

Abstract
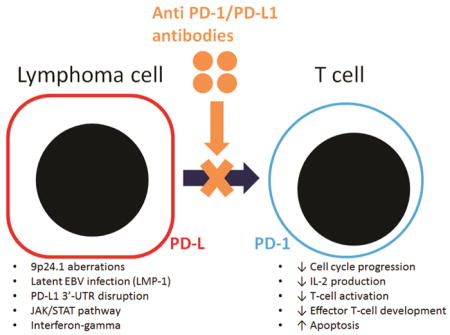
Programmed death-1 (PD-1) is a co-inhibitory molecule and is seen in CD4+ and CD8+ T cells. Upon binding to its ligands, programmed death ligand-1 (PD-L1) and -2 (PD-L2), PD-1 negatively regulates interleukin 2 (IL-2) production and T cell proliferation. Activated effector T-cells, which kill cancer cells, can be affected by PD-1 signaling in some lymphoid neoplasm that express PD-L1 or PD-L2. PD-L1 expression in tumor cells can be induced by extrinsic signal (i.e. interferon gamma) or intrinsic signals, such as genetic aberrations involving 9p24.1, latent Epstein-Barr virus infection, PD-L1 3′-untranslated region disruptions, and activated Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway. Anti-PD-1 therapy improves the overall response rate to treatment in patients with lymphoid neoplasms, particularly relapsed/refractory classical Hodgkin lymphoma. Inspired by their success in treating patients with classical Hodgkin lymphoma, medical practitioners have expanded PD-1 therapy, given as a single therapy or in combination with other drugs, to patients with other types of lymphoma. In this review, current clinical trials with anti-PD-1 or anti-PD-L1 drugs are summarized. The results of numerous clinical trials will broaden our understanding of PD-1 pathway and shall expand the list of patients who will get benefit from these agents including those who suffer from lymphoid neoplasms.
Keywords: lymphoid neoplasms, PD-1, PD-L1, PD-L2, immune checkpoint
Graphical Abstract
Introduction
T cell activation is explained by the two-signal model proposed by Bretscher and Cohn (Figure 1).[1] The first signal, which triggers T cell receptor (TCR) signaling, occurs when the major histocompatibility complex (MHC) molecule of an antigen-presenting cell (APC) presents a processed antigen to the TCR of a T cell. The second signal, which is an antigen-independent, co-stimulatory or co-inhibitory signal delivered by the APCs, modulates TCR signaling and determines the T cell’s fate. The prototypical molecule of the second signal is CD28, which constitutively expressed in resting naïve T-cells.[2] CD28’s ligands (such as B7-1 or B7-2) induce cell-cycle progression, interleukin-2 (IL-2) production, and clonal expansion. If T cells do not receive co-stimulatory second signals from molecules like CD28, they become anergic. In contrast, cytotoxic T-lymphocyte antigen-4 (CTLA-4), which shares the same ligands with CD28, is a co-inhibitory receptor on T-cells that induces T-cell tolerance.[3] If T cells do not receive co-inhibitory second signals, fatal lymphoproliferation and multiorgan autoimmunity can occur in mice.[4] Additional second-signal receptors and ligands, which are collectively called B7-CD28 family, have also been discovered and include programmed death ligand-1 (PD-L1) and PD-L2. Unlike B7-1 and B7-2, PD-L1 and PD-L2 do not bind to CD28 or CTLA-4. Instead, they bind to programmed death-1 (PD-1) on T-cells.
Figure 1.
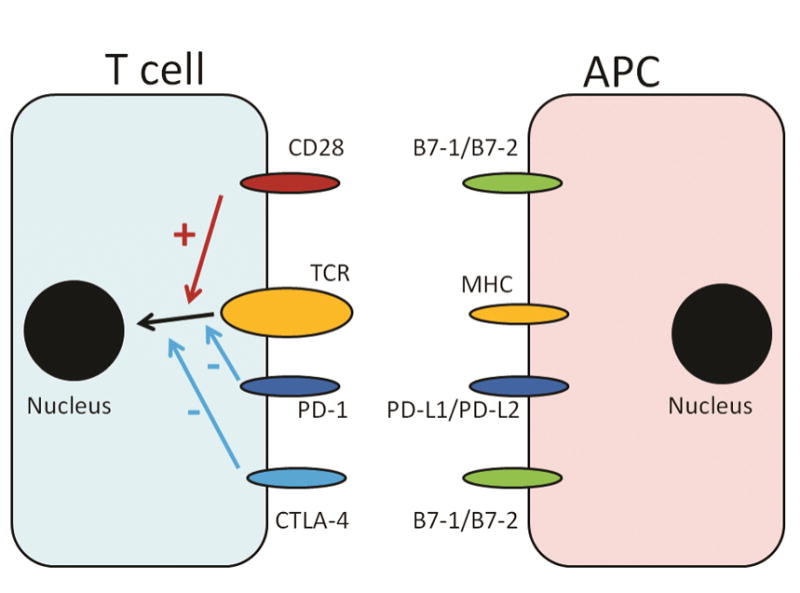
The two-signal model of T-cell activation. The first signal is a processed antigen presented by the MHC molecule of an APC to TCR of a T cells. This triggers TCR signaling, which is modulated by the antigen-independent co-stimulatory or co-inhibitory signals delivered by APCs. The T cell molecules CD28, PD-1, and CTLA-4 bind to B7-1/B7-2, PD-L1/PD-L2 and B7-1/B7-2, respectively. CD28, when engaged with its ligands, induces cell cycle progression, interleukin-2 production and clonal expansion. In contrast, PD-1 and CTLA-4 induces T cell tolerance when engaged with their respective ligands.
APC, antigen-presenting cells; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; MHC, major histocompatibility complex; TCR, T cell receptor; PD-1, programmed death 1; PD-L1/2, programmed death-ligand 1 or 2, +, activation of the pathway; −, inhibition of the pathway.
The PD-1 pathway has recently emerged as an attractive target in cancer immunotherapy. In the context of the cancer-immunity cycle, the aim of immunotherapy is to restore immune function at various steps of cancer-immunity cycle. Several clinical studies have shown that blocking the PD-1 pathway leads to significant responses in patients with various solid tumors.[5] Numerous clinical trials with PD-1 pathway blocking agents, used either alone or in combination with other therapies, are currently in progress. In this review, we discuss the functions of the PD-1 pathway in the cancer-immunity cycle, the armamentarium of PD-1 pathway blocking agents, current and prior clinical trials in patients with lymphoid malignancies and future directions in search.
The structure of PD-1, PD-L1, and PD-L2
PD-1 is a protein encoded by the 5-exon PDCD1 gene on chromosome 2q37.3. It consists of 288 amino acids, and its calculated molecular weight is 31.6 kDa. However, Agata et al’s [6] immunoprecipitation of the protein revealed broad bands with molecular weights of 50–55 kDa, suggesting that the protein is heavily glycosylated. PD-1 contains a single immunoglobulin V-like domain, a transmembrane domain, and an intracellular domain with an immunoreceptor tyrosine-based inhibitory motif (ITIM) and an immunoreceptor tyrosine-based switch motif (ITSM).[7,8]
Two PD-1ligands, PD-L1 and PD-L2, have structures similar to that of PD-1 in that they contain an immunoglobulin V-like domain, an immunoglobulin C-like domain, a transmembrane domain, and an intracellular domain.[9] These ligands interact with PD-1 via their immunoglobulin V-like domains. PD-L1 is encoded by the 8-exon CD274 gene on chromosome 9p24.1, is composed of 290 amino acids, and has a molecular weight of 33.3 kDa. Of note, it also competitively binds to B7-1, thus inhibiting the CD28-mediated co-stimulation of T-cells.[10]
PD-L2 is encoded by the 7-exon PDCD1LG2 gene, and is located 42 kilobases apart from the CD274 gene on chromosome 9p24.1. It consists of 273 amino acids, and its molecular weight is 31.0 kDa.[11]
The PD-1 signaling pathway
PD-1 is present on T-cells as a monomer and is a negative regulator of IL-2 production and T-cell proliferation.[12,13] PD-1 inhibition of antigen receptor signaling is only seen when PD-1 ligation occurs close to the site of antigen receptor engagement.[13] Indeed, it has been observed that randomly located PD-1 migrates to the immunological synapse during the interaction between T cell and APC.[14] Once PD-1 has bound to ligands, its ITIMs and ITSMs are phosphorylated by the Src-family tyrosine kinases (Figure 2). The phosphorylated tyrosine residue subsequently recruits Src homology 2 domain-containing phosphatases (SHPs), which dephosphorylate signaling intermediates and down-regulate TCR signaling. Of note, ITIM recruits only SHP-2, but ITSM recruits both SHP-1 and SHP-2.[15, 16] SHP-2 appears to be more important than SHP-1 in PD-1 signaling because T cell stimulation with PD-L2 increases the amount of SHP-2 but not of SHP-1.[17] In addition, ITSM is more important than ITIM in PD-1 signaling because PD-1’s inhibitory function is lost when ITSM is mutated but not when ITIM is mutated.[15, 18, 19]
Figure 2.
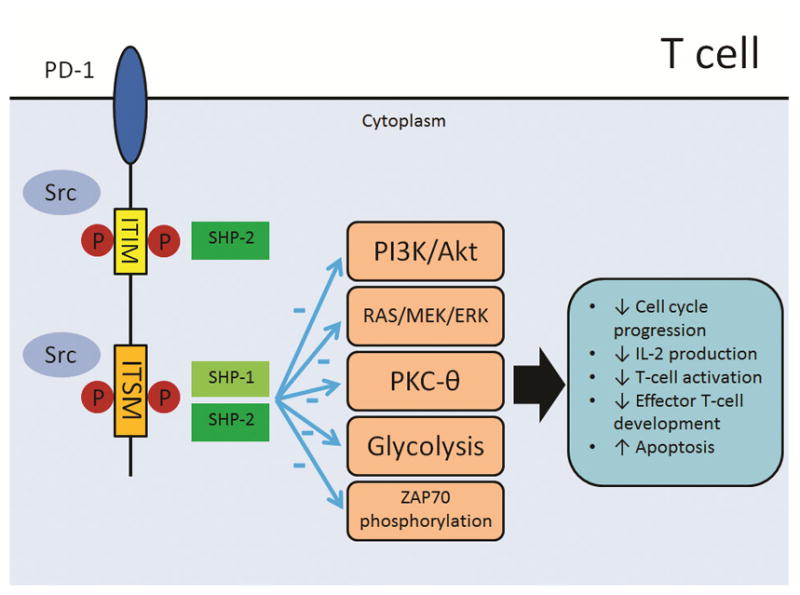
PD-1 and its downstream effect. Upon binding to ligands, PD-1’s ITIMs and ITSMs are phosphorylated by Src-family tyrosine kinases. The phosphorylated tyrosine residue subsequently recruits SHP-2 and SHP-1/SHP-2 in ITIM and ITSM, respectively. Activated PD-1 eventually hinders PI3K/Akt and RAS/MEK/ERK pathways, thwarts the function of PKC-θ and ZAP70 phosphorylation and inhibits glycolysis. The net effect is decreased cell cycle progression, IL-2 production, T-cell activation and effector T-cell development and increased apoptosis.
Src, Src-family tyrosine kinases; ITIM, immunoreceptor tyrosine-based inhibitory motif; ITSM, immunoreceptor tyrosine-based switch motif; P in red circle, phosphorylated tyrosine residues; SHP1 and SHP2, Src homology 2 domain-containing phosphatases, PI3K/Akt, Phosphatidylinositol-4,5-bisphosphate 3-kinase; Akt, Protein kinase B; PKC-θ, protein kinase C-theta; RAS/MEK/ERK, RAS/MEK/ERK pathway.
PD-1 inhibits phosphatidylinositol 3-kinase (PI3K)/Akt pathway by thwarting CD28-mediated activation of PI3K via ITSM. In contrast, CTLA-4 bypasses PI3K and instead halts Akt induction via the intracellular serine/threonine phosphatase PP2A.[20] PD-1 can also block the RAS/MEK/Erk pathway. Of interest, PD-1 inhibits the PI3K/Akt pathway within minutes, whereas it takes a few hours for it to block the RAS/MEK/Erk pathway.[17] Ultimately, PD-1’s inhibition of both of these pathways halts cell cycle progression.[21] In addition, PD-1’s inhibition of the PI3K/Akt pathway prevents T cell’s expression of the anti-apoptotic protein Bcl-xL, which depends upon PI3K.[20] PD-1 also hinders phosphorylation of ZAP70, an essential molecule for T-cell activation; inhibits activation of PKC-θ, which is critical for IL-2 production, cell cycle progression and T-cell activation; and prevents effector T-cell development by inhibiting glycolysis and promoting fatty acid oxidation.[16, 22, 23] Of note, PD-1 mediated inhibitory signals are inversely associated with the strength of the TCR signal. Furthermore, PD-1 inhibition can be overcome by T cell stimulation with CD28 or exogenous IL-2.[24]
The PD-1 pathway plays an important role in enabling tumor cells to evade the immune response. The rate of tumor lysis by cytotoxic T cells (CTLs) in vitro was lower in P815 murine mastocytoma tumor cells with transgenic expression of PD-L1 than in parental tumor cells without PD-L1 expression. Furthermore, inoculating syngenic BALB/c mice with PD-L1 expressing myeloma cells led to more rapid tumor growth, an outcome which was then suppressed by anti-PD-L1 antibodies.[25] Similarly, another study with a mouse model showed that PD-L1 expression in tumor cells confers resistance to activated CTLs but that this effect could be overcome with anti-PD-L1 or anti-PD1 antibodies.[26] In addition, Dong et al’s [27] study showed that PD-L1 expression in a melanoma cell line (624mel) enhanced apoptosis of PD-1-expressing CTLs.
Expression of PD-1, PD-L1, and PD-L2 in normal tissue
PD-1 is expressed in activated CD4+ and CD8+ T cells, naïve and activated B cells, myeloid dendritic cells, and with low intensity in monocytes. It is not expressed in resting T cells, but its expression can be induced within 24 hours of T cell activation.[28] In normal human tissue, PD-L1 expression is seen on follicular T-cells, macrophages and a subset of dendritic cells in lymphoid tissue, placental syncytiotrophoblasts, and dendritic cells/monocytes in the lung and liver, although low levels of PD-L1 mRNA are seen in almost all normal tissue.[28–30] Expression of PD-L2 is more restricted compared with PD-L1. Low levels of PD-L2 expression are seen in activated CD4+ or CD8+ T cell subsets, myeloid dendritic cells, monocytes, endothelial cells, and placental syncytiotrophoblasts.[31] Expression of PD-L1 and PD-L2 is induced by inflammatory signals such as interferon gamma (IFN-γ), granulocyte macrophage colony-stimulating factor (GM-CSF), and IL-4.[27, 32–34]
Expression of PD-L1 in lymphoid malignancies
In contrast to the infrequent PD-L1 expression in normal tissue, immunohistochemistry shows that expression of PD-L1 is often found in a variety of cancers.[5] However, the expression pattern of PD-L1 in tumor cells is not uniform. In some tumors, heterogeneous expression of PD-L1 is seen at the interface of the tumor and tumor-infiltrating lymphocytes (TILs).[35] In such cases, PD-L1 expression is likely to be induced by IFN-γ secreted from the TILs.[36] Homogeneous expression of PD-L1 is observed in other tumors, particularly in the Reed-Sternberg (RS) cells of classical Hodgkin lymphoma (CHL).[37] Homogeneous PD-L1 expression usually correlated with intrinsic signals, which are reviewed below. The underlying mechanisms of PD-L1 expression are different in different types of cancer, and antibodies targeting different domains of PD-L1 have been used in various studies (Table 1).[35, 37, 38] Furthermore, companion diagnostic assays were developed specifically for trials of particular anti-PD1 or anti-PD-L1 agents, including 22C3 for pembrolizumab, 28-8 for nivolumab, and SP142 for atezolizumab.[35, 39, 40] Therefore, PD-1 pathway investigators should pay attention to antibody types when designing a study or interpreting clinical trial results.
Table 1.
Available clones for anti-PD-L1 monoclonal antibodies
| Clone | Host | Epitope | Vendor | References |
|---|---|---|---|---|
| E1L3N | Rabbit | Intracellular | Cell Signaling | 38 |
| 405.9A11 | Mouse | Intracellular | Cell Signaling | 38 |
| SP142 | Rabbit | Intracellular | Spring Biosciences | 38 |
| SP263 | Rabbit | Intracellular | Spring Biosciences | 96 |
| E1J2J | Rabbit | Extracellular | Cell Signaling | 97 |
| 15 | Rabbit | Extracellular | Sino Biological | 38 |
| 22C3 | Mouse | Extracellular | Dako | 35 |
| 28-8 | Rabbit | Extracellular | Dako | 39 |
| 5H1 | Mouse | Extracellular | Generated in the laboratory of L. Chen, Yale School of Medicine, New Haven, CT | 98 |
| 339.7G11 | Mouse | Extracellular | Generated in the laboratory of G. Freeman, Dana–Farber Cancer Institute, Boston, MA | 38 |
| 29E.2A3 | Mouse | Extracellular | BioLegend | 99 |
| EPR1161 | Rabbit | Proprietary | Abcam | 47 |
As determined by immunohistochemistry, PD-L1 expression is present in CHL; in 87% of CHL cases, irrespective of subtype, over half of RS cells are PD-L1-positive.[41] A recent study demonstrated that almost all (97%) patients with CHL harbored either polysomy, copy-number gain or amplification of 9p24.1 detected by fluorescence in situ hybridization (FISH).[42] In approximately 40% of CHL cases of the nodular sclerosis subtype, the RS cells have copy number alterations (amplifications or gains) and/or translocations involving 9p24.1/PD-L1/PD-L2.[43] Furthermore, Green and colleagues demonstrated that amplification of 9p24.1 not only increases the genetic dosage of PD-L1/PD-L2 but also induces JAK2 amplification and, consequently, enhancement of JAK/STAT signaling.[43] Since PD-L1 has a promoter which is responsive to Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway, extra signaling for PD-L1 expression is present in CHL. Advanced stage (stages III/IV) CHL patients with 9p24.1 amplification have significantly shorter progression-free survival.[42]
Copy number alterations and/or translocations involving 9p24.1/PD-L1/PD-L2 are also frequently seen in patients with primary mediastinal large B-cell lymphoma (PMBL) (~70%), EBV-negative primary central nervous system lymphoma (PCNSL) (~60%), and primary testicular lymphoma (PTL) (~60%).[43, 44, 45] Immunohistochemistry shows that approximately 70%, 50% and 50% of PMBL, PCNSL and PTL tumors, respectively, express PD-L1, supporting the idea that PD-L1 expression is induced by cytogenetic abnormalities.[41, 44]
In DLBCL, not otherwise specified (DLBCL, NOS), PD-L1 expression is seen in 11–26% of cases by immunohistochemistry.[41, 46, 47] Interestingly, a similar percentage (~20%) of cytogenetic abnormalities in 9p24.1 (gains, amplifications or translocations) is also observed in these cases.[43, 46, 47] Gains and amplifications of 9p24.1 and translocations of PD-L1 correlate with increased PD-L1 mRNA expression and PD-L1 protein expression. Furthermore, cytogenetic abnormalities in 9p24.1 and PD-L1 protein expression are associated with activated B-cell-like or non-germinal center B cell-like phenotype of DLBCL, as determined by gene expression profiling and Hans classification, respectively.[46, 47] A Japanese study determined that PD-L1 expression in DLBCL is an independent indicator of poor prognosis.[47] Available data suggest that, in DLBCL patients, the PD-1 pathway could be an immune escape mechanism and that patients with PD-1 expression could be good candidates for anti-PD-1 or anti-PD-L1 therapies.
EBV provides an intrinsic signal to augment PD-L1 expression. EBV latent membrane protein 1 (LMP1) activates the JAK/STAT pathway and the transcription factor AP-1; this enables JAK3 to activate a PD-L1 promoter and AP-1 to stimulate a PD-L1 enhancer.[48–50] Chen et al. [41] have shown that PD-L1 expression in RS cells is more commonly present in EBV-positive CHL than in EBV-negative CHL. Similarly, PD-L1 expression is seen in all cases of EBV-positive diffuse large B-cell lymphoma (EBV+ DLBCL) and EBV-positive immunodeficiency-related DLBCL.[41] Unlike EBV-negative PCNSL, EBV-positive PCNSL frequently express PD-L1 without 9p24.1/PD-L1/PD-L2 copy number alterations, suggesting that PD-L1 expression is induced by viral proteins.[44] Other EBV-associated lymphoproliferative disorders including EBV+ post-transplant lymphoproliferative disorder (PTLD), plasmablastic lymphoma, primary effusion lymphoma, and extranodal NK-T-cell lymphoma express PD-L1 in approximately 60%, 50%, 50% and 70%, respectively.[41, 50]
Chen and colleagues showed that 91% of T-cell rich, histiocyte-rich large B-cell lymphomas (TCHRBCLs) express PD-L1, compared to only 13% of nodular lymphocyte predominant Hodgkin lymphomas (NLPHLs).[41] Since TCHRBCL and NLPHL have considerable morphologic overlap but opposite clinical behaviors, this finding is very intriguing.[51] One can speculate that acquiring PD-L1 expression is an immune escape mechanism of indolent NLPHL and that leads to more aggressive TCHRBCL. Of note, histiocytes adjacent to lymphoma cells also show strong PD-L1 expression in TCHRBCL, suggesting that both tumor cells and background inflammatory cells provide immune escape signals.[41] Since 9p24.1 aberrations and EBV infection are not generally seen in TCHRBCL, PD-L1 is thought to be induced by IFN-γ secreted from tumor-infiltrating T-cells.[52]
Another intrinsic mechanism of increased PD-L1 expression was revealed by Kataoka and colleagues, who showed that increased PD-L1 expression is associated with PD-L1 3′-untranslated region (UTR) disruption in about 30% and 10% of adult T-cell leukemia/lymphoma patients and DLBCL patients, respectively.[53] Of interest, PD-L1 expression induced by 3′-UTR disruption with truncated protein was only seen when an antibody was directed against the extracellular domain and not when an antibody was directed against the cytoplasmic domain. Whether 3′-UTR disruption in PD-L1 is present in other lymphoid neoplasms is still unknown.
PD-L1 expression also occurs through constitutive activation of the JAK/STAT pathway. In ALK+ anaplastic large cell lymphoma with NPM-ALK rearrangement, the fusion transcript has been shown to induce expression of PD-L1 via STAT3 activation.[54] Furthermore, PD-L1 expression in DLBCL correlates the ABC phenotype, which is known for its enhanced JAK/STAT pathway.[55]
PD-L1 expression in myeloma cells can be detected by flow cytometry.[56] Significant increase in copy number of PD-L1 and PD-L1 mRNA expression is seen in malignant myeloma cells, which correlates with PD-L1 expression in myeloma cells.[56] Compared with normal plasma cells, PD-L1 expression is up-regulated in non-hyperdiploid and hyperdiploid myeloma cells, with higher expression in the latter. However, PD-L1 expression is not associated with del (1q), del (13), del(17p), t(11;14), t(4;14) or t(14;16).
PD-L1 expression is not generally seen in follicular lymphoma, mantle cell lymphoma, marginal zone lymphoma, Burkitt lymphoma or chronic lymphocytic leukemia/small lymphocytic lymphoma.[57, 58]
Expression of PD-L2 in lymphoid malignancies
Lymphoid neoplasms with abnormalities in 9p24.1/PD-L1/PD-L2 generally express PD-L2. RS cells in CHL also express PD-L2, irrespective of EBV status.[59] PD-L2 expression by immunohistochemistry occurs in 72% of PMBL, and copy number gain of PDCD1LG2 gene has also been observed in most cases.[60] PD-L2 expression is also present in PTLs and PCNSLs, which are associated with 9p24.1/PD-L1/PD-L2 copy gains or translocations.[44] However, unlike PD-L1 expression, PD-L2’s expression of RNA and protein were not associated with cytogenetic abnormalities in 9p24.1 in DLBCL, NOS.[46] Also, PD-L2 expression is not associated with EBV infection or 3′-UTR disruption in the PD-L1 gene.[53, 57]
Expression of PD-1 in the lymphoid malignancy microenvironment
Although PD-1 expression is seen in a few lymphoid malignancies, particularly T cell lymphomas with the follicular helper T cell phenotype, most lymphoid neoplasms do not express PD-1.[30, 61] Thus, PD-1 expression is best examined in the microenvironment. PD-1 expression in TILs has been reported in follicular lymphoma, and NLPHL is well known for PD-1-expressing T-cell rosettes surrounding neoplastic L&H cells (LP cells or popcorn cells).[62, 63] Since both neoplasms arise from germinal center B cells, it is not surprising that their microenvironments are like those of their normal counterparts. Likewise, PD-1-expressing TILs are also correlated with DLBCL, germinal center B-cell-like (GCB) phenotype.[47] Interestingly, the high number of PD-1-expressing TILs in the DLBCL microenvironment is inversely associated with PD-L1 expression in lymphoma cells. Furthermore, contrary to observations in solid tumors, the high number of PD-1-expressing TILs in DLBCL and in follicular lymphoma (FL) is associated with a favorable prognosis; this suggests that the presence of PD-1-expressing TILs in lymphomas could simply indicate cell-of-origin, unlike tumor-mediated T-cell exhaustion in solid tumors.[47, 62, 64, 65]
Anti-PD-1 antibodies
Pembrolizumab (Keytruda®, MK-3475, SCH 900475, previously lambrolizumab)
Pembrolizumab is a fully humanized IgG4 kappa isotype anti-PD-1 monoclonal antibody. It is well tolerated and is associated with durable anti-tumor activity in multiple solid tumors.[66] In a phase 1b study (KEYNOTE-013 study, NCT01953692), pembrolizumab was administered to 31 patients with CHL every 2 weeks (10 mg/kg IV) until disease progression.[67] The median age was 32 years (range, 20–67 years). Seventeen (55%) patients had ≥ 5 lines of prior therapy, 31 (100%) failed with brentuximab vedotin (BV) treatment, 22 (71%) underwent prior autologous stem cell transplant (ASCT), and 8 (26%) were ineligible for transplantation. Examination of formalin-fixed, paraffin-embedded tissue revealed PD-L1 (clone 22C3) and PD-L2 (clone 3G2) expression in 94% and 90% of tested patients, respectively, before pembrolizumab treatment. With the median follow-up of 17.6 months (range, 10.6–22.5 months), the overall response rate (ORR) was 65%, with 5 (16%) and 15 (48%) patients achieving complete remission (CR) and partial remission (PR), respectively. Among the 20 patients, 16 (80%) achieved their best response around 12th week of therapy and 14 (70%) had ≥ 24 weeks of duration. The progression-free survival (PFS) rates were 69% and 46% at 24 weeks and 52 weeks, respectively. By flow cytometry, expansion of circulating total T cells, including CD4+ and CD8+ T cells, and NK cells was observed at cycle 7 compared to baseline. RNA profiling showed significant increases in the IFN-γ-induced, expanded immune-related, and TCR signatures.[67]
A multicohort phase 2 study is underway (KEYNOTE-087, NCT02453594) to confirm the clinical activity of pembrolizumab in CHL patients. Patients, all of whom have relapsed/refractory CHL, are in 3 cohorts of those who: (1) had an ASCT and subsequent BV therapy, (2) are ineligible for ASCT due to unresponsiveness to salvage chemotherapy and BV failure, or (3) had an ASCT without BV therapy. At the time of writing, interim analysis of cohorts 1 (n=30; median age, 36 years) and 2 (n=30; median age, 33 years) has been reported. Similar to the phase 1b study, 40 (67%) patients had ≥ 4 lines of prior therapies. In cohort 1, the ORR was 70%, with 6 (20%) and 15 (50%) patients achieving CR and PR, respectively. In cohort 2, the ORR was 80%, with 8 (27%) and 16 (53%) patients achieving CR and PR, respectively.[68]
A phase 1 study of pembrolizumab given in combination with lenalidomide and low-dose dexamethasone to patients with relapsed/refractory plasma cell myeloma determined that the maximum tolerated dose/maximum administered dose (MTD/MAD) was a 200-mg fixed dose of pembrolizumab combined with 25 mg lenalidomide and low-dose dexamethasone. In the dose determination/confirmation phase, 17 patients were evaluated. The ORR was 76% (13 patients), including 4 patients with a very good PR and 9 patients with a PR. The median duration of response was 9.7 months (range: 0–16.7 months). Updated efficacy data, including data for additional 33 patients in the expansion phase, are forthcoming (KEYNOTE-023, NCT02036502).[69, 70]
Table 2 lists numerous ongoing clinical trials of pembrolizumab given as a single therapy or in combination with other therapies.
Table 2.
Clinical trials involving anti-PD-1 or anti-PD-L1 antibodies
| Class | Agent | Intervention | Condition | Phase | Clinical Trial ID | Other title |
|---|---|---|---|---|---|---|
| Anti-PD-1 antibody | Pembrolizumab | Single therapy | R/R or disseminated malignant neoplasms including NHL or CHL in HIV-positive patients | I | NCT02595866 | |
| Single therapy | PD-L1-positive R/R lymphoma in pediatric patients | I/II | NCT02332668 | KEYNOTE-051 | ||
| Single therapy | CD19+ DLBCL, FL, or MCL who failed anti-CD19 CAR-T | I/II | NCT02650999 | |||
| Single therapy | R/R MF/SS | II | NCT02243579 | |||
| Single therapy | R/R PMBL | II | NCT02576990 | |||
| Single therapy | Recurrent PCNSL | II | NCT02779101 | |||
| Single therapy | R/R PTCL | II | NCT02535247 | |||
| Single therapy | In the lymphocyte recovery phase after high-dose chemotherapy and ASCT in PCM | II | NCT02331368 | |||
| Single therapy | Intermediate and high risk smoldering PCM | Not yet open | NCT02603887 | |||
| Pembrolizumab with dinaciclib | R/R CLL/SLL, PCM, or DLBCL | Ib | NCT02684617 | |||
| Pembrolizumab with AFM13 | R/R CHL | Ib | NCT02665650 | KEYNOTE-026 | ||
| G100 with or without pembrolizumab | FL or MZL | I/II | NCT02501473 | |||
| Pembrolizumab with INCB024360 | Cancers including DLBCL | I/II | NCT02178722 | |||
| Pembrolizumab with ACP-196 | Hematologic malignancies including NHL, PCM, Hodgkin lymphoma, CLL with or without Richter’s syndrome, and Waldenstrom macroglobulinemia | I/II | NCT02362035 | |||
| Pembrolizumab with pomalidomide | R/R PCM | I/II | NCT02289222 | |||
| Pembrolizumab with ubilituximab and TGR-1202 | R/R CLL/SLL | I/II | NCT02535286 | |||
| Pembrolizumab with idelalisib or ibrutinib | R/R low-grade B-cell lymphoma | II | NCT02332980 | |||
| Pembrolizumab with rituximab | Relapsed FL | II | NCT02446457 | |||
| Pembrolizumab as consolidation therapy | PCM with residual disease after treatment | II | NCT02636010 | |||
| Pembrolizumab as consolidation therapy after ASCT | R/R DLBCL or CHL | II | NCT02362997 | |||
| Pembrolizumab with sequential intranodal immunotherapy | Stage III/IV untreated or relapsed FL | II | NCT02677155 | |||
| Pembrolizumab with BV | R/R CHL | III | NCT02684292 | KEYNOTE-204 | ||
| Pembrolizumab with pomalidomide and low-dose dexamethasone | R/R PCM | III | NCT02576977 | KEYNOTE-183 | ||
| Pembrolizumab with lenalidomide and low-dose dexamethasone | Treatment naïve PCM | III | NCT02579863 | KEYNOTE-185 | ||
| Pembrolizumab with R-CHOP | Previously untreated DLBCL | Not described | NCT02541565 | |||
| Anti-PD-1 antibody | Nivolumab | Single therapy | Relapsed hematologic malignancies after allogeneic SCT | I | NCT01822509 | |
| Single therapy | Adult T-cell leukemia/lymphoma | II | NCT02631746 | |||
| Single therapy | R/R FL | II | NCT02038946 | CheckMate140 | ||
| Single therapy | R/R DLBCL patients who failed ASCT or were ineligible for ASCT | II | NCT02038933 | CheckMate139 | ||
| Single therapy or given with ipilimumab/lirilumab | R/R hematologic malignancies | I | NCT01592370 | |||
| Nivolumab with ipilimumab and BV | R/R CHL | I | NCT01896999 | |||
| Nivolumab with RRx-001 | Advanced lymphoma or solid tumors | I | NCT02518958 | PRIMETIME | ||
| Single therapy or with ipilimumab | R/R cancers including lymphoid neoplasms in young (12 months to 30 years) patients | I/II | NCT02304458 | |||
| Nivolumab with ibrutinib | SLL/CLL, FL, and DLBCL | I/II | NCT02329847 | |||
| Nivolumab with urelumab | Advanced B-cell NHLs | I/II | NCT02253992 | |||
| Nivolumab with INCB024360 | Cancers including DLBCL and CHL | I/II | NCT02327078 | ECHO-204 | ||
| Nivolumab with BV | R/R CHL after failure of frontline therapy | I/II | NCT02572167 | |||
| Nivolumab with BV | R/R NHLs (DLBCL, PTCL and MF/SS) with CD30 expression | I/II | NCT02581631 | |||
| Nivolumab with BMS-986016 | R/R B-cell lymphoma | I/II | NCT02061761 | |||
| Nivolumab with ipilimumab | Lymphoma or myeloma (in patients who are at high-rick for post-transplant recurrence after ASCT) | I/II | NCT02681302 | CPIT001 | ||
| Nivolumab with AVD | Newly diagnosed CHL | II | NCT02181738 | CheckMate205 cohort D | ||
| Nivolumab with ibrutinib | R/R or high-risk untreated CLL/SLL | II | NCT02420912 | |||
| Nivolumab with BV | Untreated CHL in older (≥60 years) patients | II | NCT02758717 | |||
| Nivolumab, pomalidomide, and dexamethasone | R/R PCM | III | NCT02726581 | CheckMate602 | ||
| Anti-PD-1 antibody | AMP-514 | AMP-514 with MEDI-551 | R/R aggressive B-cell lymphomas who have failed 1–2 prior lines of therapy | Ib/II | NCT02271945 | |
| Anti-PD-1 antibody | PF-06801591 | Single therapy | R/R CHL and other solid tumors | I | NCT02573259 | |
| Anti- PD-1 antibody | PDR001 | GWN323 with or without PDR001 | Advanced lymphoma or solid tumors | I | NCT02740270 | |
| Anti-PD-1 antibody | REGN2810 | Single therapy | Lymphoma | I | NCT02651662 | |
| Anti-PD-1 antibody | BGB-A317 | BGB-A317 with BGB-3111 | B-cell lymphoid malignancies | I | NCT02795182 | |
| Anti-PD-L1 antibody | Atezolizumab | Single therapy or with lenalidomide | R/R or post-ASCT PCM | Ib | NCT02431208 | |
| Atezolizumab with obinutuzumab | R/R FL or DLBCL | I | NCT02220842 | |||
| Atezolizumab with obinutuzumab and polatuzumab vedotin | R/R FL or DLBCL | Ib/II | NCT02729896 | |||
| Atezolizumab with obinutuzumab and lenalidomide | R/R FL | Ib/II | NCT02631577 | |||
| Atezolizumab, obinutuzumab and bendamustine or CHOP | Untreated or R/R FL or DLBCL | Ib/II | NCT02596971 | |||
| Anti-PD-L1 antibody | Durvalumab | Single therapy | R/R lymphoma, solid tumor, and CNS tumors in Pediatric (1 year to 17 years) patients | I | NCT02793466 | |
| Single therapy or with tremelimumab or AZD9150 | R/R DLBCL | Ib | NCT02549651 | |||
| Single therapy or with pomalidomide and with or without low- dose dexamethasone | R/R PCM | Ib | NCT02616640 | |||
| Durvalumab with anti-CD19 CAR-T | R/R DLBCL | I | NCT02706405 | |||
| Durvalumab with tremelimumab and ASCT | PCM | I | NCT02716805 | |||
| Durvalumab with tremelimumab and poly ICLC | Advanced cancers including cutaneous T-cell lymphoma | I/II | NCT02643303 | |||
| Durvalumab with ibrutinib | R/R FL or DLBCL | I/II | NCT02401048 | |||
| Durvalumab with lenalidomide with or without dexamethasone | Newly diagnosed PCM | I/II | NCT02685826 | |||
| Durvalumab with daratumumab | R/R PCM | I/II | NCT02807454 | FUSION MM-003 | ||
| Durvalumab, ibrutinib, rituximab, bendamustine, lenalidomide | CLL/SLL or other lymphoma | I/II | NCT02733042 | FUSION NHL 001 | ||
| Anti-PD-L1 antibody | Avelumab | Single therapy | Previously treated advanced stage CHL | I | NCT02603419 | JAVELIN HODGKINS |
| Anti-PD-L1 antibody | CA-170 | Single therapy | Advanced lymphoma or solid tumors | I | NCT02812875 |
Dinaciclib (MK-7965), a cyclin-dependent kinase inhibitor; AFM13, a bi-specific, tetravalent chimeric antibody construct against CD30 and CD16A; G100, a potent toll-like receptor-4 agonist; INCB024360 (Epacadostat), an indoleamine 2,3-dioxygenase 1 inhibitor; ACP-196 (Acalabrutinib), a more selective, irreversible Bruton’s Tyrosine kinase inhibitor; Pomalidomide, a derivative of thalidomide; Ubilituximab (TG-1101 or UTX), a novel, chimeric monoclonal antibody targeting a unique epitope on the CD20 antigen; TGR-1202, a PI3K δ inhibitor; Idelalisib, a PI3K δ inhibitor; Ibrutinib, a Bruton’s Tyrosine kinase inhibitor; Rituximab, a monoclonal antibody against the protein CD20; ASCT, autologous stem cell transplant; BV (Brentuximab vedotin), an antibody-drug conjugate directed to the protein CD30; lenalidomide, a derivative of thalidomide; R-CHOP; combination of rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone; ipilimumab, a monoclonal antibody targeting CTLA-4; Lirilumab, an anti-KIR monoclonal antibody; RRx-001, a pan-epigenetic anticancer agent; Urelumab, an anti-CD137 monoclonal antibody; BMS-986016, an anti-LAG-3 monoclonal antibody (an immune checkpoint inhibitor); AVD, combination of doxorubicin, vinblastine and Dacarbazine; MEDI-551, an anti-19 monoclonal antibody; GWN323, an anti-GITR antibody (GITR: glucocorticoid-induced tumor necrosis factor receptor); BGB-3111, a Bruton’s Tyrosine kinase inhibitor; Obinutuzumab, a humanized anti-CD20 monoclonal antibody; Polatuzumab vedotin, an anti-CD79b antibody-drug conjugate; Tremelimumab, an anti-CTLA-4 antibody; AZD9150, an anti-STAT3 inhibitor; CAR-T, chimeric antigen receptor T-cell therapy; Poly ICLC, a toll-like receptor-3 agonist; Daratumumab, a monoclonal anti-CD38 antibody; HIV, human immunodeficiency virus; R/R, relapsed and refractory; NHL, non-Hodgkin lymphoma; CHL, classical Hodgkin lymphoma; DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; MCL, mantle cell lymphoma; MF/SS, mycosis fungoides/Sezary syndrome; PMBL, primary mediastinal (thymic) large B-cell lymphoma; PCNSL, primary central nervous system lymphoma; PTCL, peripheral T-cell lymphoma; PCM, plasma cell myeloma; CLL/SLL, chronic lymphocytic leukemia/small lymphocytic lymphoma; MZL, marginal zone lymphoma; SCT, stem cell transplant; CNS, central nervous system.
Nivolumab (Opdivo®, BMS-936558, MDX-1106, ONO-4538)
Nivolumab is a fully human IgG4 anti-PD-1 monoclonal antibody. It has a high affinity for PD-1 and blocks it from binding to its ligands. Like pembrolizumab, it is well tolerated and is associated with durable anti-tumor activity in solid tumors.[71] In a phase 1b study, 23 patients with relapsed/refractory CHL in whom ASCT and BV was unsuccessful, received 3 mg/kg nivolumab at week 1, week 4, and then every 2 weeks until disease progression or CR or for a maximum of 2 years (NCT01592370).[37] The median age was 35 years (range, 20–54 years). Twenty (87%) patients had received ≥ 3 lines of prior therapies, 18 (78%) had received BV, and 18 (78%) had undergone ASCT. Ten (43%) patients were tested for PD-L1 and PD-L2 with FISH, and all had polysomy 9p and gain or amplification of PD-L1/PD-L2. Expression of PD-L1 (clone 405.9A11) and PD-L2 (366C.9E5) was seen in all 10 tested patients. At the median follow-up of 40 weeks (range, 0–75 weeks), the ORR was 87%, with 4 (17%) and 16 (70%) patients achieving CR and PR, respectively. Twelve (60%) patients achieved CR or PR by 8 weeks. The PFS rate was 86% at 24 weeks. The median overall survival duration had not been reached.[37]
A subsequent phase 2 study (CHECKMATE 205 cohort B, NCT02181738) recruited 80 patients with relapsed/refractory CHL who received nivolumab (3 mg/kg IV) every 2 weeks. Independent radiologic review committee (IRRC)-determined ORR was 66%, including CR and PR rates of 8.8% and 57.5%, respectively. The investigator-determined ORR was 73%, including CR and PR of 27.5% and 45%, respectively. At the median follow-up 8.9 months, the IRRC 6-month overall survival and PFS rates were 99% and 77%, respectively. Notably, 43 patients with no prior BV response showed an IRRC ORR of 72% with nivolumab treatment.[72]
A Japanese phase 2 study gave 17 patients with relapsed/refractory CHL nivolumab (3 mg/kg) on day 1 of a 14-day cycle. All patients had previously been treated with BV. The ORR in efficacy-evaluable patients was 75%, including 4 (25%) and 8 (50%) patients with CR and PR, respectively. Among 8 BV-resistant patients in the study, 3 had a CR, 4 had a PR and 1 was not evaluable at that time point were observed (JapicCTI-142755).[73]
In a phase 1 study, 81 patients with B-cell lymphoma (n=31, including DLBCL [n=11], and FL [n=10]), T cell lymphoma (n=23), and plasma cell myeloma (n=27) were treated with nivolumab alone (NCT01592370). All patients had received prior systemic treatment regimens (median 3; range, 1–12). Among the 11 patients with DLBCL, the ORR was 36%, 2 of these patients had a CR, and 2 had a PR. At the median follow-up duration of 22.7 weeks, individual response durations were 6 and 77.3+ weeks for the patients with CR and 12.1+ and 22.1 weeks for the patients with PR, respectively. The ORR of patients with FL was 40%, including 1 patient with CR and 3 patients with PR. At the median follow-up duration of 91.4 weeks, individual response durations were 81.6+ weeks for the patient with CR and 27.1+, 28.1+, and 32.1+ weeks for the patients with PR. Objective responses were not seen in the 10 patients with other B-cell lymphomas. Among the 23 patients with T cell lymphoma, the response rate was 17% (n=4), and all responding patients had PR. Individual response durations were 24.3+, 50+, 10.6, and 78.6+ weeks. For patients with plasma cell myeloma, stable disease was the best response in 17 (63%) patients. The response lasted a median of 11.4 weeks (range, 3.1–46.1 weeks).[74]
Table 2 lists numerous ongoing clinical trials of nivolumab given as a single therapy or in combination with other therapies.
Other anti-PD-1 antibodies
Clinical trials are underway with other anti-PD-1 antibodies (AMP-514, PDR001, REGN2810, BGB-A317, PF-06801591 and AMP-224), but results have not been publicized yet (Table 2).
Pidilizumab (CT-011, MDV9300, previously CT-AcTibody or BAT)
Pidilizumab was originally regarded as an anti-PD-1 antibody. However, Medivation, the company holding the right of pidilizumab, announced via a U.S. Securities and Exchange Commission filing in January 2016 that pidilizumab is not an inhibitor of PD-1.[75] The Food and Drug Administration placed a partial clinical hold on a phase 2 clinical trial of the drug in patients with relapsed or refractory DLBCL, but it lifted the hold in March 2016.[76] Although the drug showed positive results in patients with follicular lymphoma and DLBCL, its mechanism of action needs to be elucidated.[77, 78]
Anti-PD-L1 antibodies
There are several ongoing clinical trials with anti-PD-L1 antibodies (atezolizumab, durvalumab, avelumab and CA-170) in patients with lymphoid neoplasms, but results have not been published yet (Table 2).
Anti-PD-1/PD-L1 agents in combination with other treatments
Anti-PD-1/PD-L1 antibodies can be combined with other treatments in a myriad of ways. Clinical trials of anti-PD-1/PD-L1 antibodies combined with standard chemotherapy, targeted therapy (i.e., rituximab or BV) or ASCT are plentiful (Table 2). In this review, we will discuss several approaches to combination therapy with anti-PD-1/PD-L1 antibodies.
One of the strategies is to combine an anti-PD-1 antibody with an agent that blocks another co-inhibitory molecules (e.g. CTLA-4 or lymphocyte activation gene 3 [LAG-3]) in T cells. Clinical trials of nivolumab combined with ipilimumab (an anti-CTLA-4 inhibitor) or BMS-986016 (an anti-LAG-3 inhibitor) are underway in patients with hematologic malignancies (Table 2). A similar approach is to combine anti-PD-1/PD-L1 antibodies with urelumab, a cytotoxic T cell- activating drug that binds CD137. The intent of this approach is not to block two different co-inhibitory molecules; rather, it is to simultaneously enhance CTL activity and block inhibitory signals. Preclinical studies have demonstrated that this combination of anti-PD-1/PD-L1 antibodies and urelumab enhances the anti-tumor activity of T cells.[26, 79] A phase 1 clinical trial of nivolumab plus urelumab is now under way in patients with advanced B-cell non-Hodgkin lymphoma.
Combination therapy with anti-PD-1/PD-L1 antibodies can involve modulating the immunosuppressive tumor microenvironment. Epigenetic modifying agents disrupt the immunosuppressive tumor microenvironment by eradicating myeloid-derived suppressor cells or enhancing the effector function of T- and NK cells.[80, 81] Furthermore, treating a human leukemia cell line with a hypomethylating agent has been shown to augment PD-L1 and PD-L2 expression.[82] Therefore, combining epigenetic modifying agents with anti-PD/PD-L1 antibodies is a sensible approach. Indeed, preclinical and clinical studies report promising results.[83, 84] Several clinical trials are under way (Table 2).
In a melanoma mouse model, the tumor-intrinsic, active Wnt/β-catenin pathway induces T cell exclusion in the tumor microenvironment and resistance to anti-PD-L1 antibodies.[85] The kinases FAK and PYK2 augment Wnt/β-catenin pathway, and a preclinical study has shown that combining a FAK/PYK2 dual inhibitor (VS-4718) with an anti-PD-1 monoclonal antibody is more effective than anti-PD-1 therapy alone and extended survival in vivo.[86] Combining VS-4718 and an anti-PD-1 monoclonal antibody also increases the CD8+ T cell: regulatory T cells ratio, suggesting an attractive approach to modulating the tumor microenvironment to enhance the anti-tumor activity of anti-PD-1 antibodies.[87] Patients with plasma cell myeloma could be potential candidates because VS-4718 alone already has been shown to inhibit myeloma cell growth in vitro and in vivo.[88]
It is noteworthy that the administration of AFM13 and an anti-PD1 agent using an autologous PDX mouse model with donor-matched tumors and peripheral blood mononuclear cells from patients with Hodgkin lymphoma showed synergistic anti-tumor effect. AFM13 is a bi-specific, anti-CD30/CD16A, tetravalent chimeric antibody construct that targets CD30-expressing malignancies by recruiting NK cells.[89] Compared to monotherapy with anti-PD1, combined therapy with AFM13 initially enhances the infiltration of macrophages and activated NK cells and later enhances T cells and dendritic cell infiltration.[90] Based on this information, a clinical study of combined AFM13 and pembrolizumab in relapsed/refractory CHL is in preparation.
Chimeric antigen receptor (CAR) T cell therapy has been shown to be effective in patients with lymphoma, and blocking the PD-1 pathway in combination with CAR T cell therapy is an interesting approach.[91] A phase 1 study of durvalumab and anti-CD19 CAR T cell therapy will soon enroll patients with relapsed/refractory DLBCL. Oncolytic viral therapy activates innate immune responses against virally infected tumor cells and enhances adaptive anti-tumor immune responses by in vivo priming against tumor-associated antigens. A melanoma mouse model study of oncolytic vital therapy combined with anti-PD-1 or anti-CTLA-4 demonstrated significant anti-tumor activity, providing a rationale for clinical studies.[92]
Immune-related adverse events
Thanks to the clinical success of nivolumab and pembrolizumab in solid tumors, the list of tumors treatable with anti-PD-1 or anti-PD-L1 antibodies is expanding. Therefore, clinicians would observe more patients with immune related adverse events (irAEs). Overall, grade 3 or 4 irAEs are observed in 7–12% of patients with solid tumors who receive single anti-PD-1 or anti-PD-L1 antibodies.[39, 93] Of note, a predictable pattern of irAEs has been observed in such patients; dermatologic and gastrointestinal toxicities appear early, and hepatic toxicities or endocrinopathies are seen later.[94]
In patients with lymphoid neoplasms, irAEs of any grade appear in 72%–100% of patients.[37, 67, 69, 72–74] Common irAEs include thrombocytopenia, neutropenia, fatigue, infusion reaction, hypothyroidism, rash, diarrhea, nausea, pyrexia, pneumonitis, diarrhea, fatigue, back pain, decrease in platelets, dry skin, and couth.[67–69, 72] Grade 3 or higher irAEs are observed in 11–22% of patients and include interstitial pneumonia, pneumonitis, colitis, gastrointestinal inflammation, increased alanine aminotransferase/aspartate aminotransferase levels, pancreatitis, nephrotic syndrome, fulminant type 1 diabetes mellitus, myelodysplastic syndrome, leukopenia, thrombocytopenia, septic meningitis, pyrexia, infusion reaction, joint swelling, pain, stomatitis, tumor progression, and arrhythmia.[37, 67, 72–74]
The management of irAEs depends on their severity. Patients with grade 1 irAEs may continue PD-1-targeted therapy if symptoms are not observed, but it is recommended to withhold anti-PD-1 therapy from those with grade 2 irAEs and to manage symptoms with oral prednisone (1mg/kg/day) or an equivalent drug. Patients with irAEs of grade 3 or higher should discontinue anti-PD-1 therapy and be treated with intravenous methylprednisolone (2–4mg/kg/day) or an equivalent drug. Daily monitoring with liver function test is also recommended.[95]
Conclusion
Programmed death-1 (PD-1) is a co-inhibitory molecule and is seen in CD4+ and CD8+ T cells. Upon binding to its ligands, programmed death ligand-1 (PD-L1) and -2 (PD-L2), PD-1 negatively regulates interleukin 2 (IL-2) production and T cell proliferation. PD-L1 expression in normal tissue is limited, but its expression in tumor cells can be induced by extrinsic signals (e.g., IFN-γ). In various lymphoid neoplasms, PD-L1 expression can also be induced by intrinsic signals, including 1) genetic aberrations involving 9p24.1 encompassing PD-L1, PD-L2 and JAK2; 2) latent EBV infection, particularly by LMP1; 3) PD-L1 3′-UTR disruption; and 4) the activated JAK/STAT pathway. Clinical use of PD-1-pathway-blocking agents has successfully treated some lymphoid neoplasms, particularly those with PD-L1 expression induced by intrinsic signals. Currently, combination therapies involving anti-PD-1/PD-L1 agents and conventional chemotherapies, targeted therapies, or other immunotherapies are being studied, and we expect that the resulting data will broaden our understanding of the PD-1 pathway and expand the list of patients who will benefit from PD-1-pathway-blocking agents to include those who suffer from lymphoid neoplasms.
Figure 3.

Images of various lymphoid neoplasms with PD-L1 expression. A–B. Diffuse large B-cell lymphoma, not otherwise specified. Hematoxylin and eosin (A, x400) and PD-L1 stain (B, x400). C–D. Primary mediastinal large B-cell lymphoma. Hematoxylin and eosin (C, x400) and PD-L1 stain (D, x400). E–F. Epstein-Barr virus-positive diffuse large B-cell lymphoma, not otherwise specified. Hematoxylin and eosin (E, x400) and PD-L1 stain (F, x400). G–H. Primary central nervous system lymphoma. Hematoxylin and eosin (G, x400) and PD-L1 stain (H, x400). I–J. Primary testicular lymphoma. Hematoxylin and eosin (I, x400) and PD-L1 stain (J, x400). All PD-L1 stains were performed using SP142 clone (Spring Bioscience, Pleasanton, CA, USA).
Highlight points.
Immune checkpoint molecules are deregulated in hematological malignancies through diverse mechanisms
Checkpoint antagonists have shown encouraging therapeutic effects in patients with relapsed/refractory classical Hodgkin lymphoma, follicular lymphoma and diffuse large B cell lymphoma
Combined checkpoint antagonists with agents 1. reversing T cell dysfunction, 2. regulating compensatory immune pathway and 3. enhancing tumor antigen are under robust clinical evaluation and shows great promise
Checkpoint blockade may emerge as a potential agent for consolidation or salvage therapy for both autologous and allogeneic stem cell transplantation
Practical points.
Clinical value of PD-1 signaling pathway dysregulation and regimen selection
Relationship between monotherapy or combination regimen and potential complications
Development of clinical algorithm for patient evaluation
Acknowledgments
Funding Sources
The work was supported by the National Cancer Institute/National Institutes of Health (R01CA138688 and 1RC1CA146299). This work was also partially supported by the National Cancer Institute and National Institutes of Health grants P50CA136411 and P50CA142509, and by MD Anderson’s Cancer Center Support Grant CA016672. KHY is also supported by The University of Texas MD Anderson Cancer Center Institutional Research and Development Fund, an Institutional Research Grant Award, an MD Anderson Cancer Center Lymphoma Specialized Programs on Research Excellence (SPORE) Research Development Program Award, an MD Anderson Cancer Center Myeloma SPORE Research Development Program Award, a Gundersen Lutheran Medical Foundation Award, and MD Anderson Cancer Center Collaborative Funds with Roche Molecular System, Gilead Pharmaceutical, Dai Sanyo Pharmaceutical, Adaptive Biotechnology, Seattle Genetics, and HTG Molecular Diagnostics, and is partially supported by grants from the National Cancer Institute/National Institutes of Health (P50CA136411 and P50CA142509).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bretscher P, Cohn M. A theory of self-nonself discrimination. Science. 1970;169:1042–9. doi: 10.1126/science.169.3950.1042. [DOI] [PubMed] [Google Scholar]
- 2.Coyle AJ, Gutierrez-Ramos JC. The expanding B7 superfamily: increasing complexity in costimulatory signals regulating T cell function. Nat Immunol. 2001;2:203–9. doi: 10.1038/85251. [DOI] [PubMed] [Google Scholar]
- 3.Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, et al. A new member of the immunoglobulin superfamily--CTLA-4. Nature. 1987;328:267–70. doi: 10.1038/328267a0. [DOI] [PubMed] [Google Scholar]
- 4.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–8. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 5.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–7. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8:765–72. doi: 10.1093/intimm/8.5.765. [DOI] [PubMed] [Google Scholar]
- 7.Daeron M, Jaeger S, Du Pasquier L, Vivier E. Immunoreceptor tyrosine-based inhibition motifs: a quest in the past and future. Immunol Rev. 2008;224:11–43. doi: 10.1111/j.1600-065X.2008.00666.x. [DOI] [PubMed] [Google Scholar]
- 8.El Firar A, Voisin T, Rouyer-Fessard C, Ostuni MA, Couvineau A, Laburthe M. Discovery of a functional immunoreceptor tyrosine-based switch motif in a 7-transmembrane-spanning receptor: role in the orexin receptor OX1R-driven apoptosis. FASEB J. 2009;23:4069–80. doi: 10.1096/fj.09-131367. [DOI] [PubMed] [Google Scholar]
- 9.Collins M, Ling V, Carreno BM. The B7 family of immune-regulatory ligands. Genome Biol. 2005;6:223. doi: 10.1186/gb-2005-6-6-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–22. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rozali EN, Hato SV, Robinson BW, Lake RA, Lesterhuis WJ. Programmed death ligand 2 in cancer-induced immune suppression. Clin Dev Immunol. 2012;2012:656340. doi: 10.1155/2012/656340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X, Schwartz JC, Guo X, Bhatia S, Cao E, Lorenz M, et al. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity. 2004;20:337–47. doi: 10.1016/s1074-7613(04)00051-2. [DOI] [PubMed] [Google Scholar]
- 13.Bennett F, Luxenberg D, Ling V, Wang IM, Marquette K, Lowe D, et al. Program death-1 engagement upon TCR activation has distinct effects on costimulation and cytokine-driven proliferation: attenuation of ICOS, IL-4, and IL-21, but not CD28, IL-7, and IL-15 responses. J Immunol. 2003;170:711–8. doi: 10.4049/jimmunol.170.2.711. [DOI] [PubMed] [Google Scholar]
- 14.Pentcheva-Hoang T, Chen L, Pardoll DM, Allison JP. Programmed death-1 concentration at the immunological synapse is determined by ligand affinity and availability. Proc Natl Acad Sci U S A. 2007;104:17765–70. doi: 10.1073/pnas.0708767104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173:945–54. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- 16.Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004;574:37–41. doi: 10.1016/j.febslet.2004.07.083. [DOI] [PubMed] [Google Scholar]
- 17.Saunders PA, Hendrycks VR, Lidinsky WA, Woods ML. PD-L2:PD-1 involvement in T cell proliferation, cytokine production, and integrin-mediated adhesion. Eur J Immunol. 2005;35:3561–9. doi: 10.1002/eji.200526347. [DOI] [PubMed] [Google Scholar]
- 18.Boussiotis VA, Chatterjee P, Li L. Biochemical signaling of PD-1 on T cells and its functional implications. Cancer J. 2014;20:265–71. doi: 10.1097/PPO.0000000000000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U S A. 2001;98:13866–71. doi: 10.1073/pnas.231486598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patsoukis N, Sari D, Boussiotis VA. PD-1 inhibits T cell proliferation by upregulating p27 and p15 and suppressing Cdc25A. Cell Cycle. 2012;11:4305–9. doi: 10.4161/cc.22135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Isakov N, Altman A. Protein kinase C(theta) in T cell activation. Annu Rev Immunol. 2002;20:761–94. doi: 10.1146/annurev.immunol.20.100301.064807. [DOI] [PubMed] [Google Scholar]
- 23.Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692. doi: 10.1038/ncomms7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carter L, Fouser LA, Jussif J, Fitz L, Deng B, Wood CR, et al. PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur J Immunol. 2002;32:634–43. doi: 10.1002/1521-4141(200203)32:3<634::AID-IMMU634>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 25.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99:12293–7. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089–96. [PubMed] [Google Scholar]
- 27.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 28.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–9. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 30.Xerri L, Chetaille B, Serriari N, Attias C, Guillaume Y, Arnoulet C, et al. Programmed death 1 is a marker of angioimmunoblastic T-cell lymphoma and B-cell small lymphocytic lymphoma/chronic lymphocytic leukemia. Hum Pathol. 2008;39:1050–8. doi: 10.1016/j.humpath.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 31.Messal N, Serriari NE, Pastor S, Nunes JA, Olive D. PD-L2 is expressed on activated human T cells and regulates their function. Mol Immunol. 2011;48:2214–9. doi: 10.1016/j.molimm.2011.06.436. [DOI] [PubMed] [Google Scholar]
- 32.Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol. 2002;169:5538–45. doi: 10.4049/jimmunol.169.10.5538. [DOI] [PubMed] [Google Scholar]
- 33.Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9:562–7. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- 34.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–8. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 35.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–28. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 36.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5:200ra116. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372:311–9. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mahoney KM, Sun H, Liao X, Hua P, Callea M, Greenfield EA, et al. PD-L1 Antibodies to Its Cytoplasmic Domain Most Clearly Delineate Cell Membranes in Immunohistochemical Staining of Tumor Cells. Cancer Immunol Res. 2015;3:1308–15. doi: 10.1158/2326-6066.CIR-15-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:123–35. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spira AI, Park K, Mazières J, Vansteenkiste JF, Rittmeyer A, Ballinger M, et al. Efficacy, safety and predictive biomarker results from a randomized phase II study comparing MPDL3280A vs docetaxel in 2L/3L NSCLC (POPLAR) J Clin Oncol. 2015;33(suppl) abstr 8010. [Google Scholar]
- 41.Chen BJ, Chapuy B, Ouyang J, Sun HH, Roemer MG, Xu ML, et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin Cancer Res. 2013;19:3462–73. doi: 10.1158/1078-0432.CCR-13-0855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roemer MG, Advani RH, Ligon AH, Natkunam Y, Redd RA, Homer H, et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J Clin Oncol. 2016;34:2690–7. doi: 10.1200/JCO.2016.66.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O’Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116:3268–77. doi: 10.1182/blood-2010-05-282780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chapuy B, Roemer MG, Stewart C, Tan Y, Abo RP, Zhang L, et al. Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood. 2016;127:869–81. doi: 10.1182/blood-2015-10-673236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Twa DD, Chan FC, Ben-Neriah S, Woolcock BW, Mottok A, Tan KL, et al. Genomic rearrangements involving programmed death ligands are recurrent in primary mediastinal large B-cell lymphoma. Blood. 2014;123:2062–5. doi: 10.1182/blood-2013-10-535443. [DOI] [PubMed] [Google Scholar]
- 46.Georgiou K, Chen L, Berglund M, Ren W, de Miranda NF, Lisboa S, et al. Genetic basis of PD-L1 overexpression in diffuse large B-cell lymphomas. Blood. 2016;127:3026–34. doi: 10.1182/blood-2015-12-686550. [DOI] [PubMed] [Google Scholar]
- 47.Kiyasu J, Miyoshi H, Hirata A, Arakawa F, Ichikawa A, Niino D, et al. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood. 2015;126:2193–201. doi: 10.1182/blood-2015-02-629600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kieser A, Kilger E, Gires O, Ueffing M, Kolch W, Hammerschmidt W. Epstein-Barr virus latent membrane protein-1 triggers AP-1 activity via the c-Jun N-terminal kinase cascade. EMBO J. 1997;16:6478–85. doi: 10.1093/emboj/16.21.6478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ok CY, Papathomas TG, Medeiros LJ, Young KH. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328–40. doi: 10.1182/blood-2013-03-489708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Green MR, Rodig S, Juszczynski P, Ouyang J, Sinha P, O’Donnell E, et al. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: implications for targeted therapy. Clin Cancer Res. 2012;18:1611–8. doi: 10.1158/1078-0432.CCR-11-1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hartmann S, Doring C, Jakobus C, Rengstl B, Newrzela S, Tousseyn T, et al. Nodular lymphocyte predominant hodgkin lymphoma and T cell/histiocyte rich large B cell lymphoma--endpoints of a spectrum of one disease? PLoS One. 2013;8:e78812. doi: 10.1371/journal.pone.0078812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64:1140–5. doi: 10.1158/0008-5472.can-03-3259. [DOI] [PubMed] [Google Scholar]
- 53.Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature. 2016;534:402–6. doi: 10.1038/nature18294. [DOI] [PubMed] [Google Scholar]
- 54.Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1) Proc Natl Acad Sci U S A. 2008;105:20852–7. doi: 10.1073/pnas.0810958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ok CY, Chen J, Xu-Monette ZY, Tzankov A, Manyam GC, Li L, et al. Clinical implications of phosphorylated STAT3 expression in De Novo diffuse large B-cell lymphoma. Clin Cancer Res. 2014;20:5113–23. doi: 10.1158/1078-0432.CCR-14-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gorgun G, Samur MK, Cowens KB, Paula S, Bianchi G, Anderson JE, et al. Lenalidomide Enhances Immune Checkpoint Blockade-Induced Immune Response in Multiple Myeloma. Clin Cancer Res. 2015;21:4607–18. doi: 10.1158/1078-0432.CCR-15-0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Andorsky DJ, Yamada RE, Said J, Pinkus GS, Betting DJ, Timmerman JM. Programmed death ligand 1 is expressed by non-hodgkin lymphomas and inhibits the activity of tumor-associated T cells. Clin Cancer Res. 2011;17:4232–44. doi: 10.1158/1078-0432.CCR-10-2660. [DOI] [PubMed] [Google Scholar]
- 58.Gatalica Z, Bilalovic N, Vranic S, Arguello D, Reddy S, Ghosh N. PD-L1 and PD1 Expression in Lymphomas. 57th ASH annual meeting; December 7, 2015; 2015. Abstract 3899. [Google Scholar]
- 59.Yamamoto R, Nishikori M, Kitawaki T, Sakai T, Hishizawa M, Tashima M, et al. PD-1-PD-1 ligand interaction contributes to immunosuppressive microenvironment of Hodgkin lymphoma. Blood. 2008;111:3220–4. doi: 10.1182/blood-2007-05-085159. [DOI] [PubMed] [Google Scholar]
- 60.Shi M, Roemer MG, Chapuy B, Liao X, Sun H, Pinkus GS, et al. Expression of programmed cell death 1 ligand 2 (PD-L2) is a distinguishing feature of primary mediastinal (thymic) large B-cell lymphoma and associated with PDCD1LG2 copy gain. Am J Surg Pathol. 2014;38:1715–23. doi: 10.1097/PAS.0000000000000297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roncador G, Garcia Verdes-Montenegro JF, Tedoldi S, Paterson JC, Klapper W, Ballabio E, et al. Expression of two markers of germinal center T cells (SAP and PD-1) in angioimmunoblastic T-cell lymphoma. Haematologica. 2007;92:1059–66. doi: 10.3324/haematol.10864. [DOI] [PubMed] [Google Scholar]
- 62.Carreras J, Lopez-Guillermo A, Roncador G, Villamor N, Colomo L, Martinez A, et al. High numbers of tumor-infiltrating programmed cell death 1-positive regulatory lymphocytes are associated with improved overall survival in follicular lymphoma. J Clin Oncol. 2009;27:1470–6. doi: 10.1200/JCO.2008.18.0513. [DOI] [PubMed] [Google Scholar]
- 63.Nam-Cha SH, Roncador G, Sanchez-Verde L, Montes-Moreno S, Acevedo A, Dominguez-Franjo P, et al. PD-1, a follicular T-cell marker useful for recognizing nodular lymphocyte-predominant Hodgkin lymphoma. Am J Surg Pathol. 2008;32:1252–7. doi: 10.1097/PAS.0b013e318165b0d6. [DOI] [PubMed] [Google Scholar]
- 64.Thompson RH, Dong H, Lohse CM, Leibovich BC, Blute ML, Cheville JC, et al. PD-1 is expressed by tumor-infiltrating immune cells and is associated with poor outcome for patients with renal cell carcinoma. Clin Cancer Res. 2007;13:1757–61. doi: 10.1158/1078-0432.CCR-06-2599. [DOI] [PubMed] [Google Scholar]
- 65.Boussiotis VA. Cell-specific PD-L1 expression in DLBCL. Blood. 2015;126:2171–2. doi: 10.1182/blood-2015-08-663997. [DOI] [PubMed] [Google Scholar]
- 66.Patnaik A, Kang SP, Rasco D, Papadopoulos KP, Elassaiss-Schaap J, Beeram M, et al. Phase I Study of Pembrolizumab (MK-3475; Anti-PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors. Clin Cancer Res. 2015;21:4286–93. doi: 10.1158/1078-0432.CCR-14-2607. [DOI] [PubMed] [Google Scholar]
- 67.Armand P, Shipp MA, Ribrag V, Michot JM, Zinzani PL, Kuruvilla J, et al. Programmed Death-1 Blockade With Pembrolizumab in Patients With Classical Hodgkin Lymphoma After Brentuximab Vedotin Failure. J Clin Oncol. 2016 Jun 27; doi: 10.1200/JCO.2016.67.3467. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen RW, Zinzani PL, Fanale MA, Armand P, Johnson N, Ribrag V, et al. Pembrolizumab for relapsed/refractory classical Hodgkin lymphoma (R/R cHL): phase 2 KEYNOTE-087 study. J Clin Oncol. 2016;34(suppl) abstr 7555. [Google Scholar]
- 69.Mateos M-V, Orlowski RZ, Siegel DSD, Reece DE, Moreau P, Ocio EM, et al. Pembrolizumab in combination with lenalidomide and low-dose dexamethasone for relapsed/refractory multiple myeloma (RRMM): Final efficacy and safety analysis. J Clin Oncol. 2016;34(suppl) abstr 8010. [Google Scholar]
- 70.San Miguel J, Mateos M-V, Shah JJ, Ocio EM, Rodriguez-Otero P, Reece D, et al. Pembrolizumab in Combination with Lenalidomide and Low-Dose Dexamethasone for Relapsed/Refractory Multiple Myeloma (RRMM): Keynote-023. 57th ASH annual meeting; 2015;December 7; Abstract 505. [Google Scholar]
- 71.Weber JS, Kudchadkar RR, Yu B, Gallenstein D, Horak CE, Inzunza HD, et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol. 2013;31:4311–8. doi: 10.1200/JCO.2013.51.4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Younes A, Santoro A, Zinzani PL, Timmerman J, Ansell SM, Armand P, et al. Checkmate 205: Nivolumab (nivo) in classical Hodgkin lymphoma (cHL) after autologous stem cell transplant (ASCT) and brentuximab vedotin (BV)—A phase 2 study. J Clin Oncol. 2016;34(suppl) abstr 7535. [Google Scholar]
- 73.Hatake K, Kinoshita T, Fukuhara N, Choi I, Taniwaki M, Ando K, et al. Phase II study of nivolumab in Japanese patients with relapsed or refractory Hodgkin lymphoma previously treated with brentuximab vedotin (ONO-4538-15): An interim analysis. J Clin Oncol. 2016;34(suppl) abstr e19018. [Google Scholar]
- 74.Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J Clin Oncol. 2016;34:2698–704. doi: 10.1200/JCO.2015.65.9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.UNITED STATES SECURITIES AND EXCHANGE COMMISSION. [accessed 16.09.06]; accessed at < https://www.sec.gov/Archives/edgar/data/1011835/000119312516436201/d131505d8k.htm>.
- 76.U.S. FDA Lifts Partial Clinical Hold on Medivation’s Pidilizumab. [accessed 16.09.06]; accessed at < http://www.fiercebiotech.com/biotech/u-s-fda-lifts-partial-clinical-hold-on-medivation-s-pidilizumab>.
- 77.Westin JR, Chu F, Zhang M, Fayad LE, Kwak LW, Fowler N, et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: a single group, open-label, phase 2 trial. Lancet Oncol. 2014;15:69–77. doi: 10.1016/S1470-2045(13)70551-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Armand P, Nagler A, Weller EA, Devine SM, Avigan DE, Chen YB, et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: results of an international phase II trial. J Clin Oncol. 2013;31:4199–206. doi: 10.1200/JCO.2012.48.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sanmamed MF, Rodriguez I, Schalper KA, Onate C, Azpilikueta A, Rodriguez-Ruiz ME, et al. Nivolumab and Urelumab Enhance Antitumor Activity of Human T Lymphocytes Engrafted in Rag2−/−IL2Rgammanull Immunodeficient Mice. Cancer Res. 2015;75:3466–78. doi: 10.1158/0008-5472.CAN-14-3510. [DOI] [PubMed] [Google Scholar]
- 80.Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A. 2014;111:11774–9. doi: 10.1073/pnas.1410626111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang LX, Mei ZY, Zhou JH, Yao YS, Li YH, Xu YH, et al. Low dose decitabine treatment induces CD80 expression in cancer cells and stimulates tumor specific cytotoxic T lymphocyte responses. PLoS One. 2013;8:e62924. doi: 10.1371/journal.pone.0062924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. 2014;28:1280–8. doi: 10.1038/leu.2013.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kohlhof H, Hamm S, Wulff T, Baumgartner R, Vitt D. 4SC-202: Epigenetic modulation to pave the way for checkpoint inhibition. J Clin Oncol. 2016;34(suppl) abstr 11546. [Google Scholar]
- 84.Falchi L, Sawas A, Deng C, Amengual JE, Colbourn DS, Lichtenstein E, et al. Rate of complete metabolic responses to immune checkpoint inhibitors in extremely heavily pre-treated patients with classical Hodgkin’s lymphoma and immunoepigenetic priming. J Clin Oncol. 2016;34(suppl) doi: 10.1186/s13045-016-0363-1. abstr e19012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–5. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 86.Gao C, Chen G, Kuan SF, Zhang DH, Schlaepfer DD, Hu J. FAK/PYK2 promotes the Wnt/beta-catenin pathway and intestinal tumorigenesis by phosphorylating GSK3beta. Elife. 2015;4 doi: 10.7554/eLife.10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang Y, Ring JE, Sprott K, Weaver DT, Pachter JA. FAK/PYK2 inhibition enhances immune checkpoint inhibitor efficacy. AACR annual meeting; April 17, 2016; New Orleans, USA. 2016. Abstract 568. [Google Scholar]
- 88.Zhang Y, Moschetta M, Huynh D, Tai YT, Zhang Y, Zhang W, et al. Pyk2 promotes tumor progression in multiple myeloma. Blood. 2014;124:2675–86. doi: 10.1182/blood-2014-03-563981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rothe A, Sasse S, Topp MS, Eichenauer DA, Hummel H, Reiners KS, et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2015;125:4024–31. doi: 10.1182/blood-2014-12-614636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Treder M, Zhao X, Rajasekaran N, Reusch U, Marschner J-P, Kohrt HE. Anti-PD-1 to enhance NK-cell cytotoxicity towards CD30+ Hodgkin lymphoma induced by CD30/CD16A TandAb AFM13. J Clin Oncol. 2016;34(suppl) abstr e14012. [Google Scholar]
- 91.Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR, et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood. 2016;127:2980–90. doi: 10.1182/blood-2015-12-686725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Quah MY, Wong Y, Andtbacka R, Au G, Shafren DR. Elevated immune activity following an anticancer combination therapy of a novel oncolytic immunotherapeutic agent, CAVATAK (Coxsackievirus A21), and immune checkpoint blockade. AACR annual meeting; April 18, 2016; New Orleans, USA. 2016. Abstract 2341. [Google Scholar]
- 93.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372:2521–32. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 94.Weber JS, Yang JC, Atkins MB, Disis ML. Toxicities of Immunotherapy for the Practitioner. J Clin Oncol. 2015;33:2092–9. doi: 10.1200/JCO.2014.60.0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Naidoo J, Page DB, Li BT, Connell LC, Schindler K, Lacouture ME, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2016;27:1362. doi: 10.1093/annonc/mdw141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rizvi NA, Brahmer JR, Ou SI, Segal NH, Khleif S, Hwu W, et al. Safety and clinical activity of MEDI4736, an anti-programmed cell death-ligand 1 (PD-L1) antibody, in patients with non-small cell lung cancer (NSCLC) J Clin Oncol. 2015;33(suppl) abstr 8032. [Google Scholar]
- 97.McLaughlin JF, Schalper K, Carvajal-Hausdorf DE, Velcheti V, Haack H, Silver M, et al. Domain-specific PD-L1 protein measurement in non-small cell lung cancer (NSCLC) J Clin Oncol. 2014;32(suppl) abstr 8064. [Google Scholar]
- 98.Thompson RH, Gillett MD, Cheville JC, Lohse CM, Dong H, Webster WS, et al. Costimulatory B7-H1 in renal cell carcinoma patients: Indicator of tumor aggressiveness and potential therapeutic target. Proc Natl Acad Sci U S A. 2004;101:17174–9. doi: 10.1073/pnas.0406351101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol. 2003;33:3117–26. doi: 10.1002/eji.200324270. [DOI] [PubMed] [Google Scholar]