

Abstract
Purpose of the review
The complexity and heterogeneity of the clinical presentation in systemic lupus of erythematosus (SLE), combined to the inherent limitations of clinical research, have made it difficult to investigate the etiology of this disease directly in patients. Various mouse models have been developed to dissect the cellular and genetic mechanisms of SLE, as well as to identify therapeutic targets and to screen treatments. The purpose of this review is to summarize the major spontaneous and induced mouse models of SLE and to provide an update on the major advances they have contributed to the field.
Recent findings
Mouse models of SLE have continued to contribute to understand the cellular, signaling and metabolic mechanisms contributing to the disease, and how targeting these pathways can provide therapeutic targets. Whenever possible, we discuss the advantage of using one model over the others to test a specific hypothesis
Summary
Spontaneous and induced models of lupus models are useful tools for the study of the etiology of the disease, identify therapeutic targets and screen treatments in pre-clinical studies. Each model shares specific subsets of attributes with the disease observed in humans, which provides investigators a tool to tailor to their specific needs.
Keywords: Systemic lupus of erythematosus, mouse models, therapeutic targets, T cells, B cells
Introduction
Systemic lupus of erythematosus (SLE) is a chronic disorder that is characterized by the over-production of antinuclear autoantibodies (ANA) resulting in the formation of immune complexes (IC) that induce tissue inflammation and destruction in multiple organs, including the kidneys (1). The exact etiology of SLE is still unknown, but there is a strong evidence that a combination of environmental exposures, genetic predisposition, cellular dysfunctions and hormonal factors lead to the development of SLE (2). Given the high degree of clinical heterogeneity in SLE patients, preclinical mouse models summarized below (Table 1) have been very valuable to investigate the etiology of SLE as well as to identify and validate therapeutic targets.
Table 1.
Classical mouse models of lupus.
| Mouse Model | Generation/Protocol | Sex Bias | Main Clinical Manifestations |
|---|---|---|---|
| Spontaneous Models | |||
| NZB/W F1(3) | F1 hybrid between NZB and NZW strains | Female | Lymphadenopathy, splenomegaly, Anti-dsDNA IgG, IC-mediated GN |
| NZM2410/2328(4–6) | Backcross between NZB/W F1 and NZW followed by brother-sister mating | Female | Overlaps with NZB/W F1 |
| MRL/lpr(7) | lpr mutation in Fas gene on MRL background | Both | Lymphadenopathy due to accumulation of DN B220+ T cells, DNA and RNA-directed autoantibodies, IC-mediated GN and dermatitis |
| BXSB/Yaa(8) | Backcross of (B6 X SB/Le) F1 to SB/Le | Male | Lymphadenopathy, anti-DNA, RNA and gp70 autoAbs, monocytosis, IC mediated GN. |
| Induced Models | |||
| Pristane-Induced lupus(9) | i.p injection of pristane | Female | Type I interferon mediated, AutoAb, GN, arthritis, anemia, serositis (strain dependent) |
| cGVHD(10) | 1) DBA -> BDF1 (injection of spleen cells) | Female | AutoAb, GN, polyclonal B-cell and T-cell activation, proteinuria (CD8+ T cell dependent) |
| 2) B6 <-> B6.Bm12 (injection of spleen cells) | Female | AutoAb, GN, polyclonal B-cell and T-cell activation, proteinuria (donor CD4+ T cell dependent) | |
These mouse models of SLE are either spontaneous or induced, but none of them fully represents the entire clinical spectrum found in SLE patients. However, each model presents an overlapping subset of human lupus phenotypes, and offers specific features of interest to address specific preclinical purposes. In addition to polygenic models, a number of mouse models are based on a single gene knock-out or transgenic expression of genes which result in lupus-like phenotypes (11). These strains have been instrumental in delineating functional pathways in SLE as well as the involvement of specific genes in maintaining systemic immune tolerance, and preventing immune complex (IC)-induced inflammation. There have been numerous reviews of mouse models of SLE starting from the foundational work published over 30 years ago (12) that has been followed by many updates. Many reviews have focused on specific aspects of these models, such as the genetic links between human and mouse SLE (11), or the mechanisms leading to systemic autoimmunity and clinical lupus in these models (13). The present review will briefly summarize the most common mouse of SLE, stressing their unique features. We will then provide an update on the major advances they have contributed to the field, and whenever possible, we will discuss the advantage of using one model over the others to test a specific hypothesis.
Spontaneous Mouse Models of SLE
1. NZB/W F1
In the 1960s, the NZB/W F1 model of lupus refers to the F1 hybrid between the NZB and NZW strains (3). NZB mice show limited hemolytic autoimmune anemia while NZW mice are non-autoimmune. However, their F1 hybrids develop severe lupus-like phenotypes including a strong female bias, splenomegaly, elevated serum ANA mostly directed against DNA. IC-mediated nephritis develops by 5–6 months of age, leading to renal failure and death at 10–12 months of age (12). Overall, NZB/W F1 is a classic model used to study the genetic underpinning of SLE (11) as well as drug responses in many preclinical studies, including the inhibition of BAFF (14), the role of type 1 IFN (15), and the identification of biomarkers of lupus nephritis (16).
2. NZM
An accidental backcross between NZB/W F1 and NZW followed by brother-sister mating generated 27 different recombinant inbred strains of New Zealand Mixed (NZM) mice among which NZM2328 and NZM2410 are now used as lupus models (4–6). The clinical manifestations in NZM strains are similar to that of NZB/W F1 mice, although there are some differences in renal pathology (16, 17), and the response to BAFF inhibition (18). The main advantage of the NZM strains over NZB/W F1 is that they have homozygous genomes, which has facilitated genetic analyses (11). From the NZM2410 strain, a novel congenic model has been produced that combines the three susceptibility loci, Sle1, Sle2, and Sle3, that are necessary and sufficient to induce a lupus phenotype on a non-autoimmune C57BL/6 (B6) genetic background (19). The B6.NZM2410.Sle1.Sle2.Sle3 has the unique advantage to share 95% of its genome with B6, providing a robust control for immunological and genetic studies. The corresponding single (mostly Sle1) and bi-congenic (Sle1.Sle3) are well suited to breed to B6-based gene knock-outs. For instance, deletion of the plasmacytoid dendritic cells (pDC)-specific transcription factor Tcf4 in B6.Sle1.Sle3 mice provided genetic evidence that pDCs are critically involved in the development of SLE (20) *.
3. MRL/lpr
The MRL strain was developed by crossing several stains including LG/J, C3H/Di, C57BL/6, and AKR/J (12). One of the MRL substrains carrying a spontaneous mutation named lpr for lymphoproliferation developed an SLE-like phenotype characterized by accumulation of double negative (DN: CD4−CD8−) B220+ T cells. DN T cells are autoreactive (21) and expanded in SLE patients (22), making this model specifically relevant to SLE pathogenesis. Lpr corresponds to non-functional transcripts of the Fas gene, a major regulator of apoptosis in immune cells (23). Both male and female MRL/lpr mice are affected and produce of autoantibodies against dsDNA and Sm, leading to large amounts of IC that induce renal and skin pathology (7). MRL/lpr mice develop a massive lymphadenopathy that is not observed in SLE patients. However, in addition to expanded DN T cells, this model has the advantage of a rapid and severe disease development as compared to the other spontaneous models. Notably, the MRL/lpr strain has been used to dissect the role of TRL7 and TLR9 in lupus (24), to compare TLR activation in B cells and dendritic cells (25), and to dissect the development of extrafollicular autoreactive B cells (26) *. In addition, B6.lpr mice, which develop systemic autoimmunity without clinical pathology and a reduced lymphadenopathy, have been used to investigate various pathways, including the involvement of Th17 T cells in lupus (27).
4. BXSB/Yaa
A recombinant inbred strain derived from the backcross of (B6 X SB/Le) F1 to SB/Le, termed BXSB/Mp (BXSB/Yaa), develops a lupus-like disease with lymphoid hyperplasia, IC-mediated nephritis, ANA and high-serum retroviral glycoprotein gp70 titers (7, 28). Nephritis leads to the death of BXSB/Yaa males in about 5 months and BXSB females in 14 months. The rapid-onset disease in males is attributable to the Y-autoimmune accelerator (Yaa) locus, which is due to a translocation from the X to the Y chromosome, duplicates 16 genes, including TLR7 (29, 30). TLR7, regulates the activation of the type 1 IFN pathway by ribonucleic acid complexes, a critical pathway in SLE pathogenesis (31). Therefore, in spite of its presentation in males, the BXSB/Yaa strain is uniquely suited to model the consequences of an overeactive TLR7/Type 1 IFN pathway.
Induced mouse models of SLE
1. Pristane-Induced lupus
Pristane is an isoprenoid alkane found at high concentration in mineral oil. Intraperitoneal injection of pristane is a standard method to obtain ascitic fluid enriched in monoclonal antibodies. Anti-ribonucleoprotein, anti-DNA and anti-histone autoantibodies are found in BALB/c mice after pristane injections. Pristane-treated mice also have IC deposition in the kidney causing severe nephritis (32). Strain differences in the response to pristane have been observed (33), illustrating the role of gene/environment interactions in lupus susceptibility. Pristane-induced lupus is more severe in females than in males, at least in the SJL strain (34). Pristane-induced lupus is driven by a strong type 1 IFN response (35), and this model is therefore well-suited to investigate the type 1 IFN signature present in many SLE patients, but much weaker in spontaneous mouse models of this disease. This model is also useful to test the impact of a specific gene on lupus development directly in a non-autoimmune strain, such the protective effect of TLR9 evaluated in BALB/c.Tlr9−/− mice treated with pristane (36)*.
2. Chronic graft-versus-host disease (cGVHD) models
Induced cGVHD models require injections of donor lymphocytes into a semi-allogenic recipient to induce a lupus-like syndrome. In the parent → F1 model, DBA/2 strain spleen cells are injected into (C57BL/6 X DBA/2) F1 (BDF1) recipients while in the other, B6 spleen cells are injected into class II MHC-missmatched B6.bm12 recipients or reversely. In both models, donor CD4+ T cells react to host B cells triggering the polyclonal activation of autoreactive B cells, and eventually, lupus-like syndrome (10). Compared with the other models, cGVHD is easy to control, adjustable to investigator’s needs, and generally presents with a reduced inter-individual variability. In addition, autoimmune and clinical manifestations of SLE develop relatively rapidly over a period of weeks, instead of months for the other models. Finally, because the activation and expansion of donor T cells play an essential role in cGVHD response, it is easy to track them relative to host cells by flow cytometry. These models also allow the study of the effect of treatments or genetic modifications in donor cells to alter the course of the cGVHD response. The bm12 model is particularly useful to test the effect of single genes or alleles on the development of systemic autoimmunity on a B6 genetic background. This approach has been used to evaluate Slamf6 isoforms as lupus susceptibility alleles for the Sle1b locus (37, 38), and to identify the association of a naturally occurring polymorphism in the G-CSF gene with resistance to autoimmune activation (39, 40).
Recent investigations of therapeutic targets with mouse models of lupus
Table 2 lists recent treatments or genetic approaches that have been used in mouse models of lupus.
Table 2.
Treatments tested in mouse models of SLE
| Gene Target | Cell Target | Model | Treatment | Main manifestations | Ref |
|---|---|---|---|---|---|
| T cell targets | |||||
| Cellular metabolism | CD4 T cells | B6.Sle1.Sle2.Sle3BWF1B6.lpr | Metformin,2-deoxyglucose | autoAb↓, GN↓immune activation↓ | (41, 42) |
| Cellular metabolism | CD4 T cells | cGVHD | Isogarcinol | Proteinuria↓, autoAb↓, GN↓ | (43) |
| Cellular metabolism | CD4 T cells | cGVHD | Quercitrin | Proteinuria↓, autoAb↓, GN↓ | (44) |
| B7-1 | T-cell-APC interaction | Pristane-induced | B7-1 shRNA and anti-B7-1 mAb | ANA↓, anti-dsDNA IgG↓ | (45, 46) |
| ICOS-B7RP-1 | Tfh | BWF1 | Anti-ICOS-B7RP-1 | Proteinuria↓, anti-dsDNA IgG↓ | (47) |
| ICOS-B7RP-1 | Tfh | MRL/lpr | ablation of ICOS ligand in CD11c+ cells | Kidney/lung inflammation↓ | (48) |
| IL-21 | Tfh | B6.Sle1.Yaa | Anti-IL-21 MAb | GC B cells↓, CD138hi↓IgG2c↓, autoantibodies↓ | (49) |
| IL-21 | Tfh | cGVHD | IL-21 | host B cell↓, autoantibody↓, renal disease↓ | (50) |
| IL-21 | Tfh | MRL/lpr, BWF1, BXSB | IL-21R Fc | IgG↓, proteinuria↓, anti-dsDNA↓ | (51–53) |
| B cell targets | |||||
| BAFF | B cells | MRL/lpr | BAFF-R Fc | Tertiary lymphoid structures and nephritis↓ | (54) |
| BAFF | B cells | NZM2328 | KO BCR3 with TACI or BCMA | BAFF-BCMA and/or BAFF-TACI combinations contribute to SLE, | (55) |
| BTK | B cells | BWF1, MRL/lpr, pristane-induced, BXSB | Various Btk inhibition | GN↓, ANA↓, IC↓ | (56–59) |
| miR-148 | B cells | MRL/lpr | Increased miR148 | GN↓ | (60) |
| miR-155 | B cells | B6.lpr | miR155 KO | ANA↓ B cell signaling↓ | (61) |
| Proteasome | Plasma cells | BWF1, MRL/lpr | Proteasome inhibitor | ANA↓, GN↓, Survival↑ | (62) |
| Other Targets | |||||
| NLRP3 | Macrophages | BWF1 | NLRP3 inhibitor | ROS/NAPDH/COX-2↓, GN↓ | (63) |
| NLRP3 | Macrophages | Pristane-induced | NLRP3 gain-function | proteinuria↑ and GN↑ | (64) |
| IRAK4 | TLR pathway | BWF1, MRL/lpr | IRAK 4 inhibitor | proteinuria↓, dsDNA↓, GN↓ | (65) |
| Topoisomerase I | dsDNA binding | MRL/lpr | Topoisomerase I inhibitor | nephritis and skin lesions↓ | (66) |
1. T cell targets
Cellular metabolism has been identified as a major checkpoint of CD4+ T cell effector functions (67). Consequently, manipulating T cell metabolism may be a promising avenue to treat immune-related diseases (68). In lupus mice as well as SLE patients, CD4+ T cells have an elevated metabolism. Treatment with a combination of metformin and glucose inhibitor 2-deoxyglucose normalized T cell metabolism and reversed disease in several mouse models of SLE (41**, 42*). Natural compounds isogarcinol and quercitrin ameliorated disease in a cGVHD mouse model by decreasing CD4+ T cell activation as well autoantibody production (43, 44). Quercitrin is a derivative of quercetin, a glycolytic inhibitor, suggesting that metabolic inhibition was a mechanism responsible for the therapeutic effect.
The interactions between B7-1 and 2 on the B cell/antigen presenting cell side and CD28/CTLA-4 on the T cell side are cardinal regulatory pathway of the immune response, and there have been numerous attempts to target them therapeutically (69). Based on studies in mouse models, CTLA-4-Ig (abatacept) is now in clinical trial for the treatment of lupus nephritis (70). In the pristane-induced lupus model, the specific blockade of the interaction between B7-1 and CD28 decreased serum ANA and anti-dsDNA IgG (45).
T follicular help cells (Tfh) are a CD4+ helper T cell subset specialized for provision of help of B cells which plays an essential role in germinal center (GC) formation, affinity maturation and the development of most high-affinity antibodies (71). Tfh cells are expanded in mouse models of lupus, and the level of circulating Tfh cells correlates with disease severity in SLE patients (72). Consequently, therapeutic targeting of Tfh cells has been proposed for SLE patients and lupus mouse models through the IL-21, ICOS, OX40 pathways,. Genetic approaches or a soluble IL21R-Fc protein have demonstrated that blocking the IL-21 pathway prevented or greatly ameliorated disease in several mouse strains (52*, 73). A recent pre-clinical study showed that treatment of B6.Sle1.Yaa mice with an anti-IL-21 antibody reduced GC B cells, CD138hi plasmablasts, IFN-γ-dependent IgG2c production, and autoantibodies, indicating that Tfh cell-derived IL-21 is critical for pathological B cell cues in lupus (49). However, targeting the IL-21 pathway may have unintended consequences in CD8+ T cells. In BXSB.Yaa, IL-21 signaling is essential for the maintenance of CD8+ suppressor T cells (74). Moreover, in the parent → F1 cGVHD model, treatment with IL-21 strongly promoted donor CD8+ T cell expansion and rescued defective donor anti-host CTLs, resulting in host B cell elimination, decreased autoantibody levels, and attenuated renal disease, despite evidence of concurrently enhanced CD4+ T cell help for B cells (50*). Another approach to eliminate Tfh cells has been to target ICOS/B7RP-1 interactions. Treatment of NZB/WF1 mice with an anti-B7RP-1 antibody decreased the number of Tfh cells and GC B cells and ameliorated disease manifestations (47). It is also been reported that the selective ablation of ICOS ligand in CD11c+cells, but not in B cells, dramatically ameliorated kidney and lung inflammation in MRL/lpr mice (48)**.
2. B cell targets
BAFF is a cytokine that is required for B-cell development and survival. Largely based on studies in mouse models (75), BAFF blockade has been the first and only biologic treatment approved to treat lupus. BAFF also plays a previously unappreciated role in lupus nephritis by inducing renal tertiary lymphoid structures and regulating the position of T cells in the glomeruli of MRL/lpr mice (54). Moreover, genetic approaches in the NZM2328 mice demonstrated that the three BAFF/APRIL receptors (BAFF-R, TACI and BCMA) have compensatory roles, suggesting a therapeutic benefit to target multiple receptors (55).
Bruton’s tyrosine kinase (Btk) regulates signaling downstream of the B cell receptor and Fcγ receptor and it is also involved in TLR signaling. Treatment with Btk inhibitors alleviate lupus symptoms in MRL/lpr (56), NZB/WF1 (57, 58), B6.Sle1.Sle3 (76), and BXSB.Yaa mice (59) as well as in pristane-induced lupus (59). Overall, based on these pre-clinical studies, FDA-approved Btk inhibitor ibrutinib has great potential as a therapeutic agent in SLE.
Finally, two miRNAs have been identified as potent regulators of B cell tolerance. Elevated miR-148a expression impaired B cell tolerance by promoting the survival of immature B cells after engagement of the B cell receptor by suppressing the expression of the autoimmune suppressor Gadd45α, the tumor suppressor PTEN and the pro-apoptotic protein Bim. Increased expression of miR-148a, facilitated the development of lethal autoimmune disease in MRL/lpr mice (60)**. Reduction of miR-148a expression upregulated PTEN in the glomeruli and improved renal function in MRL/lpr mice. (77). Conversely, miR155 is overexpressed in B cells from B6.lpr mice, and miR155 deletion decreased B cell activation, autoantibody production and autoimmune pathology (61).
3. Other targets
Abundant ICs can trigger the activation of the NLRP3 inflammasome in macrophages in SLE patients and in mouse models, leading to cell dysfunction and tissue damage (78). In the NZB/WF1 model, a NLRP3 inhibitor termed “Citral” alleviates lupus symptoms by inhibiting levels of ROS, NAPDH and COX-2 (63). In the pristane-induced model, a more severe lupus-like syndrome developed in mice carrying the Nlrp3−R258W gain-of-function mutation, providing evidence that NLRP3 plays a role in the development of SLE (64). In a related pathway, serine/threonine kinase IL-1R–associated kinase (IRAK)4 is a regulator of innate immunity involved in TLR signaling. Treatment with an IRAK4 inhibitor ameliorate lupus symptom in NZB/WF1 and MRL/lpr mice (65). Finally, It has been proposed that topoisomerase I plays a role in anti- dsDNA antibody binding, and treatment with a topoisomerase inhibitor prevented proteinuria and prolonged survival in MRL/lpr mice (66).
Conclusions
The use of murine models has led to discovery of potential therapeutic targets in diverse signaling pathways dysregulated in SLE. Immune cells including T cells, B cells, antigen presenting cells and macrophages, are all potential targets in different models of SLE (Figure 1). Clinical lupus is an extremely complex and diverse disease, and establishment of a mouse model with all features of the disease is very difficult. Various mouse models of SLE, spontaneous, induced or genetically engineered, have been used during the past 30 years, to answer the question of how the alteration of the immune system and target organs leads to break of tolerance to self.
Figure 1.
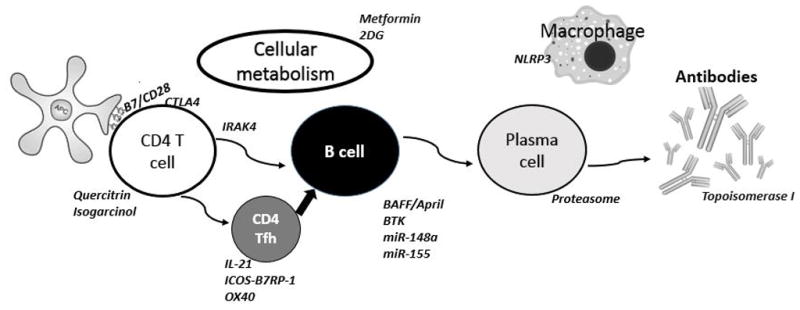
Potential therapeutic targets investigated in mouse models of SLE.
Key Points.
Spontaneous and induced models of lupus models are useful tools for the study of the etiology and mechanisms of the disease.
Mouse models of lupus have advanced the field through the identification therapeutic targets and the evaluation of corresponding treatments in pre-clinical studies.
Each model shares specific subsets of attributes with the disease observed in humans, which provides investigators a tool to tailor to their specific needs.
Acknowledgments
We would like to thank Dr. Seung-Chul Choi for helpful discussions related to this review.
Financial support and sponsorship
This review was partially supported by grants RO1 AI045050 and R01 AI058150 from the NIH to LM.
This review was supported by grants R01 AI058150 and AI045050 from the NIH to LM
Footnotes
Conflict of interest:
None.
References
- 1.Pons-Estel GJ, Alarcon GS, Scofield L, Reinlib L, Cooper GS. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39:257–268. doi: 10.1016/j.semarthrit.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365:2110–2121. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- 3.Helyer BJ, Howie JB. Renal disease associated with positive lupus erythematosus tests in a cross-bred strain of mice. Nature. 1963;197:197. doi: 10.1038/197197a0. [DOI] [PubMed] [Google Scholar]
- 4.Rudofsky UH, Evans BD, Balaban SL, Mottironi VD, Gabrielsen AE. Differences in expression of lupus nephritis in New Zealand mixed H-2z homozygous inbred strains of mice derived from New Zealand black and New Zealand white mice. Origins and initial characterization. Lab Invest. 1993;68:419–426. [PubMed] [Google Scholar]
- 5.Morel L, Rudofsky UH, Longmate JA, Schiffenbauer J, Wakeland EK. Polygenic control of susceptibility to murine systemic lupus erythematosus. Immunity. 1994;1:219–229. doi: 10.1016/1074-7613(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 6.Waters ST, Fu SM, Gaskin F, Deshmukh US, Sung SS, Kannapell CC, Tung KS, McEwen SB, McDuffie M. NZM2328: a new mouse model of systemic lupus erythematosus with unique genetic susceptibility loci. Clin Immunol. 2001;100:372–383. doi: 10.1006/clim.2001.5079. [DOI] [PubMed] [Google Scholar]
- 7.Andrews BS, Eisenberg RA, Theofilopoulos AN, Izui S, Wilson CB, McConahey PJ, Murphy ED, Roths JB, Dixon FJ. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J Exp Med. 1978;148:1198–1215. doi: 10.1084/jem.148.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy ED, Roths JB. A Y chromosome associated factor in strain BXSB producing accelerated autoimmunity and lymphoproliferation. Arthritis Rheum. 1979;22:1188–1194. doi: 10.1002/art.1780221105. [DOI] [PubMed] [Google Scholar]
- 9.Satoh M, Reeves WH. Induction of lupus-associated autoantibodies in BALB/c mice by intraperitoneal injection of pristane. J Exp Med. 1994;180:2341–2346. doi: 10.1084/jem.180.6.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eisenberg RA, Via CS. T cells, murine chronic graft-versus-host disease and autoimmunity. J Autoimmun. 2012;39:240–247. doi: 10.1016/j.jaut.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morel L. Genetics of SLE: evidence from mouse models. Nat Rev Rheumatol. 2010;6:348–357. doi: 10.1038/nrrheum.2010.63. [DOI] [PubMed] [Google Scholar]
- 12.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. 1985;37:269–390. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- 13.Sang A, Yin Y, Zheng Y-Y, Morel L. Progress in Molecular Biology and Translational Science. Vol. 105. Conn PM: Academic Press; 2012. Animal models of molecular pathology: Systemic lupus erythematosus; pp. 321–370. [DOI] [PubMed] [Google Scholar]
- 14.Ramanujam M, Wang X, Huang W, Liu Z, Schiffer L, Tao H, Frank D, Rice J, Diamond B, Yu KO, et al. Similarities and differences between selective and nonselective BAFF blockade in murine SLE. J Clin Invest. 2006;116:724–734. doi: 10.1172/JCI26385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Z, Bethunaickan R, Huang W, Ramanujam M, Madaio MP, Davidson A. IFN-alpha confers resistance of systemic lupus erythematosus nephritis to therapy in NZB/W F1 mice. J Immunol. 2011;187:1506–1513. doi: 10.4049/jimmunol.1004142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berthier CC, Kretzler M, Davidson A. From the large scale expression analysis of lupus nephritis to targeted molecular medicine. J Data Mining Genomics Proteomics. 2012:3. doi: 10.4172/2153-0602.1000123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh RR, Saxena V, Zang S, Li L, Finkelman FD, Witte DP, Jacob CO. Differential contribution of IL-4 and STAT6 vs STAT4 to the development of lupus nephritis. J Immunol. 2003;170:4818–4825. doi: 10.4049/jimmunol.170.9.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramanujam M, Bethunaickan R, Huang W, Tao H, Madaio MP, Davidson A. Selective blockade of BAFF for the prevention and treatment of systemic lupus erythematosus nephritis in NZM2410 mice. Arthritis Rheum. 2010;62:1457–1468. doi: 10.1002/art.27368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morel L. Mapping lupus susceptibility genes in the NZM2410 mouse model. Adv Immunol. 2012;115:113–139. doi: 10.1016/B978-0-12-394299-9.00004-7. [DOI] [PubMed] [Google Scholar]
- 20*.Sisirak V, Ganguly D, Lewis KL, Couillault C, Tanaka L, Bolland S, D’Agati V, Elkon KB, Reizis B. Genetic evidence for the role of plasmacytoid dendritic cells in systemic lupus erythematosus. J Exp Med. 2014;211:1969–1976. doi: 10.1084/jem.20132522. This study genetically deleted pDCs in the B6.Sle1.Sle3 mouse model of lupus to demonstrate the contribution of this cell type to disease development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez-Rodriguez N, Apostolidis SA, Fitzgerald L, Meehan BS, Corbett AJ, Martin-Villa JM, McCluskey J, Tsokos GC, Crispin JC. Pro-inflammatory self-reactive T cells are found within murine TCR-alphabeta(+) CD4(−) CD8(−) PD-1(+) cells. Eur J Immunol. 2016;46:1383–1391. doi: 10.1002/eji.201546056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crispín JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, Kyttaris VC, Juang YT, Tsokos GC. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol. 2008;181:8761–8766. doi: 10.4049/jimmunol.181.12.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen P, Eisenberg R. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Ann Rev Immunol. 1991;9:243–256. doi: 10.1146/annurev.iy.09.040191.001331. [DOI] [PubMed] [Google Scholar]
- 24.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 25.Teichmann LL, Schenten D, Medzhitov R, Kashgarian M, Shlomchik MJ. Signals via the adaptor MyD88 in B cells and DCs make distinct and synergistic contributions to immune activation and tissue damage in lupus. Immunity. 2013;38:528–540. doi: 10.1016/j.immuni.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26*.Ols ML, Cullen JL, Turqueti-Neves A, Giles J, Shlomchik MJ. Dendritic cells regulate extrafollicular autoreactive B cells via T cells expressing Fas and Fas ligand. Immunity. 2016;45:1052–1065. doi: 10.1016/j.immuni.2016.10.005. This study depleted conventional DCs in MRL/lpr mice to dissest the contribution of these antigen-presenting cells to the activation of autoreactive plasmablasts. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kyttaris VC, Zhang Z, Kuchroo VK, Oukka M, Tsokos GC. Cutting edge: IL-23 receptor deficiency prevents the development of lupus nephritis in C57BL/6-lpr/lpr mice. J Immunol. 2010;184:4605–4609. doi: 10.4049/jimmunol.0903595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maibaum MA, Haywood ME, Walport MJ, Morley BJ. Lupus susceptibility loci map within regions of BXSB derived from the SB/Le parental strain. Immunogenetics. 2000;51:370–372. doi: 10.1007/s002510050632. [DOI] [PubMed] [Google Scholar]
- 29.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–1672. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 30.Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, Liang C, Bartov G, McDaniel LD, Zhou XJ, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A. 2006;103:9970–9975. doi: 10.1073/pnas.0603912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han S, Zhuang H, Shumyak S, Yang L, Reeves WH. Mechanisms of autoantibody production in systemic lupus erythematosus. Front Immunol. 2015;6:228. doi: 10.3389/fimmu.2015.00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. 2009;30:455–464. doi: 10.1016/j.it.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Satoh M, Richards HB, Shaheen VM, Yoshida H, Shaw M, Naim JO, Wooley PH, Reeves WH. Widespread susceptibility among inbred mouse strains to the induction of lupus autoantibodies by pristane. Clin Exp Immunol. 2000;121:399–405. doi: 10.1046/j.1365-2249.2000.01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith DL, Dong X, Du S, Oh M, Singh RR, Voskuhl RR. A female preponderance for chemically induced lupus in SJL/J mice. Clin Immunol. 2007;122:101–107. doi: 10.1016/j.clim.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhuang H, Szeto C, Han S, Yang L, Reeves WH. Animal models of interferon signature positive lupus. Front Immunol. 2015;6:291. doi: 10.3389/fimmu.2015.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36*.Bossaller L, Christ A, Pelka K, Nundel K, Chiang PI, Pang C, Mishra N, Busto P, Bonegio RG, Schmidt RE, et al. TLR9 deficiency leads to accelerated renal disease and myeloid lineage abnormalities in pristane-induced murine lupus. J Immunol. 2016;197:1044–1053. doi: 10.4049/jimmunol.1501943. This study used pristane-induced lupus in BALB/c.TLR9−/− mice to investigate the protective role of TLR9 in myeloid cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keszei M, Detre C, Rietdijk ST, Munoz P, Romero X, Berger SB, Calpe S, Liao G, Castro W, Julien A, et al. A novel isoform of the Ly108 gene ameliorates murine lupus. J Exp Med. 2011;208:811–822. doi: 10.1084/jem.20101653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keszei M, Detre C, Castro W, Magelky E, O’Keeffe M, Kis-Toth K, Tsokos GC, Wang N, Terhorst C. Expansion of an osteopontin-expressing T follicular helper cell subset correlates with autoimmunity in B6. Sle1b mice and is suppressed by the H1-isoform of the Slamf6 receptor. FASEB J. 2013;27:3123–3131. doi: 10.1096/fj.12-226951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu Z, Vallurupalli A, Fuhrman C, Ostrov D, Morel L. An NZB-derived locus suppresses chronic graft versus host disease and autoantibody production through non-lymphoid bone-marrow derived cells. J Immunol. 2011;186:4130–4139. doi: 10.4049/jimmunol.1003512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lantow M, Sivakumar R, Zeumer L, Wasserfall C, Zheng YY, Atkinson MA, Morel L. The granulocyte colony stimulating factor pathway regulates autoantibody production in a murine induced model of systemic lupus erythematosus. Arthritis Res Ther. 2013;15:R49. doi: 10.1186/ar4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41**.Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, Sobel ES, Brusko TM, Morel L. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. 2015;7:274ra218. doi: 10.1126/scitranslmed.aaa0835. This article showed that normalization of T cell metabolism in mouse models of lupus through the dual inhibition of glycolysis and mitochondrial metabolism is a promising therapeutic venue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42*.Yin Y, Choi S-C, Xu Z, Zeumer L, Kanda N, Croker BP, Morel L. Glucose oxidation is critical for CD4+ T cell activation in a mouse model of systemic lupus erythematosus. J Immunol. 2016;196:80–90. doi: 10.4049/jimmunol.1501537. This paper provided new insights into how glucose metabolism regulates T cell function and autoimmunity, and revealed the importance of glucose oxidation in lupus development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li W, Li H, Zhang M, Zhong Y, Wang M, Cen J, Wu H, Yang Y, Wei Q. Isogarcinol extracted from Garcinia mangostana L ameliorates systemic lupus erythematosus-like disease in a murine model. J Agric Food Chem. 2015;63:8452–8459. doi: 10.1021/acs.jafc.5b03425. [DOI] [PubMed] [Google Scholar]
- 44.Li W, Li H, Zhang M, Wang M, Zhong Y, Wu H, Yang Y, Morel L, Wei Q. Quercitrin ameliorates the development of systemic lupus erythematosus-like disease in a chronic graft-versus-host murine model. Am J Physiol Renal Physiol. 2016;311:F217–226. doi: 10.1152/ajprenal.00249.2015. [DOI] [PubMed] [Google Scholar]
- 45.Huang L, Kong Y, Wang J, Sun J, Shi Q, Qiu YH. Reducing progression of experimental lupus nephritis via inhibition of the B7/CD28 signaling pathway. Mol Med Rep. 2015;12:4187–4195. doi: 10.3892/mmr.2015.3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi Q, Gao ZY, Xie F, Wang LF, Gu YP, Yang TJ, Huang L, Qian QH, Qiu YH. A novel monoclonal antibody against human CD80 and its immune protection in a mouse lupus-like disease. Int J Immunopathol Pharmacol. 2011;24:583–593. doi: 10.1177/039463201102400304. [DOI] [PubMed] [Google Scholar]
- 47.Hu YL, Metz DP, Chung J, Siu G, Zhang M. B7RP-1 blockade ameliorates autoimmunity through regulation of follicular helper T cells. J Immunol. 2009;182:1421–1428. doi: 10.4049/jimmunol.182.3.1421. [DOI] [PubMed] [Google Scholar]
- 48**.Teichmann LL, Cullen JL, Kashgarian M, Dong C, Craft J, Shlomchik MJ. Local triggering of the ICOS coreceptor by CD11c(+) myeloid cells drives organ inflammation in lupus. Immunity. 2015;42:552–565. doi: 10.1016/j.immuni.2015.02.015. This study showed that ICOSL+ DCs drive end organ inflammation in MRL/lpr mice independently from autoantibody production. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi JY, Seth A, Kashgarian M, Terrillon S, Fung E, Huang L, Wang LC, Craft J. Disruption of pathogenic cellular networks by IL-21 blockade leads to disease amelioration in murine lupus. J Immunol. 2017 doi: 10.4049/jimmunol.1601687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50*.Nguyen V, Rus H, Chen C, Rus V. CTL-promoting effects of IL-21 counteract murine lupus in the parent-->F1 graft-versus-host disease model. J Immunol. 2016;196:1529–1540. doi: 10.4049/jimmunol.1501824. This study used a perent --> F1 induced model of SLE to show the dual effect of IL-21 on the disease. [DOI] [PubMed] [Google Scholar]
- 51.Herber D, Brown TP, Liang S, Young DA, Collins M, Dunussi-Joannopoulos K. IL-21 has a pathogenic role in a lupus-prone mouse model and its blockade with IL-21R. Fc reduces disease progression. J Immunol. 2007;178:3822–3830. doi: 10.4049/jimmunol.178.6.3822. [DOI] [PubMed] [Google Scholar]
- 52*.Zhang M, Yu G, Chan B, Pearson JT, Rathanaswami P, Delaney J, Ching Lim A, Babcook J, Hsu H, Gavin MA. Interleukin-21 receptor blockade inhibits secondary humoral responses and halts the progression of preestablished disease in the (NZB x NZW)F1 systemic lupus erythematosus model. Arthritis Rheumatol. 2015;67:2723–2731. doi: 10.1002/art.39233. This study used the NZB/WF1 model for a preclinal demonstration of the efficacy of IL-21R blockade. [DOI] [PubMed] [Google Scholar]
- 53.Bubier JA, Sproule TJ, Foreman O, Spolski R, Shaffer DJ, Morse HC, 3rd, Leonard WJ, Roopenian DC. A critical role for IL-21 receptor signaling in the pathogenesis of systemic lupus erythematosus in BXSB-Yaa mice. Proc Natl Acad Sci U S A. 2009;106:1518–1523. doi: 10.1073/pnas.0807309106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kang S, Fedoriw Y, Brenneman EK, Truong YK, Kikly K, Vilen BJ. BAFF induces tertiary lymphoid structures and positions T cells within the glomeruli during lupus nephritis. J Immunol. 2017 doi: 10.4049/jimmunol.1600281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jacob CO, Yu N, Guo S, Jacob N, Quinn WJ, 3rd, Sindhava V, Cancro MP, Goilav B, Putterman C, Migone TS, et al. Development of systemic lupus erythematosus in NZM 2328 mice in the absence of any single BAFF receptor. Arthritis Rheum. 2013;65:1043–1054. doi: 10.1002/art.37846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010;107:13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mina-Osorio P, LaStant J, Keirstead N, Whittard T, Ayala J, Stefanova S, Garrido R, Dimaano N, Hilton H, Giron M, et al. Suppression of glomerulonephritis in lupus-prone NZB x NZW mice by RN486, a selective inhibitor of Bruton’s tyrosine kinase. Arthritis Rheum. 2013;65:2380–2391. doi: 10.1002/art.38047. [DOI] [PubMed] [Google Scholar]
- 58.Rankin AL, Seth N, Keegan S, Andreyeva T, Cook TA, Edmonds J, Mathialagan N, Benson MJ, Syed J, Zhan Y, et al. Selective inhibition of BTK prevents murine lupus and antibody-mediated glomerulonephritis. J Immunol. 2013;191:4540–4550. doi: 10.4049/jimmunol.1301553. [DOI] [PubMed] [Google Scholar]
- 59.Bender AT, Pereira A, Fu K, Samy E, Wu Y, Liu-Bujalski L, Caldwell R, Chen YY, Tian H, Morandi F, et al. Btk inhibition treats TLR7/IFN driven murine lupus. Clin Immunol. 2016;164:65–77. doi: 10.1016/j.clim.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 60**.Gonzalez-Martin A, Adams BD, Lai M, Shepherd J, Salvador-Bernaldez M, Salvador JM, Lu J, Nemazee D, Xiao C. The microRNA miR-148a functions as a critical regulator of B cell tolerance and autoimmunity. Nat Immunol. 2016;17:433–440. doi: 10.1038/ni.3385. This study showed that miR-148s regulates B cell tolernce and validates its role in SLE by showing that its overexpression in hematopoietic cells accelerates disease in MRL/lpr mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thai TH, Patterson HC, Pham DH, Kis-Toth K, Kaminski DA, Tsokos GC. Deletion of microRNA-155 reduces autoantibody responses and alleviates lupus-like disease in the Fas(lpr) mouse. Proc Natl Acad Sci U S A. 2013;110:20194–20199. doi: 10.1073/pnas.1317632110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Neubert K, Meister S, Moser K, Weisel F, Maseda D, Amann K, Wiethe C, Winkler TH, Kalden JR, Manz RA, et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat Med. 2008;14:748–755. doi: 10.1038/nm1763. [DOI] [PubMed] [Google Scholar]
- 63.Ka SM, Lin JC, Lin TJ, Liu FC, Chao LK, Ho CL, Yeh LT, Sytwu HK, Hua KF, Chen A. Citral alleviates an accelerated and severe lupus nephritis model by inhibiting the activation signal of NLRP3 inflammasome and enhancing Nrf2 activation. Arthritis Res Ther. 2015;17:331. doi: 10.1186/s13075-015-0844-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lu A, Li H, Niu J, Wu S, Xue G, Yao X, Guo Q, Wan N, Abliz P, Yang G, et al. Hyperactivation of the NLRP3 inflammasome in myeloid cells leads to severe organ damage in experimental lupus. J Immunol. 2017;198:1119–1129. doi: 10.4049/jimmunol.1600659. [DOI] [PubMed] [Google Scholar]
- 65.Dudhgaonkar S, Ranade S, Nagar J, Subramani S, Prasad DS, Karunanithi P, Srivastava R, Venkatesh K, Selvam S, Krishnamurthy P, et al. Selective IRAK4 inhibition attenuates disease in murine lupus models and demonstrates steroid sparing activity. J Immunol. 2017;198:1308–1319. doi: 10.4049/jimmunol.1600583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Keil A, Hall SR, Korner M, Herrmann M, Schmid RA, Frese S. Suppression of lupus nephritis and skin lesions in MRL/lpr mice by administration of the topoisomerase I inhibitor irinotecan. Arthritis Res Ther. 2016;18:243. doi: 10.1186/s13075-016-1144-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Ann Rev Immunol. 2013;31:259–283. doi: 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O’Sullivan D, Pearce EL. Targeting T cell metabolism for therapy. Trends Immunol. 2015;36:71–80. doi: 10.1016/j.it.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 costimulation: From mechanism to therapy. Immunity. 2016;44:973–988. doi: 10.1016/j.immuni.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.The ATG. Treatment of lupus nephritis with abatacept: The abatacept and cyclophosphamide combination efficacy and safety study. Arthritis Rheumatol. 2014;66:3096–3104. doi: 10.1002/art.38790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41:529–542. doi: 10.1016/j.immuni.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Blanco P, Ueno H, Schmitt N. T follicular helper (Tfh) cells in lupus: Activation and involvement in SLE pathogenesis. Eur J Immunol. 2016;46:281–290. doi: 10.1002/eji.201545760. [DOI] [PubMed] [Google Scholar]
- 73.Herber D, Brown TP, Liang S, Young DA, Collins M, Dunussi-Joannopoulos K. IL-21 has a pathogenic role in a lupus-prone mouse model and its blockade with IL-21R.Fc reduces disease progression. J Immunol. 2007:178. doi: 10.4049/jimmunol.178.6.3822. [DOI] [PubMed] [Google Scholar]
- 74.McPhee CG, Bubier JA, Sproule TJ, Park G, Steinbuck MP, Schott WH, Christianson GJ, Morse HC, 3rd, Roopenian DC. IL-21 is a double-edged sword in the systemic lupus erythematosus-like disease of BXSB. Yaa mice. J Immunol. 2013;191:4581–4588. doi: 10.4049/jimmunol.1300439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Davidson A. The rationale for BAFF inhibition in systemic lupus erythematosus. Curr Rheumatol Rep. 2012;14:295–302. doi: 10.1007/s11926-012-0258-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hutcheson J, Vanarsa K, Bashmakov A, Grewal S, Sajitharan D, Chang BY, Buggy JJ, Zhou XJ, Du Y, Satterthwaite AB, et al. Modulating proximal cell signaling by targeting Btk ameliorates humoral autoimmunity and end-organ disease in murine lupus. Arthritis Res Ther. 2012;14:R243–R243. doi: 10.1186/ar4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Patel V, Carrion K, Hollands A, Hinton A, Gallegos T, Dyo J, Sasik R, Leire E, Hardiman G, Mohamed SA, et al. The stretch responsive microRNA miR-148a-3p is a novel repressor of IKBKB, NF-kappaB signaling, and inflammatory gene expression in human aortic valve cells. FASEB J. 2015;29:1859–1868. doi: 10.1096/fj.14-257808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kahlenberg JM, Kaplan MJ. The inflammasome and lupus: another innate immune mechanism contributing to disease pathogenesis? Curr Opin Rheumatol. 2014;26:475–481. doi: 10.1097/BOR.0000000000000088. [DOI] [PMC free article] [PubMed] [Google Scholar]