Webster B, Ekland EH, Agle LM, Chyou S, Ruggieri R, Lu TT. Regulation of lymph node vascular growth by dendritic cells. J Exp Med. 2006;203:1903–1913. doi: 10.1084/jem.20052272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson ND, Anderson AO, Wyllie RG. Microvascular changes in lymph nodes draining skin allografts. Am J Pathol. 1975;81:131–160. [PMC free article] [PubMed] [Google Scholar]
- 8.Gaengel K, Genove G, Armulik A, Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29:630–638. doi: 10.1161/ATVBAHA.107.161521. [DOI] [PubMed] [Google Scholar]
- 9.Hellstrom M, Gerhardt H, Kalen M, Li X, Eriksson U, Wolburg H, Betsholtz C. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol. 2001;153:543–553. doi: 10.1083/jcb.153.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 11.Liao S, Ruddle NH. Synchrony of High Endothelial Venules and Lymphatic Vessels Revealed by Immunization. J Immunol. 2006;177:3369–3379. doi: 10.4049/jimmunol.177.5.3369. [DOI] [PubMed] [Google Scholar]
- 12.Herman PG, Yamamoto I, Mellins HZ. Blood microcirculation in the lymph node during the primary immune response. J Exp Med. 1972;136:697–714. doi: 10.1084/jem.136.4.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chyou S, Ekland EH, Carpenter AC, Tzeng TCJ, Tian S, Michaud M, Madri JA, Lu TT. Fibroblast-Type Reticular Stromal Cells Regulate the Lymph Node Vasculature. J Immunol. 2008;181:3887–3896. doi: 10.4049/jimmunol.181.6.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Angeli V, Ginhoux F, Llodra J, Quemeneur L, Frenette PS, Skobe M, Jessberger R, Merad M, Randolph GJ. B cell-driven lymphangiogenesis in inflamed lymph nodes enhances dendritic cell mobilization. Immunity. 2006;24:203–215. doi: 10.1016/j.immuni.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Uchimura K, Rosen SD. Sulfated L-selectin ligands as a therapeutic target in chronic inflammation. Trends in Immunology. 2006;27:559–565. doi: 10.1016/j.it.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 16.Bajenoff M, Egen JG, Koo LY, Laugier JP, Brau F, Glaichenhaus N, Germain RN. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity. 2006;25:989–1001. doi: 10.1016/j.immuni.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cyster JG. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu Rev Immunol. 2005;23:127–159. doi: 10.1146/annurev.immunol.23.021704.115628. [DOI] [PubMed] [Google Scholar]
- 18.Link A, Vogt TK, Favre S, Britschgi MR, Acha-Orbea H, Hinz B, Cyster JG, Luther SA. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol. 2007;8:1255–1265. doi: 10.1038/ni1513. [DOI] [PubMed] [Google Scholar]
- 19.Gretz JE, Anderson AO, Shaw S. Cords, channels, corridors and conduits: critical architectural elements facilitating cell interactions in the lymph node cortex. Immunol Rev. 1997;156:11–24. doi: 10.1111/j.1600-065x.1997.tb00955.x. [DOI] [PubMed] [Google Scholar]
- 20.Lammermann T, Sixt M. The microanatomy of T-cell responses. Immunol Rev. 2008;221:26–43. doi: 10.1111/j.1600-065X.2008.00592.x. [DOI] [PubMed] [Google Scholar]
- 21.Junt T, Scandella E, Ludewig B. Form follows function: lymphoid tissue microarchitecture in antimicrobial immune defence. Nat Rev Immunol. 2008;8:764–775. doi: 10.1038/nri2414. [DOI] [PubMed] [Google Scholar]
- 22.Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sapoznikov A, Pewzner-Jung Y, Kalchenko V, Krauthgamer R, Shachar I, Jung S. Perivascular clusters of dendritic cells provide critical survival signals to B cells in bone marrow niches. Nat Immunol. 2008;9:388–395. doi: 10.1038/ni1571. [DOI] [PubMed] [Google Scholar]
- 24.Darrasse-Jeze G, Deroubaix S, Mouquet H, Victora GD, Eisenreich T, Yao KH, Masilamani RF, Dustin ML, Rudensky A, Liu K, Nussenzweig MC. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J Exp Med. 2009;206:1853–1862. doi: 10.1084/jem.20090746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berger AC, Wang XQ, Zalatoris A, Cenna J, Watson JC. A murine model of ex vivo angiogenesis using aortic disks grown in fibrin clot. Microvasc Res. 2004;68:179–187. doi: 10.1016/j.mvr.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 26.Sixt M, Kanazawa N, Selg M, Samson T, Roos G, Reinhardt DP, Pabst R, Lutz MB, Sorokin L. The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the T cell area of the lymph node. Immunity. 2005;22:19–29. doi: 10.1016/j.immuni.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 27.Luther SA, Tang HL, Hyman PL, Farr AG, Cyster JG. Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc Natl Acad Sci U S A. 2000;97:12694–12699. doi: 10.1073/pnas.97.23.12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicosia RF, Ottinetti A. Modulation of microvascular growth and morphogenesis by reconstituted basement membrane gel in three-dimensional cultures of rat aorta: a comparative study of angiogenesis in matrigel, collagen, fibrin, and plasma clot. In Vitro Cell Dev Biol. 1990;26:119–128. doi: 10.1007/BF02624102. [DOI] [PubMed] [Google Scholar]
- 29.Hebel K, Griewank K, Inamine A, Chang HD, Muller-Hilke B, Fillatreau S, Manz RA, Radbruch A, Jung S. Plasma cell differentiation in T-independent type 2 immune responses is independent of CD11c(high) dendritic cells. Eur J Immunol. 2006;36:2912–2919. doi: 10.1002/eji.200636356. [DOI] [PubMed] [Google Scholar]
- 30.Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, Lew AM, Shortman K, Heath WR, Carbone FR. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 2006;25:153–162. doi: 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 31.Lopez-Bravo M, Ardavin C. In vivo induction of immune responses to pathogens by conventional dendritic cells. Immunity. 2008;29:343–351. doi: 10.1016/j.immuni.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 32.Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol. 2007;7:19–30. doi: 10.1038/nri1996. [DOI] [PubMed] [Google Scholar]
- 33.Itano AA, Jenkins MK. Antigen presentation to naive CD4 T cells in the lymph node. Nat Immunol. 2003;4:733–739. doi: 10.1038/ni957. [DOI] [PubMed] [Google Scholar]
- 34.Steinman RM. Dendritic cells in vivo: a key target for a new vaccine science. Immunity. 2008;29:319–324. doi: 10.1016/j.immuni.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 35.Plaks V, Birnberg T, Berkutzki T, Sela S, BenYashar A, Kalchenko V, Mor G, Keshet E, Dekel N, Neeman M, Jung S. Uterine DCs are crucial for decidua formation during embryo implantation in mice. J Clin Invest. 2008;118:3954–3965. doi: 10.1172/JCI36682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park SK, Yang WS, Lee SK, Ahn H, Park JS, Hwang O, Lee JD. TGF-beta(1) down-regulates inflammatory cytokine-induced VCAM-1 expression in cultured human glomerular endothelial cells. Nephrol Dial Transplant. 2000;15:596–604. doi: 10.1093/ndt/15.5.596. [DOI] [PubMed] [Google Scholar]
- 37.Gamble JR, Vadas MA. Endothelial cell adhesiveness for human T lymphocytes is inhibited by transforming growth factor-beta 1. J Immunol. 1991;146:1149–1154. [PubMed] [Google Scholar]
- 38.Kabashima K, Banks TA, Ansel KM, Lu TT, Ware CF, Cyster JG. Intrinsic lymphotoxin-beta receptor requirement for homeostasis of lymphoid tissue dendritic cells. Immunity. 2005;22:439–450. doi: 10.1016/j.immuni.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 39.Asselin-Paturel C, Brizard G, Pin JJ, Briere F, Trinchieri G. Mouse strain differences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclonal antibody. J Immunol. 2003;171:6466–6477. doi: 10.4049/jimmunol.171.12.6466. [DOI] [PubMed] [Google Scholar]
- 40.Garside P, Ingulli E, Merica RR, Johnson JG, Noelle RJ, Jenkins MK. Visualization of specific B and T lymphocyte interactions in the lymph node. Science. 1998;281:96–99. doi: 10.1126/science.281.5373.96. [DOI] [PubMed] [Google Scholar]
- 41.Okada T, Miller MJ, Parker I, Krummel MF, Neighbors M, Hartley SB, O’Garra A, Cahalan MD, Cyster JG. Antigen-engaged B cells undergo chemotaxis toward the T zone and form motile conjugates with helper T cells. PLoS Biol. 2005;3:e150. doi: 10.1371/journal.pbio.0030150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gorelik L, Gilbride K, Dobles M, Kalled SL, Zandman D, Scott ML. Normal B cell homeostasis requires B cell activation factor production by radiation-resistant cells. J Exp Med. 2003;198:937–945. doi: 10.1084/jem.20030789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pape KA, Khoruts A, Mondino A, Jenkins MK. Inflammatory cytokines enhance the in vivo clonal expansion and differentiation of antigen-activated CD4+ T cells. J Immunol. 1997;159:591–598. [PubMed] [Google Scholar]
- 44.Kojima M, Nakamura S, Morishita Y, Itoh H, Yoshida K, Ohno Y, Oyama T, Asano S, Joshita T, Mori S, Suchi T, Masawa N. Reactive follicular hyperplasia in the lymph node lesions from systemic lupus erythematosus patients: a clinicopathological and immunohistological study of 21 cases. Pathol Int. 2000;50:304–312. doi: 10.1046/j.1440-1827.2000.01052.x. [DOI] [PubMed] [Google Scholar]
- 45.Ruan J, Hyjek E, Kermani P, Christos PJ, Hooper AT, Coleman M, Hempstead B, Leonard JP, Chadburn A, Rafii S. Magnitude of stromal hemangiogenesis correlates with histologic subtype of non-Hodgkin’s lymphoma. Clin Cancer Res. 2006;12:5622–5631. doi: 10.1158/1078-0432.CCR-06-1204. [DOI] [PubMed] [Google Scholar]
- 46.Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, Xu W, Tan B, Goldschmidt N, Iqbal J, Vose J, Bast M, Fu K, Weisenburger DD, Greiner TC, Armitage JO, Kyle A, May L, Gascoyne RD, Connors JM, Troen G, Holte H, Kvaloy S, Dierickx D, Verhoef G, Delabie J, Smeland EB, Jares P, Martinez A, Lopez-Guillermo A, Montserrat E, Campo E, Braziel RM, Miller TP, Rimsza LM, Cook JR, Pohlman B, Sweetenham J, Tubbs RR, Fisher RI, Hartmann E, Rosenwald A, Ott G, Muller-Hermelink HK, Wrench D, Lister TA, Jaffe ES, Wilson WH, Chan WC, Staudt LM. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med. 2008;359:2313–2323. doi: 10.1056/NEJMoa0802885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saria A, Lundberg JM. Evans blue fluorescence: quantitative and morphological evaluation of vascular permeability in animal tissues. J Neurosci Methods. 1983;8:41–49. doi: 10.1016/0165-0270(83)90050-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.