

Abstract
In inflammatory arthritis, the dysregulation of osteoclast activity by pro-inflammatory cytokines, including TNF, interferes with bone remodeling during inflammation through Ca2+-dependent mechanisms causing pathological bone loss. Ca2+-dependent CREB/c-fos activation via CaMKIV induces transcriptional regulation of osteoclast-specific genes via NFATc1 that facilitate bone resorption. In leukocytes, Ca2+ regulation of NFAT-dependent gene expression oftentimes involves the activity of the Ca2+-activated K+ channel KCa3.1. Herein, we evaluate KCa3.1 as a modulator of Ca2+-induced NFAT-dependent osteoclast differentiation in inflammatory bone loss. Microarray analysis of RANKL-activated murine BMM cultures revealed unique up-regulation of KCa3.1 during osteoclastogenesis. The expression of KCa3.1 in vivo was confirmed by IF staining on multinucleated cells at the bone surface of inflamed mouse joints. Experiments on in vitro BMM cultures revealed that KCa3.1−/− and TRAM-34-treatment significantly reduced the expression of osteoclast-specific genes (P<0.05) alongside with decreased osteoclast formation (P<0.0001) in inflammatory (RANKL+TNF) and non-inflammatory (RANKL) conditions. In particular, live cell Ca2+ imaging and western blot analysis showed that TRAM-34 pre-treatment decreased transient RANKL-induced Ca2+ amplitudes in BMMs by approximately 50% (P<0.0001) and prevented phosphorylation of CaMKIV. KCa3.1−/− reduced RANKL+/−TNF-stimulated phosphorylation of CREB and expression of c-fos in BMMs (P<0.01) culminating in decreased NFATc1 protein expression and transcriptional activity (P<0.01). These data indicate that KCa3.1 regulates Ca2+-dependent NFATc1 expression via CaMKIV/CREB during inflammatory osteoclastogenesis in the presence of TNF, corroborating its role as a target candidate for the treatment of bone erosion in inflammatory arthritis.
Introduction
Physiological bone remodeling is a balanced process maintained by the activity of bone-resorbing osteoclasts and bone-forming osteoblasts. Osteoclasts differentiate from myeloid precursor cells into multinucleated giant cells under the influence of M-CSF and RANKL (1–5). Terminally differentiated osteoclasts are characterized by the formation of sealing zones of F-actin (“F-actin rings”), creating an isolated environment on the bone surface into which enzymatic vesicles that contain lytic enzymes including TRAP (Acp5), MMP9 (Mmp9) and CTSK (Ctsk) can be released to facilitate bone resorption (1–6). The transcriptional regulation of osteoclastogenesis by RANKL is initiated by RANK/RANKL binding which induces Ca2+-independent NF‐κB signaling, as well as c‐fos/AP-1, both resulting in the expression of NFATc1 – the master regulator of osteoclastogenesis (7–10). Simultaneously, RANKL induces c-fos/AP‐1 up-regulation through Ca2+-dependent signaling cascades involving PLCγ (11–13). Increase in intracellular Ca2+ activates the Ca2+/calmodulin-dependent CaMKIV, capable of inducing transcriptional up-regulation of c-fos/AP-1 and Nfatc1 through phosphorylation of CREB (9, 14, 15). Subsequently, NFATc1 undergoes auto-amplification by binding to its own promoter region (16). Besides CaM kinases, the Ca2+/calmodulin-dependent phosphatase CaN is also activated by increased intracellular Ca2+ concentrations (17, 18). Activated CaN de-phosphorylates NFATc1 enabling its nuclear translocation. Next, a transcriptional complex including NFATc1, c‐fos/AP‐1 and pCREB trigger the transcription of osteoclast-specific genes (13, 19). During pathological conditions as in inflammatory arthritis alternative stimuli can facilitate osteoclast formation in addition to the classic RANKL pathway (20–25). Pro-inflammatory cytokines such as TNF promote osteoclast formation and activity in rodents and humans (24, 26). TNF is released by several types of T cells as well as by synovial macrophages under inflammatory joint conditions and is involved in promoting NF-kB signaling as well as Ca2+-dependent pathways in osteoclast precursor cells (24). Hence, both RANKL- and TNF-dependent osteoclast differentiation pathways are critically dependent on Ca2+ signals (13, 14, 24).
In leukocytes, Ca2+-dependent NFAT signaling through CaN is modulated by K+ channel activity, which has been thoroughly examined in the past three decades (27). During PLCγ-induced store-operated Ca2+ entry (SOCE) through plasma membrane Ca2+ channels such as CRAC channels, K+ channels repolarize the plasma membrane by creating a positive outward current and thereby stabilize the driving force for Ca2+ influx – a prerequisite for sustained activation of CaN-dependent subsequent gene expression through NFAT (12, 27–30). However, the dependence of CaMK-pathways on K+ channel activity during leukocyte differentiation and its role in NFAT expression is much less understood at this time (31).
Recent studies have highlighted the importance of the Ca2+-dependent K+ channel KCa3.1 (Kcnn4) as a key regulator during osteoclast formation (32) and suggested that the mechanism of action of Ibandronate, a commonly used drug to treat postmenopausal osteoporosis, involves the inhibition of KCa3.1 currents (33).
In the current study we characterize the involvement of the predominant K+ channel KCa3.1 in CaMK/CREB/c-fos-dependent Ca2+-signaling and NFATc1 regulation during inflammatory compared to non-inflammatory osteoclast differentiation in the presence and absence of TNF using in vitro osteoclast assays, as well as live cell Ca2+ imaging.
Materials and Methods
Reagents and antibodies
Cells were cultured in αMEM containing 2 mM L-glutamine (12561), 10% heat-inactivated FBS, 100 IU/ml Penicillin and 100 IU/ml Streptomycin (Life Technologies). qPCR primers were either designed or used as previously described and tested for their distinctive product sizes (Integrated DNA Technologies) (34). For a detailed list of primer constructs see supplemental table 1. CMG-1412 media was generated as previously described (35). IF antibodies and reagents: anti-KCa3.1 (P4997) (Sigma-Aldrich), anti-CD68 (FA-11) (Biolegend) (36), Phalloidin-FITC (Sigma-Aldrich), goat anti-rabbit AF-594 and goat anti-rat AF488 (life technologies), goat serum (Gemini-Bio products). Recombinant cytokines (mM-CSF, mTNF and mRANKL) were purchased from R&D Systems; inhibitors: TRAM-34, KN-93, cyclosporine A (CsA) (Sigma-Aldrich); Fluo-4-AM (life technologies); Western blot antibodies: anti-CREB (06-863) and anti-phospho-CREB (Ser133) (06-519) (Millipore) (14), anti-CaMKIV (H-5) (Santa Cruz Technologies) (37), anti-phospho-CaMKIV (Thr196, Thr200) (PA5-37504) (Thermo Fisher Scientific), anti-NFATc1 (7A6) (Thermo Fisher Scientific) (14), anti-β-actin (13E5) (Cell Signaling Technology)
Animals
All animal protocols and procedures are in accordance with the Declaration of Helsinki and were approved by the UC Davis Animal Care and Use Committee (IACUC). C57Bl/6J and C57Bl/6J Kcnn4−/− mice, originally purchased from The Jackson Laboratory were subsequently bred in the UCD animal facility under standardized breeding conditions. For in vitro experiments mice were sacrificed by carbon dioxide exposure. Arthritis was induced using the IL-23 gene transfer model as previously described (38).
Osteoclast cultures and characterization
Whole bone marrow was extracted from tibiae and femora of 8- to 12-week-old wild-type (WT) C57Bl/6J and Kcnn4−/− mice, as previously described (39). Cells were plated in culture medium containing CMG medium 1:20 (v/v) in αMEM for four days until 30–40% confluence to generate macrophages. To generate osteoclasts, the culture medium was then supplemented with soluble recombinant mRANKL [30 ng/ml] for an additional three days. For inflammatory conditions, soluble recombinant mTNF was added at 10 ng/mL. Cytochemical staining was performed using Acid phosphatase, Leukocyte (TRAP) Kit according to manufacturer’s instructions (Sigma-Aldrich). For “F-actin ring” staining, cells grown on coverslips were fixed with 4% paraformaldehyde (PFA) solution and permeabilized using 0.5% Triton X-100 before incubation with Phalloidin-FITC (1:100) for one hour at RT.
IF and H&E staining
Native cells were fixed with 4% paraformaldehyde (PFA) solution and blocked with 5% donkey serum for one hour at RT. Primary KCa3.1 antibody was used (1:300) and conjugated to secondary donkey IgG affinity purified fluorescently labeled antibody (1:500). As controls for unspecific binding, secondary antibodies were used without primary. Samples were embedded using Vectashield mounting medium for IF (Vector Laboratories), containing DAPI. For staining of mouse joints, whole-ankle and -knee joints were fixed in 10% formalin decalcified in 10% EDTA and embedded in paraffin. Serial sections (4 μm) were rehydrated in alcohol gradient and stained with H&E. For IF, antigen was retrieved using 0.6 U/mL Proteinase-K solutions. Subsequent antibody-labelling and mounting for IF was performed as with native cells. H&E imaging was obtained by Olympus BX61 confocal microscope (Olympus) and analyzed with cellSens Dimension software (Olympus). IF imaging was performed with a Nikon A1 confocal microscope (Nikon Instruments) with 200× or 600× magnification. Images within one experiment were recorded under identical parameters to ensure comparability and analysed using ImageJ 1.46.
Microarray analysis
Total RNA was amplified and purified using Ambion Illumina RNA amplification kit (Ambion) to yield biotinylated cRNA and 1.5 μg of cRNA samples were hybridized to Illumina WG-6 v2.0 chips according to manufacturer’s instructions (Illumina). Arrays were scanned by Illumina iScan. Raw data were extracted using GenomeStudio (Illumina). Probe signal values were transformed by mean normalization and logarithm, and then illustrated by heat map. Data is deposited in NCBI GEO under accession number: GSE86998 http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE86998
Alamar blue assay
Alamar blue cell viability and proliferation assay was performed according to manufacturer’s instructions (invitrogen). In brief, cells were cultivated in 96-well plates. All samples were plated in triplets. Alamar blue reagent was added to each well and incubated on 37°C for seven hours before fluorescence detection with an excitation at 570 nm and emission at 590 nm.
Calcium measurements
BMMs were cultured in M-CSF (25 ng/ml) for four days. Prior to Ca2+ measurements, cells were starved from M-CSF for 6–8 hours. Directly before imaging, cells were labeled with 2.5 μM Fluo-4-AM. Cells were observed for baseline Ca2+ for 5 minutes before addition of 100 ng/mL RANKL. For KCa3.1 inhibition, cells were pre-incubated with 3 µM TRAM-34 or DMSO for 15 minutes at 37°C prior to Ca2+ measurements. Fluo-4 AM intensity was measured and tracked over time in 1 s intervals for 15 min using a Nikon Eclipse FN1 Swept-field confocal microscope with 200× magnification and NIS-Elements BR software (Nikon Instruments).
RNA isolation and qPCR
Total RNA isolation was performed using RNeasy® Mini RNA isolation kit (QIAGEN) according to manufacturer’s instructions. RNA yield was measured by photospectrometry. 500 ng of total RNA was used for cDNA synthesis using oligo(dT)15 primers and Omniscript Reverse Transcription cDNA synthesis kit following manufacturer’s recommendations (QIAGEN). qPCR was performed using QuantiTect® SYBR® Green PCR kit, see manufacturer’s instructions (QIAGEN); all cDNAs were standardized to β2M mRNA expression.
Trans-AM NFATc1 transcription factor binding assay
Transcription factor binding assay was performed according to manufacturer’s instructions (Active Motive). In brief, BMM were cultivated in 10 cm culture dishes and stimulated with RANKL [30 ng/mL] only or in combination with TNF [10 ng/mL] as described earlier. After 36 hours, nuclear lysates were extracted using Nuclear Extract kit (Active Motive). Final protein concentrations were determined using BCA protein assay kit (Pierce). Protein lysates were then incubated in an oligonucleotide pre-coated plate and detected with primary NFATc1 and secondary HRP-labelled antibodies. Absorbance was measured at 450 nm with a reference wavelength at 655 nm. Assay specificity was guaranteed by competitive binding assays using WT and control mutated consensus sequences for NFATc1.
Western blot analysis
Time-dependent phosphorylation experiments were performed in total cell lysates of BMMs and osteoclasts, respectively. For CREB phosphorylation, cells were cultivated in full culture medium and stimulated with 30 ng/mL RANKL or RANKL in combination with 10 ng/mL TNF for indicated times before cell lysates were collected. For CaMKIV phosphorylation, cells were starved for 6–8 hours before stimulation with 100 ng/mL RANKL. BMMs and osteoclasts were lysed in RIPA buffer (Cell Signaling Technologies) including phosphatase inhibitors (Pierce) and Mini EDTA-free Protease Inhibitor Cocktail (Roche). Insoluble material was removed by centrifugation at 13,000 rpm for 20 minutes at 4°C. Final protein concentrations were determined using BCA protein assay kit (Pierce). Electrophoresis was performed using 4~15% Mini-PROTEAN TGX Precast Gels (Bio-Rad). Blots were subsequently transferred to PVDF membranes and unspecific binding was blocked with 5% BSA in TBST. Immunoreactive bands were detected with the Odyssey Infrared Imaging System (LI-COR Biosciences). All specific bands were normalized to β-actin signals.
Statistical Analysis
Statistical analysis and graphical representation was performed using GraphPad Prism® Version 6.03 and Windows Excel 2010. Following a Gaussian distribution (D’Agostino and Pearson omnibus normality test) P-values were calculated for a confidence interval of 95% by paired or unpaired Student’s t-test. When calculating P-values for normalized data, one-sample t-test for a hypothetical value of 1.0 was assumed. In case of non-parametric data either Mann-Whitney U or Wilcoxon-ranked pairs test was used. For multi-parameter experiments, either one-way ANOVA preceding post-hoc Holm-Sidak’s multiple comparison test for parametric data or Kruskal-Wallis followed by Dunn’s multiple comparisons test were performed for a confidence interval of 95%, unless otherwise indicated. Bar graphs show means with SEM. n.s.=P >0.05; *P<0.05; **P<0.01; ***P<0.001; ****P< 0.0001; Calcium measurements: for population analysis, the number of responding cells was normalized to the total number of cells per frame; for assessment of the response magnitude, the maximum response after stimulation was normalized to the maximum response baseline (ΔRA/ΔBL).
Results
KCa3.1 is highly expressed during osteoclastogenesis
To investigate the regulation of K+ channel expression during osteoclast differentiation, we performed RNA microarray analysis on isolated mouse BMMs cultivated from murine M-CSF-stimulated bone marrow cells before and after RANKL-induced osteoclast differentiation (Supplemental figure 1). A comparison of a number of selected K+ channels, previously described in macrophage or osteoclast biology (32, 40), identified the up-regulation of only one Ca2+-activated K+ channel, KCa3.1 (Kcnn4), during RANKL-induced osteoclastogenesis (Supplemental figure 1A and D).
We also investigated the expression of KCa3.1 in murine inflammatory arthritis in vivo (38) and confirmed that multinucleated cells located at the bone surface of inflamed joints (Figure 1A) co-expressed KCa3.1 and CD68 (Figure 1B) (41). Similarly, qPCR analysis of BMMs stimulated with RANKL in vitro revealed up-regulation of KCa3.1 mRNA (Kcnn4) during RANKL-induced osteoclastogenesis, whereas the presence of TNF in addition to RANKL had no significant influence on Kcnn4 expression (Figure 1C). Interestingly, the expression of the voltage-gated potassium channel Kv1.3 (Kcna3), which has previously been demonstrated to play a role in pro-inflammatory macrophages/microglia differentiation, was found to be significantly underrepresented in macrophages and osteoclasts (Supplemental figure 1E) (42). By using splice variant-specific primers, we moreover detected the presence of a dominant-negative transcript of Kcnn4 (Kcnn4 b) next to the active variant (Kcnn4 a) in macrophages and osteoclasts, as suggested by a previous study in lymphocytes (34). IF staining of KCa3.1 confirmed protein expression in cultures stimulated with RANKL or TNF alone and the combination of both in multinucleated cells exhibiting “F-actin” ring structures (Figure 1D).
Figure 1. KCa3.1 is present on multinucleated cells in inflammatory arthritis.
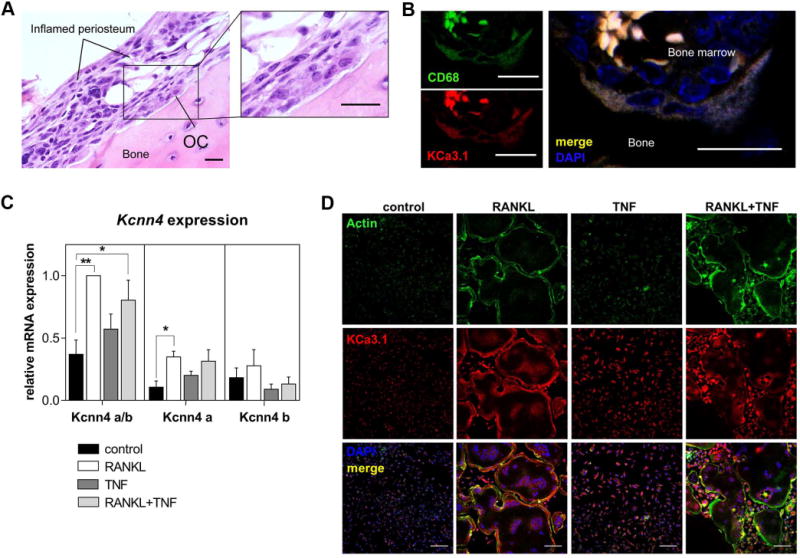
A) H&E staining of paraffin sections obtained from inflamed joints of arthritic mice 11-days after IL-23 minicircle injection showing a multinucleated cell/osteoclast (OC) at the bone surface adjacent to inflamed periosteum (scale bar = 20 µM). B) IF staining of a multinucleated cell at the bone surface showing co-expression of CD68 (green) and KCa3.1 (red) with DAPI (blue) (scale bar = 20 µM). C) qPCR analysis showing normalized expression of active (Kcnn4 a) and dominant-negative (Kcnn4 b) splice variants of the KCa3.1 gene (Kcnn4); Kcnn4 a/b primer detecting both transcripts; one-way ANOVA followed by post-hoc multiple comparison test was performed (N=4, P<0.0001), shown are means ± SEM, *P<0.05, **P<0.01; D) IF staining for KCa3.1 (red), actin (green) and DAPI (blue) in cultured mouse BMMs, unstimulated (control) and stimulated in indicated conditions (N=3, scale bar = 100 µM).
These results indicate that KCa3.1 is expressed during physiological and inflammatory osteoclastogenesis.
KCa3.1 function is required for osteoclast formation and gene expression in physiological and inflammatory osteoclast differentiation
In order to further understand the role of KCa3.1 in osteoclastogenesis during inflammation, we stimulated BMMs from WT and Kcnn4−/− mice with RANKL and RANKL in combination with TNF. Although visibly smaller in size, as previously described by Kang et al. (2014), cultures from Kcnn4−/− cells compared to WT revealed similar numbers of TRAP+, multinucleated cells in the presence of RANKL when assessed with a threshold of ≥3 nuclei (Figure 2A, B) (32). However, combining RANKL and TNF stimulation reduced those numbers significantly by 40.8%. Pharmacological inhibition of KCa3.1, using the specific KCa3.1 inhibitor TRAM-34, revealed a decrease in multinucleated cells regardless of the presence (38.9% reduction) or absence (29.4% reduction) of TNF (Figure 2A, B), suggesting potential compensation of KCa3.1 functions in Kcnn4−/− cells that partially rescues the osteoclast formation phenotype when stimulated with RANKL alone. The inhibition of KCa3.1 by TRAM‐34 was found to be concentration-dependent (Supplemental figure 2A) and cell viability and proliferation were unaltered by TRAM-34 treatment or genetic deletion of KCa3.1 before (Supplemental figure 2B) and after stimulation with RANKL, TNF or the combination of both (Supplemental figure 2C). Collectively, both, genetic deletion and pharmacological inhibition of KCa3.1 significantly reduced the number of multinucleated cells in the presence of TNF.
Figure 2. RANKL-stimulated BMMs from Kcnn4−/− mice or treated with TRAM-34 in combination with TNF show decreased osteoclast formation correlated with decreased expression of osteoclast-specific genes.
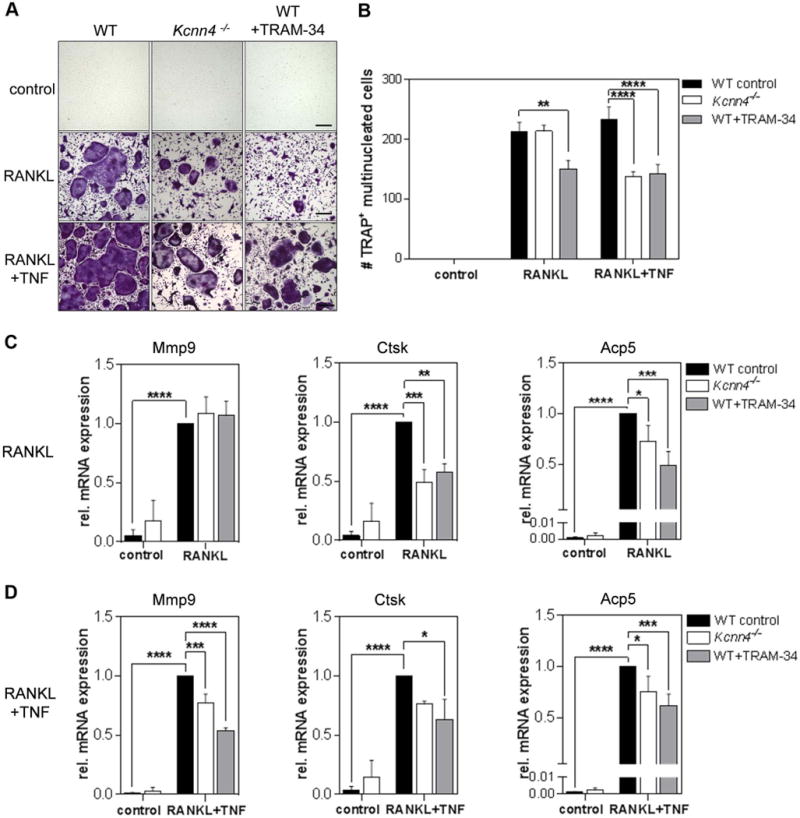
A) Representative images of cytochemical TRAP staining of murine WT, Kcnn4−/−, and WT+TRAM‐34 [10 µM] BMMs cultured in indicated condition (scale bar = 100 µM). B) Numbers of multinucleated TRAP+ cells (MNCs) (purple) (≥3 nuclei) in these cultures (N=4); C) qPCR analysis showing normalized mRNA expression of osteoclast-specific genes Mmp9, Ctsk, Acp5 in RANKL and D) RANKL+TNF stimulated BMMs; one-way ANOVA followed by multiple comparison test was performed for all experiments (N=4), shown are means ± SEM, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
These data correlated with a reduction in mRNA expression of the osteoclast-specific genes Mmp9, Ctsk and Acp5 in Kcnn4−/− and TRAM-34-treated cells compared to WT untreated controls in the presence of RANKL in combination with TNF, as evident from qPCR experiments (Figure 2D). However, in the presence of RANKL alone, only Ctsk and Acp5 were significantly reduced, and no changes in Mmp9 expression were detected (Figure 2C). Of note, genetic and pharmacological inhibition of KCa3.1 revealed to have similar effects on osteoclast-specific gene expression in the presence of RANKL, regardless of the presence of TNF (Figure 2C, D).
Inhibition of KCa3.1 reduces RANKL-induced Ca2+ responses
To elucidate, if KCa3.1 inhibition is specific to qualitative and quantitative changes in RANKL-induced Ca2+ responses, we next performed live cell Ca2+ imaging on cultured BMMs. In time-lapse videos using confocal microscopy we found that macrophages loaded with the Ca2+-sensitive dye Fluo-4-AM responded with acute Ca2+ dynamics visualized by transient increases in fluorescence intensity upon stimulation with RANKL (Fig 3A, B). Inhibiting KCa3.1 function in these cultures via pre-incubation with TRAM-34 did not significantly reduce the relative number of responsive cells (Figure 3C, left), neither induce changes in the median duration of Ca2+ transients (Figure 3C, middle). However, we found a mean reduction of 47.8% in normalized Ca2+ transient amplitudes in TRAM-34 compared to control pretreated cells (Figure 3C, right), suggesting diminished Ca2+ influx caused by the lack of KCa3.1-dependent membrane repolarization.
Figure 3. Inhibition of KCa3.1 activity changes Ca2+ signaling in BMMs.

A) Representative snapshots obtained from time-lapse videos showing Ca2+ signals during acute stimulation with RANKL [100 ng/mL] in Fluo-4-loaded BMM. B) Representative fluorescence traces of Ca2+ signals during RANKL stimulation of a control (left) and a 3 µM TRAM-34 pretreated cell (right). C) Analysis of Ca2+ responses in respective experiments; unpaired Student’s t-test and Mann-Whitney-U test were performed when applicable; overall response: control: N=13, TRAM-34: N=9; duration: control: N=37, TRAM-34: N=24; amplitude: control: N=34, TRAM-34: N=24; shown are means ± SEM, ****P<0.0001.
Changes in Ca2+ signaling can render phosphorylation and activation of Ca2+-sensitive enzymes such as CaN and CaMKIV (14, 18). We moreover, found that during inflammatory conditions, using RANKL in combination with TNF, pharmacological inhibition of both, CaMKIV by KN‐93, as well as CaN by cyclosporine A significantly decreased osteoclast formation and expression of osteoclast-specific genes (Supplemental figure 3A–C), indicating that activity of either transducer is essential for osteoclastogenesis during inflammation. To investigate a direct interference of altered Ca2+ signaling through KCa3.1 activity with CaMKIV, we stimulated BMMs with RANKL and show that an increase in CaMKIV phosphorylation (pCaMKIV) was prevented by inhibiting KCa3.1 with TRAM-34 or K+ efflux through increasing the K+ concentration in the culture medium (Supplemental figure 3D). These data suggest that both, CaN and CaMKIV are likely to be involved in effects derived from altered Ca2+ signaling through KCa3.1 inhibition during physiological and inflammatory osteoclast formation.
KCa3.1 modulates protein expression and activity of NFATc1 through CREB and c-fos
CaMKIV phosphorylates and thereby modulates transcriptional activity of CREB during RANKL-induced osteoclast differentiation (14). We next aimed to understand the consequences of KCa3.1 deletion on CREB phosphorylation in inflammatory (RANKL+TNF) compared to non-inflammatory (RANKL) osteoclastogenesis. Analysis of BMMs from WT and Kcnn4−/− mice stimulated with RANKL and RANKL in combination with TNF for 36 hours – a time period assessed to yield maximal phosphorylation of CREB in WT cells (data not shown) – revealed that both, RANKL- and RANKL-TNF co-stimulated Kcnn4−/− cells exhibited significantly decreased phosphorylation of CREB (pCREB) compared to WT cells, as demonstrated by IF (Figure 4A) and Western blot analysis (Figure 4B). Furthermore, in qPCR studies we found that the expression of c‐fos, a direct transcriptional target of phosphorylated CREB, was significantly reduced in Kcnn4−/− compared to WT cells during RANKL- and RANKL-TNF co-stimulation (Figure 4C). To evaluate the effects of reduced c‐fos expression on the activity and protein expression of its transcriptional target Nfatc1, we performed transcription factor binding assays in nuclear lysates of 36-hour-stimulated BMMs. Results revealed a significant reduction of nuclear NFATc1 activity in RANKL-stimulated Kcnn4−/− compared to WT cells at this time point (Figure 4D), whereas NFATc1 activity in RANKL+TNF-stimulated BMMs was comparable in WT and Kcnn4−/− cells. In order to understand the differences in the dynamics of NFATc1 expression during RANKL-TNF co-stimulation, we then performed Western blot studies using whole cell lysates at two different time points. Maximum NFATc1 expression in RANKL alone-stimulated cells was reached at 36 hours and slightly decreased by 60 hour-incubation (Figure 4E). When cells were incubated with RANKL and TNF, NFATc1 expression kept increasing by 60 hour-stimulation in both, WT and Kcnn4−/− cells (Figure 4F). In contrast to activity, NFATc1 protein levels were significantly lower in Kcnn4−/− cells compared to WT at both time points. In summary these data show that deletion of Kcnn4 diminished phosphorylation of CREB during RANKL- and RANKL-TNF co-stimulation, followed by a significant decrease in c-fos mRNA and NFATc1 protein expression and activity.
Figure 4. Kcnn4−/− BMMs show decreased phosphorylation of CREB and expression of c-fos followed by reduced amplification and activity of NFATc1.
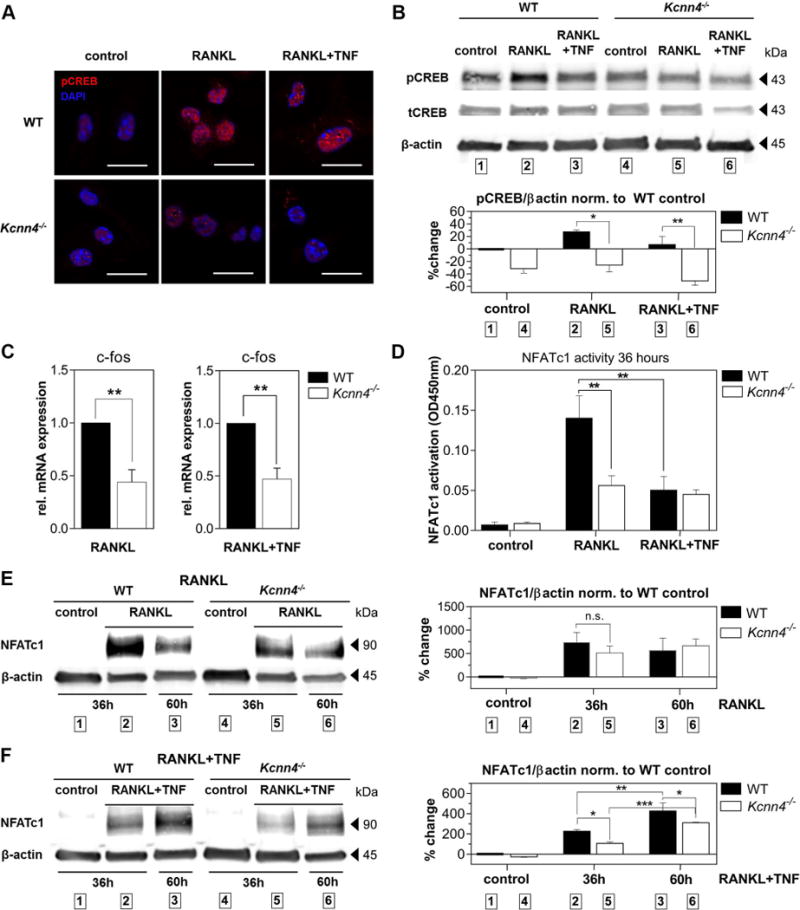
A) IF staining of phosphorylated CREB (pCREB, red) and DAPI (blue) of BMMs obtained from WT and Kcnn4−/− mice stimulated with RANKL or RANKL+TNF for 36 hours visualized with confocal microscopy (N=2, scale bar = 15 µm); B) Western blot analysis of pCREB versus total CREB (tCREB) and β-actin; top: representative blot; bottom: densitometric analysis; one-way ANOVA with post-hoc multiple comparison test was performed (N=2, P=0.004); C) qPCR analysis showing normalized c-fos mRNA expression of WT control and Kcnn4−/− BMMs cultivated in indicated conditions for 36 hours; Student’s unpaired t-test was performed (N=4); D) Transcription factor binding assays showing transcriptionally active NFATc1 protein obtained from nuclear lysates of 36-hours-stimulated WT and Kcnn4−/− BMMs in indicated conditions; one-way ANOVA with post-hoc multiple comparison test was performed (N=3, P=0.0003); E, F) Western blot analysis of NFATc1 protein expression obtained from whole cell lysates of WT and Kcnn4−/− BMMs stimulated for given time periods in indicated conditions; left: representative blots, right: densitometry analysis; one-way ANOVA with post-hoc multiple comparison test was performed (E) N=3, P=0.0164; F) N=3, P<0.0001); shown are means ± SEM, *P<0.05, **P<0.01, ***P<0.001.
Discussion
In this study, we investigated the role of the K+ channel KCa3.1 during physiological and pathological osteoclast differentiation under the influence of RANKL and the pro-inflammatory cytokine TNF. We show that KCa3.1 plays a major role in osteoclast-specific gene-expression and formation of multinucleated cells in both, RANKL and TNF-modulated osteoclastogenesis, as summarized in Figure 5.
Figure 5. Proposed mechanism of KCa3.1 activity contributing to NFATc1 expression during inflammatory osteoclastogenesis.
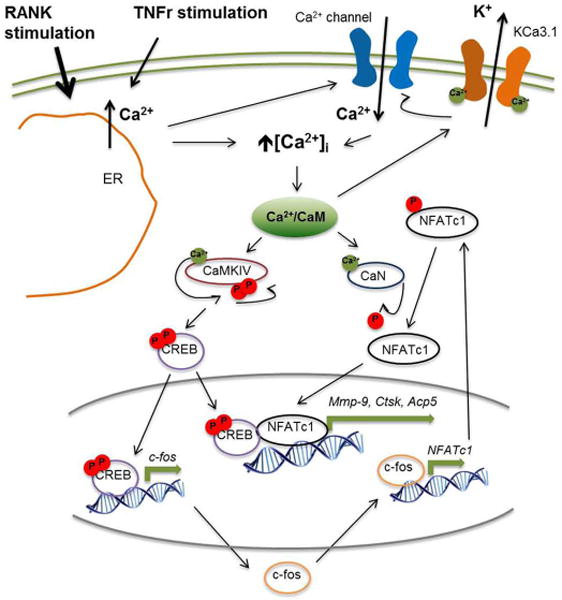
Drawing modified from (14) shows RANK and TNF receptor (TNFR) stimulation to trigger depletion of intracellular Ca2+ stores (ER) leading to an increase in intracellular Ca2+ concentration ([Ca2+]i). Increase in [Ca2+]i contributes to the formation of store-operated Ca2+ channels in the plasma membrane, as well as the activation of Ca2+/CaM-dependent proteins, such as CaMKIV, CaN and KCa3.1, as described elsewhere (14, 18, 27). Activated CaMKIV undergoes auto-phosphorylation prior to phosphorylation of CREB. Thereby, CREB becomes transcriptionally active and promotes c‐fos expression, as well as later on becomes part of a transcriptional complex including NFATc1 to transcribe osteoclast-specific genes (Mmp9, Ctsk, Acp5). Transcriptionally active c‐fos contributes to NFATc1 expression. Phosphorylated NFATc1 protein becomes activated by de-phosphorylation through CaN (18).
It has previously been reported that TNF can induce an initial increase in Kv1.3 (Kcna3) expression in murine macrophages, peaking at three hours post stimulation and decreasing afterwards (43). Our results are in agreement with this study, showing that Kv1.3 mRNA (Kcna3) levels are significantly lower than KCa3.1 (Kcnn4) at day three after stimulation. Of interest, the same study found the inwardly rectifying K+ channel Kir2.1, which we and others recently identified to be increased on anti-inflammatory macrophages/microglia, is down-regulated during TNF stimulation, (42, 43). In the present study, our microarray data reveals that also during RANKL stimulation no differences in Kir2.1 mRNA (Kcnj2) expression between macrophages and osteoclasts are apparent. Another study by Ohya and colleagues suggested the presence of a dominant-negative splice variant of Kcnn4 in mouse and human T cells which forms a non-functional N-terminally truncated protein that can polymerize with the functional protein in the cytosol, thereby interfering with membrane trafficking and formation of functional channels (34). In the present study, we detected both of these transcripts, the active (Kcnn4 a) as well as the dominant-negative variant (Kcnn4 b) to be expressed in macrophages and osteoclasts. This is to our knowledge the first time a dominant-negative variant of Kcnn4 has been described in murine myeloid cells. Furthermore, in our IL-23 gene transfer model of inflammatory arthritis we found that KCa3.1 expression was abundant on multinucleated cells on the bone surface (38). These findings extend the functional relevance of KCa3.1 expression on osteoclasts in inflammatory arthritis models in vivo. As demonstrated in a recent work by Kang and colleagues using the collagen antibody-induced arthritis (CAIA) model, Kcnn4−/− mice exhibit decreased inflammation and lower disease scores compared to WT mice (32). The same study showed that KCa3.1 deletion contributes to decreased formation of osteoclasts by diminishing cell fusion at a certain stage after RANKL stimulation. In our study, Kcnn4−/− cells, despite yielding similar numbers of osteoclasts as defined by ≥3 nuclei, they were smaller in size compared to WT cells – an observation that confirms a study by Kang and colleagues demonstrating that BMMs from Kcnn4−/− suffer from a defect in cell fusion during RANKL-induced osteoclastogenesis (32). In the same setting, when pharmacologically inhibiting KCa3.1 function, total numbers of multinucleated cells were reduced. The discrepancy in osteoclast numbers between drug treatment and genetic deletion upon RANKL-stimulation that is abolished when TNF is present may be indicative of compensation in the knock-out model during non-inflammatory as opposed to inflammatory osteoclastogenesis. The addition of TNF, mimicking differentiation in inflammatory, pathological conditions, seems to overrule this compensation, yielding a reduction in absolute numbers of multinucleated cells (≥3 nuclei) in Kcnn4−/− as well as TRAM-34-treated cells. Since TRAM-34 specificity for KCa3.1 is high, as previously demonstrated, and proved to be concentration-dependent, as shown in our supplemental data (Supplemental Figure 2), we rule out significant off-target effects of the drug (44, 45). Another aspect that seems to be puzzling at first is that we see no influence of Kcnn4 deletion or TRAM-34 treatment on Mmp9 expression when cells were stimulated with RANKL. However, in inflammatory conditions using TNF in addition to RANKL, we do see a significant decrease of Mmp9 with both, Kcnn4 deletion and TRAM‐34. One possible explanation could be that TNF in addition to RANKL initiates a signaling cascade that involves Mmp9 expression, which is to a higher degree dependent on Ca2+ and overwrites regular RANKL pathways. Another observation that supports the hypothesis that TNF renders Ca2+-dynamics during osteoclastogenesis is that interestingly, while the expression of NFATc1 in RANKL-stimulated WT cells peaked at 36 hours, WT cells stimulated with TNF in addition to RANKL showed delayed NFATc1 expression peaking at 60 hours post stimuli. This effect of TNF on temporal Ca2+ dynamics, was also reflected in NFATc1 activity assays, showing significantly less transcriptionally active NFATc1 in RANKL-TNF co-stimulated compared to RANKL only-stimulated samples at 36 hours post stimulus. Presumably because of altered temporal dynamics, differences in NFATc1 activity between Kcnn4−/− and WT cells were only detected in RANKL- but not in RANKL-TNF co-stimulation at this time point. However, when comparing total NFATc1 protein expression during RANKL-TNF co-stimulation at 36 or 60 hours, Kcnn4−/− cells consistently showed reduced NFATc1 protein expression in Kcnn4−/− compared to WT cells, supporting the role of KCa3.1 in TNF-dependent and independent Ca2+ pathways. A possibility for such TNF-dependent pathways could likely involve cytoskeletal rearrangement during osteoclastogenesis, which takes place after the initial amplification of NFATc1. TNF has been shown to recruit Ca2+-dependent GTPases, which are involved in actin rearrangement (46, 47). Although the results of this study elucidate certain aspects of the effect of RANKL and TNF on Ca2+ signaling during osteoclast differentiation, further studies are needed to identify the molecular mechanisms of Ca2+ dynamics in osteoclast precursors. One aspect we did not specifically address in this study, since it has previously been demonstrated extensively (17, 18, 27), is the particular mechanism of how alternations in Ca2+ signaling through K+ channel activity influence CaN in its ability to activate NFATc1. However, we showed that CaN activity is critical for osteoclast formation during inflammatory conditions, more so than CaMKIV, through its essential role in activating NFATc1. We thus propose that changes occurring in NFATc1 activity and osteoclast-specific gene expression by inhibition of KCa3.1 involve both, decreased activity of CaMKIV and CaN.
The important role of Ca2+ influx for osteoclast formation has previously been demonstrated (12, 13) and a number of therapeutic approaches have been attempted with regulation of Ca2+ in mind (32, 48). The inhibition of CRAC channels with 3,4-dichloropropionaniline (DCPA) has been shown to reduce bone erosion and inflammation in an inflammatory arthritis mouse model (48). However, the ubiquitous expression of CRAC channels on several immune cells including T cells challenge the therapeutic use of CRAC inhibitors because of the risk of broad immunosuppression (49). Unlike CRAC channels, type, numbers and ratios of potassium channels vary tremendously with leukocyte differentiation and activation state, allowing cell-type specific targeting (27). The availability of clinically tested small molecule modulators for K+ channels such as the KCa3.1 inhibitors TRAM-34 and ICA-17043, that exhibit high specificity alongside with low toxicity in pre-clinical and clinical testing provide incentive for their evaluation as an alternative to immunosuppressive anti-TNF therapy in inflammatory bone diseases (27, 36, 44, 50).
Supplementary Material
Acknowledgments
We thank Drs. Blythe P. Durbin Johnson and Matt Lee Settles at the Bioinformatics Core in the Genome Center at the University of California Davis for assistance with DNA microarray analysis, as well as the UC Davis Veterinary Medicine Pathology core for processing mouse joints and paraffin sections.
Abbreviations
- CaMKIV
Ca2+-calmodulin kinase IV
- RANK(L)
receptor activator of NF-κB (ligand)
- BMM
bone marrow macrophage
- TRAP
tartrate-resistant acid phosphatase
- MMP-9
matrix metalloprotease-9
- CTSK
cathepsin K
- PLCγ
phospholipase Cγ
- CaN
calcineurin
- CRAC
Ca2+-release-activated calcium
- IF
immunofluorescence
- DAPI
4,6 diamidino-2-phenylindole
- WT
wild type
References
- 1.Nombela-Arrieta C, Ritz J, Silberstein LE. The elusive nature and function of mesenchymal stem cells. Nat Rev Mol Cell Biol. 2011;12:126–131. doi: 10.1038/nrm3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh MC, Kim N, Kadono Y, Rho J, Lee SY, Lorenzo J, Choi Y. Osteoimmunology: interplay between the immune system and bone metabolism. Annu Rev Immunol. 2006;24:33–63. doi: 10.1146/annurev.immunol.24.021605.090646. [DOI] [PubMed] [Google Scholar]
- 3.Yoshida H, Hayashi S, Kunisada T, Ogawa M, Nishikawa S, Okamura H, Sudo T, Shultz LD. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442–444. doi: 10.1038/345442a0. [DOI] [PubMed] [Google Scholar]
- 4.Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 6.Teitelbaum SL. Osteoclasts: what do they do and how do they do it? Am J Pathol. 2007;170:427–435. doi: 10.2353/ajpath.2007.060834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishida N, Hayashi K, Hoshijima M, Ogawa T, Koga S, Miyatake Y, Kumegawa M, Kimura T, Takeya T. Large scale gene expression analysis of osteoclastogenesis in vitro and elucidation of NFAT2 as a key regulator. J Biol Chem. 2002;277:41147–41156. doi: 10.1074/jbc.M205063200. [DOI] [PubMed] [Google Scholar]
- 8.Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, Wagner EF, Mak TW, Kodama T, Taniguchi T. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 9.Grigoriadis AE, Wang ZQ, Cecchini MG, Hofstetter W, Felix R, Fleisch HA, Wagner EF. c-Fos: a key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science. 1994;266:443–448. doi: 10.1126/science.7939685. [DOI] [PubMed] [Google Scholar]
- 10.Kuroda Y, Hisatsune C, Nakamura T, Matsuo K, Mikoshiba K. Osteoblasts induce Ca2+ oscillation-independent NFATc1 activation during osteoclastogenesis. Proc Natl Acad Sci U S A. 2008;105:8643–8648. doi: 10.1073/pnas.0800642105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mao D, Epple H, Uthgenannt B, Novack DV, Faccio R. PLCgamma2 regulates osteoclastogenesis via its interaction with ITAM proteins and GAB2. J Clin Invest. 2006;116:2869–2879. doi: 10.1172/JCI28775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hwang SY, Putney JW. Orai1-mediated calcium entry plays a critical role in osteoclast differentiation and function by regulating activation of the transcription factor NFATc1. FASEB J. 2012;26:1484–1492. doi: 10.1096/fj.11-194399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hwang SY, Putney JW. Calcium signaling in osteoclasts. Biochim Biophys Acta. 2011;1813:979–983. doi: 10.1016/j.bbamcr.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sato K, Suematsu A, Nakashima T, Takemoto-Kimura S, Aoki K, Morishita Y, Asahara H, Ohya K, Yamaguchi A, Takai T, Kodama T, Chatila TA, Bito H, Takayanagi H. Regulation of osteoclast differentiation and function by the CaMK-CREB pathway. Nat Med. 2006;12:1410–1416. doi: 10.1038/nm1515. [DOI] [PubMed] [Google Scholar]
- 15.Ang ES, Zhang P, Steer JH, Tan JW, Yip K, Zheng MH, Joyce DA, Xu J. Calcium/calmodulin-dependent kinase activity is required for efficient induction of osteoclast differentiation and bone resorption by receptor activator of nuclear factor kappa B ligand (RANKL) J Cell Physiol. 2007;212:787–795. doi: 10.1002/jcp.21076. [DOI] [PubMed] [Google Scholar]
- 16.Asagiri M, Sato K, Usami T, Ochi S, Nishina H, Yoshida H, Morita I, Wagner EF, Mak TW, Serfling E, Takayanagi H. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med. 2005;202:1261–1269. doi: 10.1084/jem.20051150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clipstone NA, Crabtree GR. Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature. 1992;357:695–697. doi: 10.1038/357695a0. [DOI] [PubMed] [Google Scholar]
- 18.Crabtree GR, Schreiber SL. SnapShot: Ca2+-calcineurin-NFAT signaling. Cell. 2009;138:210, 210.e211. doi: 10.1016/j.cell.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishiyama K, Yashiro T, Nakano N, Kasakura K, Miura R, Hara M, Kawai F, Maeda K, Tamura N, Okumura K, Ogawa H, Takasaki Y, Nishiyama C. Involvement of PU.1 in NFATc1 promoter function in osteoclast development. Allergol Int. 2015;64:241–247. doi: 10.1016/j.alit.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 20.Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y, Cua DJ, Takayanagi H. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673–2682. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yago T, Nanke Y, Ichikawa N, Kobashigawa T, Mogi M, Kamatani N, Kotake S. IL-17 induces osteoclastogenesis from human monocytes alone in the absence of osteoblasts, which is potently inhibited by anti-TNF-alpha antibody: a novel mechanism of osteoclastogenesis by IL-17. J Cell Biochem. 2009;108:947–955. doi: 10.1002/jcb.22326. [DOI] [PubMed] [Google Scholar]
- 22.Okamoto K, Takayanagi H. Regulation of bone by the adaptive immune system in arthritis. Arthritis Res Ther. 2011;13:219. doi: 10.1186/ar3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu F, Teitelbaum SL. Osteoclasts: New Insights. Bone Res. 2013;1:11–26. doi: 10.4248/BR201301003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yarilina A, Xu K, Chen J, Ivashkiv LB. TNF activates calcium-nuclear factor of activated T cells (NFAT)c1 signaling pathways in human macrophages. Proc Natl Acad Sci U S A. 2011;108:1573–1578. doi: 10.1073/pnas.1010030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adamopoulos IE, Mellins ED. Alternative pathways of osteoclastogenesis in inflammatory arthritis. Nat Rev Rheumatol. 2015;11:189–194. doi: 10.1038/nrrheum.2014.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, Nakagawa N, Kinosaki M, Yamaguchi K, Shima N, Yasuda H, Morinaga T, Higashio K, Martin TJ, Suda T. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med. 2000;191:275–286. doi: 10.1084/jem.191.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annu Rev Immunol. 2015;33:291–353. doi: 10.1146/annurev-immunol-032414-112212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chandy KG, DeCoursey TE, Cahalan MD, McLaughlin C, Gupta S. Voltage-gated potassium channels are required for human T lymphocyte activation. J Exp Med. 1984;160:369–385. doi: 10.1084/jem.160.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- 30.Decoursey TE, Chandy KG, Gupta S, Cahalan MD. Two types of potassium channels in murine T lymphocytes. J Gen Physiol. 1987;89:379–404. doi: 10.1085/jgp.89.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferreira R, Wong R, Schlichter LC. KCa3.1/IK1 Channel Regulation by cGMP-Dependent Protein Kinase (PKG) via Reactive Oxygen Species and CaMKII in Microglia: An Immune Modulating Feedback System? Front Immunol. 2015;6:153. doi: 10.3389/fimmu.2015.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang H, Kerloc’h A, Rotival M, Xu X, Zhang Q, D’Souza Z, Kim M, Scholz JC, Ko JH, Srivastava PK, Genzen JR, Cui W, Aitman TJ, Game L, Melvin JE, Hanidu A, Dimock J, Zheng J, Souza D, Behera AK, Nabozny G, Cook HT, Bassett JH, Williams GR, Li J, Vignery A, Petretto E, Behmoaras J. Kcnn4 is a regulator of macrophage multinucleation in bone homeostasis and inflammatory disease. Cell Rep. 2014;8:1210–1224. doi: 10.1016/j.celrep.2014.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu SN, Huang YM, Liao YK. Effects of ibandronate sodium, a nitrogen-containing bisphosphonate, on intermediate-conductance calcium-activated potassium channels in osteoclast precursor cells (RAW 264.7) J Membr Biol. 2015;248:103–115. doi: 10.1007/s00232-014-9747-8. [DOI] [PubMed] [Google Scholar]
- 34.Ohya S, Niwa S, Yanagi A, Fukuyo Y, Yamamura H, Imaizumi Y. Involvement of dominant-negative spliced variants of the intermediate conductance Ca2+-activated K+ channel, K(Ca)3.1, in immune function of lymphoid cells. J Biol Chem. 2011;286:16940–16952. doi: 10.1074/jbc.M110.184192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takeshita S, Kaji K, Kudo A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J Bone Miner Res. 2000;15:1477–1488. doi: 10.1359/jbmr.2000.15.8.1477. [DOI] [PubMed] [Google Scholar]
- 36.Sfikakis PP, Tsokos GC. Towards the next generation of anti-TNF drugs. Clin Immunol. 2011;141:231–235. doi: 10.1016/j.clim.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 37.Takei Y, Tanaka T, Kent KC, Yamanouchi D. Osteoclastogenic Differentiation of Macrophages in the Development of Abdominal Aortic Aneurysms. Arterioscler Thromb Vasc Biol. 2016;36:1962–1971. doi: 10.1161/ATVBAHA.116.307715. [DOI] [PubMed] [Google Scholar]
- 38.Bouchareychas L, Grössinger EM, Kang M, Qiu H, Adamopoulos IE. Critical Role of LTB4/BLT1 in IL-23-Induced Synovial Inflammation and Osteoclastogenesis via NF-κB. J Immunol. 2016 doi: 10.4049/jimmunol.1601346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu DJ, Dixit N, Suzuki E, Nguyen T, Shin HS, Davis J, Maverakis E, Adamopoulos IE. A novel in vivo gene transfer technique and in vitro cell based assays for the study of bone loss in musculoskeletal disorders. J Vis Exp. 2014 doi: 10.3791/51810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feske S, Skolnik EY, Prakriya M. Ion channels and transporters in lymphocyte function and immunity. Nat Rev Immunol. 2012;12:532–547. doi: 10.1038/nri3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu J, Glimcher LH, Aliprantis AO. HCO3-/Cl- anion exchanger SLC4A2 is required for proper osteoclast differentiation and function. Proc Natl Acad Sci U S A. 2008;105:16934–16939. doi: 10.1073/pnas.0808763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguyen HM, Grössinger EM, Horiuchi M, Davis KW, Jin LW, Maezawa I, Wulff H. Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia. 2017;65:106–121. doi: 10.1002/glia.23078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vicente R, Escalada A, Coma M, Fuster G, Sánchez-Tilló E, López-Iglesias C, Soler C, Solsona C, Celada A, Felipe A. Differential voltage-dependent K+ channel responses during proliferation and activation in macrophages. J Biol Chem. 2003;278:46307–46320. doi: 10.1074/jbc.M304388200. [DOI] [PubMed] [Google Scholar]
- 44.Wulff H, Miller MJ, Hansel W, Grissmer S, Cahalan MD, Chandy KG. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc Natl Acad Sci U S A. 2000;97:8151–8156. doi: 10.1073/pnas.97.14.8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wulff H, Gutman GA, Cahalan MD, Chandy KG. Delineation of the clotrimazole/TRAM-34 binding site on the intermediate conductance calcium-activated potassium channel, IKCa1. J Biol Chem. 2001;276:32040–32045. doi: 10.1074/jbc.M105231200. [DOI] [PubMed] [Google Scholar]
- 46.Itzstein C, Coxon FP, Rogers MJ. The regulation of osteoclast function and bone resorption by small GTPases. Small GTPases. 2011;2:117–130. doi: 10.4161/sgtp.2.3.16453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wójciak-Stothard B, Entwistle A, Garg R, Ridley AJ. Regulation of TNF-alpha-induced reorganization of the actin cytoskeleton and cell-cell junctions by Rho, Rac, and Cdc42 in human endothelial cells. J Cell Physiol. 1998;176:150–165. doi: 10.1002/(SICI)1097-4652(199807)176:1<150::AID-JCP17>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 48.Blair HC, Soboloff J, Robinson LJ, Tourkova IL, Larrouture QC, Witt MR, Holaskova I, Schafer R, Elliott M, Hirsch R, Barnett JB. Suppression of arthritis-induced bone erosion by a CRAC channel antagonist. RMD Open. 2016;2:e000093. doi: 10.1136/rmdopen-2015-000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grundy S, Kaur M, Plumb J, Reynolds S, Hall S, House D, Begg M, Ray D, Singh D. CRAC channel inhibition produces greater anti-inflammatory effects than glucocorticoids in CD8 cells from COPD patients. Clin Sci (Lond) 2014;126:223–232. doi: 10.1042/CS20130152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ataga KI, Smith WR, De Castro LM, Swerdlow0 P, Saunthararajah Y, Castro O, Vichinsky E, Kutlar A, Orringer EP, Rigdon GC, Stocker JW, I.-.-. Investigators Efficacy and safety of the Gardos channel blocker, senicapoc (ICA-17043), in patients with sickle cell anemia. Blood. 2008;111:3991–3997. doi: 10.1182/blood-2007-08-110098. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.